Integrated Computational Approaches and Tools for Allosteric Drug Discovery

 , ,
, ,  ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Part I: Overview of Allostery and Allosteric Drugs
2.1. What Is Allostery?
2.2. Understanding Allosteric Mechanisms Using Existing Approaches
2.3. Understanding the Allosteric Effects of Disease and Drug-Resistant/Sensitive Mutations—Precision Medicine
2.4. Orthosteric versus Allosteric Drugs
2.5. FDA-Approved Allosteric Drugs
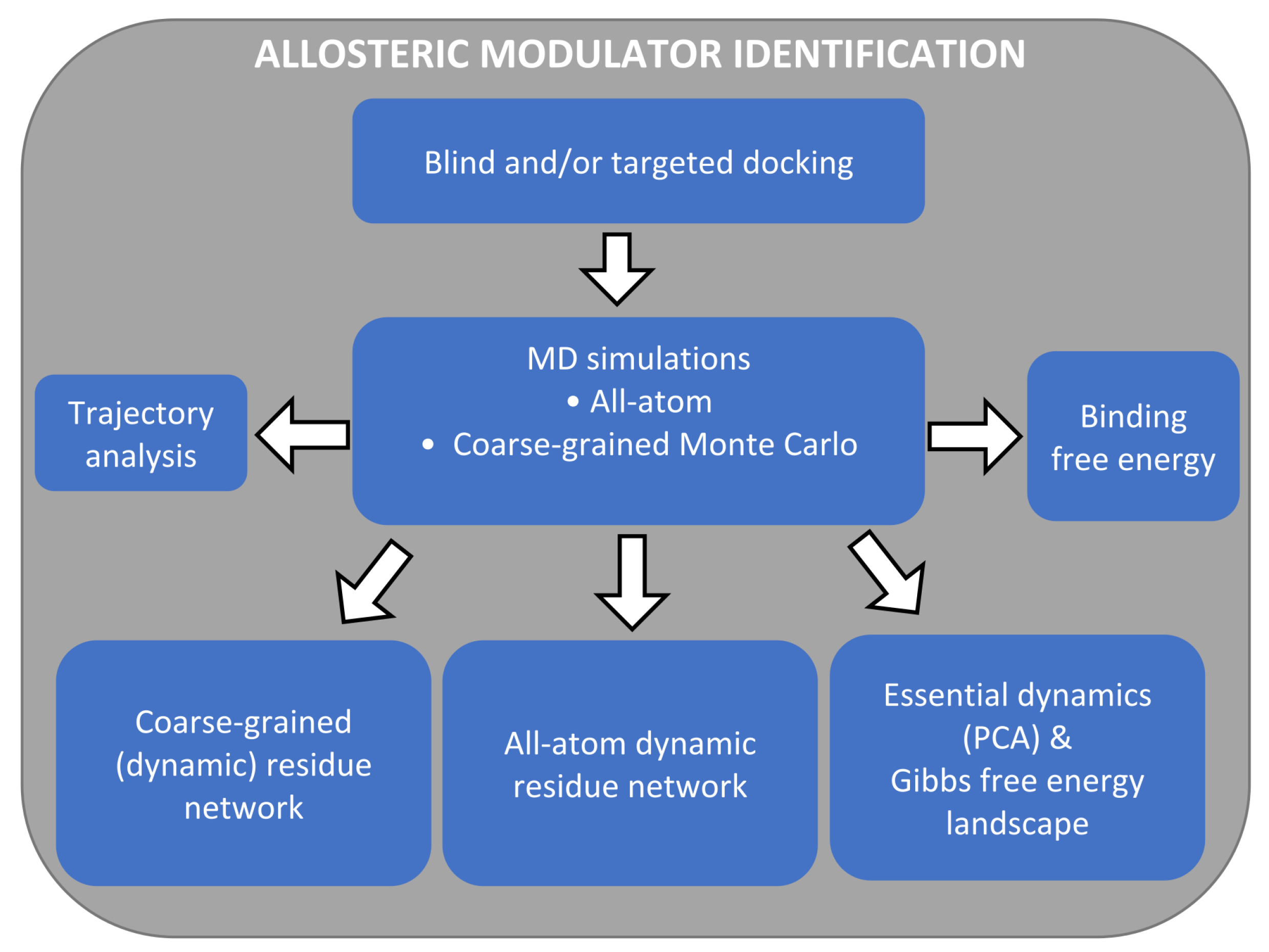
3. Part II: An Integrated In Silico Approach for Allosteric Drug Discovery
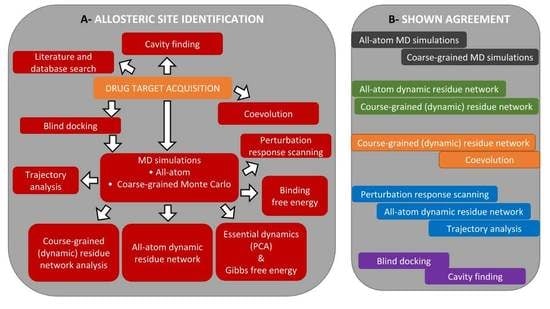
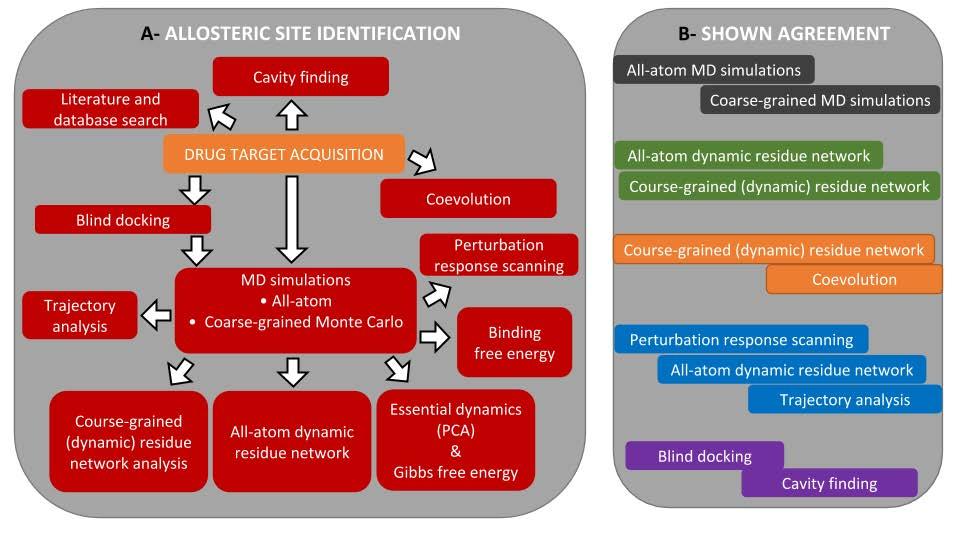
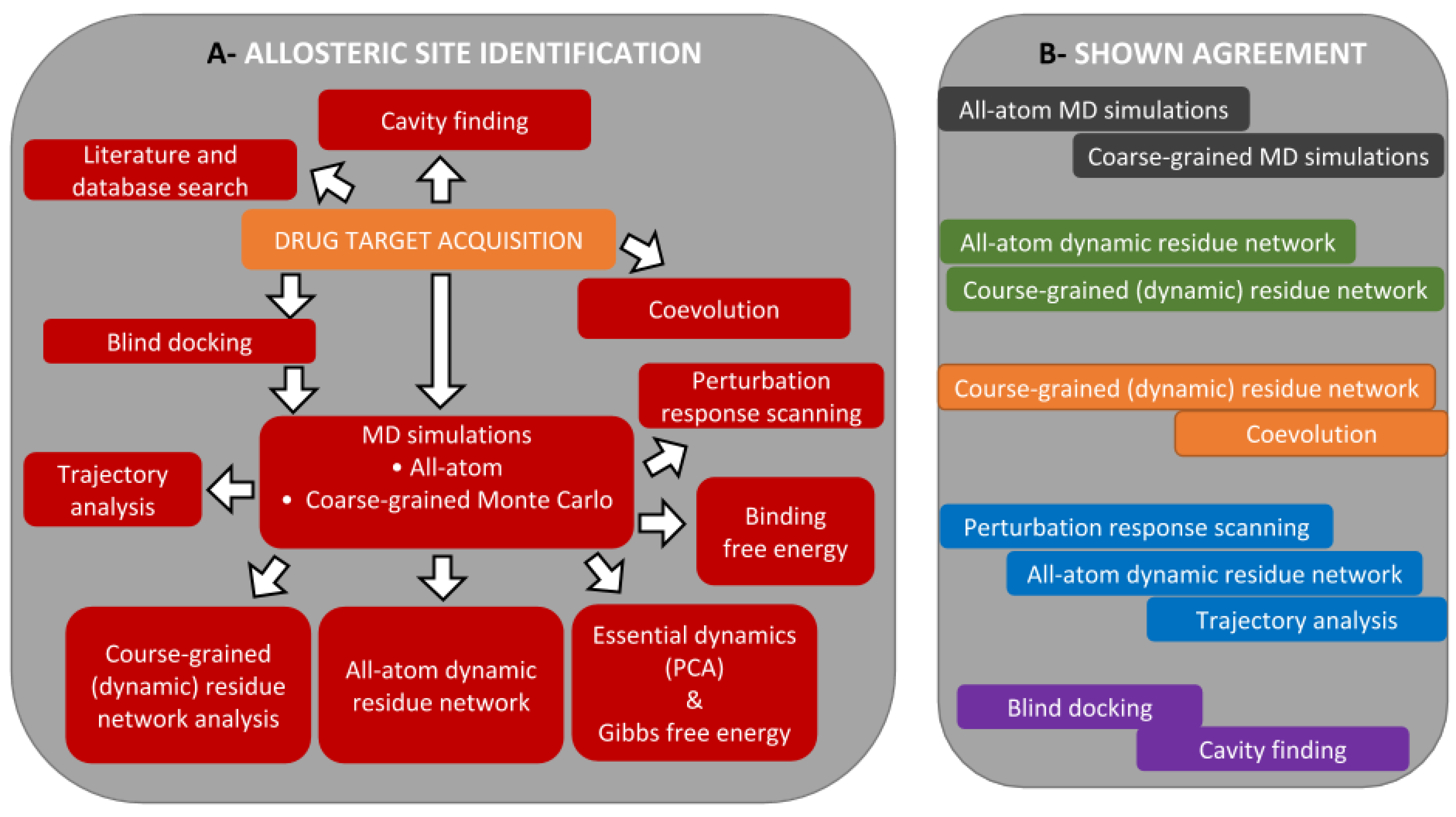
3.1. The Main Workflow
3.2. Allosteric Site and Modulator Prediction
3.3. Drug Target Acquisition
3.3.1. Mining Literature and Databases for Allostery Information
3.3.2. Cavity-Finding Approaches
3.3.3. Blind Docking
3.3.4. Perturbation Response Scanning
3.3.5. Interaction Networks in Proteins Dynamics
The Usefulness of Network Theory in Investigating Protein Dynamics and Allostery
Dynamic Residue Network Analysis
Coevolution and Residue Interaction Networks
3.3.6. Conformational Sampling
Molecular Dynamics
Coarse-Grained Simulations and Stochastic Markov State Models
3.3.7. Trajectory Analysis
RMSD
RMSF
Radius of Gyration
Dynamic Cross Correlation
Geometry Calculations
Essential Dynamics and Free-Energy Landscape
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Monod, J.; Wyman, J.; Changeux, J.P. On the nature of allosteric transitions: A plausible model. J. Mol. Biol. 1965. [Google Scholar] [CrossRef]
- Koshland, D.E.; Nemethy, J.G.; Filmer, D. Comparison of Experimental Binding Data and Theoretical Models in Proteins Containing Subunits. Biochemistry 1966. [Google Scholar] [CrossRef]
- Changeux, J.P. Allostery and the Monod-Wyman-Changeux Model After 50 Years. Annu. Rev. Biophys. 2012, 41, 103–133. [Google Scholar] [CrossRef] [PubMed]
- Changeux, J.P.; Edelstein, S.J. Allosteric mechanisms of signal transduction. Science 2005, 308, 1424–1428. [Google Scholar] [CrossRef] [PubMed]
- Popovych, N.; Sun, S.; Ebright, R.H.; Kalodimos, C.G. Dynamically driven protein allostery. Nat. Struct. Mol. Biol. 2006. [Google Scholar] [CrossRef] [PubMed]
- Boehr, D.D.; Dyson, H.J.; Wright, P.E. An NMR Perspective on Enzyme Dynamics. Chem. Rev. 2006, 106, 3055–3079. [Google Scholar] [CrossRef] [PubMed]
- Jarymowycz, V.A.; Stone, M.J. Fast time scale dynamics of protein backbones: NMR relaxation methods, applications, and functional consequences. Chem. Rev. 2006. [Google Scholar] [CrossRef] [PubMed]
- Mittermaier, A. New Tools Provide New Insights in NMR Studies of Protein Dynamics. Science 2006, 312, 224–228. [Google Scholar] [CrossRef]
- Sprangers, R.; Velyvis, A.; Kay, L.E. Solution NMR of supramolecular complexes: Providing new insights into function. Nat. Methods 2007, 4, 697–703. [Google Scholar] [CrossRef]
- Mittermaier, A.K.; Kay, L.E. Observing biological dynamics at atomic resolution using NMR. Trends Biochem. Sci. 2009, 34, 601–611. [Google Scholar] [CrossRef]
- Kay, L.E. NMR studies of protein structure and dynamics—A look backwards and forwards. J. Magn. Reson. 2011, 213, 492–494. [Google Scholar] [CrossRef] [PubMed]
- Korzhnev, D.M.; Kay, L.E. Probing invisible, low-populated states of protein molecules by relaxation dispersion NMR spectroscopy: An application to protein folding. Acc. Chem. Res. 2008, 41, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Kalodimos, C.G. NMR reveals novel mechanisms of protein activity regulation. Protein Sci. 2011, 20, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, R.; Kay, L.E. Bringing Dynamic Molecular Machines into Focus by Methyl-TROSY NMR. Annu. Rev. Biochem. 2014, 83, 291–315. [Google Scholar] [CrossRef]
- Kay, L.E. New Views of Functionally Dynamic Proteins by Solution NMR Spectroscopy. J. Mol. Biol. 2016, 428, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Lisi, G.P.; Loria, J.P. Solution NMR Spectroscopy for the Study of Enzyme Allostery. Chem. Rev. 2016, 116, 6323–6369. [Google Scholar] [CrossRef]
- Huang, C.; Kalodimos, C.G. Structures of Large Protein Complexes Determined by Nuclear Magnetic Resonance Spectroscopy. Annu. Rev. Biophys. 2017, 46, 317–336. [Google Scholar] [CrossRef]
- Jiang, Y.; Kalodimos, C.G. NMR Studies of Large Proteins. J. Mol. Biol. 2017, 429, 2667–2676. [Google Scholar] [CrossRef]
- Lisi, G.P.; Loria, J.P. Allostery in enzyme catalysis. Curr. Opin. Struct. Biol. 2017, 47, 123–130. [Google Scholar] [CrossRef]
- Gunasekaran, K.; Ma, B.; Nussinov, R. Is allostery an intrinsic property of all dynamic proteins? Proteins: Struct. Funct. Genet. 2004, 57, 433–443. [Google Scholar] [CrossRef]
- Liu, J.; Nussinov, R. Allosteric effects in the marginally stable von Hippel-Lindau tumor suppressor protein and allostery-based rescue mutant design. Proc. Natl. Acad. Sci. USA 2008, 105, 901–906. [Google Scholar] [CrossRef]
- Tsai, C.J.; del Sol, A.; Nussinov, R. Allostery: Absence of a Change in Shape Does Not Imply that Allostery Is Not at Play. J. Mol. Biol. 2008. [Google Scholar] [CrossRef]
- Tsai, C.J.; del Sol, A.; Nussinov, R. Protein allostery, signal transmission and dynamics: A classification scheme of allosteric mechanisms. Mol. BioSyst. 2009, 5, 207. [Google Scholar] [CrossRef] [PubMed]
- del Sol, A.; Tsai, C.J.; Ma, B.; Nussinov, R. The Origin of Allosteric Functional Modulation: Multiple Pre-existing Pathways. Structure 2009, 17, 1042–1050. [Google Scholar] [CrossRef] [PubMed]
- Zhuravlev, P.I.; Papoian, G.A. Protein functional landscapes, dynamics, allostery: A tortuous path towards a universal theoretical framework. Q. Rev. Biophys. 2010, 43, 295–332. [Google Scholar] [CrossRef] [PubMed]
- Blacklock, K.; Verkhivker, G.M. Computational Modeling of Allosteric Regulation in the Hsp90 Chaperones: A Statistical Ensemble Analysis of Protein Structure Networks and Allosteric Communications. PLoS Comput. Biol. 2014, 10, e1003679. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Tsai, C.J. Allostery in Disease and in Drug Discovery. Cell 2013, 153, 293–305. [Google Scholar] [CrossRef]
- Nussinov, R.; Tsai, C.J.; Csermely, P. Allo-network drugs: Harnessing allostery in cellular networks. Trends Pharmacol. Sci. 2011. [Google Scholar] [CrossRef]
- Penkler, D.L.; Tastan Bishop, Ö. Modulation of Human Hsp90α Conformational Dynamics by Allosteric Ligand Interaction at the C-Terminal Domain. Sci. Rep. 2019, 9, 1600. [Google Scholar] [CrossRef]
- Wenthur, C.J.; Gentry, P.R.; Mathews, T.P.; Lindsley, C.W. Drugs for Allosteric Sites on Receptors. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 165–184. [Google Scholar] [CrossRef]
- Szilagyi, A.; Nussinov, R.; Csermely, P. Allo-Network Drugs: Extension of the Allosteric Drug Concept to Protein- Protein Interaction and Signaling Networks. Curr. Top. Med. Chem. 2013, 13, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Amusengeri, A.; Taştan Bishop, Ö. Discorhabdin N, a South African natural compound, for Hsp72 and Hsc70 allosteric modulation: Combined study of molecular modeling and dynamic residue network analysis. Molecules 2019, 24, 188. [Google Scholar] [CrossRef] [PubMed]
- Amusengeri, A.; Astl, L.; Lobb, K.A.; Verkhivker, G.M.; Taştan Bishop, Ö. Establishing computational approaches towards identifying malarial allosteric modulators: A case study of Plasmodium falciparum Hsp70s. Int. J. Mol. Sci. 2019, 20, 5574. [Google Scholar] [CrossRef] [PubMed]
- Verkhivker, G.M. Dynamics-based community analysis and perturbation response scanning of allosteric interaction networks in the TRAP1 chaperone structures dissect molecular linkage between conformational asymmetry and sequential ATP hydrolysis. Biochim. Biophys. Acta Proteins Proteom. 2018, 1866, 899–912. [Google Scholar] [CrossRef] [PubMed]
- Penkler, D.L.; Atilgan, C.; Taştan Bishop, Ö. Allosteric Modulation of Human Hsp90α Conformational Dynamics. J. Chem. Inf. Model. 2018, 58, 383–404. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Verkhivker, G.M.; Hu, G. Integration of network models and evolutionary analysis into high-throughput modeling of protein dynamics and allosteric regulation: theory, tools and applications. Brief. Bioinform. 2019. [Google Scholar] [CrossRef]
- Changeux, J.P.; Christopoulos, A. Allosteric modulation as a unifying mechanism for receptor function and regulation. Diabetes Obes. Metab. 2017, 19, 4–21. [Google Scholar] [CrossRef]
- Greener, J.G.; Sternberg, M.J. Structure-based prediction of protein allostery. Curr. Opin. Struct. Biol. 2018, 50, 1–8. [Google Scholar] [CrossRef]
- Motlagh, H.N.; Wrabl, J.O.; Li, J.; Hilser, V.J. The ensemble nature of allostery. Nature 2014, 508, 331–339. [Google Scholar] [CrossRef]
- Nussinov, R.; Tsai, C.J. Allostery without a conformational change? Revisiting the paradigm. Curr. Opin. Struct. Biol. 2015, 30, 17–24. [Google Scholar] [CrossRef]
- Guo, J.; Zhou, H.X. Protein Allostery and Conformational Dynamics. Chem. Rev. 2016, 116, 6503–6515. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.R.; Lee, C.T.; Durrant, J.D.; Malmstrom, R.D.; Feher, V.A.; Amaro, R.E. Emerging Computational Methods for the Rational Discovery of Allosteric Drugs. Chem. Rev. 2016, 116, 6370–6390. [Google Scholar] [CrossRef]
- Lu, S.; He, X.; Ni, D.; Zhang, J. Allosteric Modulator Discovery: From Serendipity to Structure-Based Design. J. Med. Chem. 2019, 62, 6405–6421. [Google Scholar] [CrossRef]
- Hertig, S.; Latorraca, N.R.; Dror, R.O. Revealing Atomic-Level Mechanisms of Protein Allostery with Molecular Dynamics Simulations. PLoS Comput. Biol. 2016, 12, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Lam, J.; Nguyen, J.T.; O’Brien, T.; Wells, J.A. Discovery of an allosteric site in the caspases. Proc. Natl. Acad. Sci. USA 2004. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Huang, W.; Zhang, J. Recent computational advances in the identification of allosteric sites in proteins. Drug Discov. Today 2014, 19, 1595–1600. [Google Scholar] [CrossRef]
- Song, K.; Liu, X.; Huang, W.; Lu, S.; Shen, Q.; Zhang, L.; Zhang, J. Improved Method for the Identification and Validation of Allosteric Sites. J. Chem. Inf. Model. 2017, 57, 2358–2363. [Google Scholar] [CrossRef]
- Wodak, S.J.; Paci, E.; Dokholyan, N.V.; Berezovsky, I.N.; Horovitz, A.; Li, J.; Hilser, V.J.; Bahar, I.; Karanicolas, J.; Stock, G.; et al. Allostery in Its Many Disguises: From Theory to Applications. Structure 2019, 27, 566–578. [Google Scholar] [CrossRef]
- Tzeng, S.R.; Kalodimos, C.G. Dynamic activation of an allosteric regulatory protein. Nature 2009, 462, 368–372. [Google Scholar] [CrossRef]
- Tzeng, S.R.; Kalodimos, C.G. Protein dynamics and allostery: An NMR view. Curr. Opin. Struct. Biol. 2011, 21, 62–67. [Google Scholar] [CrossRef]
- Tzeng, S.R.; Kalodimos, C.G. Protein activity regulation by conformational entropy. Nature 2012, 488, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Kalodimos, C.G. Protein function and allostery: A dynamic relationship. Ann. N. Y. Acad. Sci. 2012, 1260, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Buchenberg, S.; Sittel, F.; Stock, G. Time-resolved observation of protein allosteric communication. Proc. Natl. Acad. Sci. USA 2017, 114, E6804–E6811. [Google Scholar] [CrossRef] [PubMed]
- Stock, G.; Hamm, P. A non-equilibrium approach to allosteric communication. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20170187. [Google Scholar] [CrossRef] [PubMed]
- Kalbitzer, H.R.; Rosnizeck, I.C.; Munte, C.E.; Narayanan, S.P.; Kropf, V.; Spoerner, M. Intrinsic allosteric inhibition of signaling proteins by targeting rare interaction states detected by high-pressure NMR spectroscopy. Angew. Chem. Int. Ed. 2013, 52, 14242–14246. [Google Scholar] [CrossRef] [PubMed]
- Williamson, M.P.; Kitahara, R. Characterization of low-lying excited states of proteins by high-pressure NMR. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 350–358. [Google Scholar] [CrossRef]
- Munte, C.E.; Beck-Erlach, M.; Kremer, W.; Koehler, J.; Kalbitzer, H.R. Distinct conformational states of the alzheimer β-amyloid peptide can be detected by high-pressure NMR spectroscopy. Angew. Chem. Int. Ed. 2013, 52, 8943–8947. [Google Scholar] [CrossRef]
- Cembran, A.; Kim, J.; Gao, J.; Veglia, G. NMR mapping of protein conformational landscapes using coordinated behavior of chemical shifts upon ligand binding. Phys. Chem. Chem. Phys. 2014, 16, 6508–6518. [Google Scholar] [CrossRef]
- Robustelli, P.; Stafford, K.A.; Palmer, A.G. Interpreting Protein Structural Dynamics from NMR Chemical Shifts. J. Am. Chem. Soc. 2012, 134, 6365–6374. [Google Scholar] [CrossRef]
- Selvaratnam, R.; Chowdhury, S.; VanSchouwena, B.; Melacini, G. Mapping allostery through the covariance analysis of NMR chemical shifts. Proc. Natl. Acad. Sci. USA 2011, 108, 6133–6138. [Google Scholar] [CrossRef]
- Selvaratnam, R.; VanSchouwen, B.; Fogolari, F.; Mazhab-Jafari, M.T.; Das, R.; Melacini, G. The Projection Analysis of NMR Chemical Shifts Reveals Extended EPAC Autoinhibition Determinants. Biophys. J. 2012, 102, 630–639. [Google Scholar] [CrossRef] [PubMed]
- Boulton, S.; Akimoto, M.; Selvaratnam, R.; Bashiri, A.; Melacini, G. A Tool Set to Map Allosteric Networks through the NMR Chemical Shift Covariance Analysis. Sci. Rep. 2015, 4, 7306. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, C.; Bafna, K.; Roux, L.D.; Agarwal, P.K.; Doucet, N. Applications of NMR and computational methodologies to study protein dynamics. Arch. Biochem. Biophys. 2017, 628, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Smock, R.G.; Gierasch, L.M. Sending Signals Dynamically. Science 2009, 324, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Swain, J.; Gierasch, L. The changing landscape of protein allostery. Curr. Opin. Struct. Biol. 2006, 16, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Grutsch, S.; Brüschweiler, S.; Tollinger, M. NMR Methods to Study Dynamic Allostery. PLoS Comput. Biol. 2016, 12, e1004620. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Kay, L.E. Tracing an allosteric pathway regulating the activity of the HslV protease. Proc. Natl. Acad. Sci. USA 2014, 111, 2140–2145. [Google Scholar] [CrossRef]
- Long, D.; Bouvignies, G.; Kay, L.E. Measuring hydrogen exchange rates in invisible protein excited states. Proc. Natl. Acad. Sci. USA 2014, 111, 8820–8825. [Google Scholar] [CrossRef]
- Anthis, N.J.; Clore, G.M. Visualizing transient dark states by NMR spectroscopy. Q. Rev. Biophys. 2015, 48, 35–116. [Google Scholar] [CrossRef]
- Yuwen, T.; Sekhar, A.; Kay, L.E. Separating Dipolar and Chemical Exchange Magnetization Transfer Processes in 1H-CEST. Angew. Chem. Int. Ed. 2017, 56, 6122–6125. [Google Scholar] [CrossRef]
- Boulton, S.; Melacini, G. Advances in NMR Methods To Map Allosteric Sites: From Models to Translation. Chem. Rev. 2016, 116, 6267–6304. [Google Scholar] [CrossRef] [PubMed]
- Boulton, S.; Selvaratnam, R.; Ahmed, R.; Melacini, G. Implementation of the NMR CHEmical Shift Covariance Analysis (CHESCA): A Chemical Biologist’s Approach to Allostery. In Methods in Molecular Biology; Ghose, R., Ed.; Springer: New York, NY, USA, 2018; pp. 391–405. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, D.; Rogawski, R.; Nimigean, C.M.; McDermott, A.E. Identifying coupled clusters of allostery participants through chemical shift perturbations. Proc. Natl. Acad. Sci. USA 2019, 116, 2078–2085. [Google Scholar] [CrossRef] [PubMed]
- Aoto, P.C.; Martin, B.T.; Wright, P.E. NMR Characterization of Information Flow and Allosteric Communities in the MAP Kinase p38γ. Sci. Rep. 2016, 6, 28655. [Google Scholar] [CrossRef] [PubMed]
- Burley, S.K.; Berman, H.M.; Bhikadiya, C.; Bi, C.; Chen, L.; Di Costanzo, L.; Christie, C.; Dalenberg, K.; Duarte, J.M.; Dutta, S.; et al. RCSB Protein Data Bank: Biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res. 2019, 47, D464–D474. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Ji, M.; Ni, D.; Zhang, J. Discovery of hidden allosteric sites as novel targets for allosteric drug design. Drug Discov. Today 2018, 23, 359–365. [Google Scholar] [CrossRef]
- Bowman, G.R.; Bolin, E.R.; Hart, K.M.; Maguire, B.C.; Marqusee, S. Discovery of multiple hidden allosteric sites by combining Markov state models and experiments. Proc. Natl. Acad. Sci. USA 2015, 112, 2734–2739. [Google Scholar] [CrossRef]
- Oleinikovas, V.; Saladino, G.; Cossins, B.P.; Gervasio, F.L. Understanding Cryptic Pocket Formation in Protein Targets by Enhanced Sampling Simulations. J. Am. Chem. Soc. 2016, 138, 14257–14263. [Google Scholar] [CrossRef]
- Vajda, S.; Beglov, D.; Wakefield, A.E.; Egbert, M.; Whitty, A. Cryptic binding sites on proteins: Definition, detection, and druggability. Curr. Opin. Chem. Biol. 2018, 44, 1–8. [Google Scholar] [CrossRef]
- Goncearenco, A.; Mitternacht, S.; Yong, T.; Eisenhaber, B.; Eisenhaber, F.; Berezovsky, I.N. SPACER: Server for predicting allosteric communication and effects of regulation. Nucleic Acids Res. 2013, 41, W266–W272. [Google Scholar] [CrossRef]
- Panjkovich, A.; Daura, X. PARS: A web server for the prediction of Protein Allosteric and Regulatory Sites. Bioinformatics 2014, 30, 1314–1315. [Google Scholar] [CrossRef]
- Greener, J.G.; Sternberg, M.J. AlloPred: Prediction of allosteric pockets on proteins using normal mode perturbation analysis. BMC Bioinform. 2015, 16, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chang, Y.Y.; Lee, J.Y.; Bahar, I.; Yang, L.W. DynOmics: Dynamics of structural proteome and beyond. Nucleic Acids Res. 2017, 45, W374–W380. [Google Scholar] [CrossRef] [PubMed]
- Weinkam, P.; Pons, J.; Sali, A. Structure-based model of allostery predicts coupling between distant sites. Proc. Natl. Acad. Sci. USA 2012, 109, 4875–4880. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Lu, S.; Huang, Z.; Liu, X.; Mou, L.; Luo, Y.; Zhao, Y.; Liu, Y.; Chen, Z.; Hou, T.; et al. Allosite: A method for predicting allosteric sites. Bioinformatics 2013, 29, 2357–2359. [Google Scholar] [CrossRef]
- Brown, D.K.; Sheik Amamuddy, O.; Tastan Bishop, Ö. Structure-Based Analysis of Single Nucleotide Variants in the Renin-Angiotensinogen Complex. Glob. Heart 2017. [Google Scholar] [CrossRef]
- Gallagher, M.D.; Chen-Plotkin, A.S. The Post-GWAS Era: From Association to Function. Am. J. Hum. Genet. 2018, 102, 717–730. [Google Scholar] [CrossRef]
- Brown, D.K.; Tastan Bishop, Ö. Role of Structural Bioinformatics in Drug Discovery by Computational SNP Analysis: Analyzing Variation at the Protein Level. Glob. Heart 2017, 12, 151–161. [Google Scholar] [CrossRef]
- Ng, P.C.; Levy, S.; Huang, J.; Stockwell, T.B.; Walenz, B.P.; Li, K.; Axelrod, N.; Busam, D.A.; Strausberg, R.L.; Venter, J.C. Genetic variation in an individual human exome. PLoS Genet. 2008. [Google Scholar] [CrossRef]
- Guarnera, E.; Berezovsky, I.N. On the perturbation nature of allostery: Sites, mutations, and signal modulation. Curr. Opin. Struct. Biol. 2019, 56, 18–27. [Google Scholar] [CrossRef]
- Kurochkin, I.V.; Guarnera, E.; Wong, J.H.; Eisenhaber, F.; Berezovsky, I.N. Toward allosterically increased catalytic activity of insulin-degrading enzyme against amyloid peptides. Biochemistry 2017. [Google Scholar] [CrossRef]
- Guarnera, E.; Berezovsky, I.N. Toward Comprehensive Allosteric Control over Protein Activity. Structure 2019, 27, 866–878.e1. [Google Scholar] [CrossRef] [PubMed]
- Tee, W.V.; Guarnera, E.; Berezovsky, I.N. On the Allosteric Effect of nsSNPs and the Emerging Importance of Allosteric Polymorphism. J. Mol. Biol. 2019, 431, 3933–3942. [Google Scholar] [CrossRef] [PubMed]
- Stetz, G.; Verkhivker, G.M. Computational Analysis of Residue Interaction Networks and Coevolutionary Relationships in the Hsp70 Chaperones: A Community-Hopping Model of Allosteric Regulation and Communication. PLoS Comput. Biol. 2017, 13, e1005299. [Google Scholar] [CrossRef] [PubMed]
- Doshi, U.; Holliday, M.J.; Eisenmesser, E.Z.; Hamelberg, D. Dynamical network of residue-residue contacts reveals coupled allosteric effects in recognition, catalysis, and mutation. Proc. Natl. Acad. Sci. USA 2016, 113, 4735–4740. [Google Scholar] [CrossRef]
- Sanyanga, T.A.; Nizami, B.; Taştan Bishop, Ö. Mechanism of action of non-synonymous single nucleotide variations associated with α-carbonic anhydrase II deficiency. Molecules 2019, 24, 3987. [Google Scholar] [CrossRef]
- Brown, D.K.; Penkler, D.L.; Sheik Amamuddy, O.; Ross, C.; Atilgan, A.R.; Atilgan, C.; Tastan Bishop, Ö. MD-TASK: A software suite for analyzing molecular dynamics trajectories. Bioinformatics 2017, 33, 2768–2771. [Google Scholar] [CrossRef]
- Nussinov, R.; Jang, H.; Tsai, C.J.; Cheng, F. Review: Precision medicine and driver mutations: Computational methods, functional assays and conformational principles for interpreting cancer drivers. PLoS Comput. Biol. 2019. [Google Scholar] [CrossRef]
- Garnett, M.J.; Edelman, E.J.; Heidorn, S.J.; Greenman, C.D.; Dastur, A.; Lau, K.W.; Greninger, P.; Thompson, I.R.; Luo, X.; Soares, J.; et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 2012, 483, 570–575. [Google Scholar] [CrossRef]
- Iorio, F.; Knijnenburg, T.A.; Vis, D.J.; Bignell, G.R.; Menden, M.P.; Schubert, M.; Aben, N.; Gonçalves, E.; Barthorpe, S.; Lightfoot, H.; et al. A Landscape of Pharmacogenomic Interactions in Cancer. Cell 2016, 166, 740–754. [Google Scholar] [CrossRef]
- Keshava, N.; Toh, T.S.; Yuan, H.; Yang, B.; Menden, M.P.; Wang, D. Defining subpopulations of differential drug response to reveal novel target populations. NPJ Syst. Biol. Appl. 2019, 5, 36. [Google Scholar] [CrossRef]
- Kumar, R.D.; Chang, L.W.; Ellis, M.J.; Bose, R. Prioritizing Potentially Druggable Mutations with dGene: An Annotation Tool for Cancer Genome Sequencing Data. PLoS ONE 2013, 8, e67980. [Google Scholar] [CrossRef] [PubMed]
- Niu, B.; Scott, A.D.; Sengupta, S.; Bailey, M.H.; Batra, P.; Ning, J.; Wyczalkowski, M.A.; Liang, W.W.; Zhang, Q.; McLellan, M.D.; et al. Protein-structure-guided discovery of functional mutations across 19 cancer types. Nat. Genet. 2016, 48, 827–837. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.O.; Oh, S.; Ko, G.; Park, S.J.; Kim, W.Y.; Lee, B.; Lee, S. VnD: A structure-centric database of disease-related SNPs and drugs. Nucleic Acids Res. 2011, 39, D939–D944. [Google Scholar] [CrossRef]
- Brown, D.K.; Taştan Bishop, Ö. HUMA: A platform for the analysis of genetic variation in humans. Hum. Mutat. 2018, 39, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Shuldiner, A.R. Association of Cytochrome P450 2C19 Genotype With the Antiplatelet Effect and Clinical Efficacy of Clopidogrel Therapy. JAMA 2009, 302, 849. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Chen, W.; Xu, Y.; Yi, X.; Han, Y.; Yang, Q.; Li, X.; Huang, L.; Johnston, S.C.; Zhao, X.; et al. Genetic Polymorphisms and Clopidogrel Efficacy for Acute Ischemic Stroke or Transient Ischemic Attack. Circulation 2017, 135, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Maimbo, M.; Kiyotani, K.; Mushiroda, T.; Masimirembwa, C.; Nakamura, Y. CYP2B6 genotype is a strong predictor of systemic exposure to efavirenz in HIV-infected Zimbabweans. Eur. J. Clin. Pharmacol. 2012, 68, 267–271. [Google Scholar] [CrossRef]
- Hussain, M.; Galvin, H.; Haw, T.Y.; Nutsford, A.; Husain, M. Drug resistance in influenza A virus: The epidemiology and management. Infect. Drug Resist. 2017, 10, 121–134. [Google Scholar] [CrossRef]
- Dookie, N.; Rambaran, S.; Padayatchi, N.; Mahomed, S.; Naidoo, K. Evolution of drug resistance in Mycobacterium tuberculosis: A review on the molecular determinants of resistance and implications for personalized care. J. Antimicrob. Chemother. 2018, 73, 1138–1151. [Google Scholar] [CrossRef]
- Menard, D.; Dondorp, A. Antimalarial drug resistance: A threat to malaria elimination. Cold Spring Harb. Perspect. Med. 2017. [Google Scholar] [CrossRef]
- Koigi, P.; Ngayo, M.; Khamadi, S.; Ngugi, C.; Nyamache, A. HIV type 1 drug resistance patterns among patients failing first and second line antiretroviral therapy in Nairobi, Kenya. BMC Res. Notes 2014, 7, 890. [Google Scholar] [CrossRef] [PubMed]
- Sheik Amamuddy, O.; Bishop, N.T.; Taştan Bishop, Ö. Characterizing early drug resistance-related events using geometric ensembles from HIV protease dynamics. Sci. Rep. 2018, 8, 17938. [Google Scholar] [CrossRef] [PubMed]
- Wensing, A.; Calvez, V.; Gunthard, H.; Johnson, V.; Paredes, R.; Pillay, D.; Shafer, R.; Richman, D. 2017 Update of the Drug Resistance Mutations in HIV-1. Top. Antivir. Med. 2017, 24, 132–133. [Google Scholar]
- Sheik Amamuddy, O.S.A. Application of Machine Learning, Molecular Modelling and Structural Data Mining against Antiretroviral Drug Resistance in HIV-1. Ph.D. Thesis, Rhodes University, Makhanda, South Africa, 2019. [Google Scholar]
- Yang, J.S.; Seo, S.W.; Jang, S.; Jung, G.Y.; Kim, S. Rational Engineering of Enzyme Allosteric Regulation through Sequence Evolution Analysis. PLoS Comput. Biol. 2012, 8, e1002612. [Google Scholar] [CrossRef] [PubMed]
- Dror, R.O.; Green, H.F.; Valant, C.; Borhani, D.W.; Valcourt, J.R.; Pan, A.C.; Arlow, D.H.; Canals, M.; Lane, J.R.; Rahmani, R.; et al. Structural basis for modulation of a G-protein-coupled receptor by allosteric drugs. Nature 2013, 503, 295–299. [Google Scholar] [CrossRef]
- Nussinov, R.; Tsai, C.J. The Design of Covalent Allosteric Drugs. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 249–267. [Google Scholar] [CrossRef]
- Xu, X.; Chen, Y.; Fu, Q.; Ni, D.; Zhang, J.; Li, X.; Lu, S. The chemical diversity and structure-based discovery of allosteric modulators for the PIF-pocket of protein kinase PDK1. J. Enzym. Inhib. Med. Chem. 2019, 34, 361–374. [Google Scholar] [CrossRef]
- Lin, A.; Giuliano, C.J.; Palladino, A.; John, K.M.; Abramowicz, C.; Yuan, M.L.; Sausville, E.L.; Lukow, D.A.; Liu, L.; Chait, A.R.; et al. Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials. Sci. Transl. Med. 2019, 11, eaaw8412. [Google Scholar] [CrossRef]
- Smith, R.D.; Lu, J.; Carlson, H.A. Are there physicochemical differences between allosteric and competitive ligands? PLoS Comput. Biol. 2017, 13, e1005813. [Google Scholar] [CrossRef]
- Raman, S. Systems Approaches to Understanding and Designing Allosteric Proteins. Biochemistry 2018. [Google Scholar] [CrossRef]
- Baxter, J.D.; Chasanov, W.M. An Update on HIV-1 Protease Inhibitor Resistance. J. AIDS Clin. Res. 2016, 7, 1–7. [Google Scholar] [CrossRef]
- Kagan, R.M.; Dunn, K.J.; Snell, G.P.; Nettles, R.E.; Kaufman, H.W. Trends in HIV-1 Drug Resistance Mutations from a U.S. Reference Laboratory from 2006 to 2017. AIDS Res. Hum. Retroviruses 2019, 35, 698–709. [Google Scholar] [CrossRef] [PubMed]
- Rabahi, M.F.; da Silva Júnior, J.L.R.; Ferreira, A.C.G.; Tannus-Silva, D.G.S.; Conde, M.B. Tuberculosis treatment. J. Bras. Pneumol. 2017, 43, 472–486. [Google Scholar] [CrossRef] [PubMed]
- Mega, E.R. Alarming surge in drug-resistant HIV uncovered. Nature 2019. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Antibiotic Resistance Threats in the United States; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2013.
- Zhong, W.; Cui, L.; Goh, B.C.; Cai, Q.; Ho, P.; Chionh, Y.H.; Yuan, M.; Sahili, A.E.; Fothergill-Gilmore, L.A.; Walkinshaw, M.D.; et al. Allosteric pyruvate kinase-based “logic gate” synergistically senses energy and sugar levels in Mycobacterium tuberculosis. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Mishra, A.; Mamidi, A.S.; Rajmani, R.S.; Ray, A.; Roy, R.; Surolia, A. An allosteric inhibitor of Mycobacterium tuberculosis ArgJ: Implications to a novel combinatorial therapy. EMBO Mol. Med. 2018, 10, 1–21. [Google Scholar] [CrossRef]
- Wellington, S.; Nag, P.P.; Michalska, K.; Johnston, S.E.; Jedrzejczak, R.P.; Kaushik, V.K.; Clatworthy, A.E.; Siddiqi, N.; McCarren, P.; Bajrami, B.; et al. A small-molecule allosteric inhibitor of Mycobacterium tuberculosis tryptophan synthase. Nat. Chem. Biol. 2017, 13, 943–950. [Google Scholar] [CrossRef]
- Rzomp, K.A.; Scholtes, L.D.; Briggs, B.J.; Whittaker, G.R.; Scidmore, M.A. Rab GTPases are recruited to chlamydial inclusions in both a species-dependent and species-independent manner. Infect. Immun. 2003, 71, 5855–5870. [Google Scholar] [CrossRef]
- Cortes, C.; Rzomp, K.A.; Tvinnereim, A.; Scidmore, M.A.; Wizel, B. Chlamydia pneumoniae inclusion membrane protein Cpn0585 interacts with multiple rab GTPases. Infect. Immun. 2007, 75, 5586–5596. [Google Scholar] [CrossRef]
- Bruce, E.A.; Digard, P.; Stuart, A.D. The Rab11 Pathway Is Required for Influenza A Virus Budding and Filament Formation. J. Virol. 2010, 84, 5848–5859. [Google Scholar] [CrossRef]
- Kumar, A.P.; Lukman, S. Allosteric binding sites in Rab11 for potential drug candidates. PLoS ONE 2018, 13, e0198632. [Google Scholar] [CrossRef] [PubMed]
- Hernández Alvarez, L.; Barreto Gomes, D.E.; Hernández González, J.E.; Pascutti, P.G. Dissecting a novel allosteric mechanism of cruzain: A computer-aided approach. PLoS ONE 2019, 14, e0211227. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Wang, G.; Li, S.; Liu, X.; Lu, S.; Chen, Z.; Song, K.; Yan, J.; Geng, L.; Huang, Z.; et al. ASD v3.0: Unraveling Allosteric regulation with structural mechanisms and biological networks. Nucleic Acids Res. 2016, 44, D527–D535. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Abdel-Magid, A.F. Allosteric Modulators: An Emerging Concept in Drug Discovery. ACS Med. Chem. Lett. 2015. [Google Scholar] [CrossRef]
- Van Westen, G.J.P.; Gaulton, A.; Overington, J.P. Chemical, Target, and Bioactive Properties of Allosteric Modulation. PLoS Comput. Biol. 2014, 10, e1003559. [Google Scholar] [CrossRef]
- Häberle, J. Role of carglumic acid in the treatment of acute hyperammonemia due to N-acetylglutamate synthase deficiency. Ther. Clin. Risk Manag. 2011, 327. [Google Scholar] [CrossRef]
- Van Wagenen, B.; Moe, S.; Balandrin, M.; DelMar, E.; Nemeth, E. Calcium Receptor-Active Compounds. US6211244B1, 3 April 2001. [Google Scholar]
- Adjeroud, S.; Tonon, M.C.; Leneveu, E.; Lamacz, M.; Danger, J.M.; Gouteux, L.; Cazin, L.; Vaudry, H. VI. The benzodiazepine agonist clonazepam potentiates the effects of γ-aminobutyric acid on α-MSH release from neurointermediate lobes in vitro. Life Sci. 1987. [Google Scholar] [CrossRef]
- Rice, K.D.; Aay, N.; Anand, N.K.; Blazey, C.M.; Bowles, O.J.; Bussenius, J.; Costanzo, S.; Curtis, J.K.; Defina, S.C.; Dubenko, L.; et al. Novel carboxamide-based allosteric MEK inhibitors: Discovery and optimization efforts toward XL518 (GDC-0973). ACS Med. Chem. Lett. 2012. [Google Scholar] [CrossRef]
- Desai, M.A.; Burnett, J.P.; Ornstein, P.L.; Schoepp, D.D. Cyclothiazide acts at a site on the alpha-amino-3-hydroxy-5-methyl-4- isoxazole propionic acid receptor complex that does not recognize competitive or noncompetitive AMPA receptor antagonists. J. Pharmacol. Exp. Ther. 1995, 272, 38–43. [Google Scholar] [PubMed]
- Tömösközi, Z.; Finance, O.; Arányi, P. Drotaverine interacts with the L-type Ca2+ channel in pregnant rat uterine membranes. Eur. J. Pharmacol. 2002, 449, 55–60. [Google Scholar] [CrossRef]
- Chen, J.; Yang, J.; Sun, X.; Wang, Z.; Cheng, X.; Lu, W.; Cai, X.; Hu, C.; Shen, X.; Cao, P. Allosteric inhibitor remotely modulates the conformation of the orthestric pockets in mutant IDH2/R140Q. Sci. Rep. 2017, 7, 16458. [Google Scholar] [CrossRef]
- Morlock, E.V.; Czajkowski, C. Different residues in the GABAA receptor benzodiazepine binding pocket mediate benzodiazepine efficacy and binding. Mol. Pharmacol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Krause, R.M.; Buisson, B.; Bertrand, S.; Corringer, P.J.; Galzi, J.L.; Changeux, J.P.; Bertrand, D. Ivermectin: A positive allosteric effector of the α7 neuronal nicotinic acetylcholine receptor. Mol. Pharmacol. 1998. [Google Scholar] [CrossRef] [PubMed]
- Haefely, W.; Kulcsar, A.; Mohler, H. Possible involvement of GABA in the central actions of benzodiazepines. Psychopharmacol. Bull. 1975, 14, 131–151. [Google Scholar]
- Waugh, D.J.J.; Gaivin, R.J.; Damron, D.S.; Murray, P.A.; Perez, D.M. Binding, Partial Agonism, and Potentiation of α1-Adrenergic Receptor Function by Benzodiazepines: A Potential Site of Allosteric Modulation. J. Pharmacol. Exp. Ther. 1999, 291, 1164–1171. [Google Scholar]
- Garcia-Perez, J.; Rueda, P.; Staropoli, I.; Kellenberger, E.; Alcami, J.; Arenzana-Seisdedos, F.; Lagane, B. New insights into the mechanisms whereby low molecular weight CCR5 ligands inhibit HIV-1 infection. J. Biol. Chem. 2011. [Google Scholar] [CrossRef]
- Ai, N.; Wood, R.D.; Yang, E.; Welsh, W.J. Niclosamide is a Negative Allosteric Modulator of Group I Metabotropic Glutamate Receptors: Implications for Neuropathic Pain. Pharm. Res. 2016, 33, 3044–3056. [Google Scholar] [CrossRef]
- Ahmed, A.H.; Oswald, R.E. Piracetam defines a new binding site for allosteric modulators of α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid (AMPA) receptors. J. Med. Chem. 2010. [Google Scholar] [CrossRef]
- Artsimovitch, I.; Vassylyeva, M.N.; Svetlov, D.; Svetlov, V.; Perederina, A.; Igarashi, N.; Matsugaki, N.; Wakatsuki, S.; Tahirov, T.H.; Vassylyev, D.G. Allosteric Modulation of the RNA Polymerase Catalytic Reaction Is an Essential Component of Transcription Control by Rifamycins. Cell 2005, 122, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Janssen, P.A.; Lewi, P.J.; Arnold, E.; Daeyaert, F.; De Jonge, M.; Heeres, J.; Koymans, L.; Vinkers, M.; Guillemont, J.; Pasquier, E.; et al. In search of a novel anti-HIV drug: Multidisciplinary coordination in the discovery of 4-[[4-[[4-[(1E)-2-cyanoethenyl]-2,6-dimethylphenyl]amino]-2- pyrimidinyl]amino]benzonitrile (R278474, rilpivirine). J. Med. Chem. 2005. [Google Scholar] [CrossRef] [PubMed]
- Vilella-Bach, M.; Nuzzi, P.; Fang, Y.; Chen, J. The FKBP12-rapamycin-binding domain is required for FKBP12-rapamycin- associated protein kinase activity and G1 progression. J. Biol. Chem. 1999. [Google Scholar] [CrossRef] [PubMed]
- Sinha, N. Ticagrelor: Molecular discovery to clinical evidence ticagrelor: A novel antiplatelet agent. Indian Heart J. 2012. [Google Scholar] [CrossRef]
- Gilmartin, A.G.; Bleam, M.R.; Groy, A.; Moss, K.G.; Minthorn, E.A.; Kulkarni, S.G.; Rominger, C.M.; Erskine, S.; Fisher, K.E.; Yang, J.; et al. GSK1120212 (JTP-74057) Is an Inhibitor of MEK Activity and Activation with Favorable Pharmacokinetic Properties for Sustained In Vivo Pathway Inhibition. Clin. Cancer Res. 2011, 17, 989–1000. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Nyamai, D.W.; Tastan Bishop, Ö. Aminoacyl tRNA synthetases as malarial drug targets: A comparative bioinformatics study. Malar. J. 2019, 18, 34. [Google Scholar] [CrossRef]
- Musyoka, T.M.; Kanzi, A.M.; Lobb, K.A.; Tastan Bishop, Ö. Analysis of non-peptidic compounds as potential malarial inhibitors against Plasmodial cysteine proteases via integrated virtual screening workflow. J. Biomol. Struct. Dyn. 2016, 34, 2084–2101. [Google Scholar] [CrossRef]
- Faya, N.; Penkler, D.L.; Tastan Bishop, Ö. Human, vector and parasite Hsp90 proteins: A comparative bioinformatics analysis. FEBS Open Bio 2015, 5, 916–927. [Google Scholar] [CrossRef]
- Xu, D.; Zhang, J.; Roy, A.; Zhang, Y. Automated protein structure modeling in CASP9 by I-TASSER pipeline combined with QUARK-based ab initio folding and FG-MD-based structure refinement. Proteins: Struct. Funct. Bioinform. 2011, 79, 147–160. [Google Scholar] [CrossRef]
- Pieper, U.; Webb, B.M.; Dong, G.Q.; Schneidman-Duhovny, D.; Fan, H.; Kim, S.J.; Khuri, N.; Spill, Y.G.; Weinkam, P.; Hammel, M.; et al. ModBase, a database of annotated comparative protein structure models and associated resources. Nucleic Acids Res. 2014, 42, D336–D346. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Hatherley, R.; Brown, D.K.; Glenister, M.; Tastan Bishop, Ö. PRIMO: An Interactive Homology Modeling Pipeline. PLoS ONE 2016, 11, e0166698. [Google Scholar] [CrossRef] [PubMed]
- Källberg, M.; Wang, H.; Wang, S.; Peng, J.; Wang, Z.; Lu, H.; Xu, J. Template-based protein structure modeling using the RaptorX web server. Nat. Protoc. 2012, 7, 1511–1522. [Google Scholar] [CrossRef]
- Song, Y.; DiMaio, F.; Wang, R.Y.R.; Kim, D.; Miles, C.; Brunette, T.; Thompson, J.; Baker, D. High-Resolution Comparative Modeling with RosettaCM. Structure 2013, 21, 1735–1742. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, L.; Stephens, A.; Nam, S.Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at its Core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Cassarino, T.G.; Bertoni, M.; Bordoli, L.; et al. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 42. [Google Scholar] [CrossRef]
- Šali, A.; Blundell, T.L. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Kim, D.E.; Chivian, D.; Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004, 32, W526–W531. [Google Scholar] [CrossRef]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef]
- Bradley, P.; Misura, K.M.; Baker, D. Biochemistry: Toward high-resolution de novo structure prediction for small proteins. Science 2005, 309, 1868–1871. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Freddolino, P.L.; Zhang, Y. Ab Initio Protein Structure Prediction. In From Protein Structure to Function with Bioinformatics; Springer Netherlands: Dordrecht, The Netherlands, 2017; Chapter 1; pp. 3–35. [Google Scholar] [CrossRef]
- AlQuraishi, M. End-to-End Differentiable Learning of Protein Structure. Cell Syst. 2019, 8, 292–301.e3. [Google Scholar] [CrossRef] [PubMed]
- Melo, F.; Feytmans, E. Assessing protein structures with a non-local atomic interaction energy. J. Mol. Biol. 1998, 277, 1141–1152. [Google Scholar] [CrossRef] [PubMed]
- Benkert, P.; Tosatto, S.C.E.; Schomburg, D. QMEAN: A comprehensive scoring function for model quality assessment. Proteins: Struct. Funct. Bioinform. 2008, 71, 261–277. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef]
- Lüthy, R.; Bowie, J.U.; Eisenberg, D. Assessment of protein models with three-dimensional profiles. Nature 1992, 356, 83–85. [Google Scholar] [CrossRef]
- McGuffin, L.J. The ModFOLD server for the quality assessment of protein structural models. Bioinformatics 2008, 24, 586–587. [Google Scholar] [CrossRef]
- Shen, M.Y.; Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006, 15, 2507–2524. [Google Scholar] [CrossRef]
- Sali, A. MODELLER A Program for Protein Structure Modeling Release 9v4, r6262. 2008. Available online: https://salilab.org/modeller/9v4/manual/ (accessed on 21 January 2020).
- Taştan Bishop, Ö.; De Beer, T.A.; Joubert, F. Protein homology modelling and its use in South Africa. S. Afr. J. Sci. 2008, 104, 2–6. [Google Scholar]
- Mobley, D.L.; Bannan, C.C.; Rizzi, A.; Bayly, C.I.; Chodera, J.D.; Lim, V.T.; Lim, N.M.; Beauchamp, K.A.; Slochower, D.R.; Shirts, M.R.; et al. Escaping Atom Types in Force Fields Using Direct Chemical Perception. J. Chem. Theory Comput. 2018, 14, 6076–6092. [Google Scholar] [CrossRef]
- Huang, Z.; Zhu, L.; Cao, Y.; Wu, G.; Liu, X.; Chen, Y.; Wang, Q.; Shi, T.; Zhao, Y.; Wang, Y.; et al. ASD: A comprehensive database of allosteric proteins and modulators. Nucleic Acids Res. 2011, 39, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Mou, L.; Shen, Q.; Lu, S.; Li, C.; Liu, X.; Wang, G.; Li, S.; Geng, L.; Liu, Y.; et al. ASD v2.0: Updated content and novel features focusing on allosteric regulation. Nucleic Acids Res. 2014, 42, D510–D516. [Google Scholar] [CrossRef] [PubMed]
- Astl, L.; Verkhivker, G.M. Data-driven computational analysis of allosteric proteins by exploring protein dynamics, residue coevolution and residue interaction networks. Biochim. Biophys. Acta Gen. Subj. 2019. [Google Scholar] [CrossRef] [PubMed]
- Pu, L.; Govindaraj, R.G.; Lemoine, J.M.; Wu, H.C.; Brylinski, M. DeepDrug3D: Classification of ligand-binding pockets in proteins with a convolutional neural network. PLoS Comput. Biol. 2019, 15, e1006718. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wang, S.; Hu, Q.; Gao, S.; Ma, X.; Zhang, W.; Shen, Y.; Chen, F.; Lai, L.; Pei, J. CavityPlus: A web server for protein cavity detection with pharmacophore modelling, allosteric site identification and covalent ligand binding ability prediction. Nucleic Acids Res. 2018, 46, W374–W379. [Google Scholar] [CrossRef] [PubMed]
- Hetényi, C.; Van Der Spoel, D. Blind docking of drug-sized compounds to proteins with up to a thousand residues. FEBS Lett. 2006. [Google Scholar] [CrossRef]
- Hetényi, C.; van der Spoel, D. Efficient docking of peptides to proteins without prior knowledge of the binding site. Protein Sci. 2009, 11, 1729–1737. [Google Scholar] [CrossRef]
- Hassan, N.M.; Alhossary, A.A.; Mu, Y.; Kwoh, C.K. Protein-Ligand Blind Docking Using QuickVina-W with Inter-Process Spatio-Temporal Integration. Sci. Rep. 2017. [Google Scholar] [CrossRef]
- Iorga, B.; Herlem, D.; Barré, E.; Guillou, C. Acetylcholine nicotinic receptors: Finding the putative binding site of allosteric modulators using the “blind docking” approach. J. Mol. Model. 2006, 12, 366–372. [Google Scholar] [CrossRef]
- Grant, B.J.; Lukman, S.; Hocker, H.J.; Sayyah, J.; Brown, J.H.; McCammon, J.A.; Gorfe, A.A. Novel Allosteric Sites on Ras for Lead Generation. PLoS ONE 2011, 6, e25711. [Google Scholar] [CrossRef]
- Pavlovicz, R.E.; Henderson, B.J.; Bonnell, A.B.; Boyd, R.T.; McKay, D.B.; Li, C. Identification of a Negative Allosteric Site on Human α4β2 and α3β4 Neuronal Nicotinic Acetylcholine Receptors. PLoS ONE 2011, 6, e24949. [Google Scholar] [CrossRef] [PubMed]
- Jin, T.; Yu, H.; Huang, X.F. Selective binding modes and allosteric inhibitory effects of lupane triterpenes on protein tyrosine phosphatase 1B. Sci. Rep. 2016, 6, 20766. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Jiang, H.; Li, F.; Hu, B.; Wang, Y.; Wang, M.; Wang, J.; Cheng, M. Computational insight into dengue virus NS2B-NS3 protease inhibition: A combined ligand- and structure-based approach. Comput. Biol. Chem. 2018, 77, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Hetényi, C.; Van Der Spoel, D. Toward prediction of functional protein pockets using blind docking and pocket search algorithms. Protein Sci. 2011. [Google Scholar] [CrossRef] [PubMed]
- Ghersi, D.; Sanchez, R. Improving accuracy and efficiency of blind protein-ligand docking by focusing on predicted binding sites. Proteins: Struct. Funct. Bioinform. 2009, 74, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef]
- Jain, A.N. Surflex: Fully Automatic Flexible Molecular Docking Using a Molecular Similarity-Based Search Engine. J. Med. Chem. 2003, 46, 499–511. [Google Scholar] [CrossRef]
- Venkatachalam, C.; Jiang, X.; Oldfield, T.; Waldman, M. LigandFit: A novel method for the shape-directed rapid docking of ligands to protein active sites. J. Mol. Graph. Model. 2003, 21, 289–307. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A. NIH Public Access. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Corbeil, C.R.; Williams, C.I.; Labute, P. Variability in docking success rates due to dataset preparation. J. Comput.-Aided Mol. Des. 2012, 26, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Caflisch, A. Discovery of ZAP70 inhibitors by high-throughput docking into a conformation of its kinase domain generated by molecular dynamics. Bioorgan. Med. Chem. Lett. 2013, 23, 5721–5726. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Carmona, S.; Alvarez-Garcia, D.; Foloppe, N.; Garmendia-Doval, A.B.; Juhos, S.; Schmidtke, P.; Barril, X.; Hubbard, R.E.; Morley, S.D. rDock: A Fast, Versatile and Open Source Program for Docking Ligands to Proteins and Nucleic Acids. PLoS Comput. Biol. 2014, 10, e1003571. [Google Scholar] [CrossRef]
- Allen, W.J.; Balius, T.E.; Mukherjee, S.; Brozell, S.R.; Moustakas, D.T.; Lang, P.T.; Case, D.A.; Kuntz, I.D.; Rizzo, R.C. DOCK 6: Impact of new features and current docking performance. J. Comput. Chem. 2015, 36, 1132–1156. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, H.; Yao, X.; Li, D.; Xu, L.; Li, Y.; Tian, S.; Hou, T. Comprehensive evaluation of ten docking programs on a diverse set of protein–ligand complexes: The prediction accuracy of sampling power and scoring power. Phys. Chem. Chem. Phys. 2016, 18, 12964–12975. [Google Scholar] [CrossRef]
- Lepšík, M.; Řezáč, J.; Kolář, M.; Pecina, A.; Hobza, P.; Fanfrlík, J. The Semiempirical Quantum Mechanical Scoring Function for In Silico Drug Design. ChemPlusChem 2013, 78, 921–931. [Google Scholar] [CrossRef]
- Pecina, A.; Haldar, S.; Fanfrlík, J.; Meier, R.; Řezáč, J.; Lepšík, M.; Hobza, P. SQM/COSMO Scoring Function at the DFTB3-D3H4 Level: Unique Identification of Native Protein–Ligand Poses. J. Chem. Inf. Model. 2017, 57, 127–132. [Google Scholar] [CrossRef]
- Fanfrlik, J.; Bronowska, A.K.; Rezac, J.; Prenosil, O.; Konvalinka, J.; Hobza, P. A Reliable Docking/Scoring Scheme Based on the Semiempirical Quantum Mechanical PM6-DH2 Method Accurately Covering Dispersion and H-Bonding: HIV-1 Protease with 22 Ligands. J. Phys. Chem. B 2010, 114, 12666–12678. [Google Scholar] [CrossRef]
- Brahmkshatriya, P.S.; Dobes, P.; Fanfrlik, J.; Rezac, J.; Paruch, K.; Bronowska, A.; Lepsík, M.; Hobza, P. Quantum Mechanical Scoring: Structural and Energetic Insights into Cyclin-Dependent Kinase 2 Inhibition by Pyrazolo[1,5-a]pyrimidines. Curr. Comput. Aided-Drug Des. 2013, 9, 118–129. [Google Scholar] [CrossRef]
- Dobes, P.; Rezac, J.; Fanfrlik, J.; Otyepka, M.; Hobza, P. Semiempirical Quantum Mechanical Method PM6-DH2X Describes the Geometry and Energetics of CK2-Inhibitor Complexes Involving Halogen Bonds Well, While the Empirical Potential Fails. J. Phys. Chem. B 2011, 115, 8581–8589. [Google Scholar] [CrossRef] [PubMed]
- Abdizadeh, H.; Guven, G.; Atilgan, A.R.; Atilgan, C. Perturbation response scanning specifies key regions in subtilisin serine protease for both function and stability. J. Enzym. Inhib. Med. Chem. 2015, 30, 867–873. [Google Scholar] [CrossRef] [PubMed]
- Penkler, D.L.; Sensoy, Ö.; Atilgan, C.; Tastan Bishop, Ö. Perturbation-Response Scanning Reveals Key Residues for Allosteric Control in Hsp70. J. Chem. Inf. Model. 2017, 57, 1359–1374. [Google Scholar] [CrossRef] [PubMed]
- Hatherley, R.; Brown, D.K.; Musyoka, T.M.; Penkler, D.L.; Faya, N.; Lobb, K.A.; Tastan Bishop, Ö. SANCDB: A South African natural compound database. J. Cheminform. 2015, 7, 29. [Google Scholar] [CrossRef]
- Song, K.; Li, Q.; Gao, W.; Lu, S.; Shen, Q.; Liu, X.; Wu, Y.; Wang, B.; Lin, H.; Chen, G.; et al. AlloDriver: A method for the identification and analysis of cancer driver targets. Nucleic Acids Res. 2019, 47, W315–W321. [Google Scholar] [CrossRef]
- Huang, M.; Song, K.; Liu, X.; Lu, S.; Shen, Q.; Wang, R.; Gao, J.; Hong, Y.; Li, Q.; Ni, D.; et al. AlloFinder: A strategy for allosteric modulator discovery and allosterome analyses. Nucleic Acids Res. 2018, 46, W451–W458. [Google Scholar] [CrossRef]
- Li, S.; Shen, Q.; Su, M.; Liu, X.; Lu, S.; Chen, Z.; Wang, R.; Zhang, J. Alloscore: A method for predicting allosteric ligand-protein interactions. Bioinformatics 2016, 32, 1574–1576. [Google Scholar] [CrossRef]
- Guarnera, E.; Tan, Z.W.; Zheng, Z.; Berezovsky, I.N. AlloSigMA: Allosteric signaling and mutation analysis server. Bioinformatics 2017, 33, 3996–3998. [Google Scholar] [CrossRef]
- Kaya, C.; Armutlulu, A.; Ekesan, S.; Haliloglu, T. MCPath: Monte Carlo path generation approach to predict likely allosteric pathways and functional residues. Nucleic Acids Res. 2013, 41, 249–255. [Google Scholar] [CrossRef]
- Clarke, D.; Sethi, A.; Li, S.; Kumar, S.; Chang, R.W.; Chen, J.; Gerstein, M. Identifying Allosteric Hotspots with Dynamics: Application to Inter- and Intra-species Conservation. Structure 2016, 24, 826–837. [Google Scholar] [CrossRef]
- Atilgan, C.; Atilgan, A.R. Perturbation-Response Scanning Reveals Ligand Entry-Exit Mechanisms of Ferric Binding Protein. PLoS Comput. Biol. 2009, 5. [Google Scholar] [CrossRef]
- Bahar, I.; Lezon, T.R.; Bakan, A.; Shrivastava, I.H. Normal Mode Analysis of Biomolecular Structures: Functional Mech Membrane Proteins. Chem. Rev. 2010, 110, 1463–1497. [Google Scholar] [CrossRef] [PubMed]
- Gerek, Z.N.; Ozkan, S.B. Change in allosteric network affects binding affinities of PDZ domains: Analysis through perturbation response scanning. PLoS Comput. Biol. 2011, 7, 18–25. [Google Scholar] [CrossRef]
- Vijayabaskar, M.; Vishveshwara, S. Interaction Energy Based Protein Structure Networks. Biophys. J. 2010, 99, 3704–3715. [Google Scholar] [CrossRef] [PubMed]
- Di Paola, L.; Giuliani, A. Protein contact network topology: A natural language for allostery. Curr. Opin. Struct. Biol. 2015, 31, 43–48. [Google Scholar] [CrossRef]
- Dokholyan, N.V. Controlling Allosteric Networks in Proteins. Chem. Rev. 2016. [Google Scholar] [CrossRef]
- Feher, V.A.; Durrant, J.D.; Van Wart, A.T.; Amaro, R.E. Computational approaches to mapping allosteric pathways. Curr. Opin. Struct. Biol. 2014, 25, 98–103. [Google Scholar] [CrossRef]
- Stolzenberg, S.; Michino, M.; LeVine, M.V.; Weinstein, H.; Shi, L. Computational approaches to detect allosteric pathways in transmembrane molecular machines. Biochim. Biophys. Acta Biomembr. 2016, 1858, 1652–1662. [Google Scholar] [CrossRef]
- Ricci, C.G.; Silveira, R.L.; Rivalta, I.; Batista, V.S.; Skaf, M.S. Allosteric Pathways in the PPARγ-RXRα nuclear receptor complex. Sci. Rep. 2016, 6, 19940. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Vaidehi, N. Differences in Allosteric Communication Pipelines in the Inactive and Active States of a GPCR. Biophys. J. 2014, 107, 422–434. [Google Scholar] [CrossRef]
- Guo, J.; Pang, X.; Zhou, H.X. Two Pathways Mediate Interdomain Allosteric Regulation in Pin1. Structure 2015, 23, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Blacklock, K.; Verkhivker, G.M. Allosteric Regulation of the Hsp90 Dynamics and Stability by Client Recruiter Cochaperones: Protein Structure Network Modeling. PLoS ONE 2014, 9, e86547. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Sakaguchi, R.; Liu, C.; Vishveshwara, S.; Hou, Y.M. Allosteric Communication in Cysteinyl tRNA Synthetase. J. Biol. Chem. 2011, 286, 37721–37731. [Google Scholar] [CrossRef] [PubMed]
- Sethi, A.; Eargle, J.; Black, A.A.; Luthey-Schulten, Z. Dynamical networks in tRNA:protein complexes. Proc. Natl. Acad. Sci. USA 2009, 106, 6620–6625. [Google Scholar] [CrossRef] [PubMed]
- Rivalta, I.; Sultan, M.M.; Lee, N.S.; Manley, G.A.; Loria, J.P.; Batista, V.S. Allosteric pathways in imidazole glycerol phosphate synthase. Proc. Natl. Acad. Sci. USA 2012, 109, E1428–E1436. [Google Scholar] [CrossRef] [PubMed]
- Ming, D.; Wall, M.E. Quantifying allosteric effects in proteins. Proteins: Struct. Funct. Bioinform. 2005, 59, 697–707. [Google Scholar] [CrossRef]
- Ming, D.; Wall, M.E. Interactions in Native Binding Sites Cause a Large Change in Protein Dynamics. J. Mol. Biol. 2006, 358, 213–223. [Google Scholar] [CrossRef]
- Mitternacht, S.; Berezovsky, I.N. Binding Leverage as a Molecular Basis for Allosteric Regulation. PLoS Comput. Biol. 2011, 7, e1002148. [Google Scholar] [CrossRef]
- Bowman, G.R.; Geissler, P.L. Equilibrium fluctuations of a single folded protein reveal a multitude of potential cryptic allosteric sites. Proc. Natl. Acad. Sci. USA 2012, 109, 11681–11686. [Google Scholar] [CrossRef]
- McClendon, C.L.; Friedland, G.; Mobley, D.L.; Amirkhani, H.; Jacobson, M.P. Quantifying Correlations Between Allosteric Sites in Thermodynamic Ensembles. J. Chem. Theory Comput. 2009, 5, 2486–2502. [Google Scholar] [CrossRef]
- Ausiello, G.; Firmani, D.; Laura, L. The (betweenness) centrality of critical nodes and network cores. In Proceedings of the 2013 9th International Wireless Communications and Mobile Computing Conference (IWCMC), Sardinia, Italy, 1–5 July 2013; pp. 90–95. [Google Scholar] [CrossRef]
- Kimuda, M.P.; Laming, D.; Hoppe, H.C.; Bishop, O.T. Identification of Novel Potential Inhibitors of Pteridine Reductase 1 in Trypanosoma brucei via Computational Structure-Based Approaches and in Vitro Inhibition Assays. Molecules 2019, 24, 142. [Google Scholar] [CrossRef] [PubMed]
- Süel, G.M.; Lockless, S.W.; Wall, M.A.; Ranganathan, R. Evolutionarily conserved networks of residues mediate allosteric communication in proteins. Nat. Struct. Biol. 2003, 10, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Lockless, S.W.; Ranganathan, R. Evolutionarily Conserved Pathways of Energetic Connectivity in Protein Families. Science 1999, 286, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Halabi, N.; Rivoire, O.; Leibler, S.; Ranganathan, R. Protein Sectors: Evolutionary Units of Three-Dimensional Structure. Cell 2009, 138, 774–786. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, R.N.; Poelwijk, F.J.; Raman, A.; Gosal, W.S.; Ranganathan, R. The spatial architecture of protein function and adaptation. Nature 2012. [Google Scholar] [CrossRef] [PubMed]
- Marino Buslje, C.; Teppa, E.; Di Doménico, T.; Delfino, J.M.; Nielsen, M. Networks of High Mutual Information Define the Structural Proximity of Catalytic Sites: Implications for Catalytic Residue Identification. PLoS Comput. Biol. 2010, 6, e1000978. [Google Scholar] [CrossRef]
- Simonetti, F.L.; Teppa, E.; Chernomoretz, A.; Nielsen, M.; Marino Buslje, C. MISTIC: Mutual information server to infer coevolution. Nucleic Acids Res. 2013, 41, W8–W14. [Google Scholar] [CrossRef]
- Aguilar, D.; Oliva, B.; Marino Buslje, C. Mapping the mutual information network of enzymatic families in the protein structure to unveil functional features. PLoS ONE 2012. [Google Scholar] [CrossRef]
- de Juan, D.; Pazos, F.; Valencia, A. Emerging methods in protein co-evolution. Nat. Rev. Genet. 2013, 14, 249–261. [Google Scholar] [CrossRef]
- Socolich, M.; Lockless, S.W.; Russ, W.P.; Lee, H.; Gardner, K.H.; Ranganathan, R. Evolutionary information for specifying a protein fold. Nature 2005, 437, 512–518. [Google Scholar] [CrossRef]
- Morcos, F.; Pagnani, A.; Lunt, B.; Bertolino, A.; Marks, D.S.; Sander, C.; Zecchina, R.; Onuchic, J.N.; Hwa, T.; Weigt, M. Direct-coupling analysis of residue coevolution captures native contacts across many protein families. Proc. Natl. Acad. Sci. USA 2011, 108. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Lodge, J.M.; Fierke, C.A.; Mapp, A.K. Dissecting allosteric effects of activator-coactivator complexes using a covalent small molecule ligand. Proc. Natl. Acad. Sci. USA 2014, 111, 12061–12066. [Google Scholar] [CrossRef] [PubMed]
- Nishi, H.; Hashimoto, K.; Panchenko, A.R. Phosphorylation in Protein-Protein Binding: Effect on Stability and Function. Structure 2011, 19, 1807–1815. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, S.; Panchenko, A.R. Coevolution in defining the functional specificity. Proteins: Struct. Funct. Bioinform. 2009, 75, 231–240. [Google Scholar] [CrossRef]
- Chakrabarti, S.; Panchenko, A.R. Structural and Functional Roles of Coevolved Sites in Proteins. PLoS ONE 2010, 5, e8591. [Google Scholar] [CrossRef]
- Martin, L.C.; Gloor, G.B.; Dunn, S.D.; Wahl, L.M. Using information theory to search for co-evolving residues in proteins. Bioinformatics 2005, 21, 4116–4124. [Google Scholar] [CrossRef]
- Gloor, G.B.; Martin, L.C.; Wahl, L.M.; Dunn, S.D. Mutual Information in Protein Multiple Sequence Alignments Reveals Two Classes of Coevolving Positions †. Biochemistry 2005, 44, 7156–7165. [Google Scholar] [CrossRef]
- Tillier, E.R.; Lui, T.W. Using multiple interdependency to separate functional from phylogenetic correlations in protein alignments. Bioinformatics 2003, 19, 750–755. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, Y.; Gao, Y.; Li, G.; Huang, J. Integrated Analysis of Residue Coevolution and Protein Structures Capture Key Protein Sectors in HIV-1 Proteins. PLoS ONE 2015, 10, e0117506. [Google Scholar] [CrossRef]
- Xu, P.; Duong, D.M.; Seyfried, N.T.; Cheng, D.; Xie, Y.; Robert, J.; Rush, J.; Hochstrasser, M.; Finley, D.; Peng, J. Quantitative Proteomics Reveals the Function of Unconventional Ubiquitin Chains in Proteasomal Degradation. Cell 2009, 137, 133–145. [Google Scholar] [CrossRef]
- Hsu, Y.H.; Traugh, J.A. Reciprocally Coupled Residues Crucial for Protein Kinase Pak2 Activity Calculated by Statistical Coupling Analysis. PLoS ONE 2010, 5, e9455. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.; Nam, H.J.; Choi, Y.S.; Yang, J.S.; Hwang, J.; Kim, S. Molecular Evolution of Protein Conformational Changes Revealed by a Network of Evolutionarily Coupled Residues. Mol. Biol. Evol. 2011, 28, 2675–2685. [Google Scholar] [CrossRef] [PubMed]
- Tse, A.; Verkhivker, G.M. Exploring Molecular Mechanisms of Paradoxical Activation in the BRAF Kinase Dimers: Atomistic Simulations of Conformational Dynamics and Modeling of Allosteric Communication Networks and Signaling Pathways. PLoS ONE 2016, 11, e0166583. [Google Scholar] [CrossRef] [PubMed]
- Verkhivker, G.M. Integrating genetic and structural data on human protein kinome in network-based modeling of kinase sensitivities and resistance to targeted and personalized anticancer drugs. Biocomputing 2016. [Google Scholar] [CrossRef]
- Albano, J.M.; de Paula, E.; Pickholz, M. Molecular Dynamics Simulations to Study Drug Delivery Systems. In Molecular Dynamics; InTechOpen: Melbourne, Australia, 2018; Chapter 5. [Google Scholar] [CrossRef]
- Proctor, E.A.; Kota, P.; Aleksandrov, A.A.; He, L.; Riordan, J.R.; Dokholyan, N.V. Rational coupled dynamics network manipulation rescues disease-relevant mutant cystic fibrosis transmembrane conductance regulator. Chem. Sci. 2015. [Google Scholar] [CrossRef] [PubMed]
- Kolinski, A. Protein modeling and structure prediction with a reduced representation. Acta Biochim. Pol. 2004, 51, 349–371. [Google Scholar] [CrossRef]
- Kmiecik, S.; Gront, D.; Kolinski, M.; Wieteska, L.; Dawid, A.E.; Kolinski, A. Coarse-Grained Protein Models and Their Applications. Chem. Rev. 2016, 116, 7898–7936. [Google Scholar] [CrossRef]
- Kmiecik, S.; Kouza, M.; Badaczewska-Dawid, A.; Kloczkowski, A.; Kolinski, A. Modeling of Protein Structural Flexibility and Large-Scale Dynamics: Coarse-Grained Simulations and Elastic Network Models. Int. J. Mol. Sci. 2018, 19, 3496. [Google Scholar] [CrossRef]
- Jamroz, M.; Kolinski, A.; Kmiecik, S. CABS-flex: Server for fast simulation of protein structure fluctuations. Nucleic Acids Res. 2013, 41, 427–431. [Google Scholar] [CrossRef]
- Krüger, D.M.; Ahmed, A.; Gohlke, H. NMSim web server: Integrated approach for normal mode-based geometric simulations of biologically relevant conformational transitions in proteins. Nucleic Acids Res. 2012, 40, 310–316. [Google Scholar] [CrossRef]
- Camps, J.; Carrillo, O.; Emperador, A.; Orellana, L.; Hospital, A.; Rueda, M.; Cicin-Sain, D.; D’Abramo, M.; Gelpí, J.L.; Orozco, M. FlexServ: An integrated tool for the analysis of protein flexibility. Bioinformatics 2009, 25, 1709–1710. [Google Scholar] [CrossRef] [PubMed]
- Ciemny, M.P.; Badaczewska-Dawid, A.E.; Pikuzinska, M.; Kolinski, A.; Kmiecik, S. Modeling of disordered protein structures using monte carlo simulations and knowledge-based statistical force fields. Int. J. Mol. Sci. 2019, 20, 606. [Google Scholar] [CrossRef] [PubMed]
- Tirion, M.M. Large amplitude elastic motions in proteins from a single-parameter, atomic analysis. Phys. Rev. Lett. 1996, 77, 1905–1908. [Google Scholar] [CrossRef] [PubMed]
- Bahar, I.; Atilgan, A.R.; Erman, B. Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential. Fold. Des. 1997, 2, 173–181. [Google Scholar] [CrossRef]
- Haliloglu, T.; Bahar, I.; Erman, B. Gaussian Dynamics of Folded Proteins. Phys. Rev. Lett. 1997, 79, 3090–3093. [Google Scholar] [CrossRef]
- Doruker, P.; Atilgan, A.R.; Bahar, I. Dynamics of proteins predicted by molecular dynamics simulations and analytical approaches: Application to α-amylase inhibitor. Proteins: Struct. Funct. Bioinf. 2000, 40, 512–524. [Google Scholar] [CrossRef]
- Romo, T.D.; Grossfield, A. Validating and improving elastic network models with molecular dynamics simulations. Proteins: Struct. Funct. Bioinform. 2011, 79, 23–34. [Google Scholar] [CrossRef]
- Isin, B.; Doruker, P.; Bahar, I. Functional Motions of Influenza Virus Hemagglutinin: A Structure-Based Analytical Approach. Biophys. J. 2002, 82, 569–581. [Google Scholar] [CrossRef]
- Temiz, N.A.; Bahar, I. Inhibitor binding alters the directions of domain motions in HIV-1 reverse transcriptase. Proteins: Struct. Funct. Genet. 2002, 49, 61–70. [Google Scholar] [CrossRef]
- Xu, C.; Tobi, D.; Bahar, I. Allosteric Changes in Protein Structure Computed by a Simple Mechanical Model: Hemoglobin T-R2 Transition. J. Mol. Biol. 2003, 333, 153–168. [Google Scholar] [CrossRef]
- Taly, A.; Delarue, M.; Grutter, T.; Nilges, M.; Le Novère, N.; Corringer, P.J.; Changeux, J.P. Normal Mode Analysis Suggests a Quaternary Twist Model for the Nicotinic Receptor Gating Mechanism. Biophys. J. 2005, 88, 3954–3965. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, I.H.; Bahar, I. Common Mechanism of Pore Opening Shared by Five Different Potassium Channels. Biophys. J. 2006, 90, 3929–3940. [Google Scholar] [CrossRef]
- Liu, X.; Xu, Y.; Li, H.; Wang, X.; Jiang, H.; Barrantes, F.J. Mechanics of Channel Gating of the Nicotinic Acetylcholine Receptor. PLoS Comput. Biol. 2008, 4, e19. [Google Scholar] [CrossRef] [PubMed]
- Isin, B.; Tirupula, K.C.; Oltvai, Z.N.; Klein-Seetharaman, J.; Bahar, I. Identification of motions in membrane proteins by elastic network models and their experimental validation. Methods Mol. Biol. 2012. [Google Scholar] [CrossRef]
- Ross, C.J.; Atilgan, A.R.; Tastan Bishop, Ö.; Atilgan, C. Unraveling the Motions behind Enterovirus 71 Uncoating. Biophys. J. 2018, 114, 822–838. [Google Scholar] [CrossRef]
- Zheng, W.; Brooks, B.R.; Thirumalai, D. Low-frequency normal modes that describe allosteric transitions in biological nanomachines are robust to sequence variations. Proc. Natl. Acad. Sci. USA 2006, 103, 7664–7669. [Google Scholar] [CrossRef]
- Hyeon, C.; Lorimer, G.H.; Thirumalai, D. Dynamics of allosteric transitions in GroEL. Proc. Natl. Acad. Sci. USA 2006, 103, 18939–18944. [Google Scholar] [CrossRef]
- Stan, G.; Lorimer, G.H.; Thirumalai, D.; Brooks, B.R. Coupling between allosteric transitions in GroEL and assisted folding of a substrate protein. Proc. Natl. Acad. Sci. USA 2007, 104, 8803–8808. [Google Scholar] [CrossRef]
- Chennubhotla, C.; Bahar, I. Markov propagation of allosteric effects in biomolecular systems: application to GroEL–GroES. Mol. Syst. Biol. 2006, 2, 36. [Google Scholar] [CrossRef]
- Chennubhotla, C.; Bahar, I. Signal Propagation in Proteins and Relation to Equilibrium Fluctuations. PLoS Comput. Biol. 2007, 3, e172. [Google Scholar] [CrossRef]
- Bahar, I.; Chennubhotla, C.; Tobi, D. Intrinsic dynamics of enzymes in the unbound state and relation to allosteric regulation. Curr. Opin. Struct. Biol. 2007, 17, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Chennubhotla, C.; Yang, Z.; Bahar, I. Coupling between global dynamics and signal transduction pathways: A mechanism of allostery for chaperonin GroEL. Mol. BioSyst. 2008, 4, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Májek, P.; Bahar, I. Allosteric Transitions of Supramolecular Systems Explored by Network Models: Application to Chaperonin GroEL. PLoS Comput. Biol. 2009, 5. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Tekpinar, M. Large-scale evaluation of dynamically important residues in proteins predicted by the perturbation analysis of a coarse-grained elastic model. BMC Struct. Biol. 2009, 9, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Brooks, B.; Thirumalai, D. Allosteric Transitions in Biological Nanomachines are Described by Robust Normal Modes of Elastic Networks. Curr. Protein Pept. Sci. 2009, 10, 128–132. [Google Scholar] [CrossRef]
- Yang, L.; Song, G.; Jernigan, R.L. Protein elastic network models and the ranges of cooperativity. Proc. Natl. Acad. Sci. USA 2009, 106, 12347–12352. [Google Scholar] [CrossRef]
- Pande, V.S. Understanding protein folding using Markov state models. Adv. Exp. Med. Biol. 2014, 797, 101–106. [Google Scholar] [CrossRef]
- Shukla, D.; Hernández, C.X.; Weber, J.K.; Pande, V.S. Markov state models provide insights into dynamic modulation of protein function. Acc. Chem. Res. 2015, 48, 414–422. [Google Scholar] [CrossRef]
- Shukla, S.; Shamsi, Z.; Moffett, A.S.; Selvam, B.; Shukla, D. Application of Hidden Markov Models in Biomolecular Simulations. In Hidden Markov Models; Methods in Molecular Biology; Westhead, D.R., Vijayabaskar, M.S., Eds.; Springer New York: New York, NY, USA, 2017; Volume 1552, pp. 29–41. [Google Scholar] [CrossRef]
- Husic, B.E.; Pande, V.S. Markov State Models: From an Art to a Science. J. Am. Chem. Soc. 2018, 140, 2386–2396. [Google Scholar] [CrossRef]
- McGibbon, R.T.; Schwantes, C.R.; Pande, V.S. Statistical Model Selection for Markov Models of Biomolecular Dynamics. J. Phys. Chem. B 2014, 118, 6475–6481. [Google Scholar] [CrossRef]
- Wu, H.; Paul, F.; Wehmeyer, C.; Noé, F. Multiensemble Markov models of molecular thermodynamics and kinetics. Proc. Natl. Acad. Sci. USA 2016, 113, E3221–E3230. [Google Scholar] [CrossRef] [PubMed]
- Prinz, J.H.; Wu, H.; Sarich, M.; Keller, B.; Senne, M.; Held, M.; Chodera, J.D.; Schtte, C.; Noé, F. Markov models of molecular kinetics: Generation and validation. J. Chem. Phys. 2011, 134. [Google Scholar] [CrossRef] [PubMed]
- Harrigan, M.P.; Sultan, M.M.; Hernández, C.X.; Husic, B.E.; Eastman, P.; Schwantes, C.R.; Beauchamp, K.A.; McGibbon, R.T.; Pande, V.S. MSMBuilder: Statistical Models for Biomolecular Dynamics. Biophys. J. 2017, 112, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Cronkite-Ratcliff, B.; Pande, V. MSMExplorer: Visualizing Markov state models for biomolecule folding simulations. Bioinformatics 2013, 29, 950–952. [Google Scholar] [CrossRef] [PubMed]
- Bowman, G.R.; Noé, F. An Introduction to Markov State Models and Their Application to Long Timescale Molecular Simulation. In An Introduction to Markov State Models and Their Application to Long Timescale Molecular Simulation; Bowman, G.R., Pande, V.S., Noé, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; Volume 797, Chapter 11; p. 148. [Google Scholar] [CrossRef]
- Bowman, G.R. A Tutorial on Building Markov State Models with MSMBuilder and Coarse-Graining Them with BACE. In Protein Dynamics: Methods and Protocols; Methods in Molecular Biology; Livesay, D.R., Ed.; Springer: Totowa, NJ, USA, 2014; Volume 1084, Chapter 8; pp. 141–158. [Google Scholar] [CrossRef]
- Hart, K.M.; Ho, C.M.W.; Dutta, S.; Gross, M.L.; Bowman, G.R. Modelling proteins’ hidden conformations to predict antibiotic resistance. Nat. Commun. 2016, 7, 12965. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, U.; Strodel, B. Markov models for the elucidation of allosteric regulation. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373. [Google Scholar] [CrossRef] [PubMed]
- Rosvall, M.; Bergstrom, C.T. An information-theoretic framework for resolving community structure in complex networks. Proc. Natl. Acad. Sci. USA 2007, 104, 7327–7331. [Google Scholar] [CrossRef]
- Rosvall, M.; Bergstrom, C.T. Maps of random walks on complex networks reveal community structure. Proc. Natl. Acad. Sci. USA 2008, 105, 1118–1123. [Google Scholar] [CrossRef]
- Rosvall, M.; Bergstrom, C.T. Mapping Change in Large Networks. PLoS ONE 2010, 5, e8694. [Google Scholar] [CrossRef]
- Rosvall, M.; Bergstrom, C.T. Multilevel Compression of Random Walks on Networks Reveals Hierarchical Organization in Large Integrated Systems. PLoS ONE 2011, 6, e18209. [Google Scholar] [CrossRef]
- Rosvall, M.; Esquivel, A.V.; Lancichinetti, A.; West, J.D.; Lambiotte, R. Memory in network flows and its effects on spreading dynamics and community detection. Nat. Commun. 2014, 5, 4630. [Google Scholar] [CrossRef] [PubMed]
- Delvenne, J.C.; Lambiotte, R.; Rocha, L.E.C. Diffusion on networked systems is a question of time or structure. Nat. Commun. 2015, 6, 7366. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, T.; Rosvall, M. Estimating the resolution limit of the map equation in community detection. Phys. Rev. E 2015, 91, 012809. [Google Scholar] [CrossRef] [PubMed]
- Aslak, U.; Rosvall, M.; Lehmann, S. Constrained information flows in temporal networks reveal intermittent communities. Phys. Rev. E 2018, 97, 062312. [Google Scholar] [CrossRef]
- Lambiotte, R.; Rosvall, M.; Scholtes, I. From networks to optimal higher-order models of complex systems. Nat. Phys. 2019, 15, 313–320. [Google Scholar] [CrossRef]
- Naithani, A.; Taylor, P.; Erman, B.; Walkinshaw, M.D. A Molecular Dynamics Study of Allosteric Transitions in Leishmania mexicana Pyruvate Kinase. Biophys. J. 2015, 109, 1149–1156. [Google Scholar] [CrossRef]
- Roca, C.; Requena, C.; Sebastián-Pérez, V.; Malhotra, S.; Radoux, C.; Pérez, C.; Martinez, A.; Antonio Páez, J.; Blundell, T.L.; Campillo, N.E. Identification of new allosteric sites and modulators of AChE through computational and experimental tools. J. Enzym. Inhib. Med. Chem. 2018, 33, 1034–1047. [Google Scholar] [CrossRef]
- Bowerman, S.; Wereszczynski, J. Detecting Allosteric Networks Using Molecular Dynamics Simulation. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2016; Volume 578, pp. 429–447. [Google Scholar] [CrossRef]
- Singh, B.; Bulusu, G.; Mitra, A. Understanding the thermostability and activity of bacillus subtilis lipase mutants: Insights from molecular dynamics simulations. J. Phys. Chem. B 2015, 119, 392–409. [Google Scholar] [CrossRef]
- Khan, S.; Farooq, U.; Kurnikova, M. Exploring protein stability by comparative molecular dynamics simulations of homologous hyperthermophilic, mesophilic, and psychrophilic proteins. J. Chem. Inf. Model. 2016, 56, 2129–2139. [Google Scholar] [CrossRef]
- Karamzadeh, R.; Karimi-Jafari, M.H.; Sharifi-Zarchi, A.; Chitsaz, H.; Salekdeh, G.H.; Moosavi-Movahedi, A.A. Machine Learning and Network Analysis of Molecular Dynamics Trajectories Reveal Two Chains of Red/Ox-specific Residue Interactions in Human Protein Disulfide Isomerase. Sci. Rep. 2017, 7, 3666. [Google Scholar] [CrossRef]
- David, C.C.; Jacobs, D.J. Principal component analysis: A method for determining the essential dynamics of proteins. Methods Mol. Biol. 2014, 1084, 193–226. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.; Kirschner, K.N. Principal component and clustering analysis on molecular dynamics data of the ribosomal L11·23S subdomain. J. Mol. Model. 2013, 19, 539–549. [Google Scholar] [CrossRef]
- Kumar, V.; Pandey, P.; Idrees, D.; Prakash, A.; Lynn, A. Delineating the effect of mutations on the conformational dynamics of N-terminal domain of TDP-43. Biophys. Chem. 2019, 250, 106174. [Google Scholar] [CrossRef] [PubMed]
- Mehmood, A.; Khan, M.T.; Kaushik, A.C.; Khan, A.S.; Irfan, M.; Wei, D.Q. Structural Dynamics Behind Clinical Mutants of PncA-Asp12Ala, Pro54Leu, and His57Pro of Mycobacterium tuberculosis Associated With Pyrazinamide Resistance. Front. Bioeng. Biotechnol. 2019, 7, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Ricci-López, J.; Vidal-Limon, A.; Zunñiga, M.; Jimènez, V.A.; Alderete, J.B.; Brizuela, C.A.; Aguila, S. Molecular modeling simulation studies reveal new potential inhibitors against HPV E6 protein. PLoS ONE 2019, 14, e0213028. [Google Scholar] [CrossRef]
- Tzul, F.O.; Vasilchuk, D.; Makhatadze, G.I. Evidence for the principle of minimal frustration in the evolution of protein folding landscapes. Proc. Natl. Acad. Sci. USA 2017, 114, E1627–E1632. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug/Code Name | Medical Condition | Mechanism | Enzyme Target | Discovery Method | 2D Structure |
|---|---|---|---|---|---|
| Carglumic Acid | Acute hyper- ammonaemia | Activator | Carbamoyl phosphate synthetase 1 | Experiments in rats, both in vivo and in vitro [140] |  |
| Cinacalcet | Hyper- parathyroidism | Activator | G protein- coupled receptor | Functional responses of cells regulated by calcium receptor activity: PTH secretion by parathyroid cells, calcitonin secretion by C-cells, and bone resorption by osteoclasts. [141] |  |
| Clonazepam | Epilepsy | Activator | -amino- butyric acid (GABA) | Perifused frog neuro- intermediate lobes [142] |  |
| Cobimetinib | Melanoma | Inhibitor | MAPK1, MEK1 & MEK2 | Structural insight—manipulation of previously known MEK inhibitors’ structure. Ligand- binding affinity assays [143] |  |
| Cyclothiazide | Hypertension | Activator | AMPA Receptor | AMPA- and KA-induced [3H]NE release from slices of rat hippocampus [144] |  |
| Drotaverine | Irritable bowel syndrome | Inhibitor | L-type Ca2+ channel | Saturation studies. Dissociation kinetics [145] |  |
| Enasidenib | Acute myeloid leukemia | Inhibitor | IDH2 | In silico: Binding free energy, conformational change [146] |  |
| Flurazepam | Insomnia | Activator | GABA-A receptor | Site-directed mutagenesis. Concentration-response analysis [147] |  |
| Ivermectin | Parasite infestations | Activator | Alpha7 neuronal nicotinic acetylcholine receptor | Mutagenesis. Cell line, culture, and recordings [148] |  |
| Ketazolam | Anxiety disorder | Activator | GABA-A receptor | Increase of GABA level in cat spinal cord and in the total brain of mice and rats [149] |  |
| Lorazepam | Anxiety disorder | Activator | -adrenergic receptor | Transfection. Ligand-binding affinity assays [150] |  |
| Maraviroc | HIV | Inhibitor | C-C chemokine receptor type 5 | Displacement binding assays. Dissociation kinetics [151] |  |
| Niclosamide | Neuropathic pain | Inhibitor | Group 1 metabotropic glutamate receptor | Calcium mobilization assays. Cross-receptor selectivity experiments. Computati- onal molecular modeling analysis. NP-evoked mechanical hyperalgesia model in rats [152] |  |
| Piracetam | Dementia, vertigo, cortical myoclonus, dyslexia, and sickle cell anemia | Activator | AMPA Receptor | Enzyme crystallization. Crystal structure determination. Structure analysis [153] |  |
| Rifapentine | Tuberculosis | Inhibitor | DNA- dependent RNA polymerase | Site-directed mutagenesis. In vitro transcription. RFP binding assays [154] |  |
| Rilpivirine | HIV | Inhibitor | HIV-1 reverse transcriptase | X-ray crystallo- graphy. Molecular modeling. Optimizing lead compounds [155] |  |
| Sirolimus | Immuno- suppressive | Inhibitor | FK Binding Protein-12 | Site-directed mutagenesis. FKBP12- Rapamycin (Sirolimus) binding assays [156] |  |
| Ticagrelor | Stroke; Acute coronary syndrome undergoing percutaneous coronary intervention | Inhibitor | G protein- coupled receptor | ATP analogue production. Platelet inhibition and patient outcome (PLATO) trial [157] |  |
| Trametinib | Melanoma | Inhibitor | MEK1 & MEK2 | Enzymatic and cellular studies. Pharmacokinetic analysis [158] |  |
| Web Server and URL | Functionality | Input | Output |
|---|---|---|---|
| AlloDriver [220] | Identifies potential driver mutations implicated in cancer and maps them to binding sites. | A list of annotated cancer-related mutations. | Returns a list of ranked driver mutations annotated by residue loci, scores and binding site (allosteric and orthosteric), amongst many other features. |
| AlloFinder [221] | AlloFinder identifies possible allosteric sites via dynamic perturbations and algorithms present in Allosite. It also screens for possible binders against the identified sites. Protein-ligand complexes are then scored using Alloscore algorithms. | The receptor PDB file and a ligand library. | Displays protein-ligand complex for docked ligands within the putative allosteric site. Further, a table reports the volume of the predicted allosteric site, the perturbation score, the drug-like score, the allosteric site score and the AlloScore score. Additionally, the top 100 potential allosteric ligands are ranked according to their Alloscore. Finally, the predicted site and the predicted ligands are mapped using allosterome data. |
| AlloPred [82] | Uses NMA to identify potential allosteric pockets. | The receptor PDB file and active site residues. | Displays protein structure and a list of pockets with Allopred and Fpocket rankings as well as NMA effect per residue. |
| Alloscore [222] | Uses a linear combination of non-bonded interaction terms, a deformation term and geometric features to predict the binding affinities of protein-ligand interactions. | The receptor PDB file and a pre-docked ligand MOL2 file. | File with potential ligands and their allosteric interactions (hydrogen bonds, van der Waals, hydrophobic interactions and Alloscore values). |
| AlloSigMA [223] | Calculates energetics of allosteric signalling resulting from ligand binding, mutations or a combination of the two. | The receptor PDB file. | The allosteric free energy profile, colouring residues according to difference in free energy between the ligand bound and the apo-protein. |
| Allosite 2.0 [85] | Predicts allosteric sites by means of pocket-based analysis and support vector machine (SVM) classifier algorithms. | The receptor PDB file. | Window showing the structure and identified potential allosteric sites. Pockets can be viewed on the displayed protein structure. Properties of the pocket include: (i) Its volume, (ii) Total solvent-accessible surface area (SASA), (iii) Polar SASA and (iv) Druggability score |
| AllosMod [84] | Makes use of MD simulations and energy landscapes to identify allosteric conformational changes. | The receptor PDB file and its sequence. | Returns a zipped file of further input files to be MD-run by the user via MODELLER and analysed using a provided Python script. |
| Cavity (Submodule of CavityPlus) [190] | Identifies cavities and provides their respective drug scores. | The receptor PDB file. | Displays the structure, potential cavities and constituting residues with their respective drug scores, which determine cavity druggability. |
| CorrSite (Submodule of CavityPlus) [190] | Identifies possible allosteric sites from those picked up by CavityPlus on the basis of correlated motion between allosteric and orthosteric cavities. | PDB file of a proposed orthosteric site or predetermined cavities obtained from the Cavity tool. | Displays the structure with mapped orthosteric and allosteric sites. Cavities are labelled with their corresponding correlation scores to the orthosteric site. |
| CovCys (Submodule of CavityPlus) [190] | Identifies druggable cysteine residues for covalent allosteric ligand design. | Cavities identified by the Cavity web server. | Maps any of the selected sites onto the protein structure and displays a table of Cys residues labelled by cavity ID, targetability, pKa value, exposure and their pocket binding affinity. |
| DynOmics ENM [83] | Predicts allosteric communication using ENM. | The receptor PDB file. | (i) JSmol window showing structure color-coded by the size of motions driven by the slowest two modes, lowest mobility (blue) to highest mobility (red) regions, (ii) Molecular motions animation, (iii) Mapped RMSF, (iv) 3D and 2D display of selected modes, (v) Cross correlations between residue fluctuations, and (vi) Inter-residue contact maps |
| MCPath [224] | Identifies regions in a protein structure which may function in allosteric communication using a Monte Carlo-based approach. | The receptor PDB file and pathway data (initial residue index, length and number of paths). | List of all pathways ranked according to their probabilities and populated pathways. 3D structure onto which the top three populated pathways and their residues are mapped. |
| PARS [81] | Uses NMA to identify possible allosteric pockets which, upon binding of a ligand, cause a regulatory effect in the protein. | The receptor PDB file and its sequence. | Table with identified pockets ranked according to their potential as allosteric sites. |
| SPACER [80] | Combines ENM and docking to predict allosteric communication. | The receptor PDB file. | List of ligand binding sites, for which the following can be explored: (i) Local closeness - the output structure is colored according to surface local closeness values, (ii) Binding leverage - quantifies the cost of the binding site deformation in the presence of a ligand, and (iii) Characteristics of the communication strength between a putative allosteric site and another binding site. |
| STRESS [225] | Identifies allosteric hotspot residues which result in large protein conformational changes when bound by a small ligand. | The receptor PDB file. | Ranked list of predicted sites each with an index of the binding site obtained from Monte Carlo simulations, a binding leverage score and their respective residues. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sheik Amamuddy, O.; Veldman, W.; Manyumwa, C.; Khairallah, A.; Agajanian, S.; Oluyemi, O.; Verkhivker, G.M.; Tastan Bishop, Ö. Integrated Computational Approaches and Tools for Allosteric Drug Discovery. Int. J. Mol. Sci. 2020, 21, 847. https://doi.org/10.3390/ijms21030847
Sheik Amamuddy O, Veldman W, Manyumwa C, Khairallah A, Agajanian S, Oluyemi O, Verkhivker GM, Tastan Bishop Ö. Integrated Computational Approaches and Tools for Allosteric Drug Discovery. International Journal of Molecular Sciences. 2020; 21(3):847. https://doi.org/10.3390/ijms21030847
Chicago/Turabian StyleSheik Amamuddy, Olivier, Wayde Veldman, Colleen Manyumwa, Afrah Khairallah, Steve Agajanian, Odeyemi Oluyemi, Gennady M. Verkhivker, and Özlem Tastan Bishop. 2020. "Integrated Computational Approaches and Tools for Allosteric Drug Discovery" International Journal of Molecular Sciences 21, no. 3: 847. https://doi.org/10.3390/ijms21030847
APA StyleSheik Amamuddy, O., Veldman, W., Manyumwa, C., Khairallah, A., Agajanian, S., Oluyemi, O., Verkhivker, G. M., & Tastan Bishop, Ö. (2020). Integrated Computational Approaches and Tools for Allosteric Drug Discovery. International Journal of Molecular Sciences, 21(3), 847. https://doi.org/10.3390/ijms21030847