Escherichia coli Extract-Based Cell-Free Expression System as an Alternative for Difficult-to-Obtain Protein Biosynthesis


Abstract
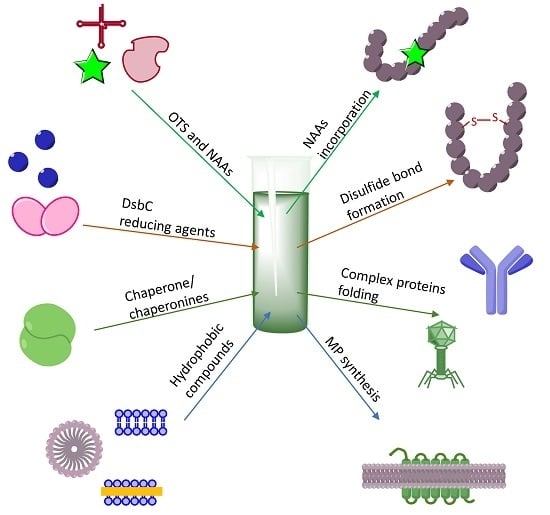
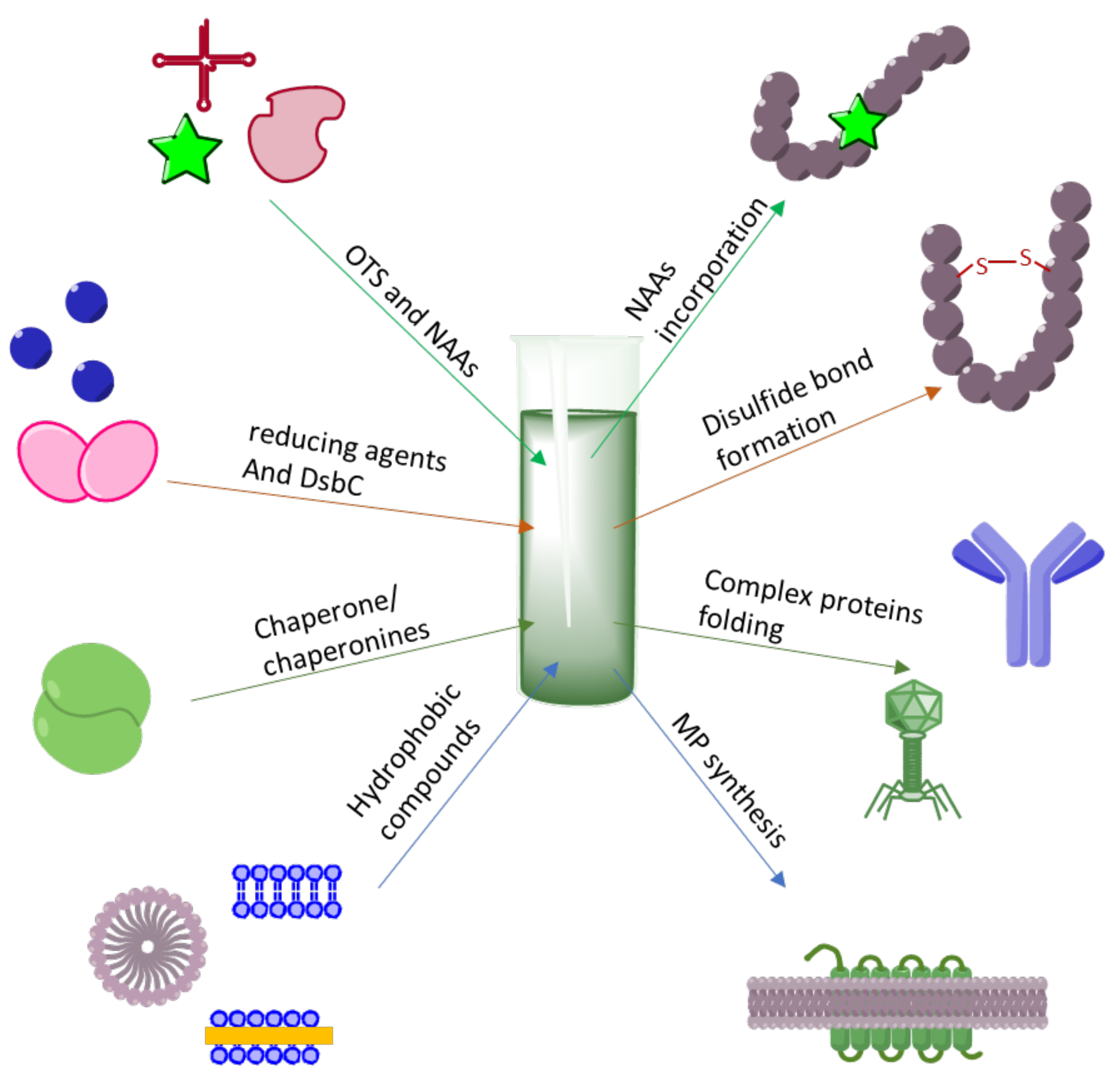
{kind=link}
{kind=link}
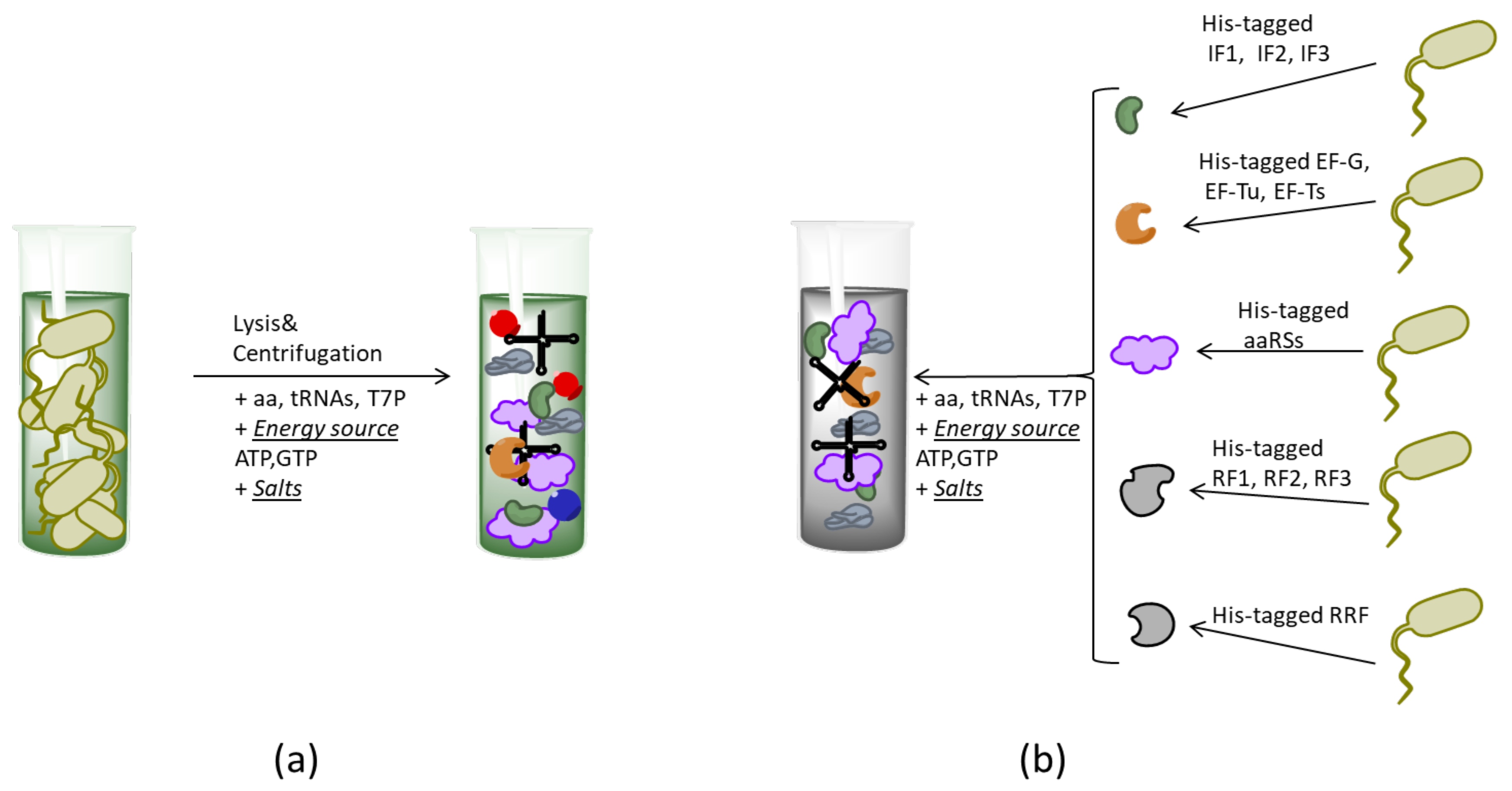{kind=link}
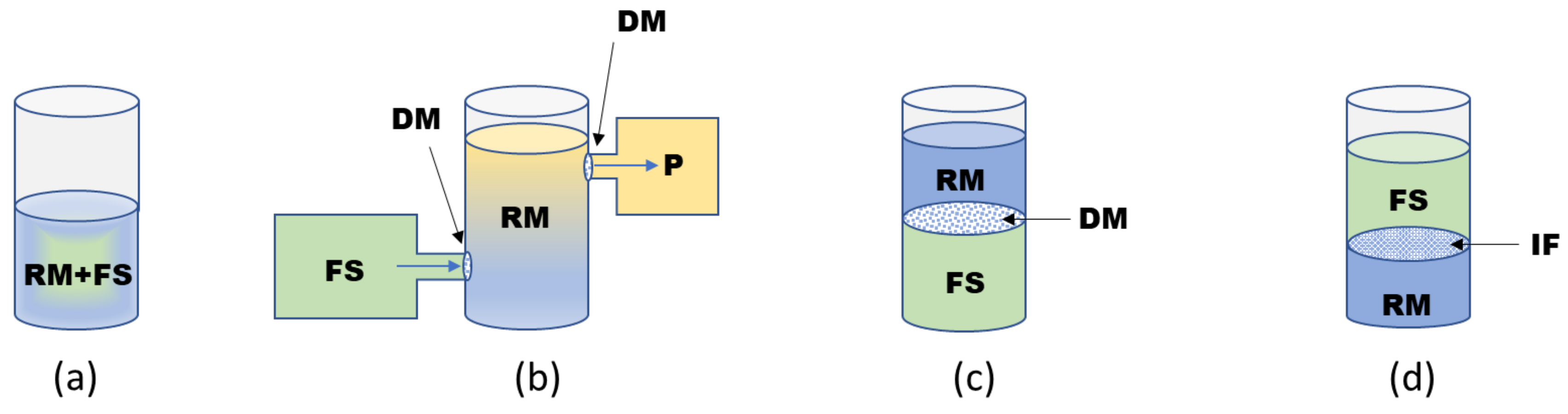{kind=link}
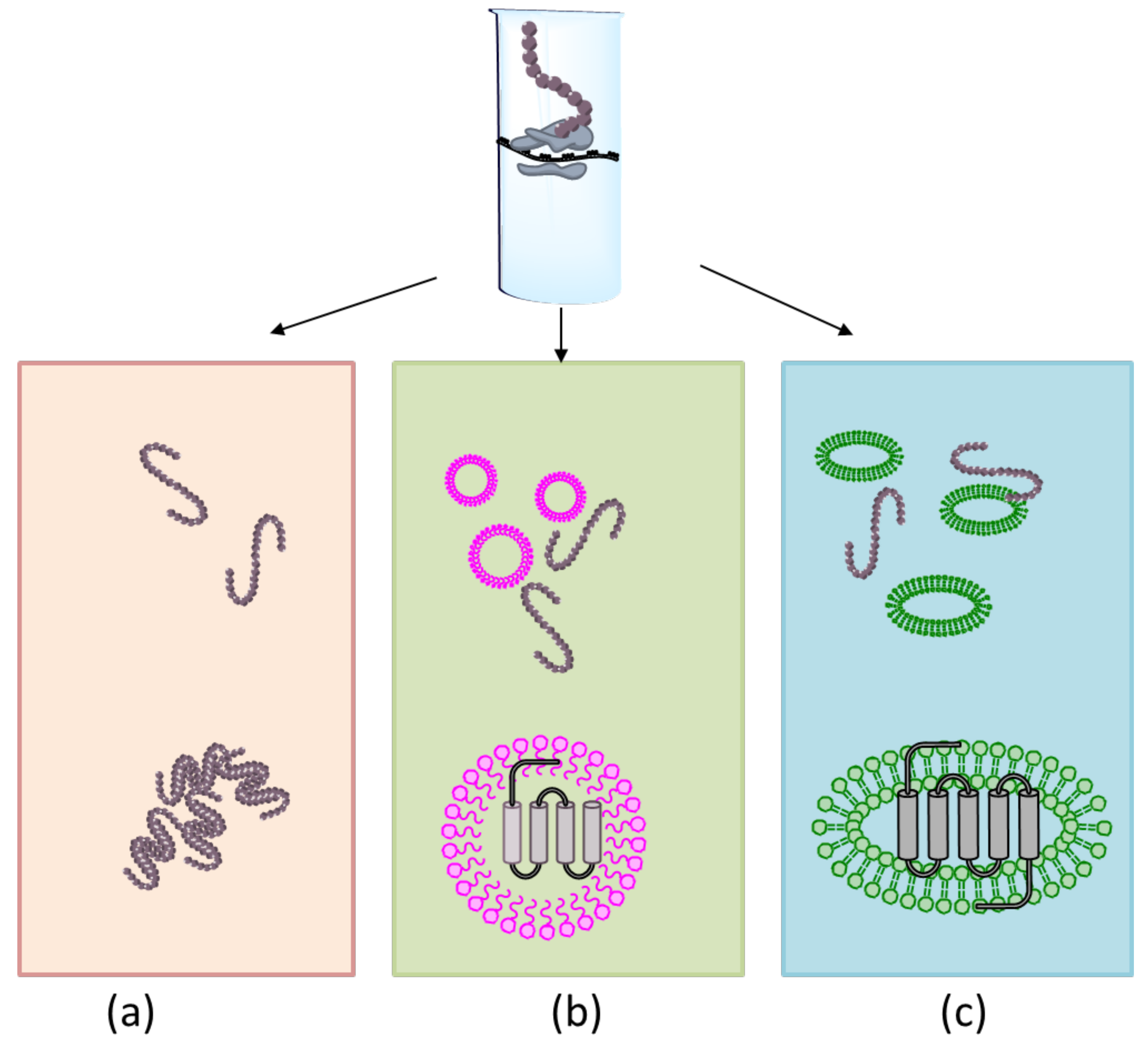{kind=link}
1. Introduction
2. History
2.1. Energy Regeneration
2.2. Formats of CF Systems
3. Making Difficult-to-Obtain Protein Synthesis Easy
3.1. Cytotoxic Protein and Peptides Therapeutics
3.2. Membrane Protein Synthesis
3.3. CF Synthesis of Folded Proteins and Peptides
3.4. Incorporation of NAAs into Proteins
3.5. Post-Translational Modifications
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CF | Cell-free |
| PURE | Protein synthesis Using Recombinant Elements |
| MP | Membrane protein |
| NLP | Nanolipoprotein |
| aaRS | aminoacyl-tRNA synthetase |
| NAA | Non-canonical or unnatural amino acid |
| OTS | Orthogonal Translation System |
| PTM | Posttranslational modification |
References
- Jia, B.; Jeon, C.O. High-throughput recombinant protein expression in Escherichia coli: Current status and future perspectives. Open Biol. 2016, 6, 160196. [Google Scholar] [CrossRef] [PubMed]
- Rosano, G.L.; Morales, E.S.; Ceccarelli, E.A. New tools for recombinant protein production in Escherichia coli: A 5-year update. Protein Sci. 2019, 28, 1412–1422. [Google Scholar] [CrossRef] [PubMed]
- Littlefield, J.W.; Keller, E.B.; Gross, J.; Zamecnik, P.C. Studies on cytoplasmic ribonucleoprotein particles from the liver of the rat. J. Biol. Chem. 1955, 217, 111–123. [Google Scholar] [PubMed]
- Winnick, T. Studies on the mechanism of protein synthesis in embryonic and tumor tissues. II. Inactivation of fetal rat liver homogenates by dialysis, and reactivation by the adenylic acid system. Arch. Biochem. 1950, 28, 338–347. [Google Scholar]
- Jewett, M.C.; Swartz, J.R. Rapid expression and purification of 100 nmol quantities of active protein using cell-free protein synthesis. Biotechnol. Prog. 2004, 20, 102–109. [Google Scholar] [CrossRef]
- Hansen, M.M.K.; Ventosa Rosquelles, M.; Yelleswarapu, M.; Maas, R.J.M.; van Vugt-Jonker, A.J.; Heus, H.A.; Huck, W.T.S. Protein Synthesis in Coupled and Uncoupled Cell-Free Prokaryotic Gene Expression Systems. ACS Synth. Biol. 2016, 5, 1433–1440. [Google Scholar] [CrossRef]
- Carlson, E.D.; Gan, R.; Hodgman, C.E.; Jewett, M.C. Cell-free protein synthesis: Applications come of age. Biotechnol. Adv. 2012, 30, 1185–1194. [Google Scholar] [CrossRef]
- Siekevitz, P. Uptake of radioactive alanine in vitro into the proteins of rat liver fractions. J. Biol. Chem. 1952, 195, 549–565. [Google Scholar]
- Zamecnik, P.C.; Frantz, I.D., Jr. Incorporation in vitro of radioactive carbon from carboxyl-labeled dl-alanine and glycine into proteins of normal and malignant rat livers. J. Biol. Chem. 1948, 175, 299–314. [Google Scholar] [PubMed]
- Hoagland, M.B.; Stephenson, M.L.; Scott, J.F.; Hecht, L.I.; Zamecnik, P.C. A soluble ribonucleic acid intermediate in protein synthesis. J. Biol. Chem. 1958, 231, 241–257. [Google Scholar] [PubMed]
- Hoagland, M.B.; Keller, E.B.; Zamecnik, P.C. Enzymatic carboxyl activation of amino acids. J. Biol. Chem. 1956, 218, 345–358. [Google Scholar] [PubMed]
- Littlefield, J.W.; Keller, E.B. Incorporation of C14-amino acids into ribonucleoprotein particles from the Ehrlich mouse ascites tumor. J. Biol. Chem. 1957, 224, 13–30. [Google Scholar]
- Lamborg, M.R.; Zamecnik, P.C. Amino acid incorporation into protein by extracts of E. coli. Biochim. Biophys. Acta 1960, 42, 206–211. [Google Scholar] [CrossRef]
- Matthaei, J.H.; Nirenberg, M.W. Characteristics and stabilization of DNAase-sensitive protein synthesis in E. coli extracts. Proc. Natl. Acad. Sci. USA 1961, 47, 1580–1588. [Google Scholar] [CrossRef]
- Nirenberg, M. Historical review: Deciphering the genetic code–a personal account. Trends Biochem. Sci. 2004, 29, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Crick, F. Central dogma of molecular biology. Nature 1970, 227, 561. [Google Scholar] [CrossRef]
- Strasser, B.J. A world in one dimension: Linus Pauling, Francis Crick and the central dogma of molecular biology. Pubbl. Stn. Zool. Napoli 2006, 491–512. [Google Scholar]
- Chen, H.; Zubay, G. Analysis of ColE1 expression in vitro after chromosome fragmentation. J. Bacteriol. 1983, 154, 650–655. [Google Scholar] [CrossRef]
- Zubay, G. In vitro synthesis of protein in microbial systems. Annu. Rev. Genet. 1973, 7, 267–287. [Google Scholar] [CrossRef]
- Krieg, P.A.; Melton, D. In vitro RNA synthesis with SP6 RNA polymerase. Methods Enzym. 1987, 155, 397–415. [Google Scholar]
- Nevin, D.E.; Pratt, J.M. A coupled in vitro transcription-translation system for the exclusive synthesis of polypeptides expressed from the T7 promoter. FEBS Lett. 1991, 291, 259–263. [Google Scholar] [CrossRef]
- Ahn, J.; Chu, H.; Kim, T.; Oh, I.; Choi, C.; Hahn, G.; Park, C.; Kim, D. Cell-free synthesis of recombinant proteins from PCR-amplified genes at a comparable productivity to that of plasmid-based reactions. Biochem. Biophys. Res. Commun. 2005, 338, 1346–1352. [Google Scholar] [CrossRef]
- Michel-Reydellet, N.; Woodrow, K.; Swartz, J. Increasing PCR fragment stability and protein yields in a cell-free system with genetically modified Escherichia coli extracts. J. Mol. Microbiol. Biotechnol. 2005, 9, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.L.; Ivashkiv, L.; Chen, H.Z.; Zubay, G.; Cashel, M. Cell-free coupled transcription-translation system for investigation of linear DNA segments. Proc. Natl. Acad. Sci. USA 1980, 77, 7029–7033. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; Matsuda, N.; Yokoyama, S.; Kigawa, T. Cell-free protein synthesis system from Escherichia coli cells cultured at decreased temperatures improves productivity by decreasing DNA template degradation. Anal. Biochem. 2008, 377, 156–161. [Google Scholar] [CrossRef]
- Marshall, R.; Maxwell, C.S.; Collins, S.P.; Beisel, C.L.; Noireaux, V. Short DNA containing χ sites enhances DNA stability and gene expression in E. coli cell-free transcription–translation systems. Biotechnol. Bioeng. 2017, 114, 2137–2141. [Google Scholar] [CrossRef]
- Wu, P.S.; Ozawa, K.; Lim, S.P.; Vasudevan, S.G.; Dixon, N.E.; Otting, G. Cell-Free Transcription/Translation from PCR-Amplified DNA for High-Throughput NMR Studies. Angew. Chem. Int. Ed. 2007, 46, 3356–3358. [Google Scholar] [CrossRef]
- Pratt, R.E.; Dzau, V.J.; Ouellette, A.J. Influence of androgen on translatable renin mRNA in the mouse submandibular gland. Hypertension 1984, 6, 605–613. [Google Scholar] [CrossRef]
- Kim, D.; Swartz, J.R. Efficient production of a bioactive, multiple disulfide-bonded protein using modified extracts of Escherichia coli. Biotechnol. Bioeng. 2004, 85, 122–129. [Google Scholar] [CrossRef]
- Kim, T.; Keum, J.; Oh, I.; Choi, C.; Park, C.; Kim, D. Simple procedures for the construction of a robust and cost-effective cell-free protein synthesis system. J. Biotechnol. 2006, 126, 554–561. [Google Scholar] [CrossRef]
- Fujiwara, K.; Doi, N. Biochemical preparation of cell extract for cell-free protein synthesis without physical disruption. PloS ONE 2016, 11, e0154614. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.; Jewett, M.C. High-throughput preparation methods of crude extract for robust cell-free protein synthesis. Sci. Rep. 2015, 5, 8663. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, P.; Holland, T.M.; Bundy, B.C. Streamlined extract preparation for Escherichia coli-based cell-free protein synthesis by sonication or bead vortex mixing. BioTechniques 2012, 53, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Inoue, A.; Tomari, Y.; Suzuki, T.; Yokogawa, T.; Nishikawa, K.; Ueda, T. Cell-free translation reconstituted with purified components. Nat. Biotechnol. 2001, 19, 751. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Kanamori, T.; Ueda, T. Protein synthesis by pure translation systems. Methods 2005, 36, 299–304. [Google Scholar] [CrossRef]
- Wang, H.H.; Huang, P.Y.; Xu, G.; Haas, W.; Marblestone, A.; Li, J.; Gygi, S.P.; Forster, A.C.; Jewett, M.C.; Church, G.M. Multiplexed in vivo His-tagging of enzyme pathways for in vitro single-pot multienzyme catalysis. ACS Synth. Biol. 2012, 1, 43–52. [Google Scholar] [CrossRef]
- Villarreal, F.; Contreras-Llano, L.; Chavez, M.; Ding, Y.; Fan, J.; Pan, T.; Tan, C. Synthetic microbial consortia enable rapid assembly of pure translation machinery. Nat. Chem. Biol. 2018, 14, 29–35. [Google Scholar] [CrossRef]
- Lee, K.; Kim, D. Recent advances in development of cell-free protein synthesis systems for fast and efficient production of recombinant proteins. FEMS Microbiol. Lett. 2018, 365, fny174. [Google Scholar] [CrossRef]
- Kim, D.; Choi, C. A semicontinuous prokaryotic coupled transcription/translation system using a dialysis membrane. Biotechnol. Prog. 1996, 12, 645–649. [Google Scholar] [CrossRef]
- Kim, D.; Swartz, J.R. Prolonging cell-free protein synthesis with a novel ATP regeneration system. Biotechnol. Bioeng. 1999, 66, 180–188. [Google Scholar] [CrossRef]
- Calhoun, K.A.; Swartz, J.R. An economical method for cell-free protein synthesis using glucose and nucleoside monophosphates. Biotechnol. Prog. 2005, 21, 1146–1153. [Google Scholar] [CrossRef] [PubMed]
- Jewett, M.C.; Calhoun, K.A.; Voloshin, A.; Wuu, J.J.; Swartz, J.R. An integrated cell-free metabolic platform for protein production and synthetic biology. Mol. Syst. Biol. 2008, 4. [Google Scholar] [CrossRef]
- Caschera, F.; Noireaux, V. A cost-effective polyphosphate-based metabolism fuels an all E. coli cell-free expression system. Metab. Eng. 2015, 27, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kim, T.; Kim, D. Prolonged production of proteins in a cell-free protein synthesis system using polymeric carbohydrates as an energy source. Process Biochem. 2011, 46, 1366–1369. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Y.P. Cell-free protein synthesis energized by slowly-metabolized maltodextrin. BMC Biotechnol. 2009, 9, 58. [Google Scholar] [CrossRef]
- Kim, H.; Kim, K.; Kang, T.; Choi, J.H.; Song, J.J.; Choi, Y.H.; Kim, B.; Kim, D. Implementing bacterial acid resistance into cell-free protein synthesis for buffer-free expression and screening of enzymes. Biotechnol. Bioeng. 2015, 112, 2630–2635. [Google Scholar] [CrossRef] [PubMed]
- Spirin, A.S.; Baranov, V.I.; Ryabova, L.A.; Ovodov, S.Y.; Alakhov, Y.B. A continuous cell-free translation system capable of producing polypeptides in high yield. Science 1988, 242, 1162–1164. [Google Scholar] [CrossRef]
- Spirin, A.S. High-throughput cell-free systems for synthesis of functionally active proteins. Trends Biotechnol. 2004, 22, 538–545. [Google Scholar] [CrossRef]
- Sawasaki, T.; Hasegawa, Y.; Tsuchimochi, M.; Kamura, N.; Ogasawara, T.; Kuroita, T.; Endo, Y. A bilayer cell-free protein synthesis system for high-throughput screening of gene products. FEBS Lett. 2002, 514, 102–105. [Google Scholar] [CrossRef]
- Damiati, S.; Mhanna, R.; Kodzius, R.; Ehmoser, E. Cell-free approaches in synthetic biology utilizing microfluidics. Genes 2018, 9, 144. [Google Scholar] [CrossRef]
- Dittrich, P.S.; Jahnz, M.; Schwille, P. A New Embedded Process for Compartmentalized Cell-Free Protein Expression and On-line Detection in Microfluidic Devices. ChemBioChem 2005, 6, 811–814. [Google Scholar] [CrossRef] [PubMed]
- Murtas, G.; Kuruma, Y.; Bianchini, P.; Diaspro, A.; Luisi, P.L. Protein synthesis in liposomes with a minimal set of enzymes. Biochem. Biophys. Res. Commun. 2007, 363, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Caschera, F.; Lee, J.W.; Ho, K.K.; Liu, A.P.; Jewett, M.C. Cell-free compartmentalized protein synthesis inside double emulsion templated liposomes with in vitro synthesized and assembled ribosomes. Chem. Commun. 2016, 52, 5467–5469. [Google Scholar] [CrossRef] [PubMed]
- Salehi, A.S.; Smith, M.T.; Bennett, A.M.; Williams, J.B.; Pitt, W.G.; Bundy, B.C. Cell-free protein synthesis of a cytotoxic cancer therapeutic: Onconase production and a just-add-water cell-free system. Biotechnol. J. 2016, 11, 274–281. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Haney, E.F.; Vogel, H.J. The expanding scope of antimicrobial peptide structures and their modes of action. Trends Biotechnol. 2011, 29, 464–472. [Google Scholar] [CrossRef]
- Martemyanov, K.A.; Shirokov, V.A.; Kurnasov, O.V.; Gudkov, A.T.; Spirin, A.S. Cell-free production of biologically active polypeptides: Application to the synthesis of antibacterial peptide cecropin. Protein Expr. Purif. 2001, 21, 456–461. [Google Scholar] [CrossRef]
- Chen, H.; Xu, Z.; Xu, N.; Cen, P. Efficient production of a soluble fusion protein containing human beta-defensin-2 in E. coli cell-free system. J. Biotechnol. 2005, 115, 307–315. [Google Scholar] [CrossRef]
- Xie, Q.; Matsunaga, S.; Wen, Z.; Niimi, S.; Kumano, M.; Sakakibara, Y.; Machida, S. In vitro system for high-throughput screening of random peptide libraries for antimicrobial peptides that recognize bacterial membranes. J. Pept. Sci. Off. Publ. Eur. Pept. Soc. 2006, 12, 643–652. [Google Scholar] [CrossRef]
- Lundstrom, K. Structural genomics for membrane proteins. Cell. Mol. Life Sci. Cmls 2006, 63, 2597–2607. [Google Scholar] [CrossRef]
- Wagner, S.; Bader, M.L.; Drew, D.; de Gier, J.W. Rationalizing membrane protein overexpression. Trends Biotechnol. 2006, 24, 364–371. [Google Scholar] [CrossRef]
- Kimura-Someya, T.; Hosaka, T.; Shinoda, T.; Shimono, K.; Shirouzu, M.; Yokoyama, S. Cell-Free Synthesis of Membrane Proteins. In Advanced Methods in Structural Biology; Senda, T., Maenaka, K., Eds.; Springer: Tokyo, Japan, 2016; pp. 123–135. [Google Scholar]
- Katzen, F.; Peterson, T.C.; Kudlicki, W. Membrane protein expression: No cells required. Trends Biotechnol. 2009, 27, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Keller, T.; Schwarz, D.; Bernhard, F.; Dotsch, V.; Hunte, C.; Gorboulev, V.; Koepsell, H. Cell free expression and functional reconstitution of eukaryotic drug transporters. Biochemistry 2008, 47, 4552–4564. [Google Scholar] [CrossRef] [PubMed]
- Klammt, C.; Lohr, F.; Schafer, B.; Haase, W.; Dotsch, V.; Ruterjans, H.; Glaubitz, C.; Bernhard, F. High level cell-free expression and specific labeling of integral membrane proteins. Eur. J. Biochem. 2004, 271, 568–580. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, G.; Goto, M.; Saeki, M.; Ito, K.; Hori, T.; Kigawa, T.; Shirouzu, M.; Yokoyama, S. Expression of G protein coupled receptors in a cell-free translational system using detergents and thioredoxin-fusion vectors. Protein Expr. Purif. 2005, 41, 27–37. [Google Scholar] [CrossRef]
- Elbaz, Y.; Steiner-Mordoch, S.; Danieli, T.; Schuldiner, S. In vitro synthesis of fully functional EmrE, a multidrug transporter, and study of its oligomeric state. Proc. Natl. Acad. Sci. USA 2004, 101, 1519–1524. [Google Scholar] [CrossRef]
- Ma, Y.; Munch, D.; Schneider, T.; Sahl, H.G.; Bouhss, A.; Ghoshdastider, U.; Wang, J.; Dotsch, V.; Wang, X.; Bernhard, F. Preparative scale cell-free production and quality optimization of MraY homologues in different expression modes. J. Biol. Chem. 2011, 286, 38844–38853. [Google Scholar] [CrossRef]
- Klammt, C.; Schwarz, D.; Lohr, F.; Schneider, B.; Dotsch, V.; Bernhard, F. Cell-free expression as an emerging technique for the large scale production of integral membrane protein. FEBS J. 2006, 273, 4141–4153. [Google Scholar] [CrossRef]
- Kai, L.; Kaldenhoff, R.; Lian, J.; Zhu, X.; Dotsch, V.; Bernhard, F.; Cen, P.; Xu, Z. Preparative scale production of functional mouse aquaporin 4 using different cell-free expression modes. PLoS ONE 2010, 5, e12972. [Google Scholar] [CrossRef]
- Klammt, C.; Schwarz, D.; Fendler, K.; Haase, W.; Dotsch, V.; Bernhard, F. Evaluation of detergents for the soluble expression of alpha-helical and beta-barrel-type integral membrane proteins by a preparative scale individual cell-free expression system. FEBS J. 2005, 272, 6024–6038. [Google Scholar] [CrossRef]
- Gourdon, P.; Alfredsson, A.; Pedersen, A.; Malmerberg, E.; Nyblom, M.; Widell, M.; Berntsson, R.; Pinhassi, J.; Braiman, M.; Hansson, Ö. Optimized in vitro and in vivo expression of proteorhodopsin: A seven-transmembrane proton pump. Protein Expr. Purif. 2008, 58, 103–113. [Google Scholar] [CrossRef]
- Kaiser, L.; Graveland-Bikker, J.; Steuerwald, D.; Vanberghem, M.; Herlihy, K.; Zhang, S. Efficient cell-free production of olfactory receptors: Detergent optimization, structure, and ligand binding analyses. Proc. Natl. Acad. Sci. USA 2008, 105, 15726–15731. [Google Scholar] [CrossRef] [PubMed]
- Henrich, E.; Hein, C.; Dötsch, V.; Bernhard, F. Membrane protein production in Escherichia coli cell-free lysates. FEBS Lett. 2015, 589, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Lyukmanova, E.; Shenkarev, Z.; Khabibullina, N.; Kopeina, G.; Shulepko, M.; Paramonov, A.; Mineev, K.; Tikhonov, R.; Shingarova, L.; Petrovskaya, L. Lipid–protein nanodiscs for cell-free production of integral membrane proteins in a soluble and folded state: Comparison with detergent micelles, bicelles and liposomes. Biochim. Et Biophys. Acta (BBA)-Biomembr. 2012, 1818, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Junge, F.; Luh, L.M.; Proverbio, D.; Schäfer, B.; Abele, R.; Beyermann, M.; Dötsch, V.; Bernhard, F. Modulation of G-protein coupled receptor sample quality by modified cell-free expression protocols: A case study of the human endothelin A receptor. J. Struct. Biol. 2010, 172, 94–106. [Google Scholar] [CrossRef]
- Berrier, C.; Park, K.; Abes, S.; Bibonne, A.; Betton, J.; Ghazi, A. Cell-free synthesis of a functional ion channel in the absence of a membrane and in the presence of detergent. Biochemistry (NY) 2004, 43, 12585–12591. [Google Scholar] [CrossRef]
- Blesneac, I.; Ravaud, S.; Juillan-Binard, C.; Barret, L.; Zoonens, M.; Polidori, A.; Miroux, B.; Pucci, B.; Pebay-Peyroula, E. Production of UCP1 a membrane protein from the inner mitochondrial membrane using the cell free expression system in the presence of a fluorinated surfactant. Biochim. Et Biophys. Acta (BBA)-Biomembr. 2012, 1818, 798–805. [Google Scholar]
- Park, K.H.; Berrier, C.; Lebaupain, F.; Pucci, B.; Popot, J.L.; Ghazi, A.; Zito, F. Fluorinated and hemifluorinated surfactants as alternatives to detergents for membrane protein cell-free synthesis. Biochem. J. 2007, 403, 183–187. [Google Scholar] [CrossRef]
- Park, K.; Billon-Denis, E.; Dahmane, T.; Lebaupain, F.; Pucci, B.; Breyton, C.; Zito, F. In the cauldron of cell-free synthesis of membrane proteins: Playing with new surfactants. New Biotechnol. 2011, 28, 255–261. [Google Scholar] [CrossRef]
- Bazzacco, P.; Billon-Denis, E.; Sharma, K.S.; Catoire, L.J.; Mary, S.; Le Bon, C.; Point, E.; Banères, J.; Durand, G.; Zito, F. Nonionic homopolymeric amphipols: Application to membrane protein folding, cell-free synthesis, and solution nuclear magnetic resonance. Biochemistry (NY) 2012, 51, 1416–1430. [Google Scholar] [CrossRef]
- Wang, X.; Corin, K.; Baaske, P.; Wienken, C.J.; Jerabek-Willemsen, M.; Duhr, S.; Braun, D.; Zhang, S. Peptide surfactants for cell-free production of functional G protein-coupled receptors. Proc. Natl. Acad. Sci. USA 2011, 108, 9049–9054. [Google Scholar] [CrossRef]
- Corin, K.; Baaske, P.; Ravel, D.B.; Song, J.; Brown, E.; Wang, X.; Wienken, C.J.; Jerabek-Willemsen, M.; Duhr, S.; Luo, Y. Designer lipid-like peptides: A class of detergents for studying functional olfactory receptors using commercial cell-free systems. PLoS ONE 2011, 6, e25067. [Google Scholar] [CrossRef]
- Sachse, R.; Dondapati, S.K.; Fenz, S.F.; Schmidt, T.; Kubick, S. Membrane protein synthesis in cell-free systems: From bio-mimetic systems to bio-membranes. FEBS Lett. 2014, 588, 2774–2781. [Google Scholar] [CrossRef] [PubMed]
- Shimono, K.; Goto, M.; Kikukawa, T.; Miyauchi, S.; Shirouzu, M.; Kamo, N.; Yokoyama, S. Production of functional bacteriorhodopsin by an Escherichia coli cell-free protein synthesis system supplemented with steroid detergent and lipid. Protein Sci. 2009, 18, 2160–2171. [Google Scholar] [CrossRef] [PubMed]
- Uhlemann, E.E.; Pierson, H.E.; Fillingame, R.H.; Dmitriev, O.Y. Cell-free synthesis of membrane subunits of ATP synthase in phospholipid bicelles: NMR shows subunit a fold similar to the protein in the cell membrane. Protein Sci. 2012, 21, 279–288. [Google Scholar] [CrossRef]
- Kalmbach, R.; Chizhov, I.; Schumacher, M.C.; Friedrich, T.; Bamberg, E.; Engelhard, M. Functional cell-free synthesis of a seven helix membrane protein: In situ insertion of bacteriorhodopsin into liposomes. J. Mol. Biol. 2007, 371, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Ando, M.; Akiyama, M.; Okuno, D.; Hirano, M.; Ide, T.; Sawada, S.; Sasaki, Y.; Akiyoshi, K. Liposome chaperon in cell-free membrane protein synthesis: One-step preparation of KcsA-integrated liposomes and electrophysiological analysis by the planar bilayer method. Biomater. Sci. 2016, 4, 258–264. [Google Scholar] [CrossRef]
- Kuruma, Y.; Stano, P.; Ueda, T.; Luisi, P.L. A synthetic biology approach to the construction of membrane proteins in semi-synthetic minimal cells. Biochim. Et Biophys. Acta (BBA)-Biomembr. 2009, 1788, 567–574. [Google Scholar] [CrossRef]
- Komar, J.; Alvira, S.; Schulze, R.J.; Martin, R.; Lycklama a Nijeholt, J.A.; Jelger, A.; Lee, S.C.; Dafforn, T.R.; Deckers-Hebestreit, G.; Berger, I.; et al. Membrane protein insertion and assembly by the bacterial holo-translocon SecYEG–SecDF–YajC–YidC. Biochem. J. 2016, 473, 3341–3354. [Google Scholar] [CrossRef]
- Kuruma, Y.; Nishiyama, K.; Shimizu, Y.; Müller, M.; Ueda, T. Development of a minimal cell-free translation system for the synthesis of presecretory and integral membrane proteins. Biotechnol. Prog. 2005, 21, 1243–1251. [Google Scholar] [CrossRef]
- Wuu, J.J.; Swartz, J.R. High yield cell-free production of integral membrane proteins without refolding or detergents. Biochim. Et Biophys. Acta (BBA)-Biomembr. 2008, 1778, 1237–1250. [Google Scholar] [CrossRef]
- Matsubayashi, H.; Kuruma, Y.; Ueda, T. Cell-free synthesis of SecYEG translocon as the fundamental protein transport machinery. Orig. Life Evol. Biosph. 2014, 44, 331–334. [Google Scholar] [CrossRef]
- Nath, A.; Atkins, W.M.; Sligar, S.G. Applications of phospholipid bilayer nanodiscs in the study of membranes and membrane proteins. Biochemistry (NY) 2007, 46, 2059–2069. [Google Scholar] [CrossRef] [PubMed]
- Chromy, B.A.; Arroyo, E.; Blanchette, C.D.; Bench, G.; Benner, H.; Cappuccio, J.A.; Coleman, M.A.; Henderson, P.T.; Hinz, A.K.; Kuhn, E.A. Different apolipoproteins impact nanolipoprotein particle formation. J. Am. Chem. Soc. 2007, 129, 14348–14354. [Google Scholar] [CrossRef] [PubMed]
- Katzen, F.; Fletcher, J.E.; Yang, J.; Kang, D.; Peterson, T.C.; Cappuccio, J.A.; Blanchette, C.D.; Sulchek, T.; Chromy, B.A.; Hoeprich, P.D. Insertion of membrane proteins into discoidal membranes using a cell-free protein expression approach. J. Proteome Res. 2008, 7, 3535–3542. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Cirico, T.; Katzen, F.; Peterson, T.C.; Kudlicki, W. Cell-free synthesis of a functional G protein-coupled receptor complexed with nanometer scale bilayer discs. BMC Biotechnol. 2011, 11, 57. [Google Scholar] [CrossRef]
- Cappuccio, J.A.; Blanchette, C.D.; Sulchek, T.A.; Arroyo, E.S.; Kralj, J.M.; Hinz, A.K.; Kuhn, E.A.; Chromy, B.A.; Segelke, B.W.; Rothschild, K.J.; et al. Cell-free co-expression of functional membrane proteins and apolipoprotein, forming soluble nanolipoprotein particles. Mol. Cell Proteom. 2008, 7, 2246–2253. [Google Scholar] [CrossRef] [PubMed]
- Shilling, P.J.; Bumbak, F.; Scott, D.J.; Bathgate, R.A.; Gooley, P.R. Characterisation of a cell-free synthesised G-protein coupled receptor. Sci. Rep. 2017, 7, 1094. [Google Scholar] [CrossRef]
- Krishnan, M.; de Leeuw, T.J.J.F.; Pandit, A. Cell-free soluble expression of the membrane protein PsbS. bioRxiv 2018, 456087. [Google Scholar] [CrossRef]
- Prinz, W.A.; Aslund, F.; Holmgren, A.; Beckwith, J. The role of the thioredoxin and glutaredoxin pathways in reducing protein disulfide bonds in the Escherichia coli cytoplasm. J. Biol. Chem. 1997, 272, 15661–15667. [Google Scholar] [CrossRef]
- Kadokura, H.; Tian, H.; Zander, T.; Bardwell, J.C.; Beckwith, J. Snapshots of DsbA in action: Detection of proteins in the process of oxidative folding. Science 2004, 303, 534–537. [Google Scholar] [CrossRef]
- Shevchik, V.E.; Condemine, G.; Robert-Baudouy, J. Characterization of DsbC, a periplasmic protein of Erwinia chrysanthemi and Escherichia coli with disulfide isomerase activity. EMBO J. 1994, 13, 2007–2012. [Google Scholar] [CrossRef]
- Frand, A.R.; Cuozzo, J.W.; Kaiser, C.A. Pathways for protein disulphide bond formation. Trends Cell Biol. 2000, 10, 203–210. [Google Scholar] [CrossRef]
- Yin, G.; Swartz, J.R. Enhancing multiple disulfide bonded protein folding in a cell-free system. Biotechnol. Bioeng. 2004, 86, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Jelenc, P.C.; Kurland, C.G. Nucleoside triphosphate regeneration decreases the frequency of translation errors. Proc. Natl. Acad. Sci. USA 1979, 76, 3174–3178. [Google Scholar] [CrossRef] [PubMed]
- Goerke, A.R.; Swartz, J.R. Development of cell-free protein synthesis platforms for disulfide bonded proteins. Biotechnol. Bioeng. 2008, 99, 351–367. [Google Scholar] [CrossRef]
- Ryabova, L.A.; Desplancq, D.; Spirin, A.S.; Pluckthun, A. Functional antibody production using cell-free translation: Effects of protein disulfide isomerase and chaperones. Nat. Biotechnol. 1997, 15, 79–84. [Google Scholar] [CrossRef]
- Jiang, X.; Ookubo, Y.; Fujii, I.; Nakano, H.; Yamane, T. Expression of Fab fragment of catalytic antibody 6D9 in an Escherichia coli in vitro coupled transcription/translation system. FEBS Lett. 2002, 514, 290–294. [Google Scholar] [CrossRef]
- Goerke, A.R.; Swartz, J.R. High-level cell-free synthesis yields of proteins containing site-specific non-natural amino acids. Biotechnol. Bioeng. 2009, 102, 400–416. [Google Scholar] [CrossRef]
- Georgiou, G.; Valax, P. Expression of correctly folded proteins in Escherichia coli. Curr. Opin. Biotechnol. 1996, 7, 190–197. [Google Scholar] [CrossRef]
- Vabulas, R.M.; Raychaudhuri, S.; Hayer-Hartl, M.; Hartl, F.U. Protein folding in the cytoplasm and the heat shock response. Cold Spring Harb Perspect. Biol. 2010, 2, a004390. [Google Scholar] [CrossRef]
- Entzminger, K.C.; Chang, C.; Myhre, R.O.; McCallum, K.C.; Maynard, J.A. The Skp chaperone helps fold soluble proteins in vitro by inhibiting aggregation. Biochemistry (NY) 2012, 51, 4822–4834. [Google Scholar] [CrossRef]
- Murakami, S.; Matsumoto, R.; Kanamori, T. Constructive approach for synthesis of a functional IgG using a reconstituted cell-free protein synthesis system. Sci. Rep. 2019, 9, 671. [Google Scholar] [CrossRef] [PubMed]
- Bundy, B.C.; Swartz, J.R. Efficient disulfide bond formation in virus-like particles. J. Biotechnol. 2011, 154, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Bundy, B.C.; Franciszkowicz, M.J.; Swartz, J.R. Escherichia coli-based cell-free synthesis of virus-like particles. Biotechnol. Bioeng. 2008, 100, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Schultz, P.G. A chemical toolkit for proteins--an expanded genetic code. Nat. Rev. Mol. Cell Biol. 2006, 7, 775–782. [Google Scholar] [CrossRef]
- Park, H.S.; Hohn, M.J.; Umehara, T.; Guo, L.T.; Osborne, E.M.; Benner, J.; Noren, C.J.; Rinehart, J.; Soll, D. Expanding the genetic code of Escherichia coli with phosphoserine. Science 2011, 333, 1151–1154. [Google Scholar] [CrossRef]
- Liu, C.C.; Schultz, P.G. Adding new chemistries to the genetic code. Annu. Rev. Biochem. 2010, 79, 413–444. [Google Scholar] [CrossRef]
- Hohsaka, T.; Sato, K.; Sisido, M.; Takai, K.; Yokoyama, S. Site-specific incorporation of photofunctional nonnatural amino acids into a polypeptide through in vitro protein biosynthesis. FEBS Lett. 1994, 344, 171–174. [Google Scholar] [CrossRef]
- Mukai, T.; Yamaguchi, A.; Ohtake, K.; Takahashi, M.; Hayashi, A.; Iraha, F.; Kira, S.; Yanagisawa, T.; Yokoyama, S.; Hoshi, H.; et al. Reassignment of a rare sense codon to a non-canonical amino acid in Escherichia coli. NAR 2015, 43, 8111–8122. [Google Scholar] [CrossRef]
- Lee, B.S.; Shin, S.; Jeon, J.Y.; Jang, K.; Lee, B.Y.; Choi, S.; Yoo, T.H. Incorporation of Unnatural Amino Acids in Response to the AGG Codon. ACS Chem. Biol. 2015, 10, 1648–1653. [Google Scholar] [CrossRef]
- Hohsaka, T.; Ashizuka, Y.; Murakami, H.; Sisido, M. Five-base codons for incorporation of nonnatural amino acids into proteins. Nucleic Acids Res. 2001, 29, 3646–3651. [Google Scholar] [CrossRef]
- Hohsaka, T.; Ashizuka, Y.; Murakami, H.; Sisido, M. Incorporation of Nonnatural Amino Acids into Streptavidin through In Vitro Frame-Shift Suppression. J. Am. Chem. Soc. 1996, 118, 9778–9779. [Google Scholar] [CrossRef]
- Sisido, M.; Hohsaka, T. Introduction of specialty functions by the position-specific incorporation of nonnatural amino acids into proteins through four-base codon/anticodon pairs. Appl. Microbiol. Biotechnol. 2001, 57, 274–281. [Google Scholar] [PubMed]
- Blattner, F.R.; Plunkett, G., 3rd; Bloch, C.A.; Perna, N.T.; Burland, V.; Riley, M.; Collado-Vides, J.; Glasner, J.D.; Rode, C.K.; Mayhew, G.F.; et al. The complete genome sequence of Escherichia coli K-12. Science 1997, 277, 1453–1462. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Morooka, N.; Yamamoto, Y.; Fujita, K.; Isono, K.; Choi, S.; Ohtsubo, E.; Baba, T.; Wanner, B.L.; Mori, H.; et al. Highly accurate genome sequences of Escherichia coli K-12 strains MG1655 and W3110. Mol. Syst. Biol. 2006, 2, 2006.0007. [Google Scholar] [CrossRef]
- Wang, L.; Magliery, T.J.; Liu, D.R.; Schultz, P.G. A New Functional Suppressor tRNA/Aminoacyl-tRNA Synthetase Pair for the in Vivo Incorporation of Unnatural Amino Acids into Proteins. J. Am. Chem. Soc. 2000, 122, 5010–5011. [Google Scholar] [CrossRef]
- Blight, S.K.; Larue, R.C.; Mahapatra, A.; Longstaff, D.G.; Chang, E.; Zhao, G.; Kang, P.T.; Green-Church, K.B.; Chan, M.K.; Krzycki, J.A. Direct charging of tRNA(CUA) with pyrrolysine in vitro and in vivo. Nature 2004, 431, 333–335. [Google Scholar] [CrossRef]
- Neumann, H.; Wang, K.; Davis, L.; Garcia-Alai, M.; Chin, J.W. Encoding multiple unnatural amino acids via evolution of a quadruplet-decoding ribosome. Nature 2010, 464, 441–444. [Google Scholar] [CrossRef]
- Neumann, H.; Peak-Chew, S.Y.; Chin, J.W. Genetically encoding N(epsilon)-acetyllysine in recombinant proteins. Nat. Chem. Biol. 2008, 4, 232–234. [Google Scholar] [CrossRef]
- Smolskaya, S.; Andreev, Y.A. Site-Specific Incorporation of Unnatural Amino Acids into Escherichia coli Recombinant Protein: Methodology Development and Recent Achievement. Biomolecules 2019, 9, 255. [Google Scholar] [CrossRef]
- Wang, L.; Xie, J.; Schultz, P.G. Expanding the genetic code. Annu. Rev. Biophys. Biomol. Struct. 2006, 35, 225–249. [Google Scholar] [CrossRef]
- Wang, L.; Schultz, P.G. A general approach for the generation of orthogonal tRNAs. Chem. Biol. 2001, 8, 883–890. [Google Scholar] [CrossRef]
- Noren, C.; Anthony-Cahill, S.; Griffith, M.; Schultz, P. A general method for site-specific incorporation of unnatural amino acids into proteins. Science 1989, 244, 182–188. [Google Scholar] [CrossRef]
- Murakami, H.; Ohta, A.; Ashigai, H.; Suga, H. A highly flexible tRNA acylation method for non-natural polypeptide synthesis. Nat. Methods 2006, 3, 357–359. [Google Scholar] [CrossRef]
- Ohuchi, M.; Murakami, H.; Suga, H. The flexizyme system: A highly flexible tRNA aminoacylation tool for the translation apparatus. Curr. Opin. Chem. Biol. 2007, 11, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Rackham, O.; Chin, J.W. A network of orthogonal ribosome x mRNA pairs. Nat. Chem. Biol. 2005, 1, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Neumann, H.; Peak-Chew, S.Y.; Chin, J.W. Evolved orthogonal ribosomes enhance the efficiency of synthetic genetic code expansion. Nat. Biotechnol. 2007, 25, 770–777. [Google Scholar] [CrossRef] [PubMed]
- Sando, S.; Ogawa, A.; Nishi, T.; Hayami, M.; Aoyama, Y. In vitro selection of RNA aptamer against Escherichia coli release factor 1. Bioorg. Med. Chem. Lett. 2007, 17, 1216–1220. [Google Scholar] [CrossRef] [PubMed]
- Agafonov, D.E.; Huang, Y.; Grote, M.; Sprinzl, M. Efficient suppression of the amber codon in E. coli in vitro translation system. FEBS Lett. 2005, 579, 2156–2160. [Google Scholar] [CrossRef] [PubMed]
- Loscha, K.V.; Herlt, A.J.; Qi, R.; Huber, T.; Ozawa, K.; Otting, G. Multiple-site labeling of proteins with unnatural amino acids. Angew. Chem. Int. Ed. Engl. 2012, 51, 2243–2246. [Google Scholar] [CrossRef]
- Mukai, T.; Hoshi, H.; Ohtake, K.; Takahashi, M.; Yamaguchi, A.; Hayashi, A.; Yokoyama, S.; Sakamoto, K. Highly reproductive Escherichia coli cells with no specific assignment to the UAG codon. Sci. Rep. 2015, 5, 9699. [Google Scholar] [CrossRef]
- Lajoie, M.J.; Rovner, A.J.; Goodman, D.B.; Aerni, H.R.; Haimovich, A.D.; Kuznetsov, G.; Mercer, J.A.; Wang, H.H.; Carr, P.A.; Mosberg, J.A.; et al. Genomically recoded organisms expand biological functions. Science 2013, 342, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.H.; Kwon, Y.; Martin, R.W.; Des Soye, B.J.; De Paz, A.M.; Swonger, K.N.; Ntai, I.; Kelleher, N.L.; Jewett, M.C. Improving cell-free protein synthesis through genome engineering of Escherichia coli lacking release factor 1. Chembiochem 2015, 16, 844–853. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.W.; Des Soye, B.J.; Kwon, Y.; Kay, J.; Davis, R.G.; Thomas, P.M.; Majewska, N.I.; Chen, C.X.; Marcum, R.D.; Weiss, M.G. Cell-free protein synthesis from genomically recoded bacteria enables multisite incorporation of noncanonical amino acids. Nat. Commun. 2018, 9, 1203. [Google Scholar] [CrossRef]
- Adachi, J.; Katsura, K.; Seki, E.; Takemoto, C.; Shirouzu, M.; Terada, T.; Mukai, T.; Sakamoto, K.; Yokoyama, S. Cell-free protein synthesis using S30 extracts from Escherichia coli RFzero strains for efficient incorporation of non-natural amino acids into proteins. Int. J. Mol. Sci. 2019, 20, 492. [Google Scholar] [CrossRef] [PubMed]
- Young, T.S.; Ahmad, I.; Yin, J.A.; Schultz, P.G. An enhanced system for unnatural amino acid mutagenesis in E. coli. J. Mol. Biol. 2010, 395, 361–374. [Google Scholar] [CrossRef]
- Ryu, Y.; Schultz, P.G. Efficient incorporation of unnatural amino acids into proteins in Escherichia coli. Nat. Methods 2006, 3, 263–265. [Google Scholar] [CrossRef]
- Bryson, D.I.; Fan, C.; Guo, L.; Miller, C.; Soll, D.; Liu, D.R. Continuous directed evolution of aminoacyl-tRNA synthetases. Nat. Chem. Biol. 2017, 13, 1253. [Google Scholar] [CrossRef]
- Amiram, M.; Haimovich, A.D.; Fan, C.; Wang, Y.S.; Aerni, H.R.; Ntai, I.; Moonan, D.W.; Ma, N.J.; Rovner, A.J.; Hong, S.H.; et al. Evolution of translation machinery in recoded bacteria enables multi-site incorporation of nonstandard amino acids. Nat. Biotechnol. 2015, 33, 1272–1279. [Google Scholar] [CrossRef]
- Wang, N.; Ju, T.; Niu, W.; Guo, J. Fine-tuning Interaction between Aminoacyl-tRNA Synthetase and tRNA for Efficient Synthesis of Proteins Containing Unnatural Amino Acids. ACS Synth. Biol. 2014. [Google Scholar] [CrossRef]
- Guo, J.; Melancon, C.E., 3rd; Lee, H.S.; Groff, D.; Schultz, P.G. Evolution of amber suppressor tRNAs for efficient bacterial production of proteins containing nonnatural amino acids. Angew. Chem. Int. Ed. Engl. 2009, 48, 9148–9151. [Google Scholar] [CrossRef]
- Fan, C.; Ip, K.; Soll, D. Expanding the genetic code of Escherichia coli with phosphotyrosine. FEBS Lett. 2016, 590, 3040–3047. [Google Scholar] [CrossRef] [PubMed]
- Smolskaya, S.; Zhang, Z.J.; Alfonta, L. Enhanced yield of recombinant proteins with site-specifically incorporated unnatural amino acids using a cell-free expression system. PLoS ONE 2013, 8, e68363. [Google Scholar] [CrossRef] [PubMed]
- Albayrak, C.; Swartz, J.R. Cell-free co-production of an orthogonal transfer RNA activates efficient site-specific non-natural amino acid incorporation. Nucleic Acids Res. 2013, 41, 5949–5963. [Google Scholar] [CrossRef] [PubMed]
- Ozer, E.; Chemla, Y.; Schlesinger, O.; Aviram, H.Y.; Riven, I.; Haran, G.; Alfonta, L. In vitro suppression of two different stop codons. Biotechnol. Bioeng. 2017, 114, 1065–1073. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuki, T.; Manabe, T.; Sisido, M. Multiple incorporation of non-natural amino acids into a single protein using tRNAs with non-standard structures. FEBS Lett. 2005, 579, 6769–6774. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Wu, Y.; Mureev, S.; Alexandrov, K. Oligonucleotide-mediated tRNA sequestration enables one-pot sense codon reassignment in vitro. Nucleic Acids Res. 2018, 46, 6387–6400. [Google Scholar] [CrossRef] [PubMed]
- Worst, E.G.; Exner, M.P.; De Simone, A.; Schenkelberger, M.; Noireaux, V.; Budisa, N.; Ott, A. Cell-free expression with the toxic amino acid canavanine. Bioorg. Med. Chem. Lett. 2015, 25, 3658–3660. [Google Scholar] [CrossRef]
- Bundy, B.C.; Swartz, J.R. Site-specific incorporation of p-propargyloxyphenylalanine in a cell-free environment for direct protein− protein click conjugation. Bioconjug. Chem. 2010, 21, 255–263. [Google Scholar] [CrossRef]
- Zemella, A.; Thoring, L.; Hoffmeister, C.; Kubick, S. Cell-Free Protein Synthesis: Pros and Cons of Prokaryotic and Eukaryotic Systems. Chembiochem 2015, 16, 2420–2431. [Google Scholar] [CrossRef]
- Lis, H.; Sharon, N. Protein glycosylation. Eur. J. Biochem. 1993, 218, 1–27. [Google Scholar] [CrossRef]
- Schwarz, F.; Aebi, M. Mechanisms and principles of N-linked protein glycosylation. Curr. Opin. Struct. Biol. 2011, 21, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Wacker, M.; Linton, D.; Hitchen, P.G.; Nita-Lazar, M.; Haslam, S.M.; North, S.J.; Panico, M.; Morris, H.R.; Dell, A.; Wren, B.W.; et al. N-Linked Glycosylation in Campylobacter jejuni and Its Functional Transfer into E. coli. Science 2002, 298, 1790–1793. [Google Scholar] [CrossRef] [PubMed]
- Guarino, C.; DeLisa, M.P. A prokaryote-based cell-free translation system that efficiently synthesizes glycoproteins. Glycobiology 2012, 22, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Jaroentomeechai, T.; Stark, J.C.; Natarajan, A.; Glasscock, C.J.; Yates, L.E.; Hsu, K.J.; Mrksich, M.; Jewett, M.C.; DeLisa, M.P. Single-pot glycoprotein biosynthesis using a cell-free transcription-translation system enriched with glycosylation machinery. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Murata, T.; Shinozuka, Y.; Obata, Y.; Yokoyama, K.K. Phosphorylation of two eukaryotic transcription factors, Jun dimerization protein 2 and activation transcription factor 2, in Escherichia coli by Jun N-terminal kinase 1. Anal. Biochem. 2008, 376, 115–121. [Google Scholar] [CrossRef]
- O’Brien, S.P.; DeLisa, M.P. Functional reconstitution of a tunable E3-dependent sumoylation pathway in Escherichia coli. PLoS ONE 2012, 7, e38671. [Google Scholar] [CrossRef]
- Magnani, R.; Chaffin, B.; Dick, E.; Bricken, M.L.; Houtz, R.L.; Bradley, L.H. Utilization of a calmodulin lysine methyltransferase co-expression system for the generation of a combinatorial library of post-translationally modified proteins. Protein Expr. Purif. 2012, 86, 83–88. [Google Scholar] [CrossRef]
- Zawada, J.F.; Yin, G.; Steiner, A.R.; Yang, J.; Naresh, A.; Roy, S.M.; Gold, D.S.; Heinsohn, H.G.; Murray, C.J. Microscale to manufacturing scale-up of cell-free cytokine production—A new approach for shortening protein production development timelines. Biotechnol. Bioeng. 2011, 108, 1570–1578. [Google Scholar] [CrossRef]
- Tinafar, A.; Jaenes, K.; Pardee, K. Synthetic Biology Goes Cell-Free. BMC Biol. 2019, 17, 64. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smolskaya, S.; Logashina, Y.A.; Andreev, Y.A. Escherichia coli Extract-Based Cell-Free Expression System as an Alternative for Difficult-to-Obtain Protein Biosynthesis. Int. J. Mol. Sci. 2020, 21, 928. https://doi.org/10.3390/ijms21030928
Smolskaya S, Logashina YA, Andreev YA. Escherichia coli Extract-Based Cell-Free Expression System as an Alternative for Difficult-to-Obtain Protein Biosynthesis. International Journal of Molecular Sciences. 2020; 21(3):928. https://doi.org/10.3390/ijms21030928
Chicago/Turabian StyleSmolskaya, Sviatlana, Yulia A. Logashina, and Yaroslav A. Andreev. 2020. "Escherichia coli Extract-Based Cell-Free Expression System as an Alternative for Difficult-to-Obtain Protein Biosynthesis" International Journal of Molecular Sciences 21, no. 3: 928. https://doi.org/10.3390/ijms21030928
APA StyleSmolskaya, S., Logashina, Y. A., & Andreev, Y. A. (2020). Escherichia coli Extract-Based Cell-Free Expression System as an Alternative for Difficult-to-Obtain Protein Biosynthesis. International Journal of Molecular Sciences, 21(3), 928. https://doi.org/10.3390/ijms21030928