Lipidated Analogs of the LL-37-Derived Peptide Fragment KR12—Structural Analysis, Surface-Active Properties and Antimicrobial Activity

 , , ,
, , , 
Abstract
1. Introduction
2. Results and Discussion
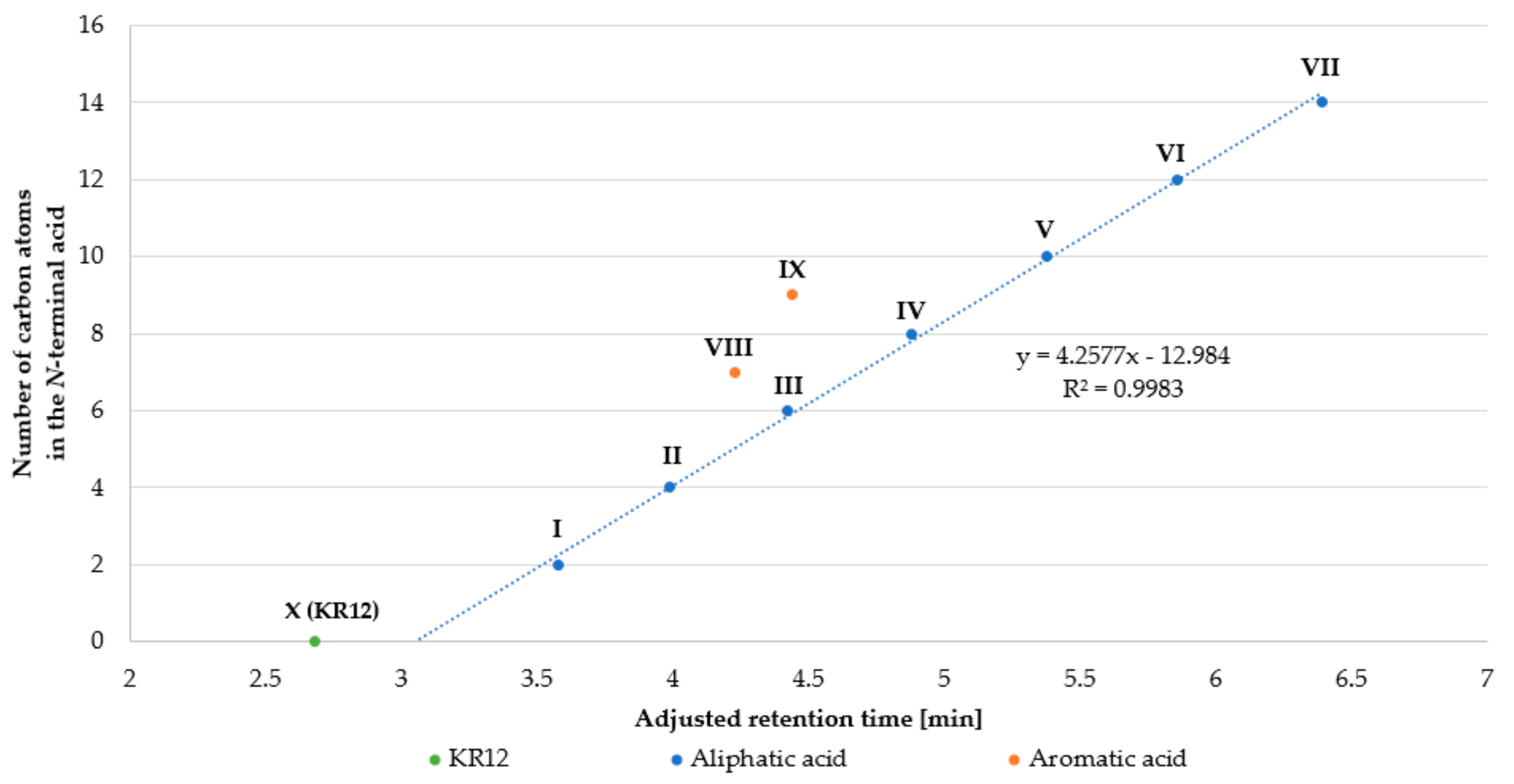
2.1. Peptide Synthesis and Purification
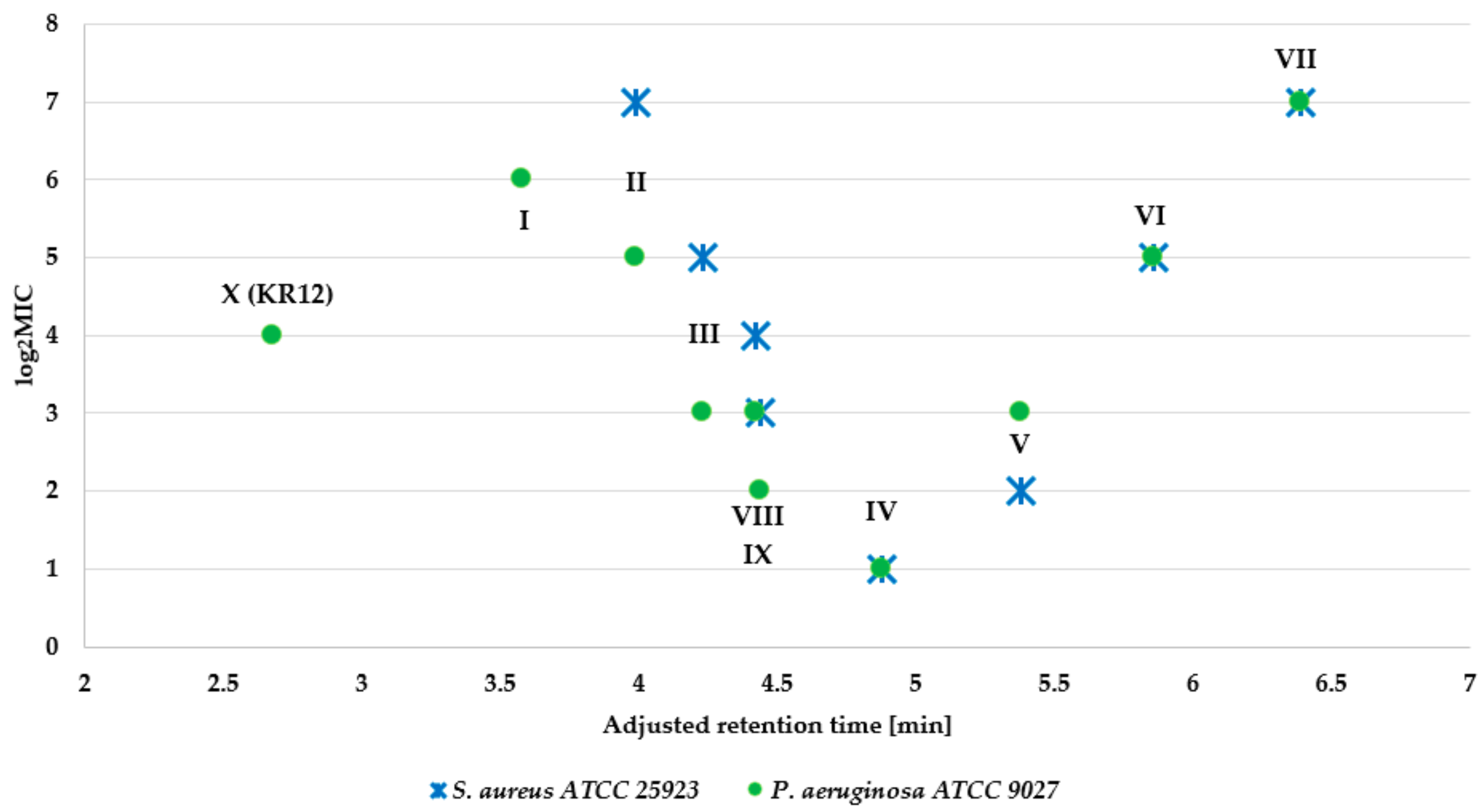
2.2. Antimicrobial Assay
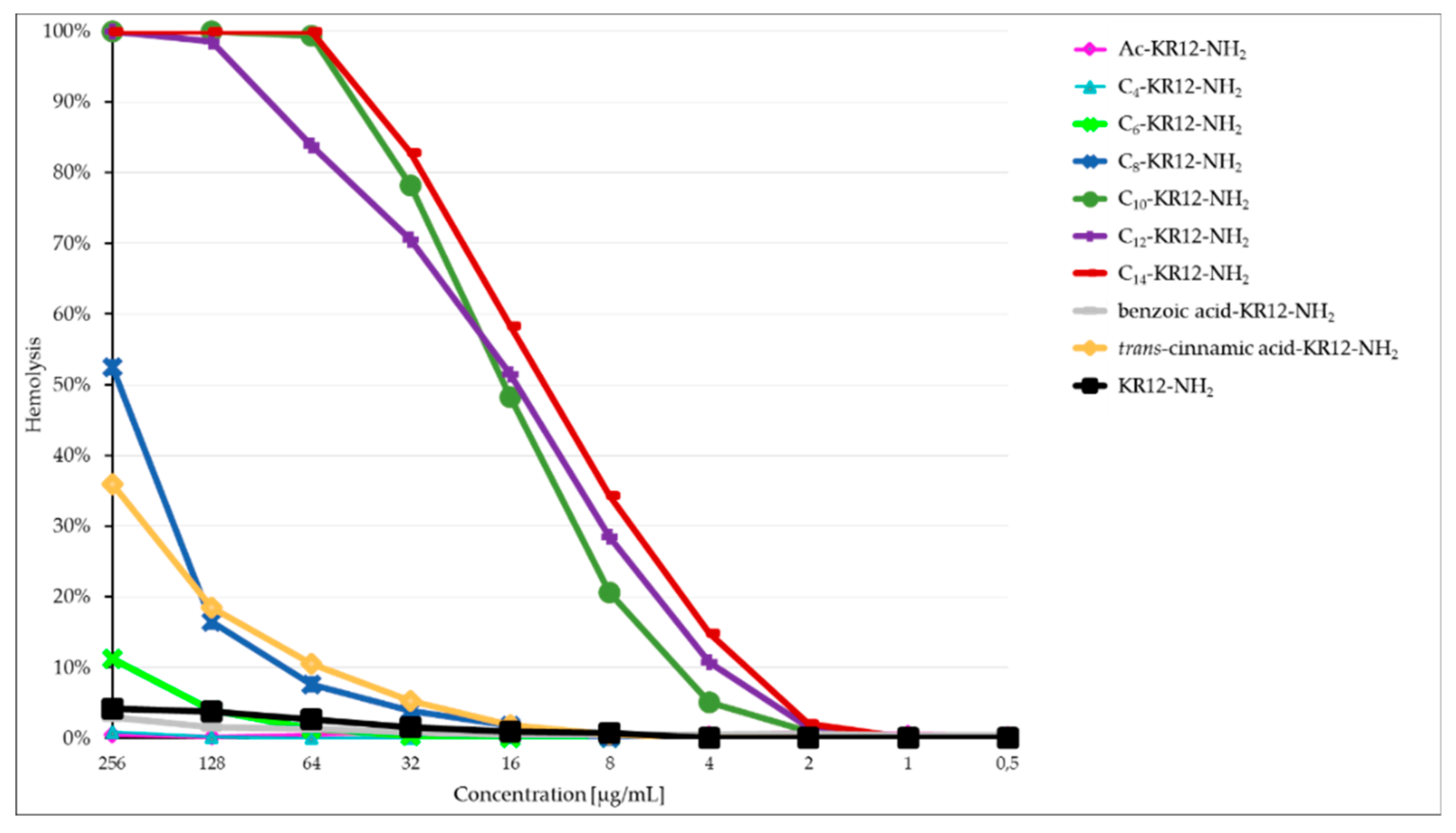
2.3. Hemolysis Assay
2.4. MTT Assay
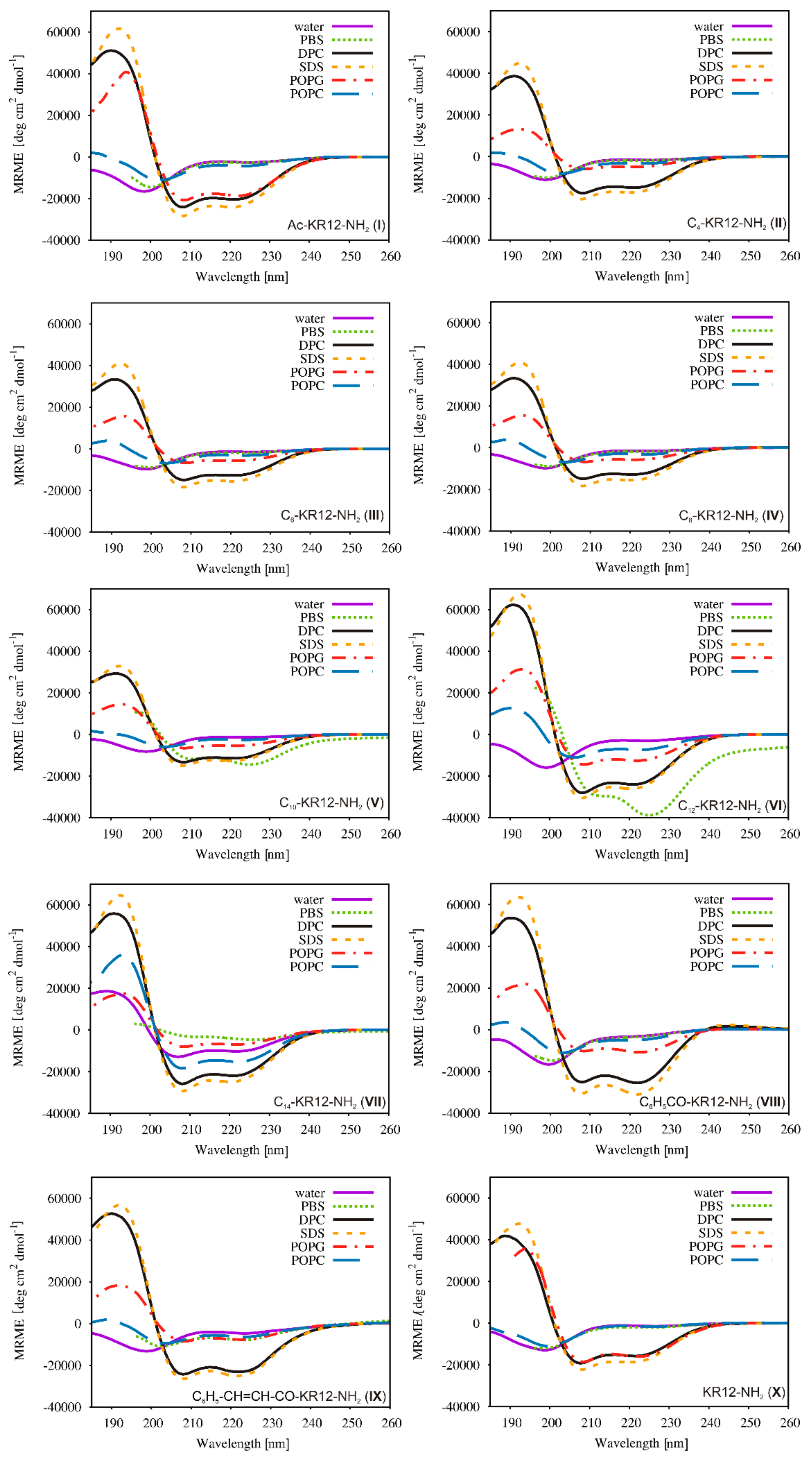
2.5. Conformational Studies
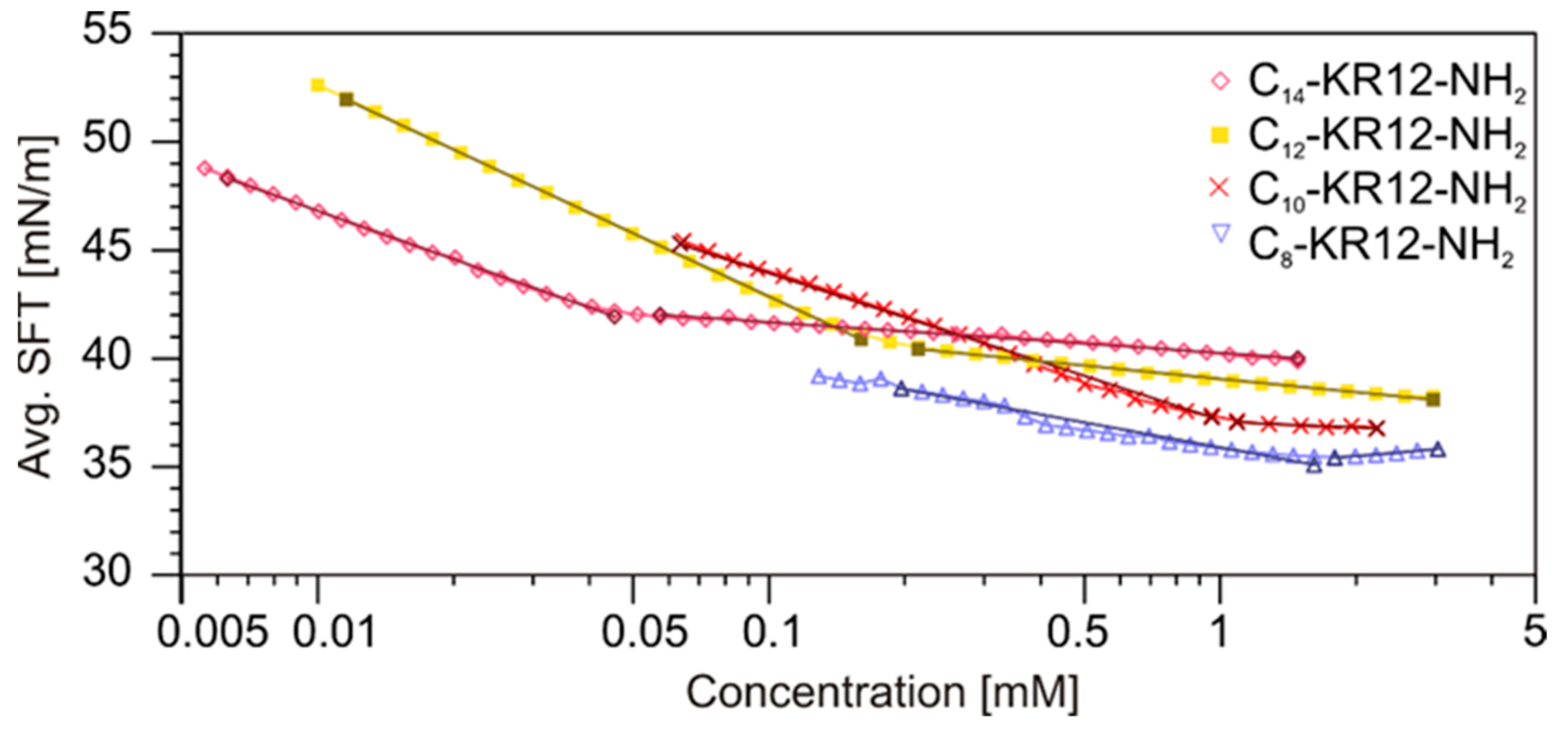
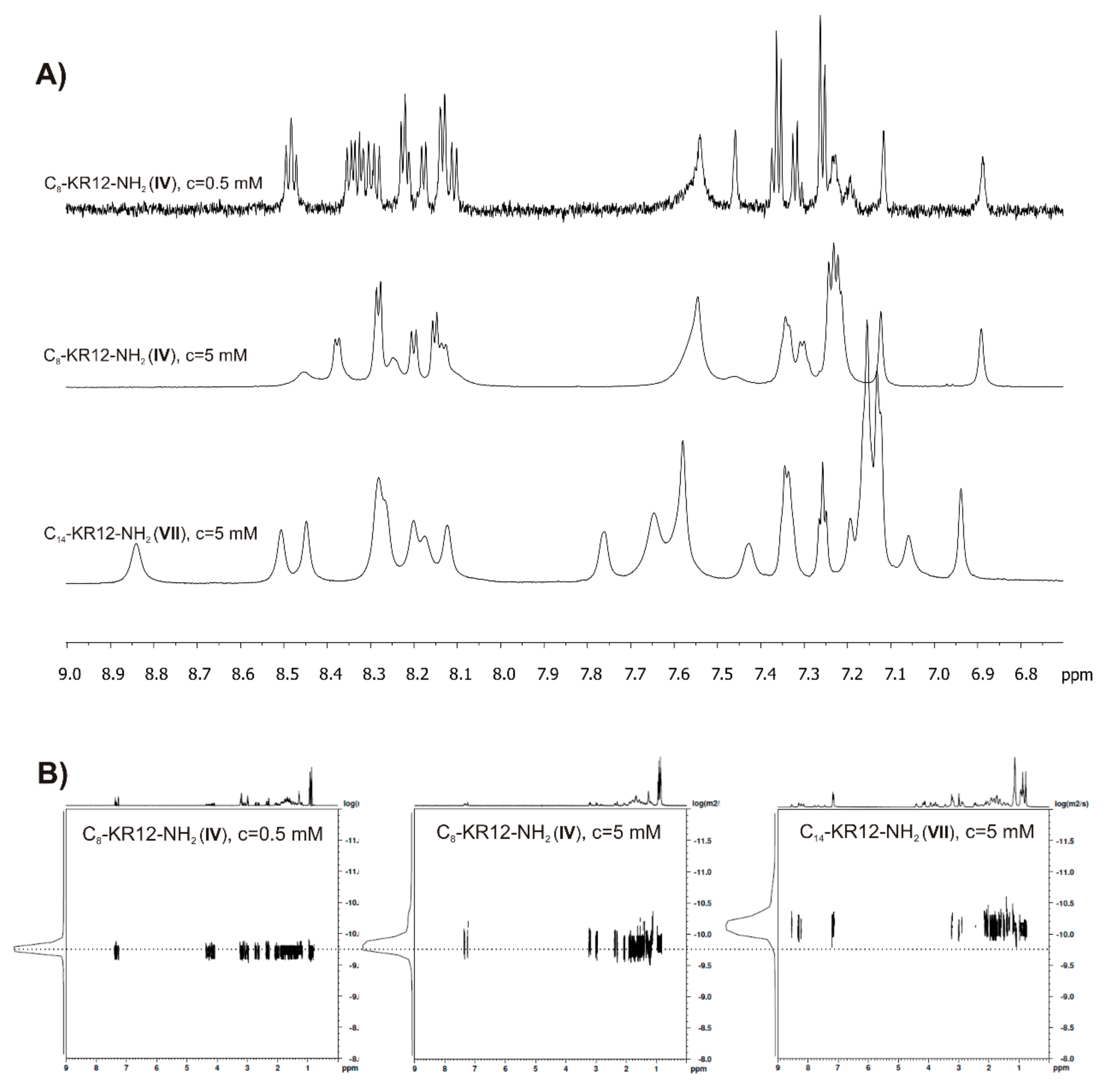
2.6. Self-Assembly Studies
3. Materials and Methods
3.1. Peptide Synthesis
3.2. Antimicrobial Assays
3.2.1. Microbial Strains and Antimicrobial Assay
3.2.2. Activity Against Staphylococcal Biofilm
3.3. The Hemolysis Assay
3.4. MTT Assay
3.5. CD Measurements
3.6. Surface Tension Measurements
3.7. NMR Measurements
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AMPs | antimicrobial peptides |
| Ac | acetyl group |
| Boc | tert-butyloxycarbonyl |
| C4 | butanoic acid residue |
| C6 | hexanoic acid residue |
| C8 | octanoic acid residue |
| C10 | decanoic acid residue |
| C12 | dodecanoic acid residue |
| C14 | tetradecanoic acid residue |
| CAC | critical aggregation concentration |
| CD | circular dichroism |
| CFU | colony forming unit |
| DCM | dichloromethane |
| DHPC | l,2-diheptanoyl-sn-glycero-3-phosphocholine |
| DIC | N,N′-diisopropylcarbodiimide |
| DMF | N,N-dimethylformamide |
| DMSO | dimethyl sulfoxide |
| DOSY | Diffusion-Ordered Spectroscopy |
| DPC | dodecylphosphocholine |
| EDTA | ethylenediaminetetraacetic acid |
| ESI MS | electrospray ionization mass spectrometry |
| ESKAPE bacteria | Enterococcus faecium, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, Klebsiella aerogenes, Staphylococcus aureus |
| Fmoc | 9-fluorenylmethoxycarbonyl |
| GM | geometric mean |
| HAIs | hospital-acquired infections |
| hRBCs | human red blood cells |
| IC50 | half maximal inhibitory concentration |
| MBEC | minimal biofilm eradication concentration |
| MDR | multidrug-resistant |
| MHC | minimal hemolytic concentration |
| MIC | minimal inhibitory concentration |
| MRSA | methicillin-resistant S. aureus |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| Pbf | 2,2,4,6,7-pentamethyl-dihydrobenzofuran-5-sulfonyl residue |
| PBS | phosphate buffered saline |
| POPC | 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine |
| POPG | 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol |
| RP-HPLC | reversed-phase high-performance liquid chromatography |
| SDS | sodium dodecyl sulfate |
| SI | selectivity index |
| SPPS | solid-phase peptide synthesis |
| TFA | trifluoroacetic acid |
| TIS | triisopropylsilane |
| WHO | World Health Organization |
References
- Mulani, M.S.; Kamble, E.E.; Kumkar, S.N.; Tawre, M.S.; Pardesi, K.R. Emerging Strategies to Combat ESKAPE Pathogens in the Era of Antimicrobial Resistance: A Review. Front. Microbiol. 2019, 10, 539. [Google Scholar] [CrossRef]
- Founou, R.C.; Founou, L.L.; Essack, S.Y. Clinical and economic impact of antibiotic resistance in developing countries: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0189621. [Google Scholar] [CrossRef] [PubMed]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef]
- Messina, G.; Ceriale, E.; Lenzi, D.; Burgassi, S.; Azzolini, E.; Manzi, P. Environmental contaminants in hospital settings and progress in disinfecting techniques. Biomed. Res. Int 2013, 2013, 429780. [Google Scholar] [CrossRef] [PubMed]
- Di Ruscio, F.; Guzzetta, G.; Bjørnholt, J.V.; Leegaard, T.M.; Moen, A.E.F.; Merler, S.; Freiesleben de Blasio, B. Quantifying the transmission dynamics of MRSA in the community and healthcare settings in a low-prevalence country. Proc. Natl. Acad. Sci. USA 2019, 116, 14599–14605. [Google Scholar] [CrossRef]
- Köck, R.; Becker, K.; Cookson, B.; van Gemert-Pijnen, J.E.; Harbarth, S.; Kluytmans, J.; Mielke, M.; Peters, G.; Skov, R.L.; Struelens, M.J.; et al. Methicillin-resistant Staphylococcus aureus (MRSA): Burden of disease and control challenges in Europe. Eurosurveillance 2010, 15, 19688. [Google Scholar] [CrossRef]
- Mah, T.-F. Biofilm-specific antibiotic resistance. Future Microbiol. 2012, 7, 1061–1072. [Google Scholar] [CrossRef]
- Sharma, D.; Misba, L.; Khan, A.U. Antibiotics versus biofilm: An emerging battleground in microbial communities. Antimicrob. Resist. Infect. Control 2019, 8, 76. [Google Scholar] [CrossRef]
- Haney, E.F.; Hancock, R.E. Peptide design for antimicrobial and immunomodulatory applications. Biopolymers 2013, 100, 572–583. [Google Scholar] [CrossRef]
- Hancock, R.E.; Haney, E.F.; Gill, E.E. The immunology of host defence peptides: Beyond antimicrobial activity. Nat. Rev. Immunol. 2016, 16, 321–334. [Google Scholar] [CrossRef]
- Li, W.; Tailhades, J.; O’Brien-Simpson, N.M.; Separovic, F.; Otvos, L., Jr.; Hossain, M.A.; Wade, J.D. Proline-rich antimicrobial peptides: Potential therapeutics against antibiotic-resistant bacteria. Amino Acids 2014, 46, 2287–2294. [Google Scholar] [CrossRef] [PubMed]
- Gudmundsson, G.H.; Agerberth, B.; Odeberg, J.; Bergman, T.; Olsson, B.; Salcedo, R. The human gene FALL39 and processing of the cathelin precursor to the antibacterial peptide LL-37 in granulocytes. Eur. J. Biochem. 1996, 238, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Dürr, U.H.; Sudheendra, U.S.; Ramamoorthy, A. LL-37, the only human member of the cathelicidin family of antimicrobial peptides. Biochim. Biophys. Acta 2006, 1758, 1408–1425. [Google Scholar] [CrossRef]
- Sørensen, O.E.; Follin, P.; Johnsen, A.H.; Calafat, J.; Tjabringa, G.S.; Hiemstra, P.S.; Borregaard, N. Human cathelicidin, hCAP-18, is processed to the antimicrobial peptide LL-37 by extracellular cleavage with proteinase 3. Blood 2001, 97, 3951–3959. [Google Scholar] [CrossRef]
- Sørensen, O.E.; Gram, L.; Johnsen, A.H.; Andersson, E.; Bangsbøll, S.; Tjabringa, G.S.; Hiemstra, P.S.; Malm, J.; Egesten, A.; Borregaard, N. Processing of seminal plasma hCAP-18 to ALL-38 by gastricsin: A novel mechanism of generating antimicrobial peptides in vagina. J. Biol. Chem. 2003, 278, 28540–28546. [Google Scholar] [CrossRef]
- Cowland, J.B.; Johnsen, A.H.; Borregaard, N. hCAP-18, a cathelin/pro-bactenecin-like protein of human neutrophil specific granules. FEBS Lett. 1995, 368, 173–176. [Google Scholar] [CrossRef]
- Burton, M.F.; Steel, P.G. The chemistry and biology of LL37. Nat. Prod. Rep. 2009, 26, 1572–1584. [Google Scholar] [CrossRef] [PubMed]
- Noore, J.; Noore, A.; Li, B. Cationic antimicrobial peptide LL-37 is effective against both extra- and intracellular Staphylococcus aureus. Antimicrob. Agents Chemother. 2013, 57, 1283–1290. [Google Scholar] [CrossRef] [PubMed]
- Haisma, E.M.; de Breij, A.; Chan, H.; van Dissel, J.T.; Drijfhout, J.W.; Hiemstra, P.S.; El Ghalbzouri, A.; Nibbering, P.H. LL-37-Derived Peptides Eradicate Multidrug-Resistant Staphylococcus aureus from Thermally Wounded Human Skin Equivalents. Antimicrob. Agents Chemother. 2014, 58, 4411–4419. [Google Scholar] [CrossRef] [PubMed]
- Saporito, P.; Vang Mouritzen, M.; Løbner-Olesen, A.; Jenssen, H. LL-37 fragments have antimicrobial activity against Staphylococcus epidermidis biofilms and wound healing potential in HaCaT cell line. J. Pept. Sci. 2018, 24, e3080. [Google Scholar] [CrossRef] [PubMed]
- Jacob, B.; Park, I.S.; Bang, J.K.; Shin, S.Y. Short KR-12 analogs designed from human cathelicidin LL-37 possessing both antimicrobial and antiendotoxic activities without mammalian cell toxicity. J. Pept. Sci. 2013, 19, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Rajasekaran, G.; Kim, E.Y.; Shin, S.Y. LL-37-derived membrane-active FK-13 analogs possessing cell selectivity, anti-biofilm activity and synergy with chloramphenicol and anti-inflammatory activity. Biochim. Biophys. Acta Biomembr. 2017, 1859, 722–733. [Google Scholar] [CrossRef] [PubMed]
- Epand, R.F.; Wang, G.; Berno, B.; Epand, R.M. Lipid Segregation Explains Selective Toxicity of a Series of Fragments Derived from the Human Cathelicidin LL-37. Antimicrob. Agents Chemother. 2009, 53, 3705–3714. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Hanke, M.; Mishra, B.; Lushnikova, T.; Heim, C.E.; Thomas, V.C.; Bayles, K.W.; Kielian, T. Transformation of Human Cathelicidin LL-37 into Selective, Stable, and Potent Antimicrobial Compounds. ACS Chem. Biol. 2014, 9, 1997–2002. [Google Scholar] [CrossRef]
- Wang, G. Structures of human host defense cathelicidin LL-37 and its smallest antimicrobial peptide KR-12 in lipid micelles. J. Biol. Chem. 2008, 283, 32637–32643. [Google Scholar] [CrossRef]
- Mishra, B.; Epand, R.F.; Epand, R.M.; Wang, G. Structural location determines functional roles of the basic amino acids of KR-12, the smallest antimicrobial peptide from human cathelicidin LL-37. RSC Adv. 2013, 3, 19560–19571. [Google Scholar] [CrossRef]
- Li, Z.; Yuan, P.; Xing, M.; He, Z.; Dong, C.; Cao, Y.; Liu, Q. Fatty acid conjugation enhances the activities of antimicrobial peptides. Recent Pat. Food Nutr. Agric. 2013, 5, 52–56. [Google Scholar] [CrossRef]
- Avrahami, D.; Shai, Y. A New Group of Antifungal and Antibacterial Lipopeptides Derived from Non-membrane Active Peptides Conjugated to Palmitic Acid. J. Biol. Chem. 2004, 279, 12277–12285. [Google Scholar] [CrossRef]
- Albada, B. Tuning Activity of Antimicrobial Peptides by Lipidation. In Health Consequence of Microbial Interactions with Hydrocarbons, Oils, and Lipids, Handbook of Hydrocarbon and Lipid Microbiology; Goldfine, H., Ed.; Springer International Publishing: Cham, Switzerland, 2019; pp. 1–18. [Google Scholar]
- Neubauer, D.; Jaśkiewicz, M.; Bauer, M.; Gołacki, K.; Kamysz, W. Ultrashort Cationic Lipopeptides–Effect of N-Terminal Amino Acid and Fatty Acid Type on Antimicrobial Activity and Hemolysis. Molecules 2020, 25, 257. [Google Scholar] [CrossRef]
- Cruz, E.; Euerby, M.R.; Johnson, C.M.; Hackett, C.A. Chromatographic classification of commercially available reverse-phase HPLC columns. Chromatographia 1997, 44, 151–161. [Google Scholar] [CrossRef]
- Kimata, K.; Iwaguchi, K.; Onishi, S.; Jinno, K.; Eksteen, R.; Hosoya, K.; Araki, M.; Tanaka, N. Chromatographic characterization of silica c18 packing materials. Correlation between a preparation method and retention behavior of stationary phase. J. Chromatogr. Sci. 1989, 27, 721–728. [Google Scholar] [CrossRef]
- Jaśkiewicz, M.; Bauer, M.; Sadowska, K.; Barańska-Rybak, W.; Kamysz, E.; Kamysz, W. Antistaphylococcal activity of the KR-12 alanine scan. In Proceedings of the 35th European Peptide Symposium, Dublin, Ireland, 26–31 August 2018. Poster, Unpublished Work. [Google Scholar]
- Dathe, M.; Nikolenko, H.; Meyer, J.; Beyermann, M.; Bienert, M. Optimization of the antimicrobial activity of magainin peptides by modification of charge. FEBS Lett. 2001, 501, 146–150. [Google Scholar] [CrossRef]
- Dathe, M.; Wieprecht, T. Structural features of helical antimicrobial peptides: Their potential to modulate activity on model membranes and biological cells. Biochim. Biophys. Acta 1999, 1462, 71–87. [Google Scholar] [CrossRef]
- Laverty, G.; McLaughlin, M.; Shaw, C.; Gorman, S.P.; Gilmore, B.F. Antimicrobial activity of short, synthetic cationic lipopeptides. Chem. Biol. Drug Des. 2010, 75, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Albada, H.B.; Prochnow, P.; Bobersky, S.; Langklotz, S.; Schriek, P.; Bandow, J.E.; Metzler-Nolte, N. Tuning the activity of a short Arg-Trp antimicrobial peptide by lipidation of a C- or N-terminal lysine side-chain. ACS Med. Chem. Lett. 2012, 3, 980–984. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.Y.; Rajasekaran, G.; Shin, S.Y. LL-37-derived short antimicrobial peptide KR-12-a5 and its D-amino acid substituted analogs with cell selectivity, anti-biofilm activity, synergistic effect with conventional antibiotics, and anti-inflammatory activity. Eur. J. Med. Chem. 2017, 136, 428–441. [Google Scholar] [CrossRef]
- Lyu, Y.; Yang, Y.; Lyu, X.; Dong, N.; Shana, A. Antimicrobial activity, improved cell selectivity and mode of action of short PMAP-36-derived peptides against bacteria and Candida. Sci. Rep. 2016, 6, 27258. [Google Scholar] [CrossRef]
- Laverty, G.; McCloskey, A.P.; Gorman, S.P.; Gilmore, B.F. Anti-biofilm activity of ultrashort cinnamic acid peptide derivatives against medical device-related pathogens. J. Pept. Sci. 2015, 21, 770–778. [Google Scholar] [CrossRef]
- Avrahami, D.; Shai, Y. Conjugation of a magainin analogue with lipophilic acids controls hydrophobicity, solution assembly, and cell selectivity. Biochemistry 2002, 41, 2254–2263. [Google Scholar] [CrossRef]
- Oren, Z.; Shai, Y. Selective lysis of bacteria but not mammalian cells by diastereomers of melittin: Structure− function study. Biochemistry 1997, 36, 1826–1835. [Google Scholar] [CrossRef]
- Shai, Y.; Oren, Z. Diastereoisomers of cytolysins, a novel class of potent antibacterial peptides. J. Biol. Chem. 1996, 271, 7305–7308. [Google Scholar] [CrossRef] [PubMed]
- Mangoni, M.L.; Carotenuto, A.; Auriemma, L.; Saviello, M.R.; Campiglia, P.; Gomez-Monterrey, I.; Malfi, S.; Marcellini, L.; Barra, D.; Novellino, E. Structure−activity relationship, conformational and biological studies of temporin L analogues. J. Med. Chem. 2011, 54, 1298–1307. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Lee, D.G. Structure-antimicrobial activity relationship between pleurocidin and its enantiomer. Exp. Mol. Med. 2008, 40, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Thongngam, M.; McClements, D.J. Influence of pH, ionic strength, and temperature on self-association and interactions of sodium dodecyl sulfate in the absence and presence of chitosan. Langmuir 2005, 21, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Manzo, G.; Carboni, M.; Rinaldi, A.C.; Casu, M.; Scorciapino, M.A. Characterization of sodium dodecylsulphate and dodecylphosphocholine mixed micelles through NMR and dynamic light scattering. Magn. Reson. Chem. 2013, 51, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Sikorska, E.; Wyrzykowski, D.; Szutkowski, K.; Greber, K.; Lubecka, E.A.; Zhukov, I. Thermodynamics, size, and dynamics of zwitterionic dodecylphosphocholine and anionic sodium dodecyl sulfate mixed micelles. J. Thermal Anal. Calor. 2016, 123, 511–523. [Google Scholar] [CrossRef]
- Jeong, J.-H.; Kim, J.-S.; Choi, S.-S.; Kim, Y. NMR structural studies of antimicrobial peptides: LPcin analogs. Biophys. J. 2016, 110, 423–430. [Google Scholar] [CrossRef]
- Khakshoor, O.; Demeler, B.; Nowick, J.S. Macrocyclic beta-sheet peptides that mimic protein quaternary structure through intermolecular beta-sheet interactions. J. Am. Chem. Soc. 2007, 129, 5558–5569. [Google Scholar] [CrossRef]
- Chang, X.; Keller, D.; O’Donoghue, S.I.; Led, J.J. NMR studies of the aggregation of glucagon-like peptide-1: Formation of a symmetric helical dimer. FEBS Lett. 2002, 515, 165–170. [Google Scholar] [CrossRef]
- Jarvet, J.; Danielsson, J.; Damberg, P.; Oleszczuk, M.; Gräslund, A. Positioning of the Alzheimer Abeta(1-40) peptide in SDS micelles using NMR and paramagnetic probes. J. Biomol. NMR 2007, 39, 63–72. [Google Scholar] [CrossRef]
- Chou, J.J.; Baber, J.L.; Bax, A. Characterization of phospholipid mixed micelles by translational diffusion. J. Biomol. NMR 2004, 29, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Sikorska, E.; Stachurski, O.; Neubauer, D.; Małuch, I.; Wyrzykowski, D.; Bauer, M.; Brzozowski, K.; Kamysz, W. Short arginine-rich lipopeptides: From self-assembly to antimicrobial activity. Biochim. Biophys. Acta Biomembr. 2018, 1860, 2242–2251. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.C.; White, P.D. Fmoc Solid Phase Peptide Synthesis: A Practical Approach; Oxford University Press: New York, NY, USA, 2000; pp. 11–74. [Google Scholar]
- Atherton, E.; Sheppard, R.C. Solid Phase Peptide Synthesis: A Practical Approach; IRL Press: Oxford, UK, 1989; pp. 28–38, 75–86, 149–162. [Google Scholar]
- Wayne, P.A. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard—Ninth Edition; Document M07-A8; Clinical and Laboratory Standards Institute (CLSI): Wayne, PA, USA, 2012; Available online: www.clsi.org (accessed on 20 January 2013).
- Andrews, J.M. Determination of minimum inhibitory concentrations. J. Antimicrob. Chemother. 2002, 49, 1049. [Google Scholar] [CrossRef]
- Maciejewska, M.; Bauer, M.; Neubauer, D.; Kamysz, W.; Dawgul, M. Influence of amphibian antimicrobial peptides and short lipopeptides on bacterial biofilms formed on contact lenses. Materials (Basel) 2016, 9, 873. [Google Scholar] [CrossRef] [PubMed]
- Migoń, D.; Jaśkiewicz, M.; Neubauer, D.; Bauer, M.; Sikorska, E.; Kamysz, E.; Kamysz, W. Alanine Scanning Studies of the Antimicrobial Peptide Aurein 1.2. Probiotics Antimicrob. Proteins 2019, 11, 1042–1054. [Google Scholar] [CrossRef] [PubMed]
- Stachurski, O.; Neubauer, D.; Małuch, I.; Wyrzykowski, D.; Bauer, M.; Bartoszewska, S.; Kamysz, W.; Sikorska, E. Effect of self-assembly on antimicrobial activity of double-chain short cationic lipopeptides. Bioorg. Med. Chem. 2019, 27, 115129. [Google Scholar] [CrossRef]
- Sikorska, E.; Dawgul, M.; Greber, K.; Iłowska, E.; Pogorzelska, A.; Kamysz, W. Self-assembly and interactions of short antimicrobial cationic lipopeptides with membrane lipids: ITC, FTIR and molecular dynamics studies. BBA-Biomembranes 2014, 1838, 2625–2634. [Google Scholar] [CrossRef]
- Sreerama, N.; Woody, R.W. Estimation of Protein Secondary Structure from Circular Dichroism Spectra: Comparison of CONTIN, SELCON, and CDSSTR Methods with an Expanded Reference Set. Anal. Biochem. 2000, 287, 252–260. [Google Scholar] [CrossRef]
- Chu-Kung, A.F.; Bozzelli, K.N.; Lockwood, N.A.; Haseman, J.R.; Mayo, K.H.; Tirrell, M.V. Promotion of peptide antimicrobial activity by fatty acid conjugation. Bioconjug. Chem. 2004, 15, 530–535. [Google Scholar] [CrossRef]
- Vaezi, Z.; Bortolotti, A.; Luca, V.; Perilli, G.; Mangoni, M.L.; Khosravi-Far, R.; Bobone, S.; Stella, L. Aggregation determines the selectivity of membrane-active anticancer and antimicrobial peptides: The case of killerFLIP. Biochim Biophys Acta Biomembr. 2020, 1862, 183107. [Google Scholar] [CrossRef]
- McCrudden, M.T.C.; McLean, D.T.F.; Zhou, M.; Shaw, J.; Linden, G.J.; Irwin, C.H.R.; Lundy, F.T. The Host Defence Peptide LL-37 is Susceptible to ProteolyticDegradation by Wound Fluid Isolated from Foot Ulcersof Diabetic Patients. Int. J. Pept. Res. Ther. 2014, 20, 457–464. [Google Scholar] [CrossRef]
- Mishra, B.; Golla, R.M.; Lau, K.; Lushnikova, T.; Wang, G. Anti-Staphylococcal Biofilm Effects of Human Cathelicidin Peptides. ACS Med. Chem. Lett. 2016, 7, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Riool, M. Novel Antibacterial Strategies to Combat Biomaterial-Associated Infections. Ph.D. Thesis, University of Amsterdam, Amsterdam, The Netherlands. Available online: https://hdl.handle.net/11245.1/db3811ec-92ce-4831-b7c7-f1c0bcc76bc9 (accessed on 27 January 2020).
- Falagas, M.E.; Kasiakou, S.K. Toxicity of polymyxins: A systematic review of the evidence from old and recent studies. Crit Care. 2006, 10, R27. [Google Scholar] [CrossRef] [PubMed]
- Pastewski, A.A.; Caruso, P.; Parris, A.R.; Dizon, R.; Kopec, R.; Sharma, S.; Mayer, S.; Ghitan, M.; Chapnick, E.K. Parenteral polymyxin B use in patients with multidrug-resistant gram-negative bacteremia and urinary tract infections: A retrospective case series. Ann. Pharmacother. 2008, 42, 1177–1187. [Google Scholar] [CrossRef]
- Steenbergen, J.N.; Alder, J.; Thorne, G.M.; Tally, F.P. Daptomycin, a New Cyclic Lipopeptide Antibiotic, for the Treatment of Resistant Gram-Positive Organisms. J. Antimicrob. Chemother. 2005, 55, 283–288. [Google Scholar] [CrossRef]
- Dunne, M.W.; Talbot, G.H.; Boucher, H.W.; Wilcox, M.; Puttagunta, S. Safety of Dalbavancin in the Treatment of Skin and Skin Structure Infections: A Pooled Analysis of Randomized, Comparative Studies. Drug Saf. 2016, 39, 147–157. [Google Scholar] [CrossRef]
- Pushkin, R.; Barriere, S.L.; Wang, W.; Corey, G.R.; Stryjewski, M.E. Telavancin for Acute Bacterial Skin and Skin Structure Infections, a Post Hoc Analysis of the Phase 3 ATLAS Trials in Light of the 2013 FDA Guidance. Antimicrob. Agents Chemother. 2015, 59, 6170–6174. [Google Scholar] [CrossRef]
- Roberts, K.D.; Sulaiman, R.M.; Rybak, M.J. Dalbavancin and Oritavancin: An Innovative Approach to the Treatment of Gram-Positive Infections. Pharmacotherapy 2015, 35, 935–948. [Google Scholar] [CrossRef]
- Zhang, L.; Bulaj, G. Converting Peptides into Drug Leads by Lipidation. Curr. Med. Chem. 2012, 19, 1602–1618. [Google Scholar] [CrossRef]
- Poupart, J.; Hou, X.; Chemtob, S.; Lubell, W.D. Application of N-Dodecyl l-Peptide to Enhance Serum Stability while Maintaining Inhibitory Effects on Myometrial Contractions Ex Vivo. Molecules 2019, 24, 4141. [Google Scholar] [CrossRef]
- Toth, I.; Flinna, N.; Hillery, A.; Gibbons, W.A.; Artursson, P. Lipidic conjugates of luteinizing hormone releasing hormone (LHRH)+ and thyrotropin releasing hormone (TRH)+ that release and protect the native hormones in homogenates of human intestinal epithelial (Caco-2) cells. Int. J. Pharm. 1994, 105, 241–247. [Google Scholar] [CrossRef]
- Zhang, L.; Robertson, C.R.; Green, B.R.; Pruess, T.H.; White, H.S.; Bulaj, G. Structural requirements for a lipoamino acid in modulating the anticonvulsant activities of systemically active galanin analogues. J. Med. Chem. 2009, 52, 1310–1316. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Name | Net Charge | HPLC t’R (min) | Average Mass (Da) | MS Analysis | ||
|---|---|---|---|---|---|---|---|
| z | m/z calc. | m/z found | |||||
| I | Ac-KR12-NH2 (C2-KR12-NH2) | +4 | 3.58 | 1612.96 | 2 | 807.49 | 807.29 |
| 3 | 538.66 | 538.69 | |||||
| 4 | 404.25 | 404.47 | |||||
| II | C4-KR12-NH2 | +4 | 3.99 | 1641.02 | 2 | 821.52 | 821.38 |
| 3 | 548.01 | 547.73 | |||||
| 4 | 411.26 | 411.36 | |||||
| III | C6-KR12-NH2 | +4 | 4.42 | 1669.07 | 2 | 835.54 | 835.16 |
| 3 | 557.36 | 556.84 | |||||
| 4 | 418.28 | 418.35 | |||||
| IV | C8-KR12-NH2 | +4 | 4.88 | 1697.12 | 2 | 849.57 | 849.46 |
| 3 | 566.71 | 566.65 | |||||
| 4 | 425.29 | 425.31 | |||||
| 5 | 340.43 | 340.56 | |||||
| V | C10-KR12-NH2 | +4 | 5.38 | 1725.18 | 2 | 863.60 | 863.43 |
| 3 | 576.07 | 575.86 | |||||
| 4 | 432.30 | 432.20 | |||||
| VI | C12-KR12-NH2 | +4 | 5.86 | 1753.23 | 2 | 877.62 | 877.03 |
| 3 | 585.42 | 585.04 | |||||
| 4 | 439.32 | 439.18 | |||||
| VII | C14-KR12-NH2 | +4 | 6.39 | 1781.28 | 2 | 891.65 | 891.42 |
| 3 | 594.77 | 594.59 | |||||
| 4 | 446.33 | 446.04 | |||||
| VIII | Benzoic acid-KR12-NH2 | +4 | 4.23 | 1675.03 | 2 | 838.52 | 838.14 |
| 3 | 559.35 | 559.00 | |||||
| 4 | 419.77 | 419.76 | |||||
| IX | trans-Cinnamic acid-KR12-NH2 | +4 | 4.44 | 1701.07 | 2 | 851.54 | 851.34 |
| 3 | 568.03 | 567.58 | |||||
| 4 | 426.28 | 425.85 | |||||
| X | KR12-NH2 (KRIVQRIKDFLR-NH2) | +5 | 2.68 | 1570.93 | 2 | 786.47 | 786.23 |
| 3 | 524.65 | 524.69 | |||||
| 4 | 393.74 | 393.94 | |||||
| 5 | 315.19 | 315.18 | |||||
| Peptide | E. faecium ATCC 700221 | K. pneumoniae ATCC 700603 | A. baumannii ATCC BAA-1605 | P. aeruginosa ATCC 9027 | K. aerogenes ATCC 13048 |
|---|---|---|---|---|---|
| I | 16 | >256 | >256 | 64 | >256 |
| II | 4 | 128 | >256 | 32 | 128 |
| III | 2 | 16 | 16 | 8 | 16 |
| IV | 1 | 2 | 2 | 2 | 2 |
| V | 2 | 16 | 8 | 8 | 16 |
| VI | 4 | 32 | 16 | 32 | 32 |
| VII | 8 | 64 | 32 | 128 | 128 |
| VIII | 1 | 16 | 16 | 8 | 16 |
| IX | 1 | 4 | 4 | 4 | 4 |
| X | 8 | >256 | 256 | 16 | >256 |
| Peptide | S. aureus ATCC 25923 | S. aureus ATCC 6538 | S. aureus ATCC 33591 | S. aureus ATCC 9144 | S. aureus ATCC 12598 |
|---|---|---|---|---|---|
| I | >256 | 256 | >256 | >256 | >256 |
| II | 128 | 32 | 128 | 64 | 64 |
| III | 16 | 4 | 16 | 8 | 8 |
| IV | 2 | 2 | 2 | 2 | 4 |
| V | 4 | 4 | 4 | 4 | 4 |
| VI | 32 | 32 | 16 | 32 | 32 |
| VII | 128 | 64 | 16 | 64 | 128 |
| VIII | 32 | 8 | 32 | 16 | 16 |
| IX | 8 | 2 | 4 | 4 | 4 |
| X | >256 | 256 | >256 | >256 | >256 |
| Peptide | S. aureus ATCC 25923 | S. aureus ATCC 6538 | S. aureus ATCC 33591 | S. aureus ATCC 9144 | S. aureus ATCC 12598 |
|---|---|---|---|---|---|
| I | >256 | >256 | >256 | >256 | >256 |
| II | 256 | 128 | 128 | 128 | 128 |
| III | 32 | 16 | 16 | 16 | 16 |
| IV | 8 | 16 | 4 | 4 | 4 |
| V | 32 | 32 | 32 | 16 | 8 |
| VI | 256 | 256 | 128 | 64 | 32 |
| VII | 256 | 256 | 256 | 128 | 64 |
| VIII | 64 | 32 | 32 | 16 | 16 |
| IX | 32 | 8 | 16 | 8 | 4 |
| X | >256 | >256 | >256 | >256 | >256 |
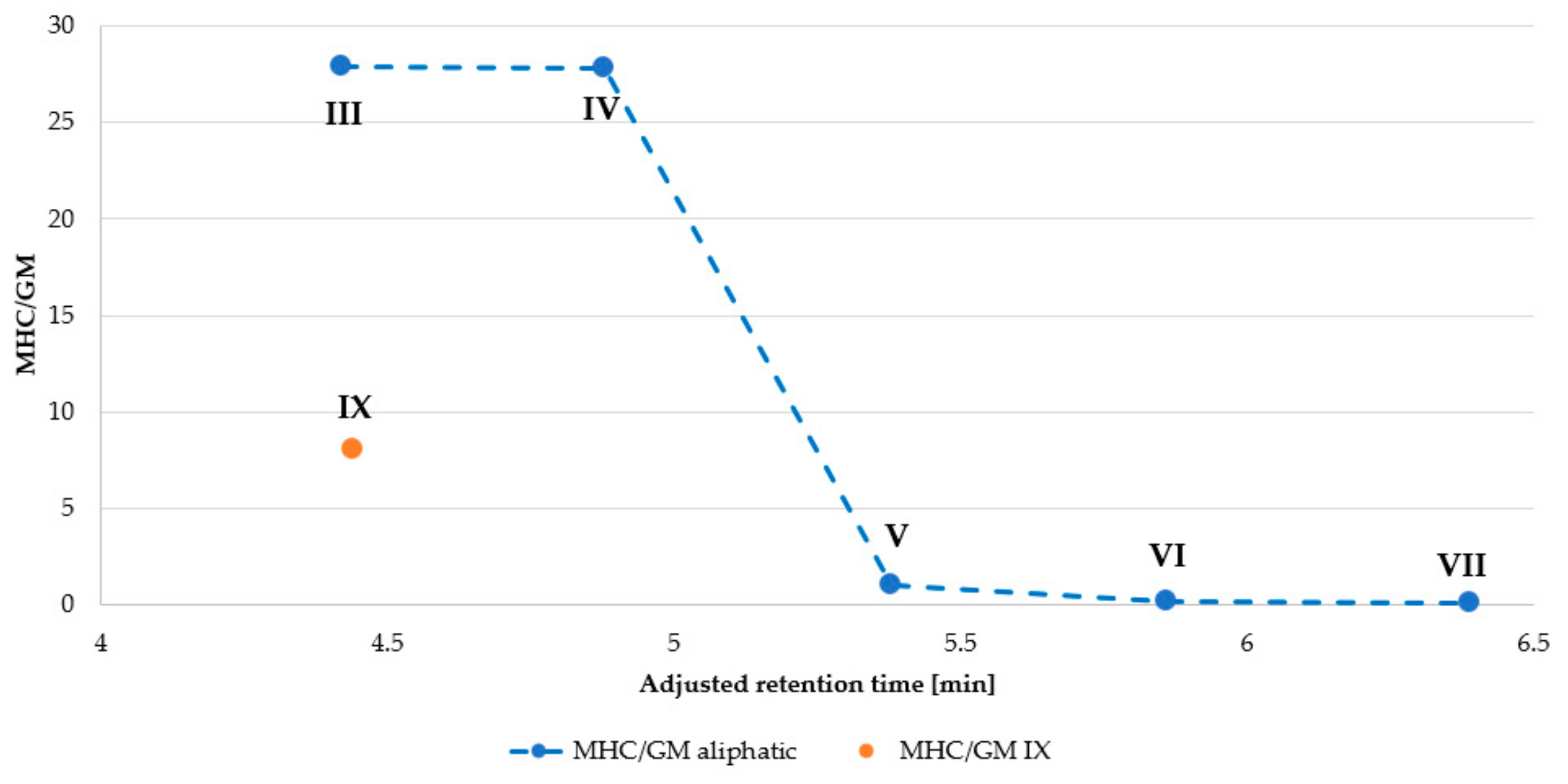
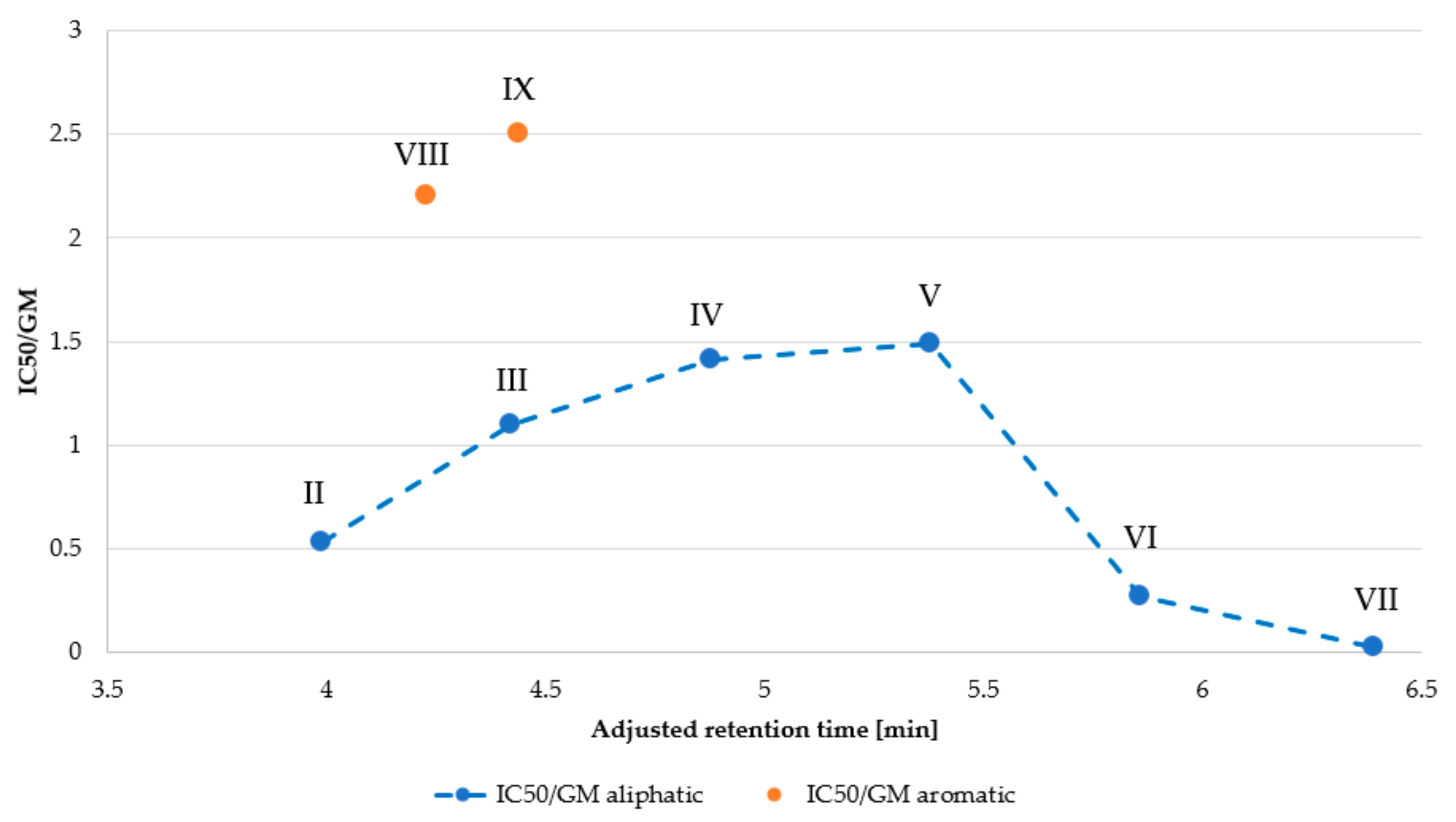
| Peptide | MHC 1 (µg/mL) | IC50 (µg/mL) | GM 2 (μg/mL) | Selectivity Index (SI) 3 | |
|---|---|---|---|---|---|
| MHC/GM | IC50/GM | ||||
| I | >256.00 | 84.20 | >256.00 | NA | NA |
| II | > 256.00 | 38.73 | 73.52 | NA | 0.53 |
| III | 256.00 | 10.13 | 9.19 | 27.86 | 1.10 |
| IV | 64.00 | 3.23 | 2.30 | 27.83 | 1.41 |
| V | 4.00 | 5.97 | 4.00 | 1.00 | 1.49 |
| VI | 4.00 | 7.60 | 27.86 | 0.14 | 0.27 |
| VII | 4.00 | 1.29 | 64.00 | 0.06 | 0.02 |
| VIII | >256.00 | 40.50 | 18.38 | NA | 2.20 |
| IX | 32.00 | 10.00 | 4.00 | 8.00 | 2.50 |
| X | >256.00 | 74.60 | >256.00 | NA | NA |
| Peptide | Helical Content % | |||||
|---|---|---|---|---|---|---|
| Water | PBS | DPC | SDS | POPG | POPC | |
| I | 8 | 8 | 72 | 82 | 69 | 16 |
| II | 7 | 6 | 69 | 71 | 67 | 9 |
| III | 7 | 7 | 65 | 67 | 24 | 16 |
| IV | 6 | 6 | 53 | 62 | 27 | 16 |
| V | 6 | 51 | 45 | 54 | 26 | 15 |
| VI | 8 | 74 | 81 | 86 | 55 | 32 |
| VII | 35 | 17 | 77 | 84 | 29 | 63 |
| VIII | 8 | 5 | 73 | 82 | 41 | 15 |
| IX | 7 | 19 | 72 | 77 | 30 | 18 |
| X | 7 | 7 | 66 | 70 | 66 | 7 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamysz, E.; Sikorska, E.; Jaśkiewicz, M.; Bauer, M.; Neubauer, D.; Bartoszewska, S.; Barańska-Rybak, W.; Kamysz, W. Lipidated Analogs of the LL-37-Derived Peptide Fragment KR12—Structural Analysis, Surface-Active Properties and Antimicrobial Activity. Int. J. Mol. Sci. 2020, 21, 887. https://doi.org/10.3390/ijms21030887
Kamysz E, Sikorska E, Jaśkiewicz M, Bauer M, Neubauer D, Bartoszewska S, Barańska-Rybak W, Kamysz W. Lipidated Analogs of the LL-37-Derived Peptide Fragment KR12—Structural Analysis, Surface-Active Properties and Antimicrobial Activity. International Journal of Molecular Sciences. 2020; 21(3):887. https://doi.org/10.3390/ijms21030887
Chicago/Turabian StyleKamysz, Elżbieta, Emilia Sikorska, Maciej Jaśkiewicz, Marta Bauer, Damian Neubauer, Sylwia Bartoszewska, Wioletta Barańska-Rybak, and Wojciech Kamysz. 2020. "Lipidated Analogs of the LL-37-Derived Peptide Fragment KR12—Structural Analysis, Surface-Active Properties and Antimicrobial Activity" International Journal of Molecular Sciences 21, no. 3: 887. https://doi.org/10.3390/ijms21030887
APA StyleKamysz, E., Sikorska, E., Jaśkiewicz, M., Bauer, M., Neubauer, D., Bartoszewska, S., Barańska-Rybak, W., & Kamysz, W. (2020). Lipidated Analogs of the LL-37-Derived Peptide Fragment KR12—Structural Analysis, Surface-Active Properties and Antimicrobial Activity. International Journal of Molecular Sciences, 21(3), 887. https://doi.org/10.3390/ijms21030887