The Cannabis Proteome Draft Map Project


Abstract
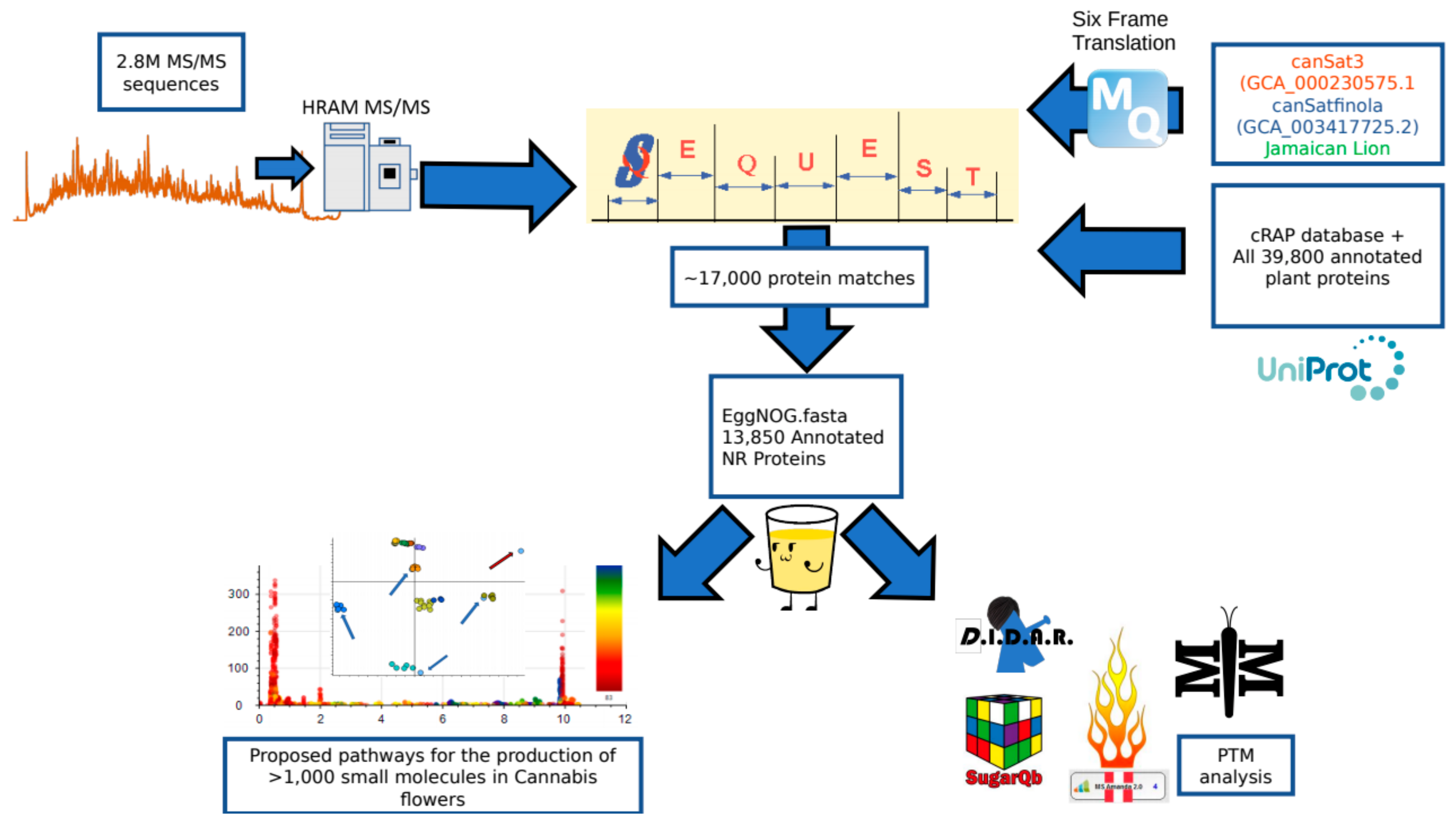
1. Introduction
2. Results and Conclusion
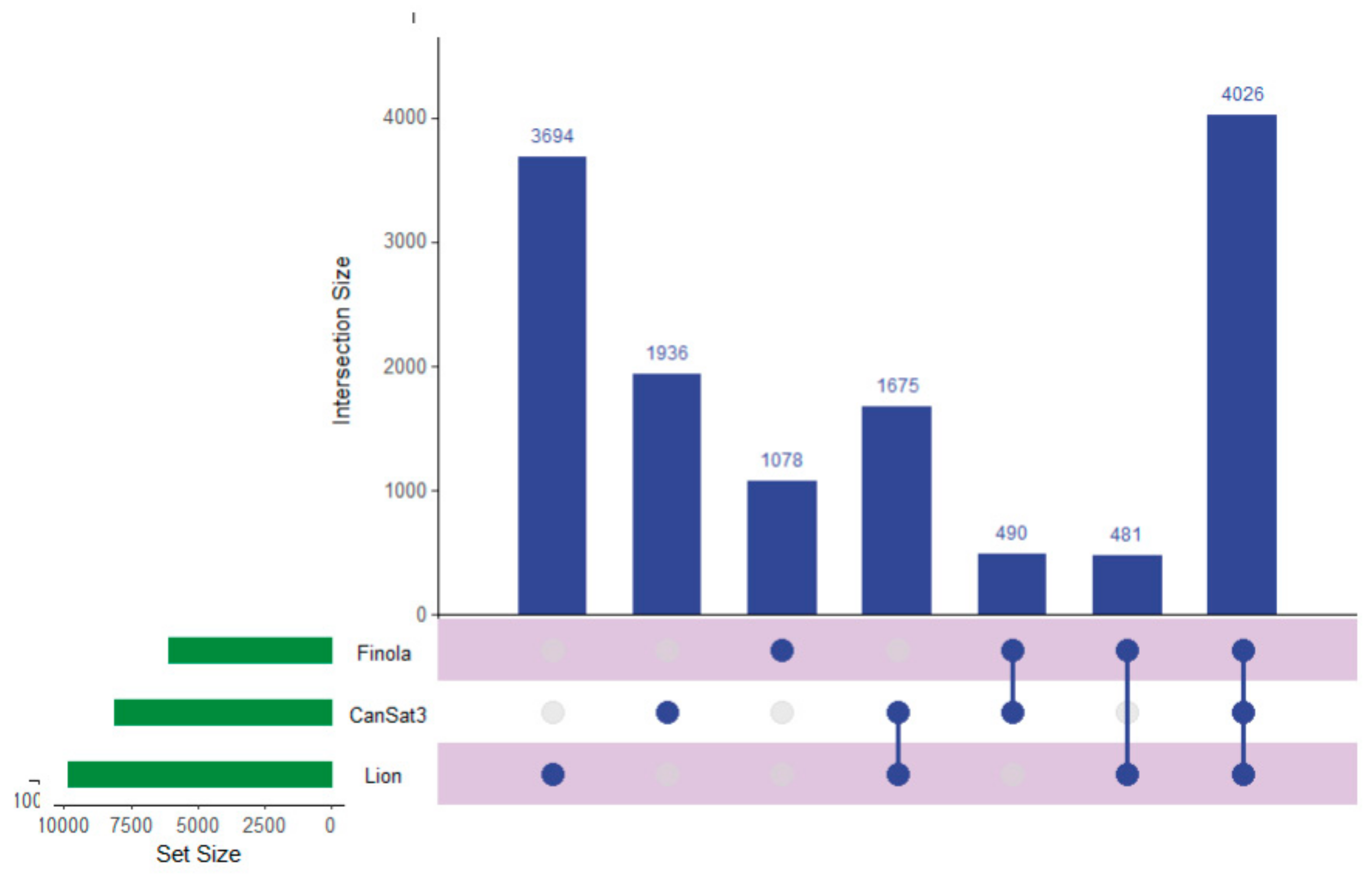
2.1. Peptide and Protein Identifications
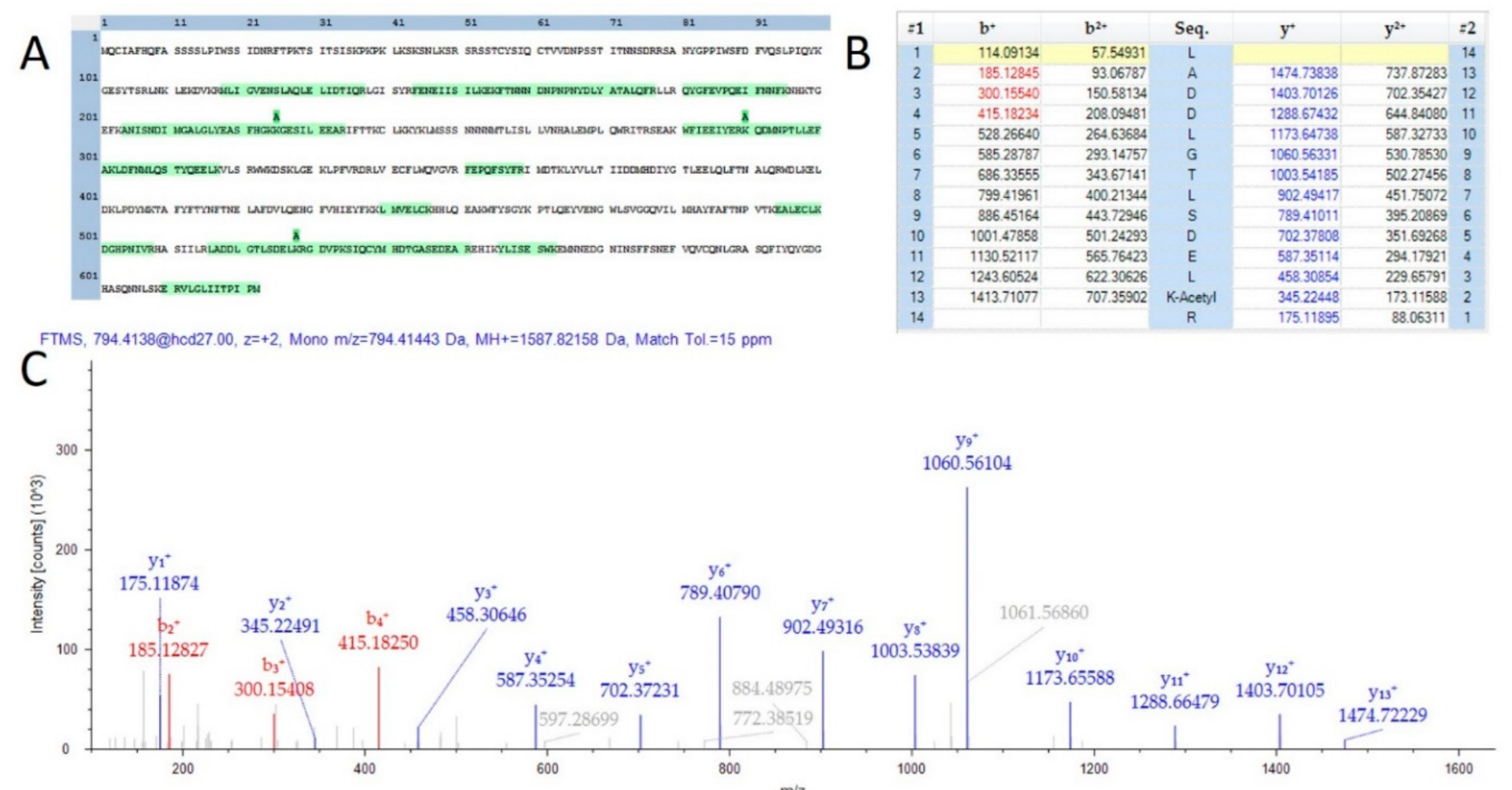
2.2. Post-Translational Modification Identification and Analysis
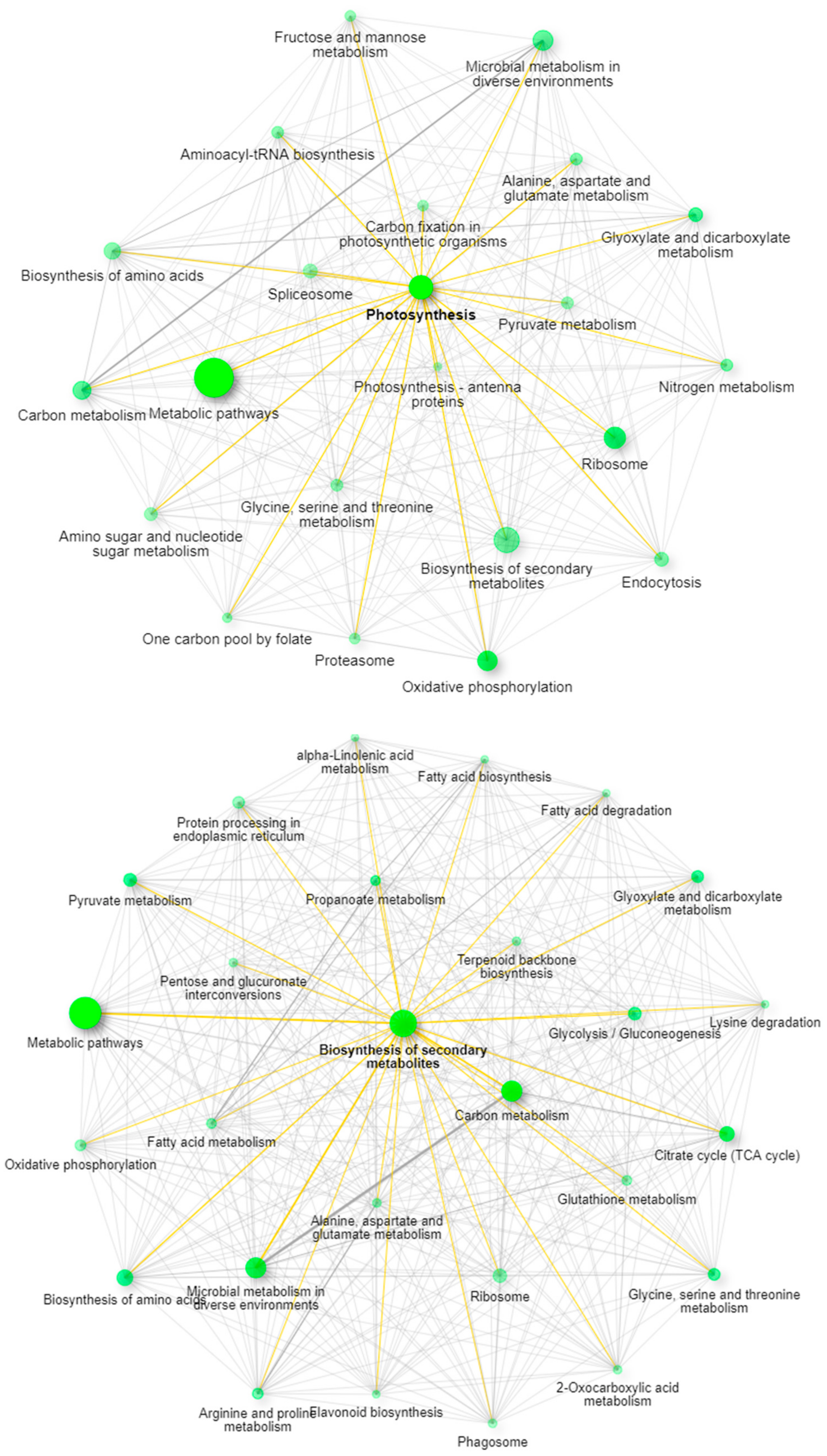
2.3. Utilizing this Resource toward the Meta-Analysis of Previous Studies
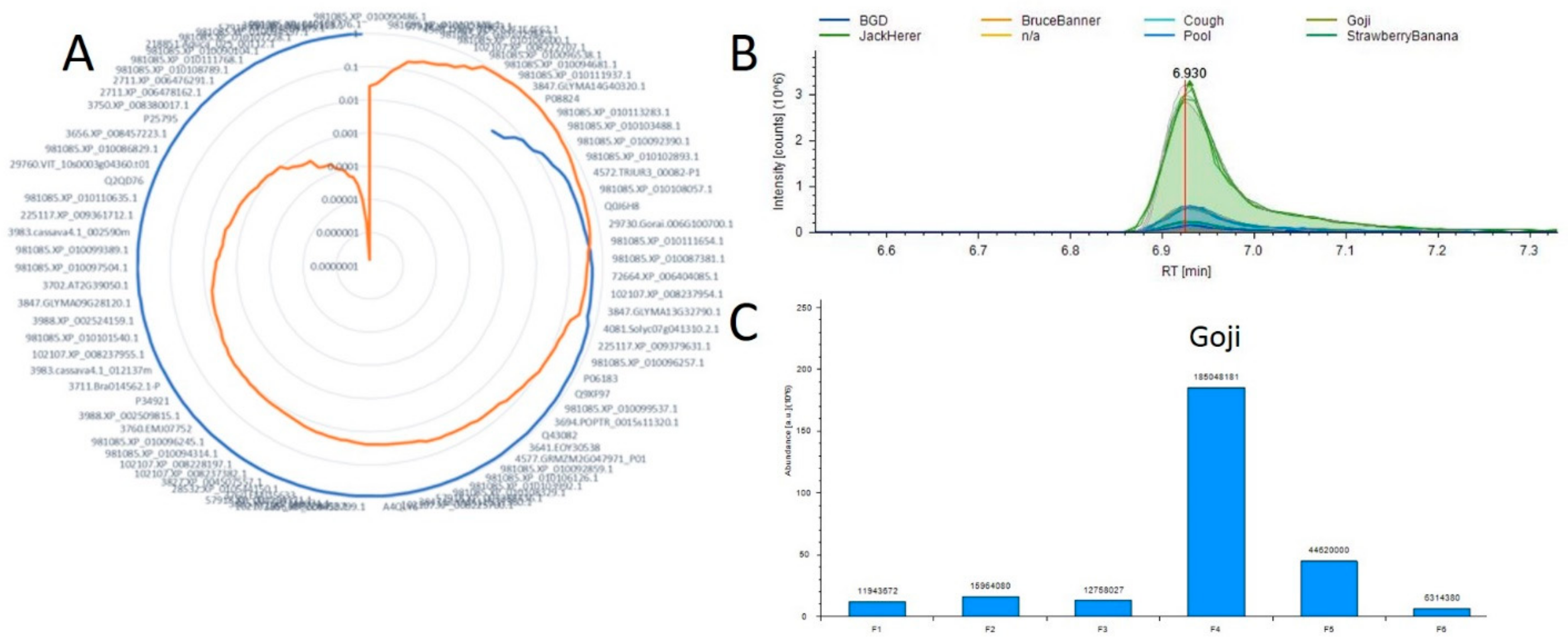
2.4. Correlation Analysis between Small Molecules and Proteins
3. Materials and Methods
3.1. Samples
3.2. Sample Preparation
3.3. Peptide Fractionation
3.4. LC-Mass Spectrometry Analysis
3.5. Peptide and Protein Identification
3.6. Generation of the EggNOG Annotated Protein FASTA
3.7. Spectral Library Generation
3.8. Chromosome Alignment
3.9. Identification and Validation of Potential Post-Translational Modifications (PTMs)
3.10. Gene Ontology Analysis for Green Plant Material
3.11. Correlation Analysis of Small Molecules and Proteins
3.12. Scaffold Files for Relative Quantification
3.13. Graph Generation
4. Conclusions
5. Future Goals
6. Significance Statement
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhang, Y.; Fonslow, B.R.; Shan, B.; Baek, M.C.; Yates, J.R. Protein analysis by shotgun/bottom-up proteomics. Chem. Rev. 2013, 113, 2343–2394. [Google Scholar] [CrossRef]
- Yates, J.R.; Eng, J.K.; McCormack, A.L.; Schieltz, D. Method to Correlate Tandem Mass Spectra of Modified Peptides to Amino Acid Sequences in the Protein Database. Anal. Chem. 1995, 67, 1426–1436. [Google Scholar] [CrossRef]
- Kim, M.S.; Pinto, S.M.; Getnet, D.; Nirujogi, R.S.; Manda, S.S.; Chaerkady, R.; Madugundu, A.K.; Kelkar, D.S.; Isserlin, R.; Jain, S.; et al. A draft map of the human proteome. Nature 2014, 509, 575–581. [Google Scholar] [CrossRef]
- Wilhelm, M.; Schlegl, J.; Hahne, H.; Gholami, A.M.; Lieberenz, M.; Savitski, M.M.; Ziegler, E.; Butzmann, L.; Gessulat, S.; Marx, H.; et al. Mass-spectrometry-based draft of the human proteome. Nature 2014, 509, 582–587. [Google Scholar] [CrossRef] [PubMed]
- Hebert, A.S.; Richards, A.L.; Bailey, D.J.; Ulbrich, A.; Coughlin, E.E.; Westphall, M.S.; Coon, J.J. The one hour yeast proteome. Mol. Cell. Proteomics 2014, 13, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Kelstrup, C.D.; Jersie-Christensen, R.R.; Batth, T.S.; Arrey, T.N.; Kuehn, A.; Kellmann, M.; Olsen, J.V. Rapid and deep proteomes by faster sequencing on a benchtop quadrupole ultra-high-field orbitrap mass spectrometer. J. Proteome Res. 2014, 13, 6187–6195. [Google Scholar] [CrossRef] [PubMed]
- Bekker-Jensen, D.B.; Kelstrup, C.D.; Batth, T.S.; Larsen, S.C.; Haldrup, C.; Bramsen, J.B.; Sorensen, K.D.; Hoyer, S.; Orntoft, T.F.; Andersen, C.L.; et al. An Optimized Shotgun Strategy for the Rapid Generation of Comprehensive Human Proteomes. Cell Syst. 2017, 4, 587. [Google Scholar] [CrossRef]
- Lössl, P.; Waterbeemd, M.; Heck, A.J. The diverse and expanding role of mass spectrometry in structural and molecular biology. EMBO J. 2016, 35, 2634–2657. [Google Scholar] [CrossRef] [PubMed]
- Witze, E.S.; Old, W.M.; Resing, K.A.; Ahn, N.G. Mapping protein post-translational modifications with mass spectrometry. Nat. Methods 2007, 4, 798–806. [Google Scholar] [CrossRef]
- Silva, A.M.N.; Vitorino, R.; Domingues, M.R.M.; Spickett, C.M.; Domingues, P. Post-translational modifications and mass spectrometry detection. Free Radic. Biol. Med. 2013, 65, 925–941. [Google Scholar] [CrossRef]
- Ghazalpour, A.; Bennett, B.; Petyuk, V.A.; Orozco, L.; Hagopian, R.; Mungrue, I.N.; Farber, C.R.; Sinsheimer, J.; Kang, H.M.; Furlotte, N.; et al. Comparative analysis of proteome and transcriptome variation in mouse. PLoS Genet. 2011, 7. [Google Scholar] [CrossRef] [PubMed]
- Raharjo, T.J.; Widjaja, I.; Roytrakul, S.; Verpoorte, R. Comparative proteomics of cannabis sativa plant tissues. J. Biomol. Tech. 2004, 15, 97–106. [Google Scholar] [PubMed]
- Sun, L.; Xu, Y.; Bai, S.; Bai, X.; Zhu, H.; Dong, H.; Wang, W.; Zhu, X.; Hao, F.; Song, C.P.; et al. Transcriptome-wide analysis of pseudouridylation of mRNA and non-coding RNAs in Arabidopsis. J. Exp. Bot. 2019, 70, 5089–5600. [Google Scholar] [CrossRef] [PubMed]
- Prasad, T.S.K.; Mohanty, A.K.; Kumar, M.; Sreenivasamurthy, S.K.; Dey, G.; Nirujogi, R.S.; Pinto, S.M.; Madugundu, A.K.; Patil, A.H.; Advani, J.; et al. Integrating transcriptomic and proteomic data for accurate assembly and annotation of genomes. Genome Res. 2017, 27, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R package for the visualization of intersecting sets and their properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.X.; Jung, D. ShinyGO: a graphical enrichment tool for animals and plants. bioRxiv 2018, 2. [Google Scholar]
- McKernan, K.J.; Helbert, Y.; Kane, L.T.; Ebling, H.; Zhang, L.; Liu, B.; Eaton, Z.; McLaughlin, S.; Kingan, S.; Baybayan, P.; et al. Sequence and annotation of 42 cannabis genomes reveals extensive copy number variation in cannabinoid synthesis and pathogen resistance genes. bioRxiv 2020. [Google Scholar] [CrossRef]
- Jenkins, C.; Orsburn, B. The First Publicly Available Annotated Genome for Cannabis plants. bioRxiv 2019, 1–12. [Google Scholar]
- Zirpel, B.; Kayser, O.; Stehle, F. Elucidation of structure-function relationship of THCA and CBDA synthase from Cannabis sativa L. J. Biotechnol. 2018, 284, 17–26. [Google Scholar] [CrossRef]
- Jenkins, C.; Norris, A.; O’Neill, M.; Das, S.; Andresson, T.; Orsburn, B. Reporter Ion Data Analysis Reduction (R.I.D.A.R) for isobaric proteomics quantification studies. bioRxiv 2018. [Google Scholar]
- Li, Q.; Shortreed, M.R.; Wenger, C.D.; Frey, B.L.; Schaffer, L.V.; Scalf, M.; Smith, L.M. Global Post-Translational Modification Discovery. J. Proteome Res. 2017, 16, 1383–1390. [Google Scholar] [CrossRef] [PubMed]
- Millikin, R.J.; Solntsev, S.K.; Shortreed, M.R.; Smith, L.M. Ultrafast Peptide Label-Free Quantification with FlashLFQ. J. Proteome Res. 2018, 17, 386–391. [Google Scholar] [CrossRef] [PubMed]
- Solntsev, S.K.; Shortreed, M.R.; Frey, B.L.; Smith, L.M. Enhanced Global Post-translational Modification Discovery with MetaMorpheus. J. Proteome Res. 2018, 17, 1844–1851. [Google Scholar] [CrossRef] [PubMed]
- Finkemeier, I.; Laxa, M.; Miguet, L.; Howden, A.J.M.; Sweetlove, L.J. Proteins of diverse function and subcellular location are lysine acetylated in Arabidopsis. Plant Physiol. 2011, 155, 1779–1790. [Google Scholar] [CrossRef] [PubMed]
- Booth, J.K.; Bohlmann, J. Terpenes in Cannabis sativa – From plant genome to humans. Plant Sci. 2019, 284, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Paul Zolg, D.; Wilhelm, M.; Schmidt, T.; Médard, G.; Zerweck, J.; Knaute, T.; Wenschuh, H.; Reimer, U.; Schnatbaum, K.; Kuster, B. Proteometools: Systematic characterization of 21 post-translational protein modifications by liquid chromatography tandem mass spectrometry (lc-ms/ms) using synthetic peptides. Mol. Cell. Proteomics 2018, 17, 1850–1863. [Google Scholar] [CrossRef] [PubMed]
- Laverty, K.U.; Stout, J.M.; Sullivan, M.J.; Shah, H.; Gill, N.; Holbrook, L.; Deikus, G.; Sebra, R.; Hughes, T.R.; Page, J.E.; et al. A physical and genetic map of Cannabis sativa identifies extensive rearrangements at the THC/CBD acid synthase loci. Genome Res. 2019, 29, 146–156. [Google Scholar] [CrossRef]
- Singh, C.; Zampronio, C.G.; Creese, A.J.; Cooper, H.J. Higher energy collision dissociation (HCD) product ion-triggered electron transfer dissociation (ETD) mass spectrometry for the analysis of N-linked glycoproteins. J. Proteome Res. 2012, 11, 4517–4525. [Google Scholar] [CrossRef]
- Hoffmann, M.; Pioch, M.; Pralow, A.; Hennig, R.; Kottler, R.; Reichl, U.; Rapp, E. The Fine Art of Destruction: A Guide to In-Depth Glycoproteomic Analyses—Exploiting the Diagnostic Potential of Fragment Ions. Proteomics 2018, 18. [Google Scholar] [CrossRef]
- Toghi Eshghi, S.; Yang, W.; Hu, Y.; Shah, P.; Sun, S.; Li, X.; Zhang, H. Classification of Tandem Mass Spectra for Identification of N- and O-linked Glycopeptides. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Vincent, D.; Rochfort, S.; Spangenberg, G. Optimisation of protein extraction from medicinal cannabis mature buds for bottom-up proteomics. Molecules 2019, 24, 659. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.A.; Winter, D.; Kirchner, M.; Chauhan, R.; Ahmed, S.; Ozlu, N.; Tzur, A.; Steen, J.A.; Steen, H. Co-regulation proteomics reveals substrates and mechanisms of APC/C-dependent degradation. EMBO J. 2014, 33, 385–399. [Google Scholar] [CrossRef]
- Jenkins, C.; Orsburn, B. Application of Global Metabolomics to the Identification of Complex Counterfeit Medicinal Products. bioRxiv 2019, 567339. [Google Scholar]
- Luo, X.; Reiter, M.A.; D’Espaux, L.; Wong, J.; Denby, C.M.; Lechner, A.; Zhang, Y.; Grzybowski, A.T.; Harth, S.; Lin, W.; et al. Complete biosynthesis of cannabinoids and their unnatural analogues in yeast. Nature 2019, 567, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Wróbel, T.; Dreger, M.; Wielgus, K.; Słomski, R. The application of plant in vitro cultures in cannabinoid production. Biotechnol. Lett. 2018, 40, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Mead, A. The legal status of cannabis (marijuana) and cannabidiol (CBD) under U.S. law. Epilepsy Behav. 2017, 70, 288–291. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; An, B.; Wang, X.; Hilchey, S.P.; Li, J.; Cao, J.; Tian, Y.; Hu, C.; Jin, L.; Ng, A.; et al. Surfactant Cocktail-Aided Extraction/Precipitation/On-Pellet Digestion Strategy Enables Efficient and Reproducible Sample Preparation for Large-Scale Quantitative Proteomics. Anal. Chem. 2018, 90, 10350–10359. [Google Scholar] [CrossRef] [PubMed]
- Navarrete-Perea, J.; Yu, Q.; Gygi, S.P.; Paulo, J.A. Streamlined Tandem Mass Tag (SL-TMT) Protocol: An Efficient Strategy for Quantitative (Phospho)proteome Profiling Using Tandem Mass Tag-Synchronous Precursor Selection-MS3. J. Proteome Res. 2018, 17, 2226–2236. [Google Scholar] [CrossRef]
- Weisser, H.; Nahnsen, S.; Grossmann, J.; Nilse, L.; Quandt, A.; Brauer, H.; Sturm, M.; Kenar, E.; Kohlbacher, O.; Aebersold, R.; et al. An automated pipeline for high-throughput label-free quantitative proteomics. J. Proteome Res. 2013, 12, 1628–1644. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Szklarczyk, D.; Jensen, L.J.; Von Mering, C.; Bork, P. Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol. Biol. Evol. 2017, 34, 2115–2122. [Google Scholar] [CrossRef]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef] [PubMed]
- Egertson, J.D.; MacLean, B.; Johnson, R.; Xuan, Y.; MacCoss, M.J. Multiplexed peptide analysis using data-independent acquisition and Skyline. Nat. Protoc. 2015, 10, 887–903. [Google Scholar] [CrossRef]
- van Bakel, H.; Stout, J.M.; Cote, A.G.; Tallon, C.M.; Sharpe, A.G.; Hughes, T.R.; Page, J.E. The draft genome and transcriptome of Cannabis sativa. Genome Biol. 2011, 12. [Google Scholar] [CrossRef] [PubMed]
- Taus, T.; Köcher, T.; Pichler, P.; Paschke, C.; Schmidt, A.; Henrich, C.; Mechtler, K. Universal and confident phosphorylation site localization using phosphoRS. J. Proteome Res. 2011, 10, 5354–5362. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2; Springer: Berlin/Heidelberg, Germany, 2009; ISBN 9780387981406. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category of Data | Number in 2019 Upload |
|---|---|
| Protein Sequenced | 17,269 |
| Protein Annotated | 13,929 |
| Proteins with homologous 3D structures | 964 |
| Acetylation sites Mapped | 584 |
| MS/MS Spectra Acquired | 1.40 × 107 |
| MS/MS Spectra Searched | 2.40 × 106 |
| MS/MS Spectra with Evidence of Glycosylation | 3.50 × 105 |
| Skyline Spectral Library | 43,612 annotated spectra |
| Gene Coding Regions Annotated | 13,850 |
| Small Molecule Features Isolated | 1050 |
| Small Molecules Identified | 535 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jenkins, C.; Orsburn, B. The Cannabis Proteome Draft Map Project. Int. J. Mol. Sci. 2020, 21, 965. https://doi.org/10.3390/ijms21030965
Jenkins C, Orsburn B. The Cannabis Proteome Draft Map Project. International Journal of Molecular Sciences. 2020; 21(3):965. https://doi.org/10.3390/ijms21030965
Chicago/Turabian StyleJenkins, Conor, and Benjamin Orsburn. 2020. "The Cannabis Proteome Draft Map Project" International Journal of Molecular Sciences 21, no. 3: 965. https://doi.org/10.3390/ijms21030965
APA StyleJenkins, C., & Orsburn, B. (2020). The Cannabis Proteome Draft Map Project. International Journal of Molecular Sciences, 21(3), 965. https://doi.org/10.3390/ijms21030965