Region-Specific Sialylation Pattern of Prion Strains Provides Novel Insight into Prion Neurotropism

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
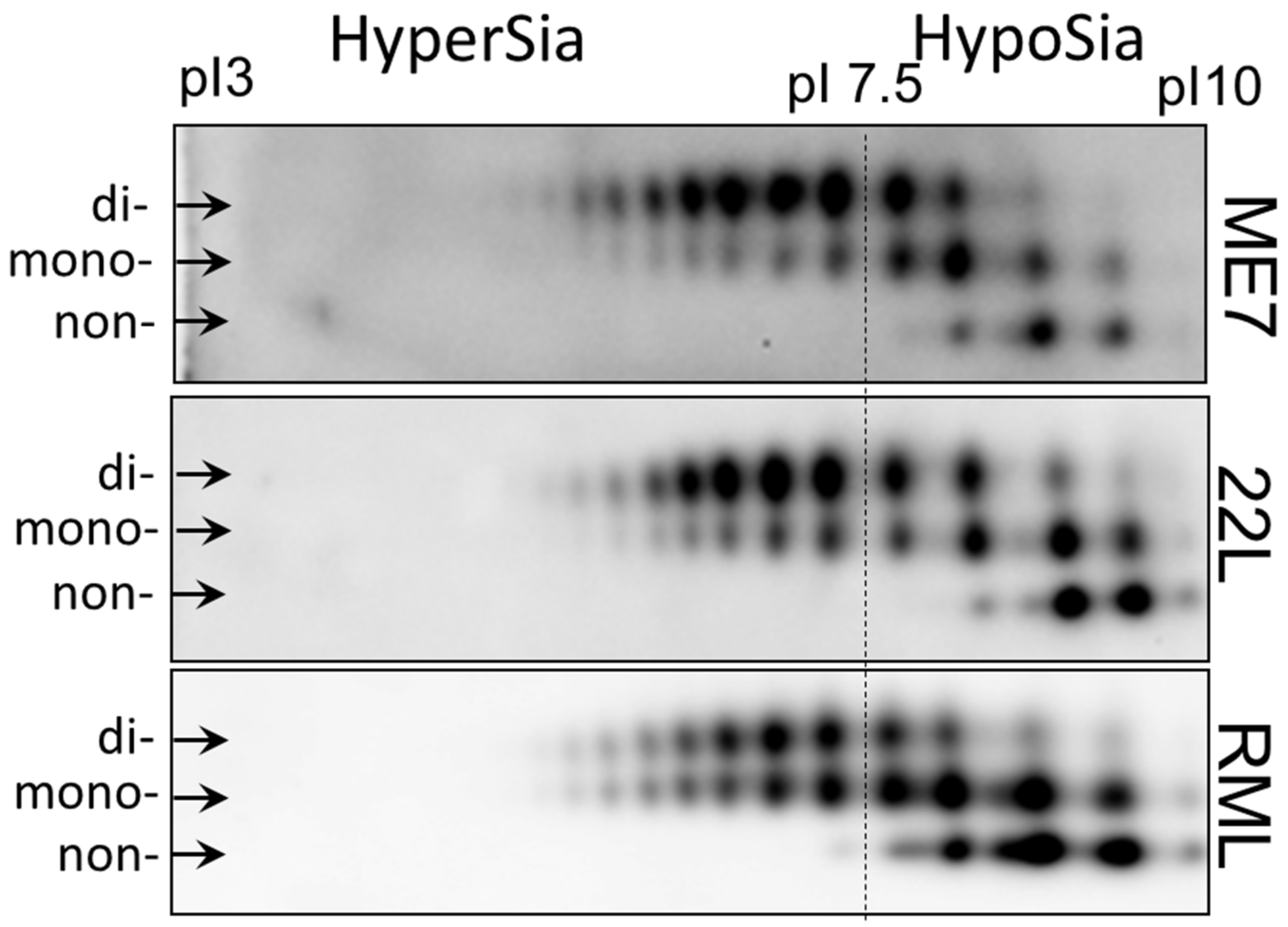
2.1. Strain-Specific Sialylation Patterns of PrPSc
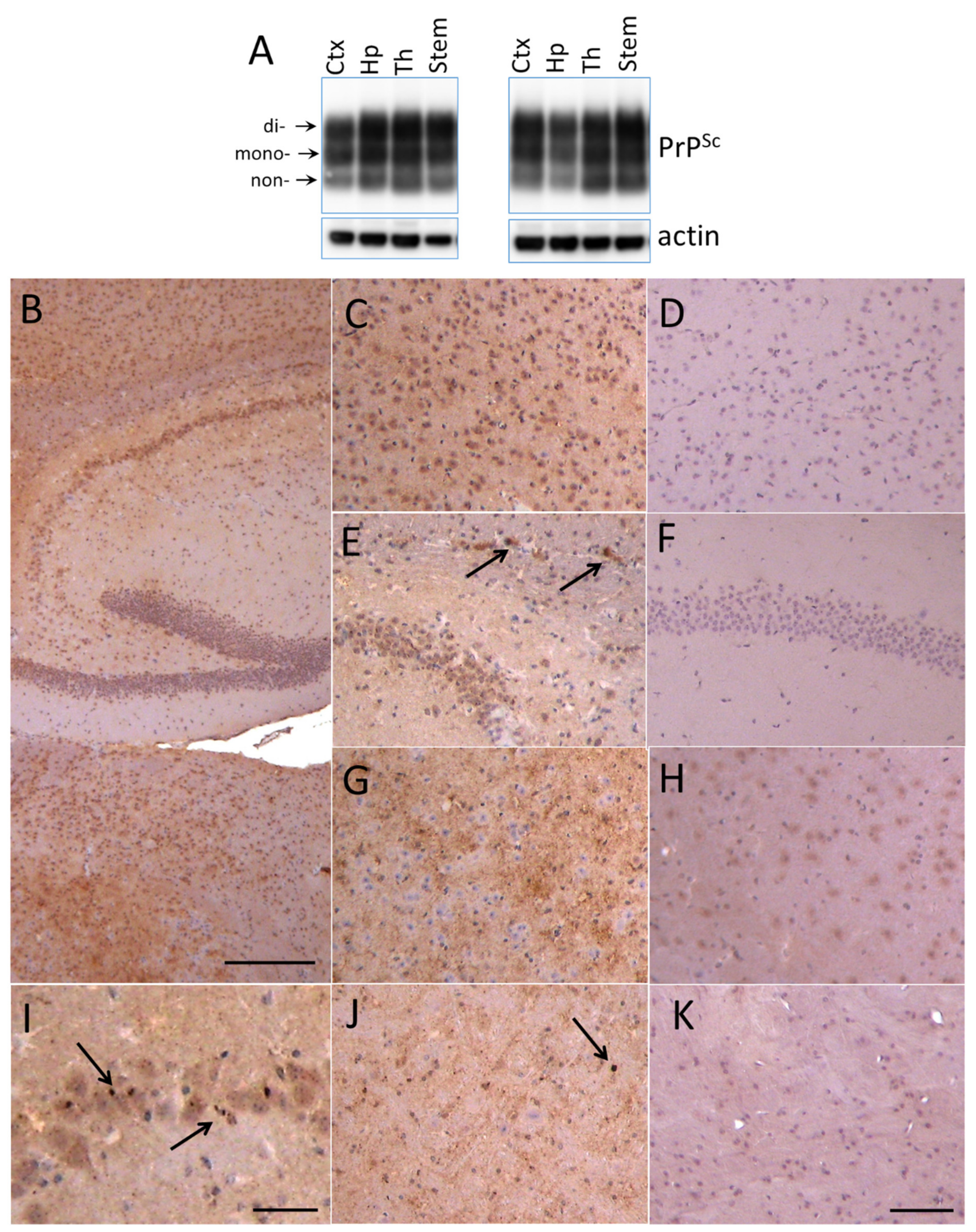
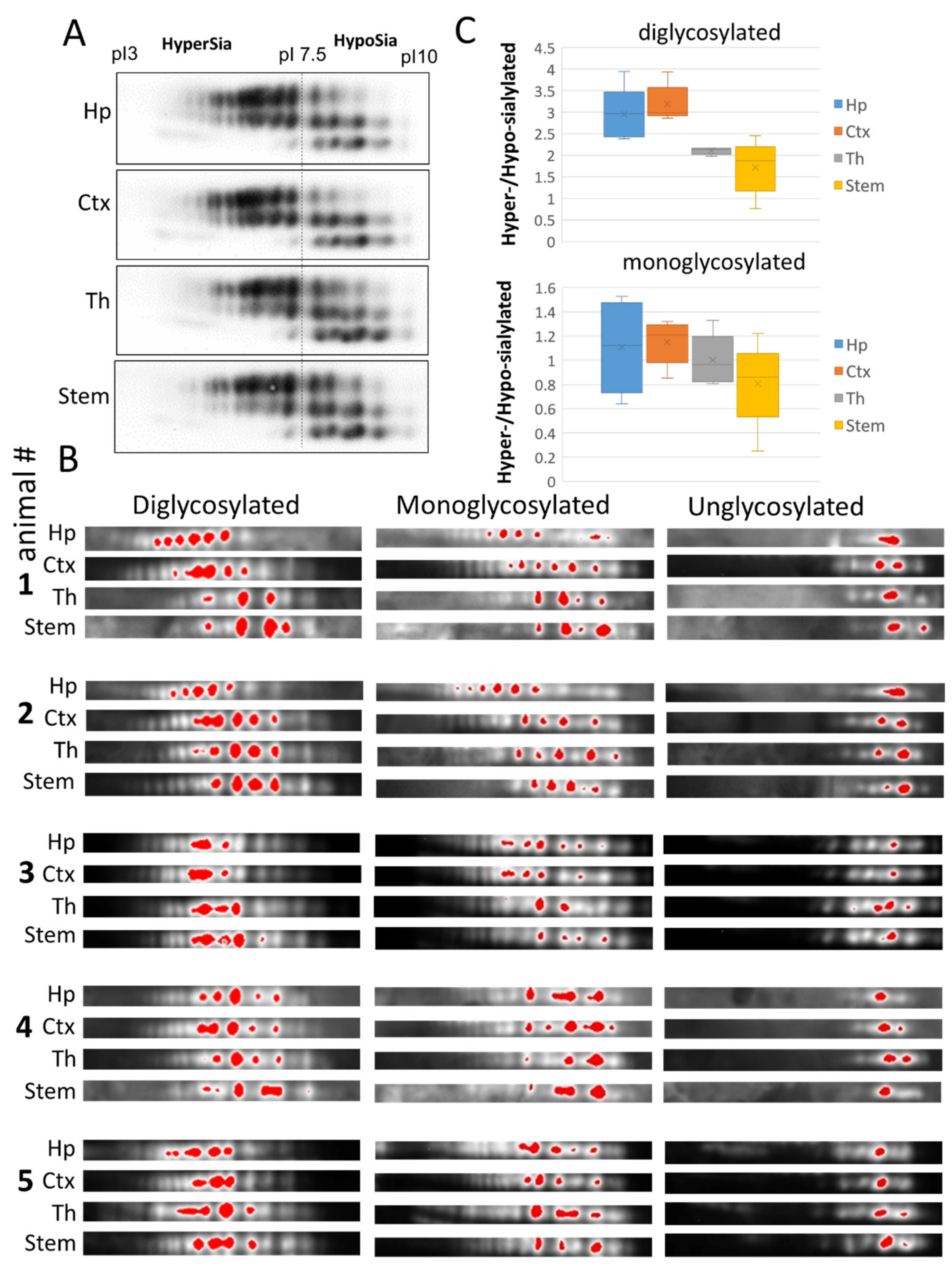
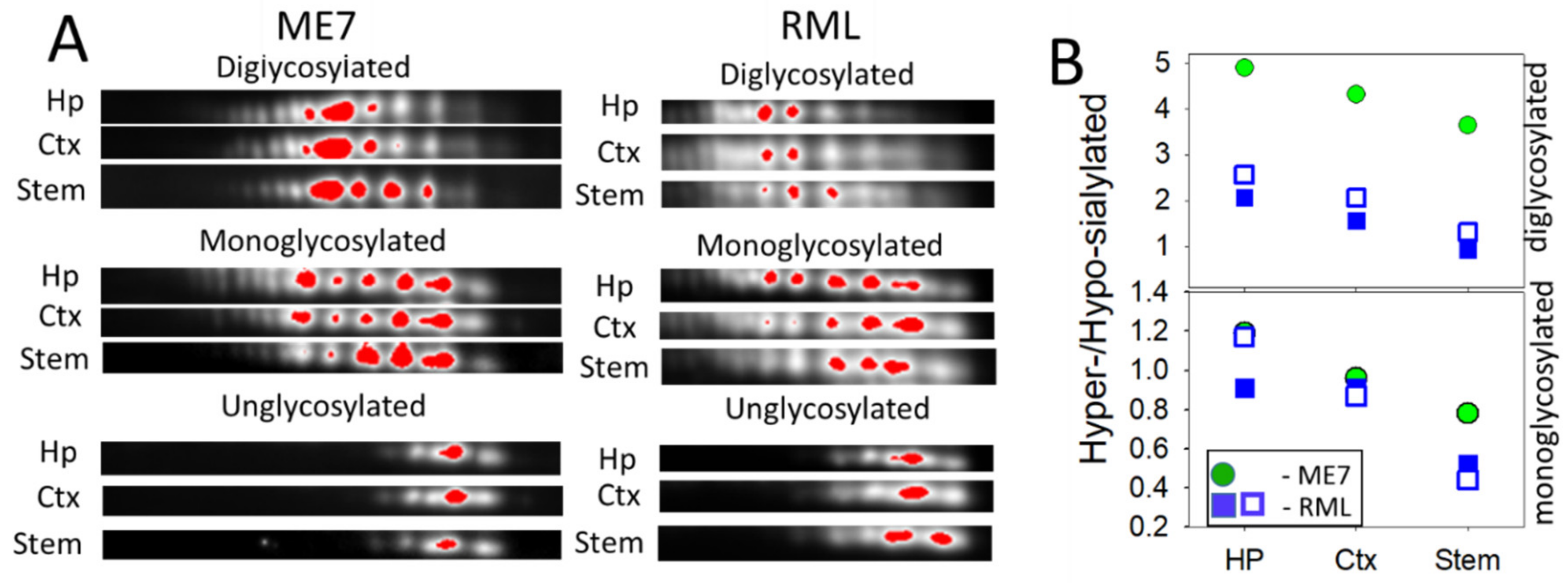
2.2. Region-Specific Sialylation of PrPSc
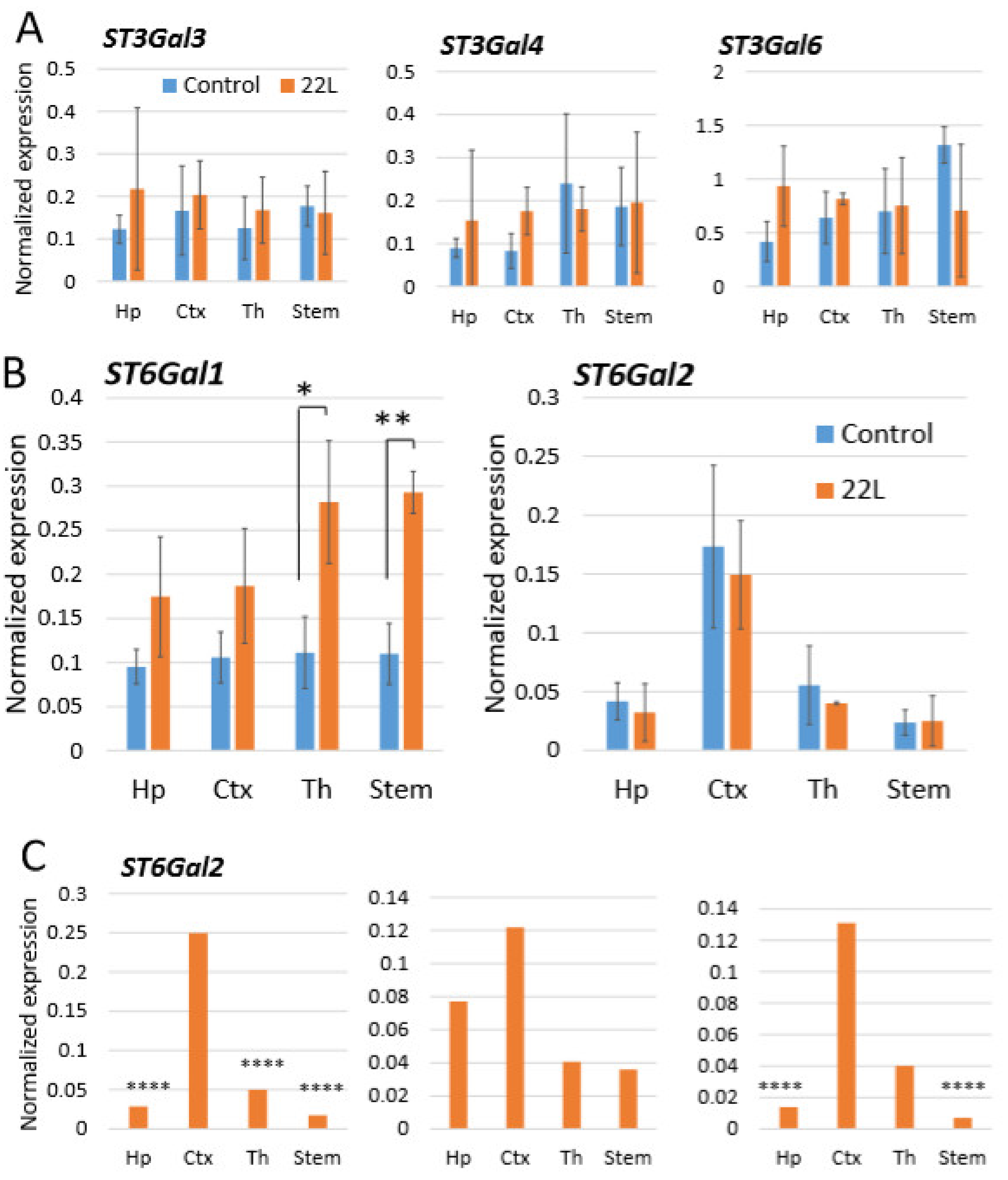
2.3. Region-Specific Expression of Sialyltransferases
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Animals
4.3. Histopathology
4.4. 2D Electrophoresis
4.5. qRT-PCR
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 22L | Mouse-adapted prion strain |
| CNS | Central nervous system |
| ME7 | Mouse-adapted prion strain |
| qRT-PCR | Quantitative reverse transcription polymerase chain reaction |
| PK | Proteinase K |
| PrPC | Cellular isoform of the prion protein |
| PrPSc | Disease-associated: transmissible isoform of the prion protein |
| RML | Mouse-adapted prion strain |
| ST | Sialyltransferase |
| ST3Gal3 | ST3 beta-galactoside alpha-2,3-sialyltransferase 3 |
| ST3Gal4 | ST3 beta-galactoside alpha-2,3-sialyltransferase 4 |
| ST3Gal6 | ST3 beta-galactoside alpha-2,3-sialyltransferase 6 |
| ST6Gal1 | ST6 beta-galactoside alpha-2,6-sialyltransferase 1 |
| ST6Gal2 | ST6 beta-galactoside alpha-2,6-sialyltransferase 2 |
References
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Legname, G.; Baskakov, I.V.; Nguyen, H.O.B.; Riesner, D.; Cohen, F.E.; DeArmond, S.J.; Prusiner, S.B. Synthetic mammalian prions. Science 2004, 305, 673–676. [Google Scholar] [CrossRef] [PubMed]
- Cohen, F.E.; Prusiner, S.B. Pathologic conformations of prion proteins. Annu. Rev. Biochem. 1998, 67, 793–819. [Google Scholar] [CrossRef] [PubMed]
- Baskakov, I.V.; Breydo, L. Converting the prion protein: What makes the protein infectious. Biochim. Biophys Acta. 2007, 1772, 692–703. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.Y.; Baker, C.A.; Manuelidis, L. New Molecular Markers of Early and Progressive CJD Brain Infection. J. Cell Biochem. 2004, 93, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Baker, C.A.; Manuelidis, L. Unique inflammatory RNA profiles of microglia in Creutzfeldt-Jakob disease. Proc. Natl. Acad. Sci. USA 2003, 100, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.A.; Striebel, J.F.; Rangel, A.; Woods, T.; Phillips, K.; Peterson, K.E.; Race, B.; Chesebro, B. Prion Strain Differences in Accumulation of PrPSc on Neurons and Glia Are Associated with Similar Expression Profiles of Neuroinflammatory Genes: Comparison of Three Prion Strains. PLoS Pathog. 2016, 12, e1005551. [Google Scholar] [CrossRef]
- Collinge, J.; Clarke, A.R. A General Model of Prion Strains and Their Pathogenicity. Science 2007, 318, 930–936. [Google Scholar] [CrossRef]
- Kimberlin, R.H.; Cole, S.; Walker, C.A. Temporary and permanent modifications to a single strain of mouse scrapie on transmission to rats and hamsters. J. Gen. Virol. 1987, 68, 1875–1881. [Google Scholar] [CrossRef]
- Kimberlin, R.H.; Walker, C.A.; Fraser, H. The genomic identity of different strains of mouse scrapie is expressed in hamsters and preserved on reisolation in mice. J. Gen. Virol. 1989, 70, 2017–2025. [Google Scholar] [CrossRef]
- Bruce, M.E.; Dickinson, A.G. Biological evidence that the scrapie agent has an independent genome. J. Gen. Virol. 1987, 68, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Karapetyan, Y.E.; Saa, P.; Mahal, S.P.; Sferrazza, G.F.; Sherman, A.; Sales, N.; Weissmann, C.; Lasmezas, C.I. Prion strain discrimination based on rapid in vivo amplification and analysis by the cell panel assay. PLoS ONE 2009, 4, e5730. [Google Scholar] [CrossRef] [PubMed]
- Bessen, R.A.; Marsh, R.F. Identification of two biologically distinct strains of transmissible mink encephalopathy in hamsters. J. Gen. Virol. 1992, 73, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Peretz, D.; Scott, M.; Groth, D.; Williamson, A.; Burton, D.; Cohen, F.E.; Prusiner, S.B. Strain-specified relative conformational stability of the scrapie prion protein. Protein Sci. 2001, 10, 854–863. [Google Scholar] [CrossRef]
- Safar, J.; Wille, H.; Itri, V.; Groth, D.; Serban, H.; Torchia, M.; Cohen, F.E.; Prusiner, S.B. Eight prion strains have PrP Sc molecules with different conformations. Nat. Med. 1998, 4, 1157–1165. [Google Scholar] [CrossRef]
- Ayers, J.L.; Schutt, C.R.; Shikiya, R.A.; Aguzzi, A.; Kincaid, A.E.; Bartz, J.C. The strain-encoded relationship between PrP replication, stability and processing in neurons is predictive of the incubation period of disease. PLOS Pathog. 2011, 7, e1001317. [Google Scholar] [CrossRef]
- Gonzalez-Montalban, N.; Makarava, N.; Savtchenko, R.; Baskakov, I.V. Relationship between Conformational Stability and Amplification Efficiency of Prions. Biochemistry 2011, 50, 7933–7940. [Google Scholar] [CrossRef]
- Klimova, N.; Makarava, N.; Baskakov, I.V. The diversity and relationship of prion protein self-replicating states. Virus Res. 2015, 207, 113–119. [Google Scholar] [CrossRef]
- Caughey, B.; Raymond, G.J.; Bessen, R.A. Strain-dependent differences in b-sheet conformations of abnormal prion protein. J. Biol. Chem. 1998, 273, 32230–32235. [Google Scholar] [CrossRef]
- Thomzig, A.; Spassov, S.; Friedrich, M.; Naumann, D.; Beekes, M. Discriminating Scrapie and Bovine Spongiform Encephalopathy Isolates by Infrared Spectroscopy of Pathological Prion Protein. J. Biol. Chem. 2004, 279, 33854. [Google Scholar] [CrossRef]
- Spassov, S.; Beekes, M.; Naumann, D. Structural differences between TSEs strains investigated by FT-IR spectroscopy. Biochim. Biophys Acta 2006, 1760, 1138–1149. [Google Scholar] [CrossRef] [PubMed]
- Tixador, P.; Herzog, L.; Reine, F.; Jaumain, E.; Chapuis, J.; Le Dur, A.; Laude, H.; Beringue, V. The physical relationship between infectivity and prion protein aggregates is strain-dependent. PLOS Pathog. 2010, 6, e1000859. [Google Scholar] [CrossRef] [PubMed]
- Morales, R.; Hu, P.P.; Duran-Aniotz, C.; Moda, F.; Diaz-Espinoza, R.; Chen, B.; Bravo-Alegria, J.; Makarava, N.; Baskakov, I.V.; Soto, C. Strain-dependent profile of misfolded prion protein aggregates. Sci. Rep. 2016, 6, 20526. [Google Scholar] [CrossRef] [PubMed]
- Stahl, N.; Borchelt, D.R.; Hsiao, K.; Prusiner, S.B. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 1987, 51, 229–240. [Google Scholar] [CrossRef]
- Endo, T.; Groth, D.; Prusiner, S.B.; Kobata, A. Diversity of oligosaccharide structures linked to asparagines of the scrapie prion protein. Biochemistry 1989, 28, 8380–8388. [Google Scholar] [CrossRef]
- Turk, E.; Teplow, D.B.; Hood, L.E.; Prusiner, S.B. Purification and properties of the cellular and scrapie hamster prion proteins. Eur. J. Biochem. 1988, 176, 21–30. [Google Scholar] [CrossRef]
- Katorcha, E.; Makarava, N.; Savtchenko, R.; Baskakov, I.V. Sialylation of the prion protein glycans controls prion replication rate and glycoform ratio. Sci. Rep. 2015, 5, 16912. [Google Scholar] [CrossRef]
- Stimson, E.; Hope, J.; Chong, A.; Burlingame, A.L. Site-specific characterization of the N-linked glycans of murine prion protein by high-performance liquid chromatography/electrospray mass spectrometry and exoglycosidase digestions. Biochemistry 1999, 38, 4885–4895. [Google Scholar] [CrossRef]
- Katorcha, E.; Baskakov, I.V. Analyses of N-linked glycans of PrPSc revealed predominantly 2,6-linked sialic acid residues. FEBS J. 2017, 284, 3727–3738. [Google Scholar] [CrossRef] [PubMed]
- Stahl, N.; Baldwin, M.A.; Teplow, D.B.; Hood, L.; Gibson, B.W.; Burlingame, A.L.; Prusiner, S.B. Structural studies of the scrapie prion protein using mass spectrometry and amino acid sequencing. Biochemistry 1993, 32, 1991–2002. [Google Scholar] [CrossRef]
- Rudd, P.M.; Endo, T.; Colominas, C.; Groth, D.; Wheeler, S.F.; Harvey, D.J.; Wormald, M.R.; Serban, H.; Prusiner, S.B.; Kobata, A.; et al. Glycosylation differences between the normal and pathogenic prion protein isoforms. Proc. Natl. Acad. Sci. U S A 1999, 96, 13044–13049. [Google Scholar] [CrossRef] [PubMed]
- Bolton, D.C.; Meyer, R.K.; Prusiner, S.B. Scrapie PrP 27-30 is a sialoglycoprotein. J. Virol. 1985, 53, 596–606. [Google Scholar] [CrossRef] [PubMed]
- Baskakov, I.V.; Katorcha, E. Multifaceted role of sialylation in prion diseases. Front. Neurosci. 2016, 10, 358. [Google Scholar] [CrossRef] [PubMed]
- Baskakov, I.V.; Katorcha, E.; Makarava, N. Prion Strain-Specific Structure and Pathology: A View from the Perspective of Glycobiology. Viruses 2018, 10, 723. [Google Scholar] [CrossRef]
- Baskakov, I.V. Limited understanding of the functional diversity of N-linked glycans as a major gap of prion biology. Prion 2017, 11, 82–88. [Google Scholar] [CrossRef]
- Kooyk, Y.; Rabinovich, G.A. Protein-glycan interactions in the control of innate and adaptive immune responses. Nat. Immunol. 2008, 9, 593–601. [Google Scholar] [CrossRef]
- Varki, A.; Gagneux, P. Multifarious roles of sialic acid in immunity. Ann. New York Acad. Sci. 2012, 1253, 16–36. [Google Scholar] [CrossRef]
- Linnartz-Gerlach, B.; Mathews, M.; Neumann, H. Sensing the neuronal glycocalyx by glial sialic acid binding immunoglobulin-like lectins. Neuroscience 2014, 275, 113. [Google Scholar] [CrossRef]
- Linnartz-Gerlach, B.; Schuy, C.; Shahraz, A.; Tenner, A.J.; Neumann, H. Sialylation of neurites inhibits complement-mediated macrophage removal in a human macrophage-neuron Co-Culture System. Glia 2016, 64, 35–47. [Google Scholar] [CrossRef]
- Srivastava, S.; Katorcha, E.; Makarava, N.; Barrett, J.P.; Loane, D.J.; Baskakov, I.V. Inflammatory response of microglia to prions is controlled by sialylation of PrPSc. Sci. Rep. 2018, 8, e11326. [Google Scholar] [CrossRef]
- Varki, A. Sialic acids in human health and disease. Trends Mol. Med. 2008, 14, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.C.; Neher, J.J. Microglial phagocytosis of live neurons. Nat. Rev. Neurosc. 2014, 15, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Aminoff, D.; Bruegge, W.F.; Bell, W.C.; Sarpolis, K.; Williams, R. Role of sialic acid in survival of erythrocytes in the circulation: interaction of neuraminidase-treated and untreated erythrocytes with spleen and liver at the cellular level. Proc. Acad. Natl. Sci. U S A 1977, 74, 1521–1524. [Google Scholar] [CrossRef] [PubMed]
- Jansen, A.J.G.; Josefsson, E.C.; Rumjantseva, V.; Liu, Q.P.; Falet, H.; Bergmeier, W.; Cifuni, S.; Sackstein, R.; von Andrian, U.H.; Wagner, D.D.; et al. Desialylation accelerates platelet clearance after refrigeration and initiates GPIba metalloproteinase-mediated cleavage in mice. Blood 2012, 119, 1263–1273. [Google Scholar] [CrossRef] [PubMed]
- Katorcha, E.; Makarava, N.; Savtchenko, R.; D’Azzo, A.; Baskakov, I.V. Sialylation of prion protein controls the rate of prion amplification, the cross-species barrier, the ratio of PrPSc glycoform and prion infectivity. PLOS Pathog. 2014, 10, e1004366. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Makarava, N.; Katorcha, E.; Savtchenko, R.; Brossmer, R.; Baskakov, I.V. Post-conversion sialylation of prions in lymphoid tissues. Proc. Acad. Natl. Sci. U S A 2015, 112, E6654–E6662. [Google Scholar] [CrossRef]
- Katorcha, E.; Daus, M.L.; Gonzalez-Montalban, N.; Makarava, N.; Lasch, P.; Beekes, M.; Baskakov, I.V. Reversible off and on switching of prion infectivity via removing and reinstalling prion sialylation. Sci. Rep. 2016, 6, 33119. [Google Scholar] [CrossRef]
- Srivastava, S.; Katorcha, E.; Daus, M.L.; Lasch, P.; Beekes, M.; Baskakov, I.V. Sialylation controls prion fate in vivo. J. Biol. Chem. 2017, 292, 2359–2368. [Google Scholar] [CrossRef]
- Collinge, J. Prion strain mutation and selection. Science 2010, 328, 1111–1112. [Google Scholar] [CrossRef]
- Piro, J.R.; Harris, B.T.; Nishina, K.; Soto, C.; Morales, R.; Rees, J.R.; Supattapone, S. Prion Protein Glycosylation Is Not Requiered for Strain-Specific Neurotropism. J. Virol. 2009, 83, 5321–5328. [Google Scholar] [CrossRef]
- Katorcha, E.; Baskakov, I.V. Analysis of Covalent Modifications of Amyloidogenic Proteins Using Two-Dimensional Electrophoresis: Prion Protein and Its Sialylation. Methods Mol. Biol 2018, 1779, 241–255. [Google Scholar] [PubMed]
- Zou, W.Q.; Capellari, S.; Parchi, P.; Sy, M.S.; Gambetti, P.; Chen, S.G. Identification of Novel Proteinase K-resistant C-terminal Fragments of PrP in Creutzfeldt-Jakob Disease. J. Biol. Chem. 2003, 278, 40429–40436. [Google Scholar] [CrossRef] [PubMed]
- Monaco, S.; Fiorini, M.; Farinazzo, A.; Ferrari, S.; Gelati, M.; Piccardo, P.; Zanusso, G.; Ghetti, B. Allelic origin of protease-sensitive and protease-resistant prion protein isoforms in Gerstmann-Sträussler-Scheinker disease with the P102L mutation. PLoS One 2012, 7, e32382. [Google Scholar] [CrossRef]
- Stahl, N.; Baldwin, M.A.; Hecker, R.; Pan, K.M.; Burlingame, A.L.; Prusiner, S.B. Glycosylinositol phospholipid anchors of the scrapie and cellular prion proteins contain sialic acid. Biochemistry 1992, 31, 5043–5053. [Google Scholar] [CrossRef]
- Katorcha, E.; Srivastava, S.; Klimova, N.; Baskakov, I.V. Sialylation of GPI Anchors of Mammalian Prions is Regulated in a Host-, Tissue- and Cell-Specific Manner. J. Biol. Chem. 2016, 291, 17009–17019. [Google Scholar] [CrossRef] [PubMed]
- Miyagi, T.; Yamaguchi, K. Mammalian sialidases: physiological and pathological roles in cellular functions. Glycobiology 2012, 22, 880–896. [Google Scholar] [CrossRef]
- Audry, M.; Jeanneau, C.; Imberty, A.; Harduin-Lepers, A.; Delannoy, P.; Breton, C. Current trend in the structure-activity relationships of sialylatransferases. Glycobiology 2011, 21, 716–726. [Google Scholar] [CrossRef]
- Katorcha, E.; Klimova, N.; Makarava, N.; Savtchenko, R.; Pan, X.; Annunziata, I.; Takahashi, K.; Miyagi, T.; Pshezhetsky, A.V.; d’Azzo, A.; et al. Knocking out of cellular neuraminidases Neu1, Neu3 or Neu4 does not affect sialylation status of the prion protein. PLoS ONE 2015, 10, e0143218. [Google Scholar]
- Takashima, S. Characterization of Mouse Sialylatransferase Gene: their Evolution and Diversity. Biosci. Biotechnol. Biochem. 2008, 72, 1155–1167. [Google Scholar] [CrossRef]
- Guillerme-Bosselut, F.; Forestier, L.; Jayat-Vignoles, C.; Vilotte, J.L.; Popa, I.; Portoukalian, J.; Dur, A.L.; Laude, H.; Julien, R.; Gallet, P.F. Glycosylation-related gene expression profiling in the brain and spleen of scrapie-affected mouse. Glycobiology 2009, 19, 879–889. [Google Scholar] [CrossRef][Green Version]
- Cancellotti, E.; Bradford, B.M.; Tuzi, N.L.; Hickey, R.D.; Brown, D.; Brown, K.L.; Barron, R.M.; Kisielewski, D.; Piccardo, P.; Manson, J.C. Glycosylation of PrPC determines timing of neuroinvasion and targeting in the brain following transmissible spongiform encephalopathy infection by a peripheral route. J. Virol. 2010, 84, 3464–3475. [Google Scholar] [CrossRef]
- Lehoux, S.; Groux-Degroote, S.; Cazet, A.; Dhaenens, C.-M.; Maurage, C.-A.; Caillet-Boudin, M.-L.; Delannoy, P.; Krzewinski-Recchi, M.-A. Transcriptional regulation of the human ST6GAL2 gene in cerebral cortex and neuronal cells. Glycoconj. J. 2010, 27, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Takashima, S.; Tsuji, S.; Tsujimoto, M. Comparison of the enzymatic properties of mouse beta-galactoside alpha2,6-sialyltransferases, ST6Gal I and II. J.Biochem. 2003, 134, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Takashima, S.; Truji, S.; Tsujimoto, M. Characterization of the second type of human beta-galactoside alpha 2,6-sialyltransferase (ST6Gal II), which sialylates Galbeta 1,4GlcNAc structures on oligosaccharides preferentially. Genomic analysis of human sialyltransferase genes. J. Biol. Chem. 2002, 277, 45719–45728. [Google Scholar] [CrossRef] [PubMed]
- Cobb, B.A. The history of IgG glycosylation and where we are now. Glycobiology 2019. [Google Scholar] [CrossRef] [PubMed]
- Makarava, N.; Chang, J.C.-Y.; Kushwaha, R.; Baskakov, I.V. Region-Specific Response of Astrocytes to Prion Infection. Front. Neurosci. 2019, 13, 1048. [Google Scholar] [CrossRef]
- Bradford, B.M.; Wijaya, C.A.W.; Mabbott, N.A. Discrimination of Prion Strain Targeting in the Central Nervous System via Reactive Astrocyte Heterogeneity in CD44 Expression. Front. Cell. Neurosci. 2019, 13, 411. [Google Scholar] [CrossRef]
- Mathys, H.; Adaikkan, C.; Gao, F.; Young, J.Z.; Manet, E.; Hemberg, M.; De Jager, P.L.; Ransohoff, R.M.; Regev, A.; Tsai, L.-H. Temporal Tracking of Microglia Activation in Neurodegeneration at Single-Cell Resolution. Cell Reports 2017, 21, 366–380. [Google Scholar] [CrossRef]
- Grabert, K.; Michoel, T.; Karavolos, M.H.; Clohisey, S.; Baillie, J.K.; Stevens, M.P.; Freeman, T.C.; Summers, K.M.; McColl, B.W. Microglial brain region−dependent diversity and selective regional sensitivities to aging. Nat. Neurosci. 2016, 19, 504. [Google Scholar] [CrossRef] [PubMed]
- Soreq, L.; Consortium, U.B.E.; Consortium, N.A.B.E.; Rose, J.; Soreq, E.; Hardy, J.; Trabzuni, D.; Cookson, M.R.; Smith, C.; Ryten, M.; et al. Major Shifts in Glial Regional Identity Are a Transcriptional Hallmark of Human Brain Aging. Cell Rep. 2017, 18, 557–570. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.E.; Liddelow, S.A.; Chakraborty, C.; Munich, A.E.; Heiman, M.; Barres, B.A. Normal aging induces A1-like astrocyte reactivity. Proc. Natl. Acad. Sci. USA 2018, 115, E1896–E1905. [Google Scholar] [CrossRef] [PubMed]
- Boisvert, M.M.; Erikson, G.A.; Shokhirev, M.N.; Allen, N.J. The Aging Astrocyte Transcriptome from Multiple Regions of the Mouse Brain. Cell Rep. 2018, 22, 269–285. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, A.; Hochgerner, H.; Lonnerberg, P.; Johnsson, A.; Memic, F.; van der Zwan, J.; Harig, M.; Braun, E.; Borm, L.E.; La Manno, G.; et al. Molecular Architecture of the Mouse Nervous System. Cell 2018, 174, 999–1014. [Google Scholar] [CrossRef] [PubMed]
- Makarava, N.; Chang, J.C.-Y.; Molesworth, K.; Baskakov, I.V. Loss of region-specific glial homeostatic signature in prion diseases. BioRxiv 2019, 823732. [Google Scholar]
- Sandberg, M.K.; Al-Doujaily, H.; Sharps, B.; De Oliveira, M.W.; Schmidt, C.; Richard-Londt, A.; Lyall, S.; Linehan, J.M.; Brandner, S.; Wadsworth, J.D.; et al. Prion neuropathology follows the accumulation of alternate prion protein isoforms after infective titre has peaked. Nat. Commun. 2014, 5, e4347. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Makarava, N.; Chang, J.C.-Y.; Baskakov, I.V. Region-Specific Sialylation Pattern of Prion Strains Provides Novel Insight into Prion Neurotropism. Int. J. Mol. Sci. 2020, 21, 828. https://doi.org/10.3390/ijms21030828
Makarava N, Chang JC-Y, Baskakov IV. Region-Specific Sialylation Pattern of Prion Strains Provides Novel Insight into Prion Neurotropism. International Journal of Molecular Sciences. 2020; 21(3):828. https://doi.org/10.3390/ijms21030828
Chicago/Turabian StyleMakarava, Natallia, Jennifer Chen-Yu Chang, and Ilia V. Baskakov. 2020. "Region-Specific Sialylation Pattern of Prion Strains Provides Novel Insight into Prion Neurotropism" International Journal of Molecular Sciences 21, no. 3: 828. https://doi.org/10.3390/ijms21030828
APA StyleMakarava, N., Chang, J. C.-Y., & Baskakov, I. V. (2020). Region-Specific Sialylation Pattern of Prion Strains Provides Novel Insight into Prion Neurotropism. International Journal of Molecular Sciences, 21(3), 828. https://doi.org/10.3390/ijms21030828