Ketone Bodies Promote Amyloid-β1–40 Clearance in a Human in Vitro Blood–Brain Barrier Model

 ,
,  , , ,
, , ,
Abstract
1. Introduction
2. Results
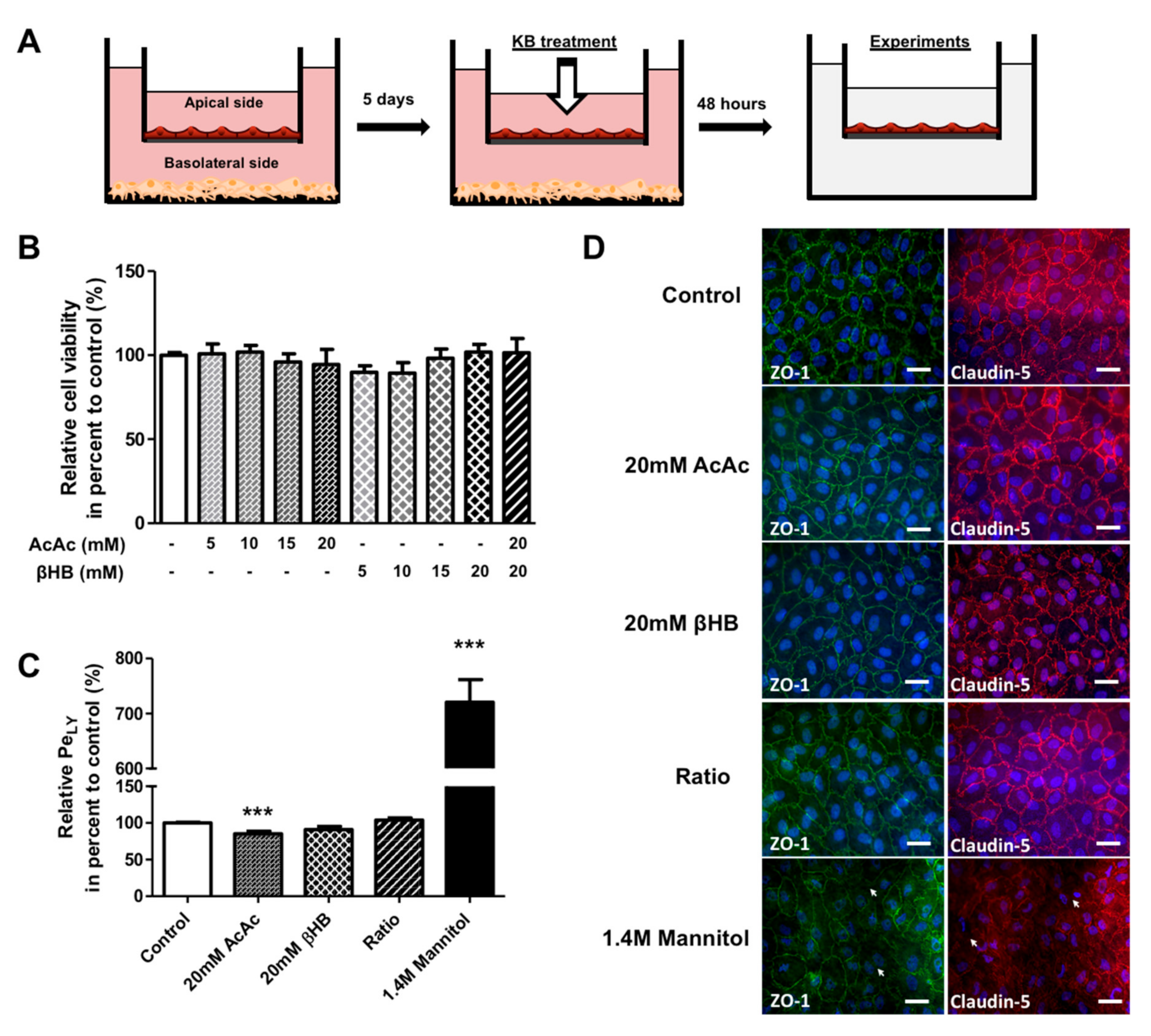
2.1. KBs do not Affect the Viability and Integrity of BLECs
2.2. KBs Increase MCT1 and GLUT1 Protein Levels in BLECs
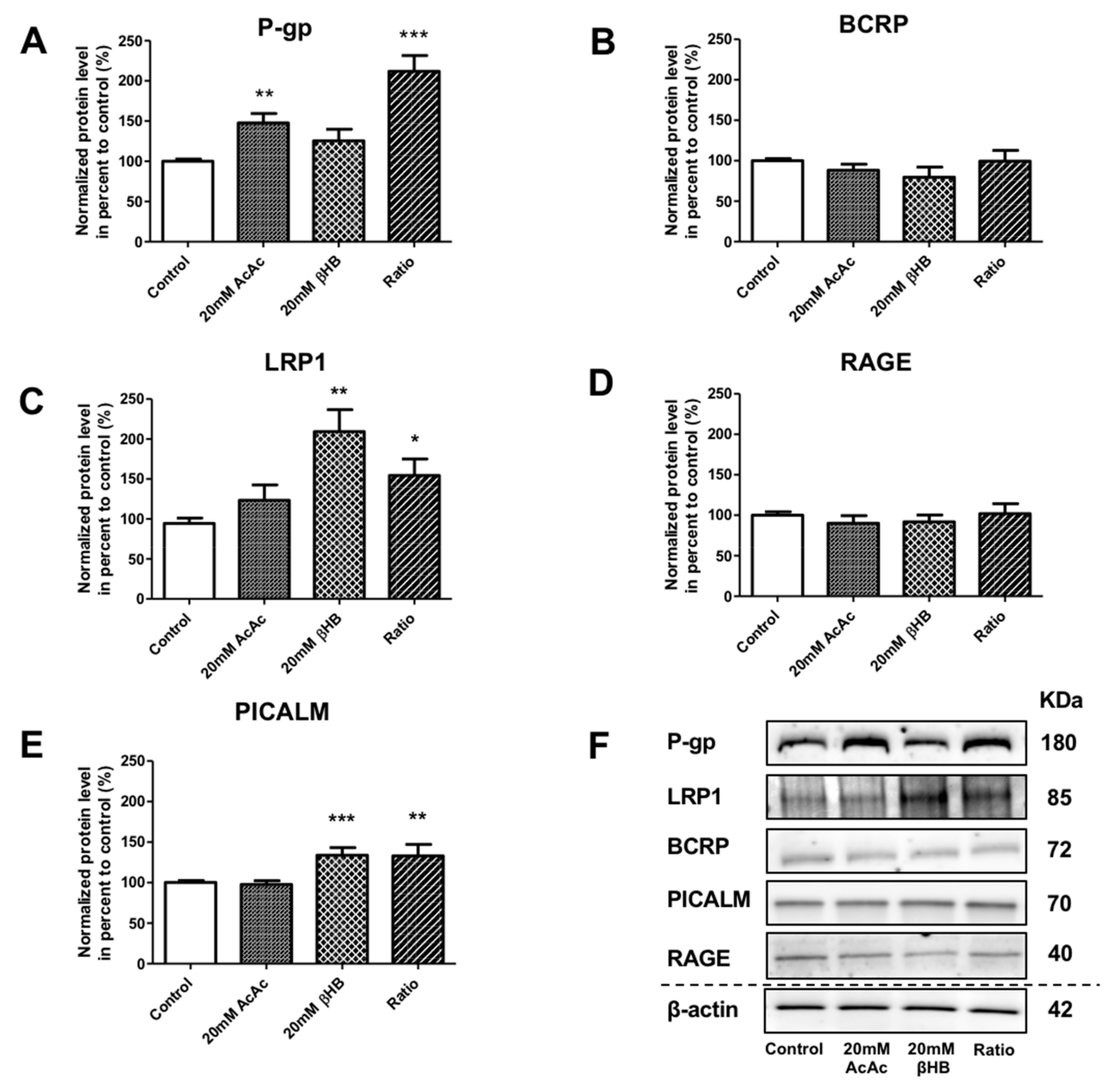
2.3. KBs Modify the Levels of Proteins Involved in Aβ1–40 Peptide Transport Across the BBB
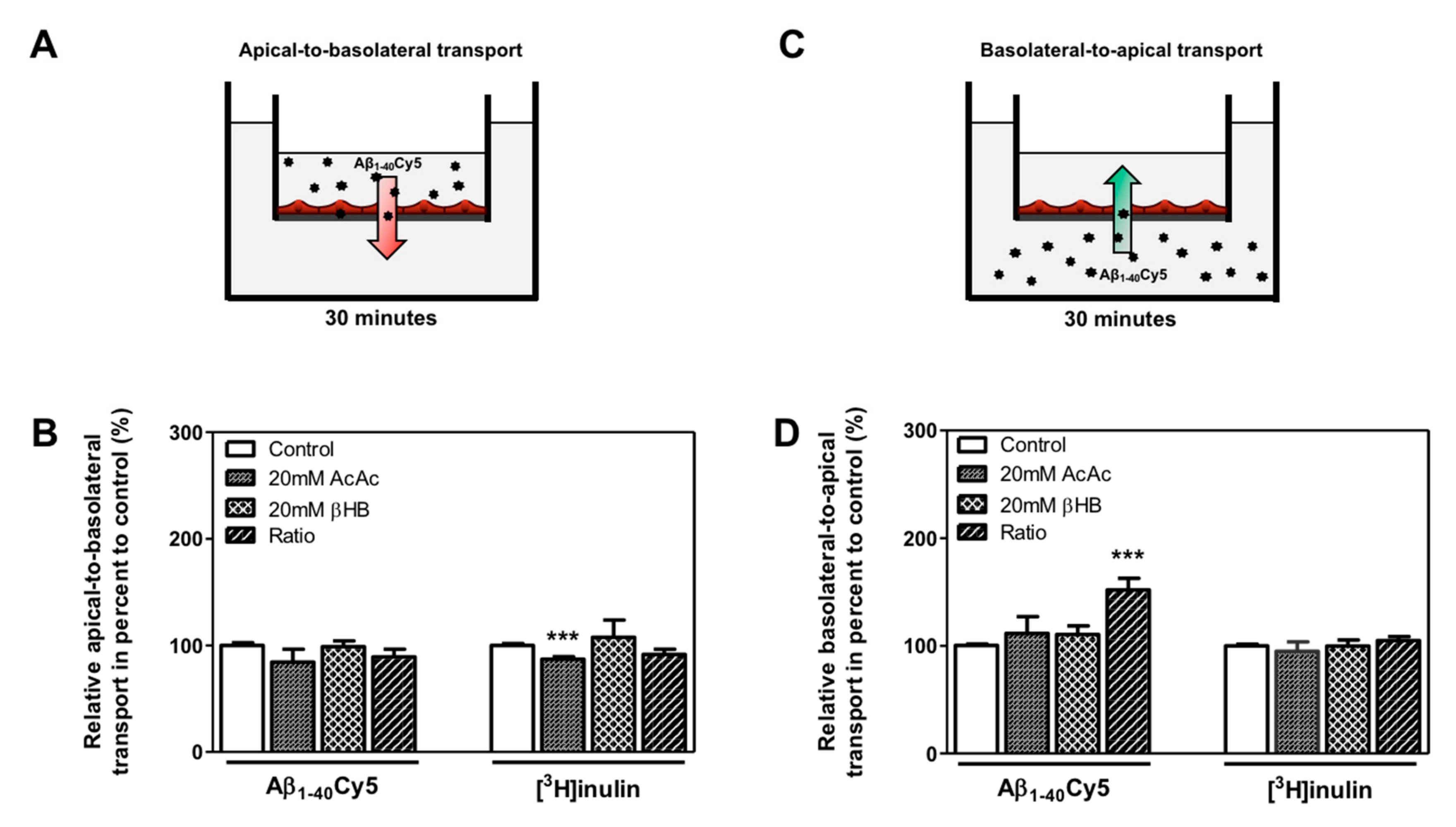
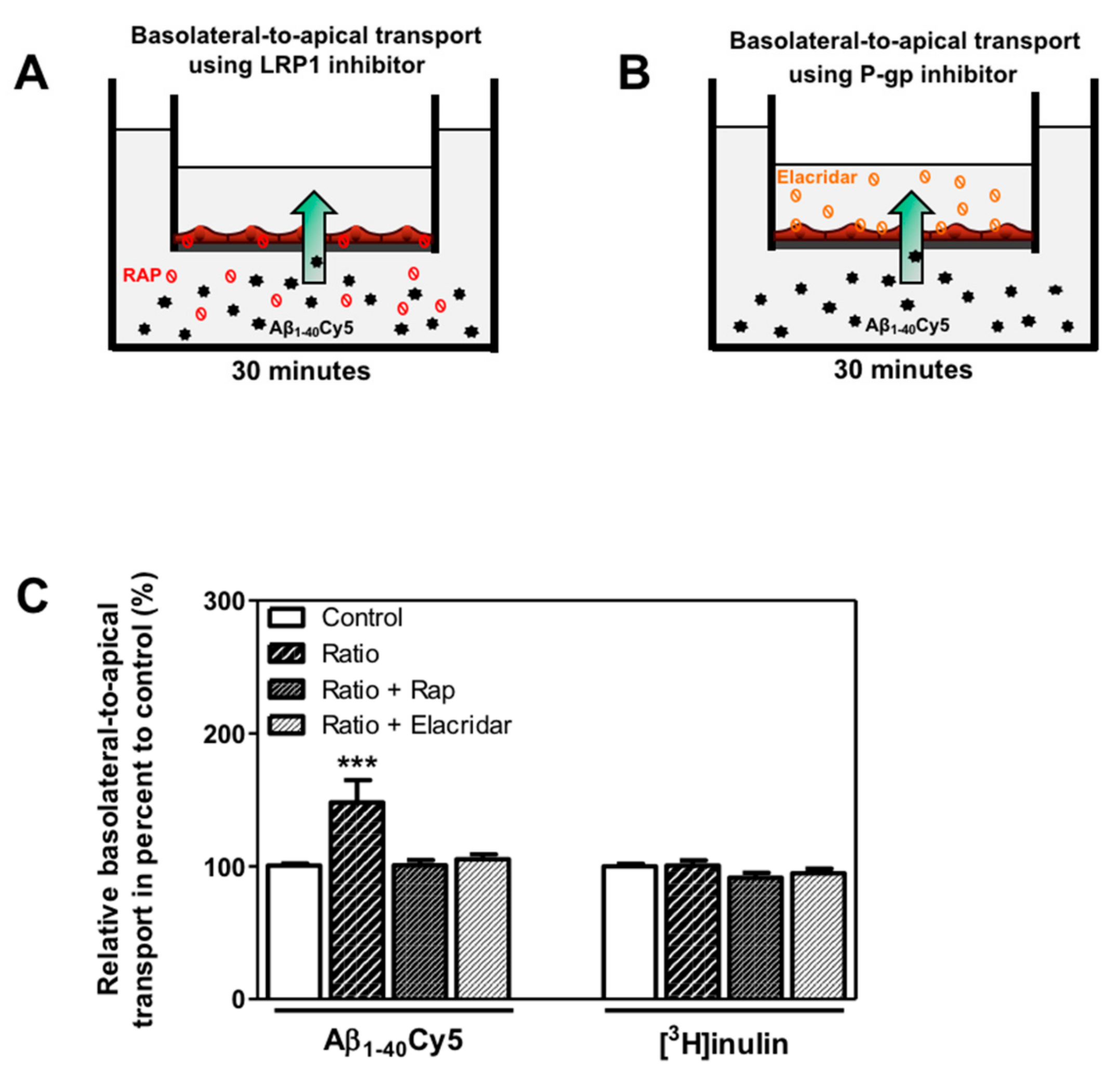
2.4. KBs Increase Basolateral-to-Apical Aβ Peptide Transport Through the BBB with the Involvement of LRP-1 and P-gp
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. The Human in Vitro BBB Model
4.3. Treatment with KBs
4.4. Cell Viability Assay
4.5. Measurement of glucose and βHB levels in culture medium
4.6. Evaluation of BLEC Monolayer Permeability
4.7. Immunostaining
4.8. Protein Extraction and Immunoblots
4.9. Amyloid-β (1-40) Peptide Transport Studies
4.10. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Aa | Amino acid |
| AcAc | Acetoacetate |
| AD | Alzheimer’s disease |
| Aβ | Amyloid β |
| BBB | Blood-brain barrier |
| BCRP | Breast cancer resistance protein |
| βHB | β-hydroxybutyrate |
| BLECs | Brain-like endothelial cells |
| CNS | Central nervous system |
| DMSO | Dimethyl sulfoxide |
| ECM | Endothelial cell medium |
| EGM-2 | Endothelial cell growth medium-2 |
| FCS | Foetal calf serum |
| GLUT1 | Glucose transporter 1 |
| HSA | Human serum albumin |
| KBs | Ketone bodies |
| KD | Ketogenic diet |
| LRP1 | Low-density lipoprotein receptor-related protein 1 |
| LY | Lucifer yellow |
| MCT1 | Monocarboxylate transporter 1 |
| NGS | Normal goat serum |
| Pe | Endothelial permeability |
| PBS-CMF | Phosphate-buffered saline calcium– and magnesium–free |
| P-gp | P-glycoprotein |
| RAGE | Receptor for advanced glycation endproducts |
| RAP | Receptor–associated protein |
| RH | Ringer-HEPES |
| RT | Room temperature |
| SEM | Standard error of the mean |
| TBST | Tris-buffered saline 0.1% Tween 20 |
| ZO–1 | Zonula occludens–1 |
References
- Castellani, R.J.; Perry, G. Tau Biology, Tauopathy, Traumatic Brain Injury, and Diagnostic Challenges. J. Alzheimers Dis. 2019, 67, 447–467. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a004457. [Google Scholar] [CrossRef]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. 1984. Biochem. Biophys. Res. Commun. 2012, 425, 534–539. [Google Scholar] [CrossRef]
- Zlokovic, B.V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 2011, 12, 723–738. [Google Scholar] [CrossRef]
- Gosselet, F.; Saint-Pol, J.; Candela, P.; Fenart, L. Amyloid-beta Peptides, Alzheimer’s Disease and the Blood-brain Barrier. Curr. Alzheimer Res. 2013, 10, 1015–1033. [Google Scholar] [CrossRef] [PubMed]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef] [PubMed]
- Gosselet, F.; Saint-Pol, J.; Fenart, L. Effects of oxysterols on the blood-brain barrier: Implications for Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2014, 446, 687–691. [Google Scholar] [CrossRef] [PubMed]
- Cecchelli, R.; Berezowski, V.; Lundquist, S.; Culot, M.; Renftel, M.; Dehouck, M.P.; Fenart, L. Modelling of the blood-brain barrier in drug discovery and development. Nat. Rev. Drug Discov. 2007, 6, 650–661. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Bell, R.D.; Sagare, A.P.; Friedman, A.E.; Bedi, G.S.; Holtzman, D.M.; Deane, R.; Zlokovic, B.V. Transport pathways for clearance of human Alzheimer’s amyloid beta-peptide and apolipoproteins E and J in the mouse central nervous system. J. Cereb. Blood Flow Metab. 2007, 27, 909–918. [Google Scholar] [CrossRef]
- Shibata, M.; Yamada, S.; Kumar, S.R.; Calero, M.; Bading, J.; Frangione, B.; Holtzman, D.M.; Miller, C.A.; Strickland, D.K.; Ghiso, J.; et al. Clearance of Alzheimer’s amyloid-ss(1-40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J. Clin. Invest. 2000, 106, 1489–1499. [Google Scholar] [CrossRef] [PubMed]
- Deane, R.; Singh, I.; Sagare, A.P.; Bell, R.D.; Ross, N.T.; LaRue, B.; Love, R.; Perry, S.; Paquette, N.; Deane, R.J.; et al. A multimodal RAGE-specific inhibitor reduces amyloid beta-mediated brain disorder in a mouse model of Alzheimer disease. J. Clin. Invest. 2012, 122, 1377–1392. [Google Scholar] [CrossRef] [PubMed]
- Candela, P.; Gosselet, F.; Saint-Pol, J.; Sevin, E.; Boucau, M.C.; Boulanger, E.; Cecchelli, R.; Fenart, L. Apical-to-basolateral transport of amyloid-beta peptides through blood-brain barrier cells is mediated by the receptor for advanced glycation end-products and is restricted by P-glycoprotein. J. Alzheimers Dis 2010, 22, 849–859. [Google Scholar] [CrossRef] [PubMed]
- Tai, L.M.; Loughlin, A.J.; Male, D.K.; Romero, I.A. P-glycoprotein and breast cancer resistance protein restrict apical-to-basolateral permeability of human brain endothelium to amyloid-beta. J. Cereb. Blood Flow. Metab. 2009, 29, 1079–1083. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef]
- Cirrito, J.R.; Deane, R.; Fagan, A.M.; Spinner, M.L.; Parsadanian, M.; Finn, M.B.; Jiang, H.; Prior, J.L.; Sagare, A.; Bales, K.R.; et al. P-glycoprotein deficiency at the blood-brain barrier increases amyloid-beta deposition in an Alzheimer disease mouse model. J. Clin. Invest. 2005, 115, 3285–3290. [Google Scholar] [CrossRef]
- Storck, S.E.; Hartz, A.M.S.; Bernard, J.; Wolf, A.; Kachlmeier, A.; Mahringer, A.; Weggen, S.; Pahnke, J.; Pietrzik, C.U. The concerted amyloid-beta clearance of LRP1 and ABCB1/P-gp across the blood-brain barrier is linked by PICALM. Brain Behav. Immun. 2018, 73, 21–33. [Google Scholar] [CrossRef]
- Harold, D.; Abraham, R.; Hollingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; Dowzell, K.; Williams, A.; et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1088–1093. [Google Scholar] [CrossRef]
- Xu, W.; Tan, L.; Wang, H.F.; Jiang, T.; Tan, M.S.; Tan, L.; Zhao, Q.F.; Li, J.Q.; Wang, J.; Yu, J.T. Meta-analysis of modifiable risk factors for Alzheimer’s disease. J. Neurol. Neurosurg. Psych. 2015, 86, 1299–1306. [Google Scholar] [CrossRef]
- Bomben, V.; Holth, J.; Reed, J.; Cramer, P.; Landreth, G.; Noebels, J. Bexarotene reduces network excitability in models of Alzheimer’s disease and epilepsy. Neurobiol. Aging 2014, 35, 2091–2095. [Google Scholar] [CrossRef]
- Ota, M.; Matsuo, J.; Ishida, I.; Takano, H.; Yokoi, Y.; Hori, H.; Yoshida, S.; Ashida, K.; Nakamura, K.; Takahashi, T.; et al. Effects of a medium-chain triglyceride-based ketogenic formula on cognitive function in patients with mild-to-moderate Alzheimer’s disease. Neurosci. Lett. 2019, 690, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Baranano, K.W.; Hartman, A.L. The ketogenic diet: uses in epilepsy and other neurologic illnesses. Curr. Treat Options Neurol. 2008, 10, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Paleologou, E.; Ismayilova, N.; Kinali, M. Use of the Ketogenic Diet to Treat Intractable Epilepsy in Mitochondrial Disorders. J. Clin. Med. 2017, 6, 56. [Google Scholar] [CrossRef] [PubMed]
- Pawlosky, R.J.; Kemper, M.F.; Kashiwaya, Y.; King, M.T.; Mattson, M.P.; Veech, R.L. Effects of a dietary ketone ester on hippocampal glycolytic and tricarboxylic acid cycle intermediates and amino acids in a 3xTgAD mouse model of Alzheimer’s disease. J. Neurochem. 2017, 141, 195–207. [Google Scholar] [CrossRef]
- Branco, A.F.; Ferreira, A.; Simoes, R.F.; Magalhaes-Novais, S.; Zehowski, C.; Cope, E.; Silva, A.M.; Pereira, D.; Sardao, V.A.; Cunha-Oliveira, T. Ketogenic diets: from cancer to mitochondrial diseases and beyond. Eur. J. Clin. Invest. 2016, 46, 285–298. [Google Scholar] [CrossRef]
- Robinson, A.M.; Williamson, D.H. Physiological roles of ketone bodies as substrates and signals in mammalian tissues. Physiol. Rev. 1980, 60, 143–187. [Google Scholar] [CrossRef]
- Wlodarek, D. Role of Ketogenic Diets in Neurodegenerative Diseases (Alzheimer’s Disease and Parkinson’s Disease). Nutrients 2019, 11, 169. [Google Scholar] [CrossRef]
- Morris, A.A. Cerebral ketone body metabolism. J. Inherit. Metab. Dis. 2005, 28, 109–121. [Google Scholar] [CrossRef]
- Puchalska, P.; Crawford, P.A. Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef]
- Cunnane, S.; Nugent, S.; Roy, M.; Courchesne-Loyer, A.; Croteau, E.; Tremblay, S.; Castellano, A.; Pifferi, F.; Bocti, C.; Paquet, N.; et al. Brain fuel metabolism, aging, and Alzheimer’s disease. Nutrition 2011, 27, 3–20. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Mukherjee, P. Targeting energy metabolism in brain cancer: review and hypothesis. Nutr. Metab. 2005, 2, 30. [Google Scholar] [CrossRef] [PubMed]
- Gasior, M.; Rogawski, M.A.; Hartman, A.L. Neuroprotective and disease-modifying effects of the ketogenic diet. Behav. Pharmacol. 2006, 17, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Kashiwaya, Y.; Bergman, C.; Lee, J.H.; Wan, R.; King, M.T.; Mughal, M.R.; Okun, E.; Clarke, K.; Mattson, M.P.; Veech, R.L. A ketone ester diet exhibits anxiolytic and cognition-sparing properties, and lessens amyloid and tau pathologies in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2013, 34, 1530–1539. [Google Scholar] [CrossRef] [PubMed]
- Stafstrom, C.E.; Rho, J.M. The ketogenic diet as a treatment paradigm for diverse neurological disorders. Front. Pharmacol. 2012, 3, 59. [Google Scholar] [CrossRef]
- Ari, C.; Kovacs, Z.; Juhasz, G.; Murdun, C.; Goldhagen, C.R.; Koutnik, A.P.; Poff, A.M.; Kesl, S.L.; D'Agostino, D.P. Exogenous Ketone Supplements Reduce Anxiety-Related Behavior in Sprague-Dawley and Wistar Albino Glaxo/Rijswijk Rats. Front. Mol. Neurosci. 2016, 9, 137. [Google Scholar] [CrossRef] [PubMed]
- Van der Auwera, I.; Wera, S.; Van Leuven, F.; Henderson, S.T. A ketogenic diet reduces amyloid beta 40 and 42 in a mouse model of Alzheimer’s disease. Nutr. Metab. 2005, 2, 28. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.X.; Maalouf, M.; Han, P.; Zhao, M.; Gao, M.; Dharshaun, T.; Ryan, C.; Whitelegge, J.; Wu, J.; Eisenberg, D.; et al. Ketones block amyloid entry and improve cognition in an Alzheimer’s model. Neurobiol. Aging 2016, 39, 25–37. [Google Scholar] [CrossRef]
- Cecchelli, R.; Aday, S.; Sevin, E.; Almeida, C.; Culot, M.; Dehouck, L.; Coisne, C.; Engelhardt, B.; Dehouck, M.P.; Ferreira, L. A Stable and Reproducible Human Blood-Brain Barrier Model Derived from Hematopoietic Stem Cells. PLoS ONE 2014, 9, e99733. [Google Scholar] [CrossRef]
- Kuntz, M.; Candela, P.; Saint-Pol, J.; Lamartiniere, Y.; Boucau, M.C.; Sevin, E.; Fenart, L.; Gosselet, F. Bexarotene Promotes Cholesterol Efflux and Restricts Apical-to-Basolateral Transport of Amyloid-beta Peptides in an In Vitro Model of the Human Blood-Brain Barrier. J. Alzheimers Dis. 2015, 48, 849–862. [Google Scholar] [CrossRef]
- Praca, C.; Rai, A.; Santos, T.; Cristovao, A.C.; Pinho, S.L.; Cecchelli, R.; Dehouck, M.P.; Bernardino, L.; Ferreira, L.S. A nanoformulation for the preferential accumulation in adult neurogenic niches. J. Control. Release 2018, 284, 57–72. [Google Scholar] [CrossRef]
- Cornford, E.M.; Hyman, S.; Black, K.L.; Cornford, M.E.; Vinters, H.V.; Pardridge, W.M. High expression of the Glut1 glucose transporter in human brain hemangioblastoma endothelium. J. Neuropathol. Exp. Neurol. 1995, 54, 842–851. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Campos, C.; Elizondo, R.; Llanos, P.; Uranga, R.M.; Nualart, F.; Garcia, M.A. MCT expression and lactate influx/efflux in tanycytes involved in glia-neuron metabolic interaction. PLoS ONE 2011, 6, e16411. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, A.; Nelson, A.R.; Sagare, A.P.; Zlokovic, B.V. Impaired vascular-mediated clearance of brain amyloid beta in Alzheimer’s disease: the role, regulation and restoration of LRP1. Front. Aging Neurosci. 2015, 7, 136. [Google Scholar] [CrossRef] [PubMed]
- Candela, P.; Saint-Pol, J.; Kuntz, M.; Boucau, M.C.; Lamartiniere, Y.; Gosselet, F.; Fenart, L. In vitro discrimination of the role of LRP1 at the BBB cellular level: focus on brain capillary endothelial cells and brain pericytes. Brain Res. 2015, 1594, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V.; Martel, C.L.; Matsubara, E.; McComb, J.G.; Zheng, G.; McCluskey, R.T.; Frangione, B.; Ghiso, J. Glycoprotein 330/megalin: probable role in receptor-mediated transport of apolipoprotein J alone and in a complex with Alzheimer disease amyloid beta at the blood-brain and blood-cerebrospinal fluid barriers. Proc. Natl. Acad. Sci. USA 1996, 93, 4229–4234. [Google Scholar] [CrossRef] [PubMed]
- Saint-Pol, J.; Candela, P.; Boucau, M.C.; Fenart, L.; Gosselet, F. Oxysterols decrease apical-to-basolateral transport of Abeta peptides via an ABCB1-mediated process in an in vitro Blood-brain barrier model constituted of bovine brain capillary endothelial cells. Brain Res. 2013, 1517, 1–15. [Google Scholar] [CrossRef]
- Storck, S.E.; Meister, S.; Nahrath, J.; Meissner, J.N.; Schubert, N.; Di Spiezio, A.; Baches, S.; Vandenbroucke, R.E.; Bouter, Y.; Prikulis, I.; et al. Endothelial LRP1 transports amyloid-beta(1-42) across the blood-brain barrier. J. Clin. Invest. 2016, 126, 123–136. [Google Scholar] [CrossRef]
- Gameiro, M.; Silva, R.; Rocha-Pereira, C.; Carmo, H.; Carvalho, F.; Bastos, M.L.; Remiao, F. Cellular Models and In Vitro Assays for the Screening of modulators of P-gp, MRP1 and BCRP. Molecules 2017, 22. [Google Scholar] [CrossRef]
- Andersen, O.M.; Christensen, L.L.; Christensen, P.A.; Sorensen, E.S.; Jacobsen, C.; Moestrup, S.K.; Etzerodt, M.; Thogersen, H.C. Identification of the minimal functional unit in the low density lipoprotein receptor-related protein for binding the receptor-associated protein (RAP). A conserved acidic residue in the complement-type repeats is important for recognition of RAP. J. Biol. Chem. 2000, 275, 21017–21024. [Google Scholar] [CrossRef]
- Deane, R.; Wu, Z.; Sagare, A.; Davis, J.; Du Yan, S.; Hamm, K.; Xu, F.; Parisi, M.; LaRue, B.; Hu, H.W.; et al. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron 2004, 43, 333–344. [Google Scholar] [CrossRef]
- Deane, R.; Sagare, A.; Zlokovic, B.V. The role of the cell surface LRP and soluble LRP in blood-brain barrier Abeta clearance in Alzheimer’s disease. Curr. Pharm. Des. 2008, 14, 1601–1605. [Google Scholar] [CrossRef] [PubMed]
- Jedlitschky, G.; Grube, M.; Mosyagin, I.; Kroemer, H.K.; Vogelgesang, S. Targeting CNS transporters for treatment of neurodegenerative diseases. Curr. Pharm. Des. 2014, 20, 1523–1533. [Google Scholar] [CrossRef] [PubMed]
- Hertz, L.; Chen, Y.; Waagepetersen, H.S. Effects of ketone bodies in Alzheimer’s disease in relation to neural hypometabolism, beta-amyloid toxicity, and astrocyte function. J. Neurochem. 2015, 134, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Reger, M.A.; Henderson, S.T.; Hale, C.; Cholerton, B.; Baker, L.D.; Watson, G.S.; Hyde, K.; Chapman, D.; Craft, S. Effects of beta-hydroxybutyrate on cognition in memory-impaired adults. Neurobiol. Aging 2004, 25, 311–314. [Google Scholar] [CrossRef]
- Kong, G.; Huang, Z.; Ji, W.; Wang, X.; Liu, J.; Wu, X.; Huang, Z.; Li, R.; Zhu, Q. The Ketone Metabolite beta-Hydroxybutyrate Attenuates Oxidative Stress in Spinal Cord Injury by Suppression of Class I Histone Deacetylases. J. Neurotrauma 2017, 34, 2645–2655. [Google Scholar] [CrossRef] [PubMed]
- Isales, C.M.; Min, L.; Hoffman, W.H. Acetoacetate and beta-hydroxybutyrate differentially regulate endothelin-1 and vascular endothelial growth factor in mouse brain microvascular endothelial cells. J. Diabetes Complications 1999, 13, 91–97. [Google Scholar] [CrossRef]
- Mullin, G.E. Issues in complementary and alternative nutrition treatments. Nutr. Clin. Pract. 2009, 24, 543–548. [Google Scholar] [CrossRef]
- Hashim, S.A.; VanItallie, T.B. Ketone body therapy: from the ketogenic diet to the oral administration of ketone ester. J. Lipid Res. 2014, 55, 1818–1826. [Google Scholar] [CrossRef]
- Ballaban-Gil, K.; Callahan, C.; O'Dell, C.; Pappo, M.; Moshe, S.; Shinnar, S. Complications of the ketogenic diet. Epilepsia 1998, 39, 744–748. [Google Scholar] [CrossRef]
- Wood, T.R.; Stubbs, B.J.; Juul, S.E. Exogenous Ketone Bodies as Promising Neuroprotective Agents for Developmental Brain Injury. Dev. Neurosci. 2018, 40, 451–462. [Google Scholar] [CrossRef]
- Henderson, S.T.; Vogel, J.L.; Barr, L.J.; Garvin, F.; Jones, J.J.; Costantini, L.C. Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer’s disease: a randomized, double-blind, placebo-controlled, multicenter trial. Nutr. Metab. 2009, 6, 31. [Google Scholar] [CrossRef] [PubMed]
- D'Agostino, D.P.; Pilla, R.; Held, H.E.; Landon, C.S.; Puchowicz, M.; Brunengraber, H.; Ari, C.; Arnold, P.; Dean, J.B. Therapeutic ketosis with ketone ester delays central nervous system oxygen toxicity seizures in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 304, R829–R836. [Google Scholar] [CrossRef] [PubMed]
- Stubbs, B.J.; Cox, P.J.; Evans, R.D.; Santer, P.; Miller, J.J.; Faull, O.K.; Magor-Elliott, S.; Hiyama, S.; Stirling, M.; Clarke, K. On the Metabolism of Exogenous Ketones in Humans. Front. Physiol. 2017, 8, 848. [Google Scholar] [CrossRef] [PubMed]
- Vannucci, S.J.; Simpson, I.A. Developmental switch in brain nutrient transporter expression in the rat. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E1127–E1134. [Google Scholar] [CrossRef]
- Leino, R.L.; Gerhart, D.Z.; Duelli, R.; Enerson, B.E.; Drewes, L.R. Diet-induced ketosis increases monocarboxylate transporter (MCT1) levels in rat brain. Neurochem. Int. 2001, 38, 519–527. [Google Scholar] [CrossRef]
- Puchowicz, M.A.; Xu, K.; Sun, X.; Ivy, A.; Emancipator, D.; LaManna, J.C. Diet-induced ketosis increases capillary density without altered blood flow in rat brain. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E1607–E1615. [Google Scholar] [CrossRef]
- Ma, D.; Wang, A.C.; Parikh, I.; Green, S.J.; Hoffman, J.D.; Chlipala, G.; Murphy, M.P.; Sokola, B.S.; Bauer, B.; Hartz, A.M.S.; et al. Ketogenic diet enhances neurovascular function with altered gut microbiome in young healthy mice. Sci. Rep. 2018, 8, 6670. [Google Scholar] [CrossRef]
- Pifferi, F.; Tremblay, S.; Croteau, E.; Fortier, M.; Tremblay-Mercier, J.; Lecomte, R.; Cunnane, S.C. Mild experimental ketosis increases brain uptake of 11C-acetoacetate and 18F-fluorodeoxyglucose: a dual-tracer PET imaging study in rats. Nutr. Neurosci. 2011, 14, 51–58. [Google Scholar] [CrossRef]
- Tanegashima, K.; Sato-Miyata, Y.; Funakoshi, M.; Nishito, Y.; Aigaki, T.; Hara, T. Epigenetic regulation of the glucose transporter gene Slc2a1 by beta-hydroxybutyrate underlies preferential glucose supply to the brain of fasted mice. Genes Cells 2017, 22, 71–83. [Google Scholar] [CrossRef]
- Studzinski, C.M.; MacKay, W.A.; Beckett, T.L.; Henderson, S.T.; Murphy, M.P.; Sullivan, P.G.; Burnham, W.M. Induction of ketosis may improve mitochondrial function and decrease steady-state amyloid-beta precursor protein (APP) levels in the aged dog. Brain Res. 2008, 1226, 209–217. [Google Scholar] [CrossRef]
- Gillette-Guyonnet, S.; Secher, M.; Vellas, B. Nutrition and neurodegeneration: epidemiological evidence and challenges for future research. Br. J. Clin. Pharmacol. 2013, 75, 738–755. [Google Scholar] [CrossRef] [PubMed]
- Hu, N.; Yu, J.T.; Tan, L.; Wang, Y.L.; Sun, L.; Tan, L. Nutrition and the risk of Alzheimer’s disease. BioMed Res. Int. 2013, 2013, 524820. [Google Scholar] [CrossRef] [PubMed]
- Fuso, A.; Nicolia, V.; Cavallaro, R.A.; Ricceri, L.; D'Anselmi, F.; Coluccia, P.; Calamandrei, G.; Scarpa, S. B-vitamin deprivation induces hyperhomocysteinemia and brain S-adenosylhomocysteine, depletes brain S-adenosylmethionine, and enhances PS1 and BACE expression and amyloid-beta deposition in mice. Mol. Cell Neurosci. 2008, 37, 731–746. [Google Scholar] [CrossRef] [PubMed]
- Qosa, H.; Batarseh, Y.S.; Mohyeldin, M.M.; El Sayed, K.A.; Keller, J.N.; Kaddoumi, A. Oleocanthal Enhances Amyloid-beta Clearance from the Brains of TgSwDI Mice and in Vitro across a Human Blood-Brain Barrier Model. ACS Chem. Neurosci. 2015, 6, 1849–1859. [Google Scholar] [CrossRef]
- Qosa, H.; Abuznait, A.H.; Hill, R.A.; Kaddoumi, A. Enhanced Brain Amyloid-beta Clearance by Rifampicin and Caffeine as a Possible Protective Mechanism Against Alzheimer’s Disease. J. Alzheimers Dis. 2012, 31, 151–165. [Google Scholar] [CrossRef]
- Deane, R.; Du Yan, S.; Submamaryan, R.K.; LaRue, B.; Jovanovic, S.; Hogg, E.; Welch, D.; Manness, L.; Lin, C.; Yu, J.; et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat. Med. 2003, 9, 907–913. [Google Scholar] [CrossRef]
- Zhao, Z.; Nelson, A.R.; Betsholtz, C.; Zlokovic, B.V. Establishment and Dysfunction of the Blood-Brain Barrier. Cell 2015, 163, 1064–1078. [Google Scholar] [CrossRef]
- Do, T.M.; Noel-Hudson, M.S.; Ribes, S.; Besengez, C.; Smirnova, M.; Cisternino, S.; Buyse, M.; Calon, F.; Chimini, G.; Chacun, H.; et al. ABCG2- and ABCG4-mediated efflux of amyloid-beta peptide 1-40 at the mouse blood-brain barrier. J. Alzheimers Dis. 2012, 30, 155–166. [Google Scholar] [CrossRef]
- Krohn, M.; Lange, C.; Hofrichter, J.; Scheffler, K.; Stenzel, J.; Steffen, J.; Schumacher, T.; Bruning, T.; Plath, A.S.; Alfen, F.; et al. Cerebral amyloid-beta proteostasis is regulated by the membrane transport protein ABCC1 in mice. J. Clin. Invest. 2011, 121, 3924–3931. [Google Scholar] [CrossRef]
- Guo, Y.X.; He, L.Y.; Zhang, M.; Wang, F.; Liu, F.; Peng, W.X. 1,25-Dihydroxyvitamin D3 regulates expression of LRP1 and RAGE in vitro and in vivo, enhancing Abeta1-40 brain-to-blood efflux and peripheral uptake transport. Neuroscience 2016, 322, 28–38. [Google Scholar] [CrossRef]
- Cheng, S.; Wu, Q.; Yang, F.; Xu, M.; Leski, M.; Chen, G.Q. Influence of DL-beta-hydroxybutyric acid on cell proliferation and calcium influx. Biomacromolecules 2005, 6, 593–597. [Google Scholar] [CrossRef] [PubMed]
- Kanikarla-Marie, P.; Jain, S.K. Hyperketonemia (acetoacetate) upregulates NADPH oxidase 4 and elevates oxidative stress, ICAM-1, and monocyte adhesivity in endothelial cells. Cell Physiol. Biochem. 2015, 35, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.K.; Kannan, K.; Lim, G.; McVie, R.; Bocchini, J.A., Jr. Hyperketonemia increases tumor necrosis factor-alpha secretion in cultured U937 monocytes and Type 1 diabetic patients and is apparently mediated by oxidative stress and cAMP deficiency. Diabetes 2002, 51, 2287–2293. [Google Scholar] [CrossRef] [PubMed]
- Anderson, O.S.; Sant, K.E.; Dolinoy, D.C. Nutrition and epigenetics: An interplay of dietary methyl donors, one-carbon metabolism and DNA methylation. J. Nutr. Biochem. 2012, 23, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.C.; Verdin, E. β-Hydroxybutyrate: A Signaling Metabolite. Annu. Rev. Nutr. 2017, 37, 51–76. [Google Scholar] [CrossRef]
- Pedroso, D.C.; Tellechea, A.; Moura, L.; Fidalgo-Carvalho, I.; Duarte, J.; Carvalho, E.; Ferreira, L. Improved survival, vascular differentiation and wound healing potential of stem cells co-cultured with endothelial cells. PLoS ONE 2011, 6, e16114. [Google Scholar] [CrossRef]
- Vandenhaute, E.; Dehouck, L.; Boucau, M.C.; Sevin, E.; Uzbekov, R.; Tardivel, M.; Gosselet, F.; Fenart, L.; Cecchelli, R.; Dehouck, M.P. Modelling the neurovascular unit and the blood-brain barrier with the unique function of pericytes. Curr. Neurovasc. Res. 2011, 8, 258–269. [Google Scholar] [CrossRef]
- Paoli, A.; Bosco, G.; Camporesi, E.M.; Mangar, D. Ketosis, ketogenic diet and food intake control: a complex relationship. Front. Psych. 2015, 6, 27. [Google Scholar] [CrossRef]
- Laffel, L. Ketone bodies: a review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes Metab. Res. Rev. 1999, 15, 412–426. [Google Scholar] [CrossRef]
- Dehouck, M.P.; Jolliet-Riant, P.; Bree, F.; Fruchart, J.C.; Cecchelli, R.; Tillement, J.P. Drug transfer across the blood-brain barrier: correlation between in vitro and in vivo models. J. Neurochem. 1992, 58, 1790–1797. [Google Scholar] [CrossRef]
- Sevin, E.; Dehouck, L.; Fabulas-da Costa, A.; Cecchelli, R.; Dehouck, M.P.; Lundquist, S.; Culot, M. Accelerated Caco-2 cell permeability model for drug discovery. J. Pharmacol. Toxicol. Methods 2013, 68, 334–339. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Target | Antibody Reference | Fixation/Permeabilization | Antibody Dilution |
|---|---|---|---|
| Claudin-5 | 34-1600 (Invitrogen) | Ice-methanol 30’’ | 1:100 |
| GLUT1 | 07-1401 (Merck Millipore) | Ice-methanol 30’’ | 1:100 |
| MCT1 | AB3538P (Merck Millipore) | Ice-methanol 30’’ | 1:100 |
| ZO1 | 61-7300 (Invitrogen) | Paraformaldehyde 1% 10’/Triton X100 0,1% 10’ | 1:200 |
| Protein Target | Antibody Reference | Lysis Buffer | Special Condition | Antibody Dilution | Size (KDa) |
|---|---|---|---|---|---|
| β-actin | A5541 (Sigma Aldrich) | RIPA | - | 1:20000 | 42 |
| BCRP | Ab3380 (Abcam) | RIPA | Without heat denaturation | 1:1000 | 72 |
| GLUT1 | 07-1401 (Merck millipore) | RIPA | - | 1:1000 | 55 |
| LRP1 | 5A6 (Santa Cruz) | UT4 | Without reduction / β-mercaptoethanol | 1:200 | 85 |
| MCT1 | Ab179832 (Abcam) | RIPA | - | 1:1000 | 54 |
| P-gp | C219 (Gene Tex) | RIPA | Without heat denaturation | 1:500 | 180 |
| PICALM | HPA019053 (Sigma Aldrich) | RIPA | - | 1:1250 | 70 |
| RAGE | Ab37647 (Abcam) | RIPA | - | 1:1000 | 40 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Versele, R.; Corsi, M.; Fuso, A.; Sevin, E.; Businaro, R.; Gosselet, F.; Fenart, L.; Candela, P. Ketone Bodies Promote Amyloid-β1–40 Clearance in a Human in Vitro Blood–Brain Barrier Model. Int. J. Mol. Sci. 2020, 21, 934. https://doi.org/10.3390/ijms21030934
Versele R, Corsi M, Fuso A, Sevin E, Businaro R, Gosselet F, Fenart L, Candela P. Ketone Bodies Promote Amyloid-β1–40 Clearance in a Human in Vitro Blood–Brain Barrier Model. International Journal of Molecular Sciences. 2020; 21(3):934. https://doi.org/10.3390/ijms21030934
Chicago/Turabian StyleVersele, Romain, Mariangela Corsi, Andrea Fuso, Emmanuel Sevin, Rita Businaro, Fabien Gosselet, Laurence Fenart, and Pietra Candela. 2020. "Ketone Bodies Promote Amyloid-β1–40 Clearance in a Human in Vitro Blood–Brain Barrier Model" International Journal of Molecular Sciences 21, no. 3: 934. https://doi.org/10.3390/ijms21030934
APA StyleVersele, R., Corsi, M., Fuso, A., Sevin, E., Businaro, R., Gosselet, F., Fenart, L., & Candela, P. (2020). Ketone Bodies Promote Amyloid-β1–40 Clearance in a Human in Vitro Blood–Brain Barrier Model. International Journal of Molecular Sciences, 21(3), 934. https://doi.org/10.3390/ijms21030934