Mass Spectrometry-Based Multivariate Proteomic Tests for Prediction of Outcomes on Immune Checkpoint Blockade Therapy: The Modern Analytical Approach


Abstract
1. Introduction
2. Multivariate Serum-Based Tests: Challenges and Solutions
3. BDX008 Test and ICB Test
4. PIR Test
5. The VeriStrat Test
6. Biological Mechanisms Associated with the Tests
7. Comparative Characteristics of the Tests
8. Independence of Proteomic Tests from Clinical Characteristics and Biomarkers for Immunotherapy
9. Discussion
10. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Sosman, J.A.; Atkins, M.B.; Leming, P.D.; et al. Five-Year Survival and Correlates Among Patients With Advanced Melanoma, Renal Cell Carcinoma, or Non–Small Cell Lung Cancer Treated With Nivolumab. JAMA Oncol. 2019, 5, 1411–1420. [Google Scholar] [CrossRef]
- Zimmermann, S.; Peters, S. Appraising the Tail of the Survival Curve in the Era of PD-1/PD-L1 Checkpoint Blockade Editorial. JAMA Oncol. 2019, 5, 1403–1405, [Epub ahead of print]. [Google Scholar] [CrossRef]
- Music, M.; Prassas, I.; Diamandis, E.P. Optimizing cancer immunotherapy: Is it time for personalized predictive biomarkers? Crit. Rev. Clin. Lab. Sci. 2018, 55, 466–479. [Google Scholar] [CrossRef] [PubMed]
- Pitteri, S.J.; Kelly-Spratt, K.S.; Gurley, K.E.; Kennedy, J.; Buson, T.B.; Chin, A.; Wang, H.; Zhang, Q.; Wong, C.H.; Chodosh, L.A.; et al. Tumor Microenvironment-Derived Proteins Dominate the Plasma Proteome Response during Breast Cancer Induction and Progression. Cancer Res. 2011, 71, 5090–5100. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Buttner, R.; Gosney, J.R.; Skov, B.G.; Adam, J.; Motoi, N.; Bloom, K.J.; Dietel, M.; Longshore, J.W.; Lopez-Rios, F.; Penault-Llorca, F.; et al. Programmed Death-Ligand 1 Immunohistochemistry Testing: A Review of Analytical Assays and Clinical Implementation in Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2017, 35, 3867–3876. [Google Scholar] [CrossRef] [PubMed]
- Dempke, W.C.M.; Fenchel, K.; Dale, S.P. Programmed cell death ligand-1 (PD-L1) as a biomarker for non-small cell lung cancer (NSCLC) treatment-are we barking up the wrong tree? Transl. Lung Cancer Res. 2018, 7, S275–S279. [Google Scholar] [CrossRef] [PubMed]
- Rittmeyer, A.; Barlesi, F.; Waterkamp, D.; Park, K.; Ciardiello, F.; von Pawel, J.; Gadgeel, S.M.; Hida, T.; Kowalski, D.M.; Dols, M.C.; et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet 2017, 389, 255–265. [Google Scholar] [CrossRef]
- Chan, T.A.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.A.; Stenzinger, A.; Peters, S. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Annal. Oncol. 2018, 30, 44–56, [Epub ahead of print]. [Google Scholar] [CrossRef]
- Lu, S.; Stein, J.E.; Rimm, D.L.; Wang, D.W.; Bell, J.M.; Johnson, D.B.; Sosman, J.A.; Schalper, K.A.; Anders, R.A.; Wang, H.; et al. Comparison of Biomarker Modalities for Predicting Response to PD-1/PD-L1 Checkpoint Blockade: A Systematic Review and Meta-analysis. JAMA Oncol. 2019, 5, 1195–1240. [Google Scholar] [CrossRef]
- Lemery, S.; Keegan, P.; Pazdur, R. First FDA Approval Agnostic of Cancer Site — When a Biomarker Defines the Indication. New Engl. J. Med. 2017, 377, 1409–1412. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Zhang, W. Precision medicine becomes reality-tumor type-agnostic therapy. Cancer Commun. 2018, 38, 6. [Google Scholar] [CrossRef] [PubMed]
- Galuppini, F.; Dal Pozzo, C.A.; Deckert, J.; Loupakis, F.; Fassan, M.; Baffa, R. Tumor mutation burden: From comprehensive mutational screening to the clinic. Cancer Cell. Int. 2019, 19, 209. [Google Scholar] [CrossRef] [PubMed]
- Garassino, M.; Rodriguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; Speranza, G.; Reck, M.; Hui, R.; Boyer, M.; Cristescu, R.; et al. OA04.06 Evaluation of TMB in KEYNOTE-189: Pembrolizumab Plus Chemotherapy vs. Placebo Plus Chemotherapy for Nonsquamous NSCLC. J. Thorac. Oncol. 2019, 14, S216–S217. [Google Scholar] [CrossRef]
- Masucci, G.V.; Cesano, A.; Hawtin, R.; Janetzki, S.; Zhang, J.; Kirsch, I.; Dobbin, K.K.; Alvarez, J.; Robbins, P.B.; Selvan, S.R.; et al. Validation of biomarkers to predict response to immunotherapy in cancer: Volume I - pre-analytical and analytical validation. J. Immunother. Cancer. 2016, 4, 76. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Mlecnik, B.; Bindea, G.; Angell, H.K.; Berger, A.; Lagorce, C.; Lugli, A.; Zlobec, I.; Hartmann, A.; Bifulco, C.; et al. Towards the introduction of the ‘Immunoscore’ in the classification of malignant tumours. J. Pathol. 2014, 232, 199–209. [Google Scholar] [CrossRef]
- Angell, H.K.; Bruni, D.; Barrett, J.C.; Herbst, R.; Galon, J. The Immunoscore: Colon Cancer and Beyond. Clin. Cancer Res. 2020, 26, 332–339. [Google Scholar] [CrossRef]
- Cristescu, R.; Mogg, R.; Ayers, M.; Albright, A.; Murphy, E.; Yearley, J.; Sher, X.; Liu, X.Q.; Lu, H.; Nebozhyn, M.; et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade–based immunotherapy. Science 2018, 362, eaar3593. [Google Scholar] [CrossRef]
- Yu, Y.; Zeng, D.; Ou, Q.; Liu, S.; Li, A.; Chen, Y.; Lin, D.; Gao, Q.; Zhou, H.; Liao, W.; et al. With Immunotherapy in Patients With Non–Small Cell Lung Cancer: A Meta-analysis and Individual Patient–Level Analysis. JAMA Network Open. 2019, 2, e196879. [Google Scholar] [CrossRef]
- Lee, J.S.; Ruppin, E. Multiomics Prediction of Response Rates to Therapies to Inhibit Programmed Cell Death 1 and Programmed Cell Death 1 Ligand 1. JAMA Oncol. 2019, 5, 1614–1618, [Epub ahead of print]. [Google Scholar] [CrossRef]
- Altorki, N.K.; Markowitz, G.J.; Gao, D.; Port, J.L.; Saxena, A.; Stiles, B.; McGraw, T.; Mittal, V. The lung microenvironment: an important regulator of tumour growth and metastasis. Nat Revs. Cancer. 2019, 19, 9–31. [Google Scholar] [CrossRef]
- McAllister, S.S.; Weinberg, R.A. Tumor–host interactions: A far-reaching relationship. J. Clin. Oncol. 2010, 28, 4022–4028. [Google Scholar] [CrossRef] [PubMed]
- Zahorec, R. Ratio of neutrophil to lymphocyte counts--rapid and simple parameter of systemic inflammation and stress in critically ill. Bratislavske lekarske Listy 2001, 102, 5–14. [Google Scholar] [PubMed]
- Capone, M.; Giannarelli, D.; Mallardo, D.; Madonna, G.; Festino, L.; Grimaldi, A.M.; Vanella, V.; Simeone, E.; Paone, M.; Palmieri, G.; et al. Baseline neutrophil-to-lymphocyte ratio (NLR) and derived NLR could predict overall survival in patients with advanced melanoma treated with nivolumab. J. Immunother. Cancer. 2018, 6, 74. [Google Scholar] [CrossRef] [PubMed]
- Kazandjian, D.; Gong, Y.; Keegan, P.; Pazdur, R.; Blumenthal, G.M. Prognostic Value of the Lung Immune Prognostic Index for Patients Treated for Metastatic Non–Small Cell Lung Cancer. JAMA Oncol. 2019, 5, 1481–1485, [Epub ahead of print]. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.L.; Anderson, N.G. The human plasma proteome: history, character, and diagnostic prospects. Mol. Cell Proteom. 2002, 1, 845–867. [Google Scholar] [CrossRef]
- Gautam, P.; Nair, S.C.; Ramamoorthy, K.; Swamy, C.V.; Nagaraj, R. Analysis of human blood plasma proteome from ten healthy volunteers from Indian population. PLoS ONE 2013, 8, e72584. [Google Scholar] [CrossRef]
- Kelly-Spratt, K.S.; Pitteri, S.J.; Gurley, K.E.; Liggitt, D.; Chin, A.; Kennedy, J.; Wong, C.H.; Zhang, Q.; Buson, T.B.; Wang, H.; et al. Plasma proteome profiles associated with inflammation, angiogenesis, and cancer. PLoS ONE 2011, 6, e19721. [Google Scholar] [CrossRef]
- Ye, R.D.; Sun, L. Emerging functions of serum amyloid A in inflammation. J. Leukoc. Biol. 2015, 98, 923–929. [Google Scholar] [CrossRef]
- Berraondo, P.; Minute, L.; Ajona, D.; Corrales, L.; Melero, I.; Pio, R. Innate immune mediators in cancer: between defense and resistance. Immunol. Rev. 2016, 274, 290–306. [Google Scholar] [CrossRef]
- Diamandis, E.P. The failure of protein cancer biomarkers to reach the clinic: Why, and what can be done to address the problem? BMC Med. 2012, 10, 87. [Google Scholar] [CrossRef] [PubMed]
- Hernández, B.; Parnell, A.; Pennington, S.R. Why have so few proteomic biomarkers “survived” validation? (Sample size and independent validation considerations). Proteomics 2014, 14, 1587–1592. [Google Scholar] [CrossRef] [PubMed]
- Goodfellow, I.; Bengio, Y.; Courville, A. Deep Learning; The MIT Press: Cambridge, MA, USA, 2016. [Google Scholar]
- Roder, J.; Oliveira, C.; Net, L.; Tsypin, M.; Linstid, B.; Roder, H. A dropout-regularized classifier development approach optimized for precision medicine test discovery from omics data. BMC Bioinformat. 2019, 20, 325. [Google Scholar] [CrossRef] [PubMed]
- Roder, H.; Oliveira, C.; Net, L.; Linstid, B.; Tsypin, M.; Roder, J. Robust identification of molecular phenotypes using semi-supervised learning. BMC Bioinformat. 2019, 20, 273. [Google Scholar] [CrossRef] [PubMed]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Srivastava, N.; Hinton, G.; Krizhevsky, A.; Sutskever, I.; Salakhutdinov, R. Dropout: A Simple Way to Prevent Neural Networks from Overfitting. J. Mach. Learn. Res. 2014, 15, 1929–1958. [Google Scholar]
- Breiman, L. Bagging predictors. Mach. Learn. 1996, 24, 123–140. [Google Scholar] [CrossRef]
- Breiman, L. Out-of-bag estimation. Technical Report. 1996. Available online: https://www.stat.berkeley.edu/~breiman/OOBestimation.pdf (accessed on 30 November 2019).
- Shaked, Y. The pro-tumorigenic host response to cancer therapies. Nat. Rev. Cancer. 2019, 19, 667–685. [Google Scholar] [CrossRef]
- Hortin, G.L. The MALDI-TOF mass spectrometric view of the plasma proteome and peptidome. Clin. Chem. 2006, 52, 1223–1237. [Google Scholar] [CrossRef]
- Tsypin, M.; Asmellash, S.; Meyer, K.; Touchet, B.; Roder, H. Extending the information content of the MALDI analysis of biological fluids via multi-million shot analysis. PLOS ONE 2019, 14, e0226012. [Google Scholar] [CrossRef]
- Weber, J.S.; Sznol, M.; Sullivan, R.J.; Blackmon, S.; Boland, G.; Kluger, H.M.; Halaban, R.; Bacchiocchi, A.; Ascierto, P.A.; Capone, M.; et al. A Serum Protein Signature Associated with Outcome after Anti-PD-1 Therapy in Metastatic Melanoma. Cancer Immunol. Res. 2018, 6, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, F.; Solomon, B.; Gregorc, V.; Roder, H.; Gray, R.; Kasahara, K.; Nishio, M.; Brahmer, J.; Spreafico, A.; Ludovini, V. Mass spectrometry to classify non-small-cell lung cancer patients for clinical outcome after treatment with epidermal growth factor receptor tyrosine kinase inhibitors: a multicohort cross-institutional study. J. Natl. Cancer Inst. 2007, 99, 838–846. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.S.; Kudchadkar, R.R.; Yu, B.; Gallenstein, D.; Horak, C.E.; Inzunza, H.D.; Zhao, X.; Martinez, A.J.; Wang, W.; Gibney, G.; et al. Safety, efficacy, and biomarkers of nivolumab with vaccine in ipilimumab-refractory or -naive melanoma. J. Clin. Oncol. 2013, 31, 4311–4318. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Capone, M.; Grimaldi, A.M.; Mallardo, D.; Simeone, E.; Madonna, G.; Roder, H.; Meyer, K.; Asmellash, S.; Oliveira, C.; et al. Proteomic test for anti-PD-1 checkpoint blockade treatment of metastatic melanoma with and without BRAF mutations. J. Immunother. Cancer 2019, 7, 91. [Google Scholar] [CrossRef] [PubMed]
- Aerts, J.; Smit, E.; Muller, M.; Niemeijer, A.; Oliveira, C.; Roder, H.; Roder, J. Prediction of primary resistance to anti-PD1 therapy in 2nd line NSCLC. In Proceedings of the IASLC 19th World Conference on Lung Cancer, Toronto, Canada, 23–26 September 2018. [Google Scholar]
- Gregorc, V.; Novello, S.; Lazzari, C.; Barni, S.; Aieta, M.; Mencoboni, M.; Grossi, F.; De Pas, T.; de Marinis, F.; Bearz, A.; et al. Predictive value of a proteomic signature in patients with non-small-cell lung cancer treated with second-line erlotinib or chemotherapy (PROSE): a biomarker-stratified, randomised phase 3 trial. Lancet Oncol. 2014, 15, 713–721. [Google Scholar] [CrossRef]
- Rich, P.; Roder, J.; Dubay, J.; Oubre, D.; Pauli, E.K.; Orsini, J.M.; Santos, E.S.; Coleman, M.; Khan, W.; Akerley, W.; et al. Real-world Performance of Blood-Based Proteomic Profiling in Frontline Immunotherapy Treatment in Advanced stage NSCLC. Int. J. Radiat. Oncol. 2019, 104, 236. [Google Scholar] [CrossRef]
- Grossi, F.; Rijavec, E.; Biello, F.; Rossi, G.; Barletta, G.; Maggioni, C.; Genova, C.; Dal Bello, M.G.; Distefano, R.; Roder, J.; et al. Evaluation of a pretreatment serum tests for nivolumab benefit in patients with non-small cell lung cancer. J. Thorac. Oncol. 2017, 12, S1322. [Google Scholar] [CrossRef]
- Grigorieva, J.; Asmellash, S.; Oliveira, C.; Roder, H.; Net, L.; Roder, J. Application of Protein Set Enrichment Analysis to Correlation of Protein Functional Sets with Mass Spectral Features and Multivariate Proteomic Tests. Clin. Mass Spectromet 2019. [Google Scholar] [CrossRef]
- Weber, J.; Martinez, A.; Roder, H.; Roder, J.; Meyer, K.; Asmellash, S.; Grigorieva, J.; Tsypin, M.; Oliveira, C.; Steingrimsson, A. Pre-treatment patient selection for nivolumab benefit based on serum mass spectra. J. Immunother. Cancer 2015, 3, P103. [Google Scholar] [CrossRef]
- Fidler, M.J.; Fhied, C.L.; Roder, J.; Basu, S.; Sayidine, S.; Fughhi, I.; Pool, M.; Batus, M.; Bonomi, P.; Borgia, J.A. The serum-based VeriStrat(R) test is associated with proinflammatory reactants and clinical outcome in non-small cell lung cancer patients. BMC Cancer 2018, 18, 310. [Google Scholar] [CrossRef]
- Carbone, D.P.; Ding, K.; Roder, H.; Grigorieva, J.; Roder, J.; Tsao, M.S.; Seymour, L.; Shepherd, F. Prognostic and Predictive Role of the VeriStrat Plasma Test in Patients with Advanced Non-Small-Cell Lung Cancer Treated with Erlotinib or Placebo in the NCIC Clinical Trials Group BR.21 Trial. J. Thorac. Oncol. 2012, 7, 1653–1660. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.; Upadhyay, S.; Lewanski, C.; Falk, S.; Skailes, G.; Woll, P.J.; Hatton, M.; Lal, R.; Jones, R.; Toy, E.; et al. The clinical role of VeriStrat testing in patients with advanced non-small cell lung cancer considered unfit for first-line platinum-based chemotherapy. Eur. J. Cancer. 2019, 120, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Grossi, F.; Genova, C.; Rijavec, E.; Barletta, G.; Biello, F.; Dal Bello, M.G.; Meyer, K.; Roder, J.; Roder, H.; Grigorieva, J. Prognostic role of the VeriStrat test in first line patients with non-small cell lung cancer treated with platinum-based chemotherapy. Lung Cancer 2018, 117, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Roder, J.; Linstid, B.; Oliveira, C. Improving the power of gene set enrichment analyses. BMC Bioinformat. 2019, 20, 257. [Google Scholar] [CrossRef]
- Pio, R.; Ajona, D.; Ortiz-Espinosa, S.; Mantovani, A.; Lambris, J.D. Complementing the Cancer-Immunity Cycle. Front. Immunol. 2019, 10, 774. [Google Scholar] [CrossRef] [PubMed]
- Mackiewicz, A.; Speroff, T.; Ganapathi, M.K.; Kushner, I. Effects of cytokine combinations on acute phase protein production in two human hepatoma cell lines. J. Immunol. 1991, 146, 3032–3037. [Google Scholar]
- Malle, E.; Sodin-Semrl, S.; Kovacevic, A. Serum amyloid A: An acute-phase protein involved in tumour pathogenesis. Cell. Mol. Life Sci. 2009, 66, 9–26. [Google Scholar] [CrossRef]
- Benci, J.L.; Xu, B.; Qiu, Y.; Wu, T.J.; Dada, H.; Twyman-Saint Victor, C.; Cucolo, L.; Lee, D.S.M.; Pauken, K.E.; Huang, A.C.; et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 2016, 167, 1540–1554. [Google Scholar] [CrossRef]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: a dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef]
- Mushtaq, M.U.; Papadas, A.; Pagenkopf, A.; Flietner, E.; Morrow, Z.; Chaudhary, S.G.; Asimakopoulos, F. Tumor matrix remodeling and novel immunotherapies: the promise of matrix-derived immune biomarkers. J. Immunother. Cancer 2018, 6, 65. [Google Scholar] [CrossRef]
- Shalapour, S.; Karin, M. Immunity, inflammation, and cancer: an eternal fight between good and evil. J. Clin. Invest. 2015, 125, 3347–3355. [Google Scholar] [CrossRef] [PubMed]
- DeNardo, D.G.; Coussens, L.M. Inflammation and breast cancer. Balancing immune response: crosstalk between adaptive and innate immune cells during breast cancer progression. Breast Cancer Res. 2007, 9, 212. [Google Scholar] [CrossRef] [PubMed]
- Buttigliero, C.; Shepherd, F.A.; Barlesi, F.; Schwartz, B.; Orlov, S.; Favaretto, A.G.; Santoro, A.; Hirsh, V.; Ramlau, R.; Blackler, A.R.; et al. Retrospective Assessment of a Serum Proteomic Test in a Phase III Study Comparing Erlotinib plus Placebo with Erlotinib plus Tivantinib (MARQUEE) in Previously Treated Patients with Advanced Non-Small Cell Lung Cancer. Oncologist 2018, 23, e251–e259. [Google Scholar] [CrossRef] [PubMed]
- Smit, E.F.; Aerts, J.G.; Muller, M.; Niemeijer, A.N.; Roder, H.; Oliveira, C.; Roder, J. Prediction of primary resistance to anti-PD1 therapy (APD1) in second-line NSCLC. Ann. Oncol. 2018, 29 (Suppl. 8), viii14–viii57. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: the next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Crusz, S.M.; Balkwill, F.R. Inflammation and cancer: advances and new agents. Nat. Rev. Clin. Oncol. 2015, 12, 584–596. [Google Scholar] [CrossRef]
- Danaher, P.; Warren, S.; Lu, R.; Samayoa, J.; Sullivan, A.; Pekker, I.; Wallden, B.; Marincola, F.M.; Cesano, A. Pan-cancer adaptive immune resistance as defined by the Tumor Inflammation Signature (TIS): results from The Cancer Genome Atlas (TCGA). J. Immunother. Cancer 2018, 6, 63. [Google Scholar] [CrossRef]
- Duruisseaux, M.; Martinez-Cardus, A.; Calleja-Cervantes, M.E.; Moran, S.; Castro de Moura, M.; Davalos, V.; Pineyro, D.; Sanchez-Cespedes, M.; Girard, N.; Brevet, M.; et al. Epigenetic prediction of response to anti-PD-1 treatment in non-small-cell lung cancer: a multicentre, retrospective analysis. Lancet Respir. Med. 2018, 6, 771–781. [Google Scholar] [CrossRef]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef]
- Jamieson, N.B.; Maker, A.V. Gene-expression profiling to predict responsiveness to immunotherapy. Cancer Gene Ther. 2017, 24, 134–140. [Google Scholar] [CrossRef]




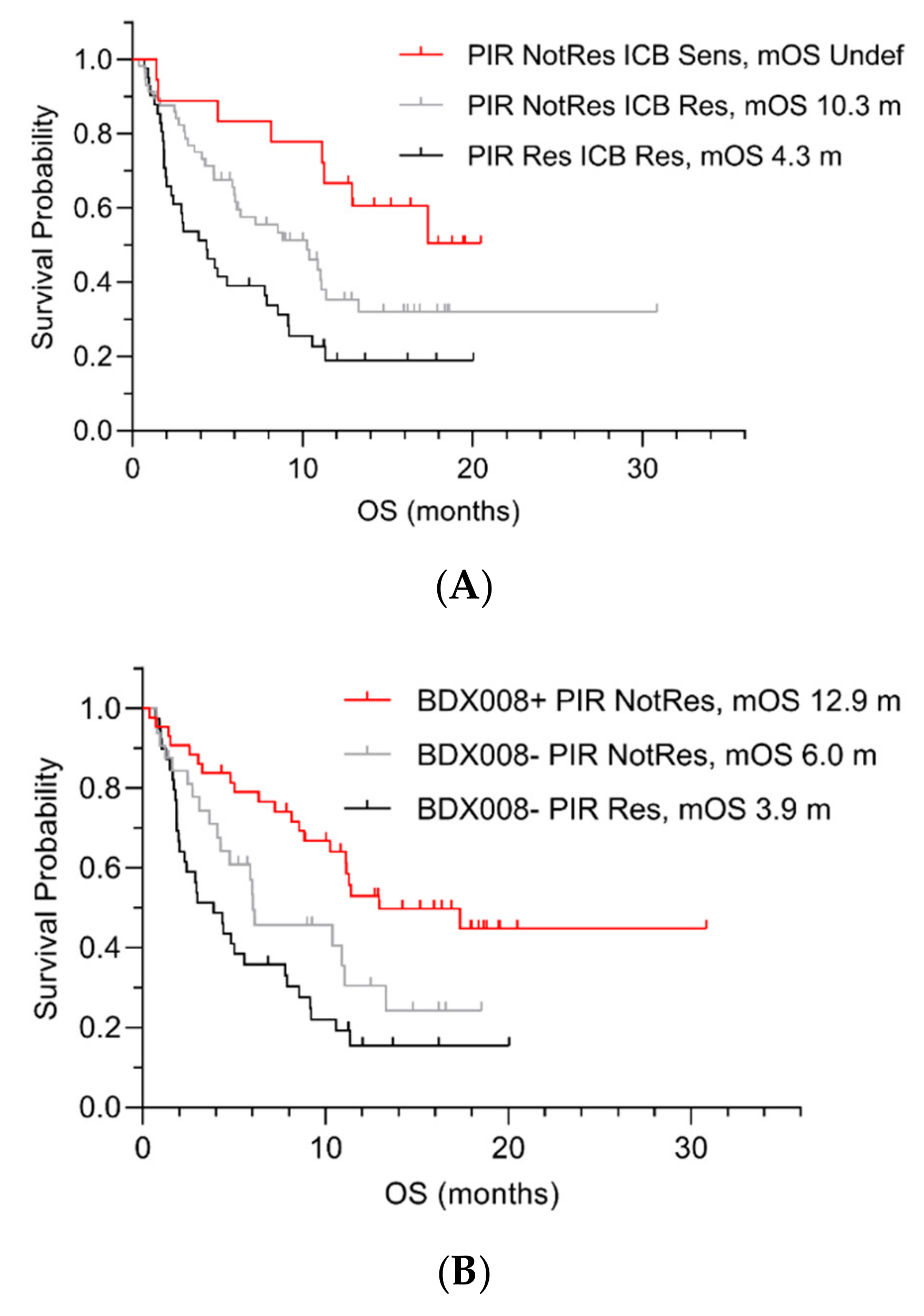{kind=link}
{kind=link}
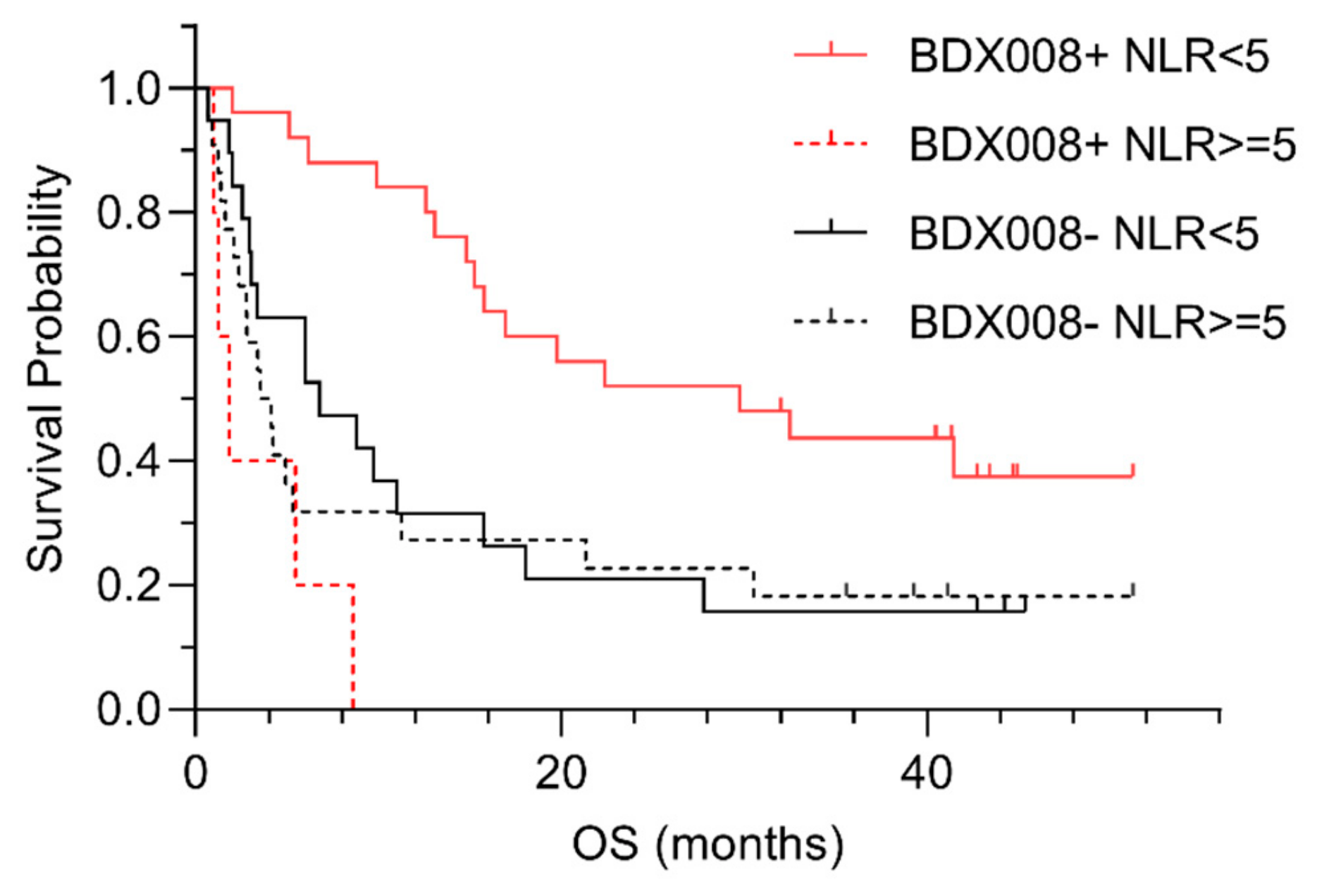{kind=link}
| Cohort | Patients (pts) | N pts | Reference |
|---|---|---|---|
| Melanoma development set | Unresectable melanoma patients treated with nivolumab in the NCT01176461 clinical trial (74% prior ipilimumab therapy) | 119 | Weber et al., 2013 [45] |
| Melanoma validation set | Unresectable melanoma patients treated with nivolumab or pembrolizumab in 2nd and higher lines (35% prior targeted therapy) | 71 | Ascierto et al., 2019 [46] |
| PIR development set | NSCLC patients treated with nivolumab in 2nd line (prior platinum-based chemotherapy) | 116 | Aerts et al. 2018 [47] |
| PIR control set | NSCLC patients treated with docetaxel in 2nd line (prior platinum-based chemotherapy) | 68 | Gregorc et al., 2014 [48] |
| INSIGHT validation sets | NSCLC patients treated with ICI in scope of INSIGHT registry study (NCT03289780) | Rich et al., 2019 [49] | |
| Monotherapy ICI 1st line | 46 | ||
| ICI plus chemotherapy 1st line | 33 | ||
| Lung cancer validation set | NSCLC patients treated with nivolumab in 2nd and higher lines enrolled in a single-institutional translational research study (prior platinum-based chemotherapy) | 60 | Grossi et al., 2017 [50] |
| PSEA reference set | NSCLC patients; samples obtained from commercial biobanks Conversant Bio (Huntsville, AL) and Oncology Metrix (Fort Worth, TX) | 100 | Grigorieva et al., 2019 [51] |
| Test | BDX008 | ICB | ||
|---|---|---|---|---|
| Classification | BDX008+ | BDX008– | ICB Sensitive | ICB Resistant |
| n (%) | 72 (61%) | 47 (39%) | 34 (29%) | 85 (71%) |
| 2-year survival | 55% | 21% | 67% | 33% |
| 3-year survival | 51% | 14% | 58% | 28% |
| OS curves comparison | HR = 0.38 (0.19–0.55), p < 0.001 | HR = 0.37 (0.19–0.71), p = 0.002 | ||
| Biological Processes | BDX008 | ICB | PIR (Resistant /Not Resistant) | VeriStrat |
|---|---|---|---|---|
| Acute inflammatory response | x | x | x | x |
| Acute phase reaction | x | x | x | x |
| Angiogenesis | ||||
| B cell-mediated immunity | ||||
| Chronic inflammatory response | x | |||
| Complement activation | x | x | x | x |
| Cytokine production in immune response | ||||
| Epithelial-mesenchymal transition | ||||
| Extracellular matrix organization | x | |||
| Glycolysis activation | ||||
| IFN type 1 signaling/response | x | x | ||
| IFN γ signaling/response | x | x | ||
| Immune tolerance/suppression | x | x | x | |
| Innate immune response | x | x | ||
| NK cell-meditated immunity | ||||
| Response to hypoxia | ||||
| T cell-mediated immunity | ||||
| Type 1 immune response | ||||
| Type 17 immune response | x | |||
| Type 2 immune response | ||||
| Wound healing | x | x |
| Test 1 | Test 2 | Melanoma Development set | Lung Cancer Development Set | PSEA Reference Set | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| BDX008 | BDX008+ | BDX008– | Total | BDX008+ | BDX008– | Total | BDX008+ | BDX008– | Total | |
| ICB | Sensitive | 34 | 0 | 34 | 18 | 0 | 18 | 24 | 0 | 24 |
| Resistant | 38 | 47 | 85 | 27 | 71 | 98 | 19 | 57 | 76 | |
| Total | 72 | 47 | 119 | 45 | 71 | 116 | 43 | 57 | 100 | |
| BDX008 | BDX008+ | BDX008– | Total | BDX008+ | BDX008– | Total | BDX008+ | BDX008– | Total | |
| PIR | NotResist | 64 | 4 | 68 | 43 | 32 | 75 | 42 | 28 | 70 |
| Resistant | 8 | 43 | 51 | 2 | 39 | 41 | 1 | 29 | 30 | |
| Total | 72 | 47 | 119 | 45 | 71 | 116 | 43 | 57 | 100 | |
| BDX008 | BDX008+ | BDX008– | Total | BDX008+ | BDX008– | Total | BDX008+ | BDX008– | Total | |
| VeriStrat | VS Good | 72 | 0 | 72 | 45 | 0 | 45 | 43 | 35 | 78 |
| VS Poor | 26 | 21 | 47 | 43 | 28 | 71 | 0 | 22 | 22 | |
| Total | 98 | 21 | 119 | 88 | 28 | 116 | 43 | 57 | 100 | |
| ICB | Sensitive | Resistant | Total | Sensitive | Resistant | Total | Sensitive | Resistant | Total | |
| PIR | NotResist | 34 | 34 | 68 | 18 | 57 | 75 | 23 | 47 | 70 |
| Resistant | 0 | 51 | 51 | 0 | 41 | 41 | 1 | 29 | 30 | |
| Total | 34 | 85 | 119 | 18 | 98 | 116 | 24 | 76 | 100 | |
| ICB | Sensitive | Resistant | Total | Sensitive | Resistant | Total | Sensitive | Resistant | Total | |
| VeriStrat | VS Good | 34 | 64 | 98 | 18 | 70 | 88 | 24 | 54 | 78 |
| VS Poor | 0 | 21 | 21 | 0 | 28 | 28 | 0 | 22 | 22 | |
| Total | 34 | 85 | 119 | 18 | 98 | 116 | 24 | 76 | 100 | |
| VeriStrat | VS Good | VS Poor | Total | VS Good | VS Poor | Total | VS Good | VS Poor | Total | |
| PIR | NotResist | 68 | 0 | 68 | 56 | 3 | 59 | 64 | 6 | 70 |
| Resistant | 30 | 21 | 51 | 32 | 25 | 57 | 14 | 16 | 30 | |
| Total | 98 | 21 | 119 | 88 | 28 | 116 | 78 | 22 | 100 | |
| # | Test Stratification | Subgroup (n) | Process | p |
|---|---|---|---|---|
| 1 | PIR Not resistant/resistant | BDX008– (57) | Wound healing | 0.048 |
| ICB Resistant (76) | Innate immune response | 0.012 | ||
| Wound healing | 0.013 | |||
| VeriStrat Good (78) | Innate immune response | 0.011 | ||
| Wound healing | 0.014 | |||
| 2 | ICB sensitive/resistant | BDX008+ (43) | Complement | 0.026 * |
| Type 1 immune response | 0.049 * | |||
| PIR Not Resistant (70) | Complement | <0.001 | ||
| Acute inflammatory response | <0.001 | |||
| Acute phase reaction | 0.001 | |||
| Immune tolerance/suppression | 0.007 | |||
| IFN γ signaling/response | 0.011 | |||
| 3 | BDX008 +/− | ICB Resistant (76) | Acute phase reaction | <0.001 |
| Innate immune response | 0.031 | |||
| Acute inflammatory response | 0.031 | |||
| Type 17 immune response | 0.032 | |||
| IFN γ signaling/response | 0.049 | |||
| PIR Not Resistant (70) | Acute phase reaction | <0.001 | ||
| IFN γ signaling/response | 0.001 | |||
| Acute inflammatory response | 0.003 | |||
| Immune tolerance/suppression | 0.013 | |||
| Type 17 immune response | 0.013 | |||
| Complement | 0.020 | |||
| 4 | VeriStrat (VSG/VSP) | PIR Not Resistant (70) | Immune tolerance/suppression | <0.001 * |
| Acute phase reaction | 0.001 * | |||
| Type 17 immune response | 0.007 * | |||
| Complement | 0.008 * |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grigorieva, J.; Asmellash, S.; Net, L.; Tsypin, M.; Roder, H.; Roder, J. Mass Spectrometry-Based Multivariate Proteomic Tests for Prediction of Outcomes on Immune Checkpoint Blockade Therapy: The Modern Analytical Approach. Int. J. Mol. Sci. 2020, 21, 838. https://doi.org/10.3390/ijms21030838
Grigorieva J, Asmellash S, Net L, Tsypin M, Roder H, Roder J. Mass Spectrometry-Based Multivariate Proteomic Tests for Prediction of Outcomes on Immune Checkpoint Blockade Therapy: The Modern Analytical Approach. International Journal of Molecular Sciences. 2020; 21(3):838. https://doi.org/10.3390/ijms21030838
Chicago/Turabian StyleGrigorieva, Julia, Senait Asmellash, Lelia Net, Maxim Tsypin, Heinrich Roder, and Joanna Roder. 2020. "Mass Spectrometry-Based Multivariate Proteomic Tests for Prediction of Outcomes on Immune Checkpoint Blockade Therapy: The Modern Analytical Approach" International Journal of Molecular Sciences 21, no. 3: 838. https://doi.org/10.3390/ijms21030838
APA StyleGrigorieva, J., Asmellash, S., Net, L., Tsypin, M., Roder, H., & Roder, J. (2020). Mass Spectrometry-Based Multivariate Proteomic Tests for Prediction of Outcomes on Immune Checkpoint Blockade Therapy: The Modern Analytical Approach. International Journal of Molecular Sciences, 21(3), 838. https://doi.org/10.3390/ijms21030838