The Role of Calmodulin in Tumor Cell Migration, Invasiveness, and Metastasis


Abstract
1. Introduction
2. Calmodulin
3. Calmodulin and Cell Migration
3.1. CaM and Cytoskeleton Dynamics
3.2. CaM-Dependent Phosphorylation
3.3. The Role of Calcineurin
3.4. CaM-Regulated Small G Proteins
4. Calmodulin-Regulated Proteins in Cell Invasion and Metastasis
4.1. CaM-Dependent Protein Kinases
4.2. Calcineurin
4.3. CaM and Matrix Metalloproteases
4.4. CaM-Regulated Scaffold/Adaptor Proteins
4.5. Other CaM-Binding Proteins
5. Targeting Calmodulin-Dependent Systems to Inhibit Tumor Cell Invasion and Metastasis
6. Perspectives
Funding
Conflicts of Interest
References
- Brundage, R.; Fogarty, K.; Tuft, R.; Fay, F. Calcium gradients underlying polarization and chemotaxis of eosinophils. Science 1991, 254, 703–706. [Google Scholar] [CrossRef]
- Tsai, F.C.; Kuo, G.H.; Chang, S.W.; Tsai, P.J. Ca2+ signaling in cytoskeletal reorganization, cell migration, and cancer metastasis. Biomed. Res. Int. 2015, 2015, 409245. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Wang, X.; Chen, M.; Ouyang, K.; Song, L.-S.; Cheng, H. Calcium flickers steer cell migration. Nature 2009, 457, 901–905. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Wang, X.; Zheng, M.; Cheng, H. Calcium gradients underlying cell migration. Curr. Opin. Cell Biol. 2012, 24, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, I.; Mccollum, D. Control of cellular responses to mechanical cues through YAP/TAZ regulation. J. Biol. Chem. 2019, 294, 17693–17706. [Google Scholar] [CrossRef]
- Oancea, E.; Wolfe, J.T.; Clapham, D.E. Functional TRPM7 channels accumulate at the plasma membrane in response to fluid flow. Circ. Res. 2006, 98, 245–253. [Google Scholar] [CrossRef]
- Inoue, R.; Jensen, L.J.; Shi, J.; Morita, H.; Nishida, M.; Honda, A.; Ito, Y. Transient receptor potential channels in cardiovascular function and disease. Circ. Res. 2006, 99, 119–131. [Google Scholar] [CrossRef]
- Hamill, O.P.; Martinac, B. Molecular basis of mechanotransduction in living cells. Physiol. Rev. 2001, 81, 685–740. [Google Scholar] [CrossRef]
- Martino, F.; Perestrelo, A.R.; Vinarsky, V.; Pagliari, S.; Forte, G. Cellular mechanotransduction: From tension to function. Front. Physiol. 2018, 9, 824. [Google Scholar] [CrossRef]
- Suyama, E.; Wadhwa, R.; Kaur, K.; Miyagishi, M.; Kaul, S.C.; Kawasaki, H.; Taira, K. Identification of metastasis-related genes in a mouse model using a library of randomized ribozymes. J. Biol. Chem. 2004, 279, 38083–38086. [Google Scholar] [CrossRef]
- Zhu, H.; Zhang, H.; Jin, F.; Fang, M.; Huang, M.; Yang, C.S.; Chen, T.; Fu, L.; Pan, Z. Elevated Orai1 expression mediates tumor-promoting intracellular Ca2+ oscillations in human esophageal squamous cell carcinoma. Oncotarget 2014, 5, 3455–3471. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhang, J.J.; Huang, X.-Y. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell 2009, 15, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, J.; Iwabuchi, K.; Matsui, T.; Ishibashi, T.; Masuoka, T.; Nishio, M. Knockdown of stromal interaction molecule 1 (STIM1) suppresses store-operated calcium entry, cell proliferation and tumorigenicity in human epidermoid carcinoma A431 cells. Biochem. Pharmacol. 2012, 84, 1592–1603. [Google Scholar] [CrossRef] [PubMed]
- Middelbeek, J.; Kuipers, A.J.; Henneman, L.; Visser, D.; Eidhof, I.; Van Horssen, R.; Wieringa, B.; Canisius, S.V.; Zwart, W.; Wessels, L.F.; et al. TRPM7 is required for breast tumor cell metastasis. Cancer Res. 2012, 72, 4250–4261. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, L.; Dong, C. [Ca2+]i as a potential downregulator of α2β1-integrin-mediated A2058 tumor cell migration to type IV collagen. Am. J. Physiol. Cell Physiol. 2001, 281, C106–C113. [Google Scholar] [CrossRef]
- O’Neil, K.T.; DeGrado, W.F. How calmodulin binds its targets: Sequence independent recognition of amphiphilic α-helices. Trends Biochem. Sci. 1990, 15, 59–64. [Google Scholar] [CrossRef]
- Rhoads, A.R.; Friedberg, F. Sequence motifs for calmodulin recognition. FASEB J. 1997, 11, 331–340. [Google Scholar] [CrossRef]
- Yap, K.L.; Kim, J.; Truong, K.; Sherman, M.; Yuan, T.; Ikura, M. Calmodulin target database. J. Struct. Funct. Genom. 2000, 1, 8–14. [Google Scholar] [CrossRef]
- Bähler, M.; Rhoads, A. Calmodulin signaling via the IQ motif. FEBS Lett. 2002, 513, 107–113. [Google Scholar] [CrossRef]
- O’Day, D.H.; Mathavarajah, S.; Myre, M.A.; Huber, R.J. Calmodulin-mediated events during the life cycle of the amoebozoan Dictyostelium discoideum. Biol. Rev. 2019. [Google Scholar] [CrossRef]
- Villalobo, A.; Ishida, H.; Vogel, H.J.; Berchtold, M.W. Calmodulin as a protein linker and a regulator of adaptor/scaffold proteins. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 507–521. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.; Ouyang, H.; Vogel, H.J. Surface exposure of the methionine side chains of calmodulin in solution. A nitroxide spin label and two-dimensional NMR study. J. Biol. Chem. 1999, 274, 8411–8420. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.; Vogel, H.J. Substitution of the methionine residues of calmodulin with the unnatural amino acid analogs ethionine and norleucine: Biochemical and spectroscopic studies. Protein Sci 1999, 8, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Chin, D.; Means, A.R. Calmodulin: A prototypical calcium sensor. Trends Cell Biol. 2000, 10, 322–328. [Google Scholar] [CrossRef]
- Hoeflich, K.P.; Ikura, M. Calmodulin in action: Diversity in target recognition and activation mechanisms. Cell 2002, 108, 739–742. [Google Scholar] [CrossRef]
- Vetter, S.W.; Leclerc, E. Novel aspects of calmodulin target recognition and activation. Eur. J. Biochem. 2003, 270, 404–414. [Google Scholar] [CrossRef]
- Berchtold, M.W.; Villalobo, A. The many faces of calmodulin in cell proliferation, programmed cell death, autophagy, and cancer. Biochim. Biophys. Acta 2014, 1843, 398–435. [Google Scholar] [CrossRef]
- Benaim, G.; Villalobo, A. Phosphorylation of calmodulin. Functional implications. Eur. J. Biochem. 2002, 269, 3619–3631. [Google Scholar] [CrossRef]
- Villalobo, A. The multifunctional role of phospho-calmodulin in pathophysiological processes. Biochem. J. 2018, 475, 4011–4023. [Google Scholar] [CrossRef]
- Wehrle-Haller, B. Structure and function of focal adhesions. Curr. Opin. Cell Biol. 2012, 24, 116–124. [Google Scholar] [CrossRef]
- Ciobanasu, C.; Faivre, B.; Le Clainche, C. Integrating actin dynamics, mechanotransduction and integrin activation: The multiple functions of actin binding proteins in focal adhesions. Eur. J. Cell Biol. 2013, 92, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Burridge, K.; Guilluy, C. Focal adhesions, stress fibers and mechanical tension. Exp. Cell Res. 2016, 343, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Burridge, K. Focal adhesions: A personal perspective on a half century of progress. FEBS J. 2017, 284, 3355–3361. [Google Scholar] [CrossRef] [PubMed]
- Veillat, V.; Spuul, P.; Daubon, T.; Egaña, I.; Kramer, I.; Genot, E. Podosomes: Multipurpose organelles? Int. J. Biochem. Cell Biol. 2015, 65, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Dries, K.V.D.; Bolomini-Vittori, M.; Cambi, A. Spatiotemporal organization and mechanosensory function of podosomes. Cell Adhes. Migr. 2014, 8, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Seano, G.; Daubon, T.; Genot, E.; Primo, L. Podosomes as novel players in endothelial biology. Eur. J. Cell Biol. 2014, 93, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Linder, S.; Aepfelbacher, M. Podosomes: Adhesion hot-spots of invasive cells. Trends Cell Biol. 2003, 13, 376–385. [Google Scholar] [CrossRef]
- Siddiqui, T.A.; Lively, S.; Vincent, C.; Schlichter, L.C. Regulation of podosome formation, microglial migration and invasion by Ca2+-signaling molecules expressed in podosomes. J. Neuroinflamm. 2012, 9, 250. [Google Scholar] [CrossRef]
- Zhang, M.; Tanaka, T.; Ikura, M. Calcium-induced conformational transition revealed by the solution structure of apo calmodulin. Nat. Struct. Mol. Biol. 1995, 2, 758–767. [Google Scholar] [CrossRef]
- Chattopadhyaya, R.; Meador, W.E.; Means, A.R.; Quiocho, F.A. Calmodulin structure refined at 1.7 Å resolution. J. Mol. Biol. 1992, 228, 1177–1192. [Google Scholar] [CrossRef]
- Hong, F.; Haldeman, B.D.; Jackson, D.; Carter, M.; Baker, J.E.; Cremo, C.R. Biochemistry of smooth muscle myosin light chain kinase. Arch. Biochem. Biophys. 2011, 510, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Berchtold, M.; Egli, R.; Rhyner, J.; Hameister, H.; Strehler, E. Localization of the human bona fide calmodulin genes CALM1, CALM2, and CALM3 to chromosomes 14q24–q31, 2p21.1–p21.3, and 19q13.2–q13.3. Genomoics 1993, 16, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Toutenhoofd, S.; Strehler, E. The calmodulin multigene family as a unique case of genetic redundancy: Multiple levels of regulation to provide spatial and temporal control of calmodulin pools? Cell Calcium 2000, 28, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Saragai, S.; Naito, A.; Ichio, K.; Kawauchi, D.; Murakami, F. Calm1 signaling pathway is essential for the migration of mouse precerebellar neurons. J. Cell Sci. 2015, 128, 375–384. [Google Scholar] [CrossRef]
- Connor, C.G.; Brady, R.C.; Brownstein, B.L. Trifluoperazine inhibits spreading and migration of cells in culture. J. Cell. Physiol. 1981, 108, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Nakao, J.; Ito, H.; Ooyama, T.; Chang, W.C.; Murota, S. Calcium dependency of aortic smooth muscle cell migration induced by 12-L-hydroxy-5,8,10,14-eicosatetraenoic acid. Effects of A23187, nicardipine and trifluoperazine. Atherosclerosis 1983, 46, 309–319. [Google Scholar] [CrossRef]
- Matthews, N.; Franklin, R.J.; Kendrick, D.A. Structure-activity relationships of phenothiazines in inhibiting lymphocyte motility as determined by a novel flow cytometric assay. Biochem. Pharmacol. 1995, 50, 1053–1061. [Google Scholar] [CrossRef]
- Saito, H.; Minamiya, Y.; Kitamura, M.; Saito, S.; Enomoto, K.; Terada, K.; Ogawa, J. Endothelial myosin light chain kinase regulates neutrophil migration across human umbilical vein endothelial cell monolayer. J. Immunol. 1998, 161, 1533–1540. [Google Scholar]
- Yanase, M.; Ikeda, H.; Ogata, I.; Matsui, A.; Noiri, E.; Tomiya, T.; Arai, M.; Inoue, Y.; Tejima, K.; Nagashima, K.; et al. Functional diversity between Rho-kinase- and MLCK-mediated cytoskeletal actions in a myofibroblast-like hepatic stellate cell line. Biochem. Biophys. Res. Commun. 2003, 305, 223–228. [Google Scholar] [CrossRef]
- Kumada, T.; Komuro, H. Completion of neuronal migration regulated by loss of Ca2+ transients. Proc. Natl. Acad. Sci. USA 2004, 101, 8479–8484. [Google Scholar] [CrossRef]
- Guan, C.X.; Cui, Y.R.; Zhang, M.; Bai, H.B.; Khunkhun, R.; Fang, X. Intracellular signaling molecules involved in vasoactive intestinal peptide-mediated wound healing in human bronchial epithelial cells. Peptides 2007, 28, 1667–1673. [Google Scholar] [CrossRef] [PubMed]
- Lugnier, C.; Follenius, A.; Gérard, D.; Stoclet, J. Bepridil and flunarizine as calmodulin inhibitors. Eur. J. Pharmacol. 1984, 98, 157–158. [Google Scholar] [CrossRef]
- Sezzi, M.L.; De Luca, G.; Materazzi, M.; Bellelli, L. Effects of a calcium-antagonist (flunarizine) on cancer cell movement and phagocytosis. Anticancer. Res. 1985, 5, 265–271. [Google Scholar] [PubMed]
- Fink-Puches, R.; Helige, C.; Kerl, H.; Smolle, J.; Tritthart, H.A. Inhibition of melanoma cell directional migration in vitro via different cellular targets. Exp. Dermatol. 1993, 2, 17–24. [Google Scholar] [CrossRef]
- Shen, W.; Peng, W.; Dai, G.; Xu, J.; Zhang, Y.; Li, C. Calmodulin is essential for angiogenesis in response to hypoxic stress in endothelial cells. Cell Biol. Int. 2007, 31, 126–134. [Google Scholar] [CrossRef]
- Naito, M.; Hayashi, T.; Kuzuya, M.; Funaki, C.; Asai, K.; Kuzuya, F. Vascular endothelial cell migration in vitro roles of cyclic nucleotides, calcium ion and cytoskeletal system. Artery 1989, 17, 21–31. [Google Scholar]
- Kielbassa, K.; Schmitz, C.; Gerke, V. Disruption of endothelial microfilaments selectively reduces the transendothelial migration of monocytes. Exp. Cell Res. 1998, 243, 129–141. [Google Scholar] [CrossRef]
- Rosen, E.M.; Meromsky, L.; Goldberg, I.; Bhargava, M.; Setter, E. Studies on the mechanism of scatter factor. Effects of agents that modulate intracellular signal transduction, macromolecule synthesis and cytoskeleton assembly. J. Cell Sci. 1990, 96, 639–649. [Google Scholar]
- Linxweiler, M.; Schorr, S.; Schäuble, N.; Jung, M.; Linxweiler, J.; Langer, F.; Schäfers, H.-J.; Cavalié, A.; Zimmermann, R.; Greiner, M. Targeting cell migration and the endoplasmic reticulum stress response with calmodulin antagonists: A clinically tested small molecule phenocopy of SEC62 gene silencing in human tumor cells. BMC Cancer 2013, 13, 574. [Google Scholar] [CrossRef]
- Linxweiler, M.; Schick, B.; Zimmermann, R. Let’s talk about Secs: Sec61, Sec62 and Sec63 in signal transduction, oncology and personalized medicine. Signal Transduct. Target. Ther. 2017, 2, 17002. [Google Scholar] [CrossRef]
- Ranta-Knuuttila, T.; Kiviluoto, T.; Mustonen, H.; Puolakkainen, P.; Watanabe, S.; Sato, N.; Kivilaakso, E. Migration of primary cultured rabbit gastric epithelial cells requires intact protein kinase C and Ca2+/calmodulin activity. Dig. Dis. Sci. 2002, 47, 1008–1014. [Google Scholar] [CrossRef] [PubMed]
- Hadeed, J.G.; Bond, J.E.; Selim, M.A.; Bergeron, A.; Levin, L.S.; Levinson, H. Calcium-dependent signaling in Dupuytren’s disease. Hand (N. Y.) 2011, 6, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Levinson, H.; Moyer, K.E.; Saggers, G.C.; Ehrlich, H.P. Calmodulin-myosin light chain kinase inhibition changes fibroblast-populated collagen lattice contraction, cell migration, focal adhesion formation, and wound contraction. Wound Repair Regen. 2004, 12, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Soong, H.K.; Cintron, C. Different corneal epithelial healing mechanisms in rat and rabbit: Role of actin and calmodulin. Investig. Ophthalmol. Vis. Sci. 1985, 26, 838–848. [Google Scholar]
- Ehrlich, H.P.; Rajaratnam, J.B.M.; Griswold, T.R. ATP-induced cell contraction in dermal fibroblasts: Effects of cAMP and myosin light-chain kinase. J. Cell. Physiol. 1986, 128, 223–230. [Google Scholar] [CrossRef]
- Watanabe, S.; Hirose, M.; Yasuda, T.; Miyazaki, A.; Sato, N. Role of actin and calmodulin in migration and proliferation of rabbit gastric mucosal cells in culture. J. Gastroenterol. Hepatol. 1994, 9, 325–333. [Google Scholar] [CrossRef]
- Verploegen, S.; van Leeuwen, C.M.; van Deutekom, H.W.; Lammers, J.W.; Koenderman, L.; Coffer, P.J. Role of Ca2+/calmodulin regulated signaling pathways in chemoattractant induced neutrophil effector functions. Comparison with the role of phosphotidylinositol-3 kinase. Eur. J. Biochem. 2002, 269, 4625–4634. [Google Scholar] [CrossRef]
- Freed, D.H.; Chilton, L.; Li, Y.; Dangerfield, A.L.; Raizman, J.E.; Rattan, S.G.; Visen, N.; Hryshko, L.V.; Dixon, I.M.C. Role of myosin light chain kinase in cardiotrophin-1-induced cardiac myofibroblast cell migration. Am. J. Physiol. Circ. Physiol. 2011, 301, H514–H522. [Google Scholar] [CrossRef]
- Usui, T.; Morita, T.; Okada, M.; Yamawaki, H. Histone deacetylase 4 controls neointimal hyperplasia via stimulating proliferation and migration of vascular smooth muscle cells. Hypertension 2014, 63, 397–403. [Google Scholar] [CrossRef]
- Mohri, T.; Kameshita, I.; Suzuki, S.; Hioki, K.; Tokunaga, R.; Takatani, S. Rapid adhesion and spread of non-adherent colon cancer Colo201 cells induced by the protein kinase inhibitors, K252a and KT5720 and suppression of the adhesion by the immunosuppressants FK506 and cyclosporin A. Cell Struct. Funct. 1998, 23, 255–264. [Google Scholar] [CrossRef][Green Version]
- Saito, N.; Mine, N.; Kufe, D.W.; Von Hoff, D.D.; Kawabe, T. CBP501 inhibits EGF-dependent cell migration, invasion and epithelial-to–mesenchymal transition of non-small cell lung cancer cells by blocking K-Ras to calmodulin binding. Oncotarget 2017, 8, 74006–74018. [Google Scholar] [CrossRef] [PubMed]
- Mine, N.; Yamamoto, S.; Saito, N.; Sato, T.; Sakakibara, K.; Kufe, D.W.; Vonhoff, D.D.; Kawabe, T. CBP501 suppresses macrophage induced cancer stem cell like features and metastases. Oncotarget 2017, 8, 64015–64031. [Google Scholar] [CrossRef] [PubMed]
- Grabski, R.; Dewit, J.; De Braekeleer, J.; Malicka-Blaskiewicz, M.; De Baetselier, P.; Verschueren, H. Inhibition of T-cell invasion across cultured fibroblast monolayers by phenothiazine-related calmodulin inhibitors: Impairment of lymphocyte motility by trifluoperazine and chlorpromazine, and alteration of the monolayer by pimozide. Biochem. Pharmacol. 2001, 61, 1313–1317. [Google Scholar] [CrossRef]
- Dewhurst, L.; Gee, J.; Rennie, I.; MacNeil, S. Tamoxifen, 17β-oestradiol and the calmodulin antagonist J8 inhibit human melanoma cell invasion through fibronectin. Br. J. Cancer 1997, 75, 860–868. [Google Scholar] [CrossRef][Green Version]
- MacNeil, S.; Wagner, M.; Rennie, I. Tamoxifen inhibition of ocular melanoma cell attachment to matrix proteins. Pigment. Cell Res. 1994, 7, 222–226. [Google Scholar] [CrossRef]
- Pietras, R.J.; Weinberg, O.K. Antiangiogenic steroids in human cancer therapy. Evid. Based Complement. Alternat. Med. 2005, 2, 49–57. [Google Scholar] [CrossRef]
- Iwakawa, M.; Ando, K.; Ohkawa, H.; Koike, S.; Chen, Y.-J. A murine model for bone marrow metastasis established by an i.v. injection of C-1300 neuroblastoma in A/J mice. Clin. Exp. Metastasis 1994, 12, 231–237. [Google Scholar] [CrossRef]
- Mac Neil, S.; Wagner, M.; Kirkham, P.R.; Blankson, E.A.; Lennard, M.S.; Goodall, T.; Rennie, I.G. Inhibition of melanoma cell/matrix interaction by tamoxifen. Melanoma Res. 1993, 3, 67–74. [Google Scholar] [CrossRef]
- Roos, E.; Middelkoop, O.P.; Van De Pavert, I.V. Adhesion of tumor cells to hepatocytes: Different mechanisms for mammary carcinoma compared with lymphosarcoma cells. J. Natl. Cancer Inst. 1984, 73, 963–969. [Google Scholar]
- Qian, K.; Sun, L.; Zhou, G.; Ge, H.; Meng, Y.; Li, J.; Li, X.; Fang, X. Trifluoperazine as an alternative strategy for the inhibition of tumor growth of colorectal cancer. J. Cell. Biochem. 2019, 120, 15756–15765. [Google Scholar] [CrossRef]
- Shen, W.-G.; Peng, W.-X.; Shao, Y.; Xu, J.-F.; Dai, G.; Zhang, Y.; Pan, F.-Y.; Li, C.-J. Localization and activity of calmodulin is involved in cell–cell adhesion of tumor cells and endothelial cells in response to hypoxic stress. Cell Biol. Toxicol. 2007, 23, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Wang, J.Z.; Shimura, K. Inhibition of lung metastasis by a calmodulin antagonist, N-(6-aminohexyl)-5-chloro-1-naphthalenesulfonamide (W-7), in mice bearing Lewis lung carcinoma. Anticancer. Res. 1991, 11, 249–252. [Google Scholar] [PubMed]
- Hwang, Y.P.; Jeong, H.G. Metformin blocks migration and invasion of tumour cells by inhibition of matrix metalloproteinase-9 activation through a calcium and protein kinase Cα-dependent pathway: Phorbol-12-myristate-13-acetate-induced/extracellular signal-regulated kinase/activator protein-1. Br. J. Pharmacol. 2010, 160, 1195–1211. [Google Scholar] [PubMed]
- Xu, B.; Chelikani, P.; Bhullar, R.P. Characterization and functional analysis of the calmodulin-binding domain of Rac1 GTPase. PLoS ONE 2012, 7, e42975. [Google Scholar] [CrossRef]
- Xu, B.; Bhullar, R.P. Regulation of Rac1 and Cdc42 activation in thrombin- and collagen-stimulated CHRF-288-11 cells. Mol. Cell. Biochem. 2011, 353, 73–79. [Google Scholar] [CrossRef]
- Parker, C.; Sherbet, G.V. Modulators of intracellular Ca2+ and the calmodulin inhibitor W-7 alter the expression of metastasis-associated genes MTS1 and NM23 in metastatic variants of the B16 murine melanoma. Melanoma Res. 1992, 2, 337–343. [Google Scholar] [CrossRef]
- Eguchi, H.; Horikoshi, T. The expression of integrin α2β1 and attachment to type I collagen of melanoma cells are preferentially induced by tumour promoter, TPA (12-O-tetradecanoyl phorbol-13-acetate). Br. J. Dermatol. 1996, 134, 33–39. [Google Scholar] [CrossRef]
- Soong, K.; Cintron, C.; Soong, H. Disparate effects of calmodulin inhibitors on corneal epithelial migration in rabbit and rat. Ophthalmic Res. 1985, 17, 27–33. [Google Scholar] [CrossRef]
- Poggi, A.; Zocchi, M.R.; Carosio, R.; Ferrero, E.; Angelini, D.F.; Galgani, S.; Caramia, M.D.; Bernardi, G.; Borsellino, G.; Battistini, L. Transendothelial migratory pathways of Vδ1+TCRγδ+ and Vδ2+TCRγδ+ T lymphocytes from healthy donors and multiple sclerosis patients: Involvement of phosphatidylinositol 3 kinase and calcium calmodulin-dependent kinase II. J. Immunol. 2002, 168, 6071–6077. [Google Scholar] [CrossRef]
- Yeon, J.-T.; Choi, S.-W.; Ryu, B.J.; Kim, K.-J.; Lee, J.Y.; Byun, B.J.; Son, Y.-J.; Kim, S.H. Praeruptorin A inhibits in vitro migration of preosteoclasts and in vivo bone erosion, possibly due to its potential to target calmodulin. J. Nat. Prod. 2015, 78, 776–782. [Google Scholar] [CrossRef]
- Hendey, B.; Klee, C.; Maxfield, F. Inhibition of neutrophil chemokinesis on vitronectin by inhibitors of calcineurin. Science 1992, 258, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Rzeniewicz, K.; Newe, A.; Gallardo, A.R.; Davies, J.; Holt, M.R.; Patel, A.; Charras, G.T.; Stramer, B.; Molenaar, C.; Tedder, T.F.; et al. L-selectin shedding is activated specifically within transmigrating pseudopods of monocytes to regulate cell polarity in vitro. Proc. Natl. Acad. Sci. USA 2015, 112, E1461–E1470. [Google Scholar] [CrossRef] [PubMed]
- Rhyner, J.A.; Koller, M.; Durussel-Gerber, I.; Cox, J.A.; Strehler, E.E. Characterization of the human calmodulin-like protein expressed in Escherichia coli. Biochemistry 1992, 31, 12826–12832. [Google Scholar] [CrossRef] [PubMed]
- Durussel, I.; Rhyner, J.A.; Strehler, E.E.; Cox, J.A. Cation binding and conformation of human calmodulin-like protein. Biochemistry 1993, 32, 6089–6094. [Google Scholar] [CrossRef]
- Han, B.-G.; Han, M.; Sui, H.; Yaswen, P.; Walian, P.J.; Jap, B.K. Crystal structure of human calmodulin-like protein: Insights into its functional role. FEBS Lett. 2002, 521, 24–30. [Google Scholar] [CrossRef]
- Bennett, R.D.; Mauer, A.S.; Strehler, E.E. Calmodulin-like protein increases filopodia-dependent cell motility via up-regulation of myosin-10. J. Biol. Chem. 2007, 282, 3205–3212. [Google Scholar] [CrossRef]
- Bennett, R.D.; Mauer, A.S.; Pittelkow, M.R.; Strehler, E.E. Calmodulin-like protein upregulates myosin-10 in human keratinocytes and is regulated during epidermal wound healing in vivo. J. Investig. Dermatol. 2009, 129, 765–769. [Google Scholar] [CrossRef]
- Balasubramaniam, S.L.; Gopalakrishnapillai, A.; Gangadharan, V.; Duncan, R.L.; Barwe, S.P. Sodium-calcium exchanger 1 regulates epithelial cell migration via calcium-dependent extracellular signal-regulated kinase signaling. J. Biol. Chem. 2015, 290, 12463–12473. [Google Scholar] [CrossRef]
- Means, A.R.; Tash, J.S.; Chafouleas, J.G.; Lagace, L.; Guerriero, V. Regulation of the cytoskeleton by Ca2+-calmodulin and cAMP. Ann. N. Y. Acad. Sci. 1982, 383, 69–84. [Google Scholar] [CrossRef]
- Stull, J.T.; Kamm, K.E.; Vandenboom, R. Myosin light chain kinase and the role of myosin light chain phosphorylation in skeletal muscle. Arch. Biochem. Biophys. 2011, 510, 120–128. [Google Scholar] [CrossRef]
- Tsukamoto, O.; Kitakaze, M. Biochemical and physiological regulation of cardiac myocyte contraction by cardiac-specific myosin light chain kinase. Circ. J. 2013, 77, 2218–2225. [Google Scholar] [CrossRef] [PubMed]
- Grazina, R.; Teixeira, S.; Ramos, R.; Sousa, H.; Ferreira, A.; Lemos, R. Dupuytren’s disease: Where do we stand? EFORT Open Rev. 2019, 4, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Klemke, R.L.; Cai, S.; Giannini, A.L.; Gallagher, P.J.; De Lanerolle, P.; Cheresh, D.A. Regulation of cell motility by mitogen-activated protein kinase. J. Cell Biol. 1997, 137, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Munnich, S.; Taft, M.H.; Manstein, D.J. Crystal structure of human myosin 1c—The motor in GLUT4 exocytosis: Implications for Ca2+ regulation and 14-3-3 binding. J. Mol. Biol. 2014, 426, 2070–2081. [Google Scholar] [CrossRef]
- Bastian, P.; Lang, K.; Niggemann, B.; Zaenker, K.S.; Entschladen, F. Myosin regulation in the migration of tumor cells and leukocytes within a three-dimensional collagen matrix. Cell. Mol. Life Sci. 2005, 62, 65–76. [Google Scholar] [CrossRef]
- Yang, S.; Huang, X.Y. Ca2+ influx through L-type Ca2+ channels controls the trailing tail contraction in growth factor-induced fibroblast cell migration. J. Biol. Chem. 2005, 280, 27130–27137. [Google Scholar] [CrossRef]
- Totsukawa, G.; Wu, Y.; Sasaki, Y.; Hartshorne, D.J.; Yamakita, Y.; Yamashiro, S.; Matsumura, F. Distinct roles of MLCK and ROCK in the regulation of membrane protrusions and focal adhesion dynamics during cell migration of fibroblasts. J. Cell Biol. 2004, 164, 427–439. [Google Scholar] [CrossRef]
- Mayanagi, T.; Sobue, K. Diversification of caldesmon-linked actin cytoskeleton in cell motility. Cell Adhes. Migr. 2011, 5, 150–159. [Google Scholar] [CrossRef]
- Lin, J.J.; Li, Y.; Eppinga, R.D.; Wang, Q.; Jin, J.P. Chapter 1: Roles of caldesmon in cell motility and actin cytoskeleton remodeling. Int. Rev. Cell Mol. Biol. 2009, 274, 1–68. [Google Scholar]
- Li, Y.; Lin, J.L.; Reiter, R.S.; Daniels, K.; Soll, D.R.; Lin, J.J. Caldesmon mutant defective in Ca2+-calmodulin binding interferes with assembly of stress fibers and affects cell morphology, growth and motility. J. Cell Sci. 2004, 117, 3593–3604. [Google Scholar] [CrossRef]
- Mirzapoiazova, T.; Kolosova, I.A.; Romer, L.; Garcia, J.G.; Verin, A.D. The role of caldesmon in the regulation of endothelial cytoskeleton and migration. J. Cell. Physiol. 2005, 203, 520–528. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Patton, W.F.; Hechtman, H.B.; Shepro, D. A novel anti-inflammatory peptide inhibits endothelial cell cytoskeletal rearrangement, nitric oxide synthase translocation, and paracellular permeability increases. J. Cell. Physiol. 1997, 172, 171–182. [Google Scholar] [CrossRef]
- Ivaska, J.; Pallari, H.-M.; Nevo, J.; Eriksson, J.E. Novel functions of vimentin in cell adhesion, migration, and signaling. Exp. Cell Res. 2007, 313, 2050–2062. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, R.A.; Delic, S.; Herrmann, H.; Snider, N.T. Vimentin on the move: New developments in cell migration. F1000Research 2018, 7, 1796. [Google Scholar] [CrossRef] [PubMed]
- Spruill, W.A.; Zysk, J.R.; Tres, L.L.; Kierszenbaum, A.L. Calcium/calmodulin-dependent phosphorylation of vimentin in rat Sertoli cells. Proc. Natl. Acad. Sci. USA 1983, 80, 760–764. [Google Scholar] [CrossRef] [PubMed]
- Kitani, A.; Nakashima, N.; Izumihara, T.; Inagaki, M.; Baoui, X.; Yu, S.; Matsuda, T.; Matsuyama, T. Soluble VCAM-1 induces chemotaxis of Jurkat and synovial fluid T cells bearing high affinity very late antigen-4. J. Immunol. 1998, 161, 4931–4938. [Google Scholar]
- Goldenring, J.R.; Vallano, M.L.; DeLorenzo, R.J. Phosphorylation of microtubule-associated protein 2 at distinct sites by calmodulin-dependent and cyclic-AMP-dependent kinases. J. Neurochem. 1985, 45, 900–905. [Google Scholar] [CrossRef]
- Teng, J.; Takei, Y.; Harada, A.; Nakata, T.; Chen, J.; Hirokawa, N. Synergistic effects of MAP2 and MAP1B knockout in neuronal migration, dendritic outgrowth, and microtubule organization. J. Cell Biol. 2001, 155, 65–76. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Brent, G.A. A complex deoxyribonucleic acid response element in the rat Ca2+/calmodulin-dependent protein kinase IV gene 5’-flanking region mediates thyroid hormone induction and chicken ovalbumin upstream promoter transcription factor 1 repression. Mol. Endocrinol. 2002, 16, 2439–2451. [Google Scholar] [CrossRef][Green Version]
- Morte, B.; Diez, D.; Auso, E.; Belinchon, M.M.; Gil-Ibanez, P.; Grijota-Martinez, C.; Navarro, D.; de Escobar, G.M.; Berbel, P.; Bernal, J. Thyroid hormone regulation of gene expression in the developing rat fetal cerebral cortex: Prominent role of the Ca2+/calmodulin-dependent protein kinase IV pathway. Endocrinology 2010, 151, 810–820. [Google Scholar] [CrossRef]
- He, L.; Hou, Z.; Qi, R.Z. Calmodulin binding and Cdk5 phosphorylation of p35 regulate its effect on microtubules. J. Biol. Chem. 2008, 283, 13252–13260. [Google Scholar] [CrossRef] [PubMed]
- Saddouk, F.Z.; Ginnan, R.; Singer, H.A. Ca2+/calmodulin-dependent protein kinase II in vascular smooth muscle. Adv. Pharmacol. 2017, 78, 171–202. [Google Scholar] [PubMed]
- Cheyou, E.R.; Bouallegue, A.; Srivastava, A.K. Ca2+/calmodulin-dependent protein kinase- II in vasoactive peptide-induced responses and vascular biology. Curr. Vasc. Pharmacol. 2014, 12, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Pauly, R.R.; Bilato, C.; Sollott, S.J.; Monticone, R.; Kelly, P.T.; Lakatta, E.G.; Crow, M.T. Role of calcium/calmodulin-dependent protein kinase II in the regulation of vascular smooth muscle cell migration. Circulation 1995, 91, 1107–1115. [Google Scholar] [CrossRef]
- Lundberg, M.S.; Curto, K.A.; Bilato, C.; Monticone, R.E.; Crow, M.T. Regulation of vascular smooth muscle migration by mitogen-activated protein kinase and calcium/calmodulin-dependent protein kinase II signaling pathways. J. Mol. Cell. Cardiol. 1998, 30, 2377–2389. [Google Scholar] [CrossRef]
- Mercure, M.Z.; Ginnan, R.; Singer, H.A. CaM kinase IIδ2-dependent regulation of vascular smooth muscle cell polarization and migration. Am. J. Physiol. Cell Physiol. 2008, 294, C1465–C1475. [Google Scholar] [CrossRef]
- Liu, Y.; Sun, L.Y.; Singer, D.V.; Ginnan, R.; Singer, H.A. CaMKIIδ-dependent inhibition of cAMP-response element-binding protein activity in vascular smooth muscle. J. Biol. Chem. 2013, 288, 33519–33529. [Google Scholar] [CrossRef]
- Sun, P.; Enslen, H.; Myung, P.S.; Maurer, R.A. Differential activation of CREB by Ca2+/calmodulin-dependent protein kinases type II and type IV involves phosphorylation of a site that negatively regulates activity. Genes Dev. 1994, 8, 2527–2539. [Google Scholar] [CrossRef]
- Ginnan, R.; Zou, X.; Pfleiderer, P.J.; Mercure, M.Z.; Barroso, M.; Singer, H.A. Vascular smooth muscle cell motility is mediated by a physical and functional interaction of Ca2+/calmodulin-dependent protein kinase IIδ2 and Fyn. J. Biol. Chem. 2013, 288, 29703–29712. [Google Scholar] [CrossRef]
- Blystone, S.D.; Slater, S.E.; Williams, M.P.; Crow, M.T.; Brown, E.J. A molecular mechanism of integrin crosstalk: αvβ3 suppression of calcium/calmodulin-dependent protein kinase II regulates α5β1 function. J. Cell Biol. 1999, 145, 889–897. [Google Scholar] [CrossRef]
- Millon-Fremillon, A.; Brunner, M.; Abed, N.; Collomb, E.; Ribba, A.S.; Block, M.R.; Albiges-Rizo, C.; Bouvard, D. Calcium and calmodulin-dependent serine/threonine protein kinase type II (CaMKII)-mediated intramolecular opening of integrin cytoplasmic domain-associated protein-1 (ICAP-1alpha) negatively regulates β1 integrins. J. Biol. Chem. 2013, 288, 20248–20260. [Google Scholar] [CrossRef] [PubMed]
- Haws, H.J.; McNeil, M.A.; Hansen, M.D.H. Control of cell mechanics by RhoA and calcium fluxes during epithelial scattering. Tissue Barriers 2016, 4, e1187326. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Li, B.; Dong, Q.; Qian, C.; Cheng, J.; Wang, Y. Repetitive transient ischemia-induced cardiac angiogenesis is mediated by CaMKII activation. Cell Physiol. Biochem. 2018, 47, 914–924. [Google Scholar] [CrossRef] [PubMed]
- Toescu, E.C. Hypoxia sensing and pathways of cytosolic Ca2+ increases. Cell Calcium 2004, 36, 187–199. [Google Scholar] [CrossRef]
- Banumathi, E.; O’Connor, A.; Gurunathan, S.; Simpson, D.A.; McGeown, J.G.; Curtis, T.M. VEGF-induced retinal angiogenic signaling is critically dependent on Ca2+ signaling by Ca2+/calmodulin-dependent protein kinase II. Investig. Ophthalmol. Vis. Sci. 2011, 52, 3103–3111. [Google Scholar] [CrossRef]
- Küry, S.; van Woerden, G.M.; Besnard, T.; Onori, M.P.; Latypova, X.; Towne, M.C.; Cho, M.T.; Prescott, T.E.; Ploeg, M.A.; Sanders, S.; et al. De novo mutations in protein kinase genes CAMK2A and CAMK2B cause intellectual disability. Am. J. Hum. Genet. 2017, 101, 768–788. [Google Scholar] [CrossRef]
- Lewis, C.A.; Townsend, P.A.; Isacke, C.M. Ca2+/calmodulin-dependent protein kinase mediates the phosphorylation of CD44 required for cell migration on hyaluronan. Biochem. J. 2001, 357, 843–850. [Google Scholar] [CrossRef]
- Bourguignon, L.Y.; Gilad, E.; Brightman, A.; Diedrich, F.; Singleton, P. Hyaluronan-CD44 interaction with leukemia-associated RhoGEF and epidermal growth factor receptor promotes Rho/Ras co-activation, phospholipase Cε-Ca2+ signaling, and cytoskeleton modification in head and neck squamous cell carcinoma cells. J. Biol. Chem. 2006, 281, 14026–14040. [Google Scholar] [CrossRef]
- Mishra, J.P.; Mishra, S.; Gee, K.; Kumar, A. Differential involvement of calmodulin-dependent protein kinase II-activated AP-1 and c-Jun N-terminal kinase-activated EGR-1 signaling pathways in tumor necrosis factor-α and lipopolysaccharide-induced CD44 expression in human monocytic cells. J. Biol. Chem. 2005, 280, 26825–26837. [Google Scholar] [CrossRef]
- Qin, X.; Liang, Y.; Guo, Y.; Liu, X.; Zeng, W.; Wu, F.; Lin, Y.; Zhang, Y. Eukaryotic initiation factor 5A and Ca2+/calmodulin-dependent protein kinase 1D modulate trophoblast cell function. Am. J. Reprod. Immunol. 2018, 80, e12845. [Google Scholar] [CrossRef]
- Ha, C.H.; Jin, Z.G. Protein kinase D1, a new molecular player in VEGF signaling and angiogenesis. Mol. Cells 2009, 28, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Yokokura, H.; Picciotto, M.R.; Nairn, A.C.; Hidaka, H. The regulatory region of calcium/calmodulin-dependent protein kinase I contains closely associated autoinhibitory and calmodulin-binding domains. J. Biol. Chem. 1995, 270, 23851–23859. [Google Scholar] [CrossRef] [PubMed]
- Verploegen, S.; Ulfman, L.; Van Deutekom, H.W.M.; Van Aalst, C.; Honing, H.; Lammers, J.-W.J.; Koenderman, L.; Coffer, P.J. Characterization of the role of CaMKI-like kinase (CKLiK) in human granulocyte function. Blood 2005, 106, 1076–1083. [Google Scholar] [CrossRef] [PubMed]
- Means, A.R. The year in basic science: Calmodulin kinase cascades. Mol. Endocrinol. 2008, 22, 2759–2765. [Google Scholar] [CrossRef]
- Kokubo, M.; Nishio, M.; Ribar, T.J.; Anderson, K.A.; West, A.E.; Means, A.R. BDNF-mediated cerebellar granule cell development is impaired in mice null for CaMKK2 or CaMKIV. J. Neurosci. 2009, 29, 8901–8913. [Google Scholar] [CrossRef]
- Khodosevich, K.; Seeburg, P.H.; Monyer, H. Major signaling pathways in migrating neuroblasts. Front. Mol. Neurosci. 2009, 2, 7. [Google Scholar] [CrossRef]
- Hutchins, B.I.; Klenke, U.; Wray, S. Calcium release-dependent actin flow in the leading process mediates axophilic migration. J. Neurosci. 2013, 33, 11361–11371. [Google Scholar] [CrossRef]
- Kou, R.; Sartoretto, J.; Michel, T. Regulation of Rac1 by simvastatin in endothelial cells: Differential roles of AMP-activated protein kinase and calmodulin-dependent kinase kinase-β. J. Biol. Chem. 2009, 284, 14734–14743. [Google Scholar] [CrossRef]
- A Reihill, J.; Ewart, M.-A.; Salt, I.P. The role of AMP-activated protein kinase in the functional effects of vascular endothelial growth factor-A and -B in human aortic endothelial cells. Vasc. Cell 2011, 3, 9. [Google Scholar] [CrossRef]
- Baruzzi, A.; Caveggion, E.; Berton, G. Regulation of phagocyte migration and recruitment by Src-family kinases. Cell. Mol. Life Sci. 2008, 65, 2175–2190. [Google Scholar] [CrossRef]
- Anguita, E.; Villalobo, A. Ca2+ signaling and Src-kinases-controlled cellular functions. Arch. Biochem. Biophys. 2018, 650, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Stateva, S.R.; Salas, V.; Anguita, E.; Benaim, G.; Villalobo, A. Ca2+/calmodulin and apo-calmodulin both bind to and enhance the tyrosine kinase activity of c-Src. PLoS ONE 2015, 10, e0128783. [Google Scholar] [CrossRef] [PubMed]
- Anguita, E.; Villalobo, A. Src-family tyrosine kinases and the Ca2+ signal. Biochim. Biophys. Acta 2017, 1864, 915–932. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, P.; Rosenbaum, M.A.; Sinharoy, P.; Damron, D.S.; Birnbaumer, L.; Graham, L.M. Membrane translocation of TRPC6 channels and endothelial migration are regulated by calmodulin and PI3 kinase activation. Proc. Natl. Acad. Sci. USA 2016, 113, 2110–2115. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhang, M.; Jang, H.; Lu, S.; Lin, S.; Chen, G.-Q.; Nussinov, R.; Zhang, J.; Gaponenko, V. Interaction of calmodulin with the cSH2 domain of the p85 regulatory subunit. Biochemistry 2018, 57, 1917–1928. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.; Julsgart, J.; Berchtold, M.W. High affinity calmodulin target sequence in the signalling molecule PI3-kinase. FEBS Lett. 1998, 425, 175–177. [Google Scholar] [CrossRef]
- Will, N.; Lee, K.; Hajredini, F.; Giles, D.H.; Abzalimov, R.R.; Clarkson, M.; Dalby, K.N.; Ghose, R. Structural dynamics of the activation of elongation factor 2 kinase by Ca2+-calmodulin. J. Mol. Biol. 2018, 430, 2802–2821. [Google Scholar] [CrossRef]
- Proud, C.G. Regulation and roles of elongation factor 2 kinase. Biochem. Soc. Trans. 2015, 43, 328–332. [Google Scholar] [CrossRef]
- Usui, T.; Nijima, R.; Sakatsume, T.; Otani, K.; Kameshima, S.; Okada, M.; Yamawaki, H. Eukaryotic elongation factor 2 kinase controls proliferation and migration of vascular smooth muscle cells. Acta Physiol. (Oxf.) 2015, 213, 472–480. [Google Scholar] [CrossRef]
- Rusnak, F.; Mertz, P. Calcineurin: Form and function. Physiol. Rev. 2000, 80, 1483–1521. [Google Scholar] [CrossRef]
- Shibasaki, F.; Hallin, U.; Uchino, H. Calcineurin as a multifunctional regulator. J. Biochem. 2002, 131, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Zaninetti, R.; Tacchi, S.; Erriquez, J.; DiStasi, C.; Maggi, R.; Cariboni, A.; Condorelli, F.; Canonico, P.L.; Genazzani, A.A. Calcineurin primes immature gonadotropin-releasing hormone-secreting neuroendocrine cells for migration. Mol. Endocrinol. 2008, 22, 729–736. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lawson, M.A.; Maxfield, F.R. Ca2+- and calcineurin-dependent recycling of an integrin to the front of migrating neutrophils. Nature 1995, 377, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Cherfils, J.; Zeghouf, M. Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol. Rev. 2013, 93, 269–309. [Google Scholar] [CrossRef]
- Pantarelli, C.; Welch, H.C.E. Rac-GTPases and Rac-GEFs in neutrophil adhesion, migration and recruitment. Eur. J. Clin. Investig. 2018, 48, e12939. [Google Scholar] [CrossRef]
- Mishra, A.K.A.K.; Lambright, D.G. Invited review: Small GTPases and their GAPs. Biopolym. 2016, 105, 431–448. [Google Scholar] [CrossRef]
- Debant, A.; Serra-Pages, C.; Seipel, K.; O’Brien, S.; Tang, M.; Park, S.H.; Streuli, M. The multidomain protein Trio binds the LAR transmembrane tyrosine phosphatase, contains a protein kinase domain, and has separate rac-specific and rho-specific guanine nucleotide exchange factor domains. Proc. Natl. Acad. Sci. USA 1996, 93, 5466–5471. [Google Scholar] [CrossRef]
- Sarhan, A.R.; Patel, T.R.; Cowell, A.R.; Tomlinson, M.G.; Hellberg, C.; Heath, J.K.; Cunningham, D.L.; Hotchin, N.A. LAR protein tyrosine phosphatase regulates focal adhesions through CDK. J. Cell Sci. 2016, 129, 2962–2971. [Google Scholar] [CrossRef]
- Diring, J.; Mouilleron, S.; McDonald, N.Q.; Treisman, R. RPEL-family rhoGAPs link Rac/Cdc42 GTP loading to G-actin availability. Nature 2019, 21, 845–855. [Google Scholar] [CrossRef]
- Rikitake, Y.; Takai, Y. Directional cell migration regulation by small G proteins, nectin-like molecule-5, and afadin. Int. Rev. Cell Mol. Biol. 2011, 287, 97–143. [Google Scholar]
- Ridley, A.J. Rho GTPase signalling in cell migration. Curr. Opin. Cell Biol. 2015, 36, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Lawson, C.D.; Ridley, A.J. Rho GTPase signaling complexes in cell migration and invasion. J. Cell Biol. 2018, 217, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Kale, V.P.; Hengst, J.A.; Desai, D.H.; Amin, S.G.; Yun, J.K. The regulatory roles of ROCK and MRCK kinases in the plasticity of cancer cell migration. Cancer Lett. 2015, 361, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Bishop, A.L.; Hall, A. Rho GTPases and their effector proteins. Biochem. J. 2000, 348, 241–255. [Google Scholar] [CrossRef]
- Ridley, A.J. Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends. Cell Biol. 2006, 16, 522–529. [Google Scholar] [CrossRef]
- Parri, M.; Chiarugi, P. Rac and Rho GTPases in cancer cell motility control. Cell Commun. Signal. 2010, 8, 23. [Google Scholar] [CrossRef]
- Stengel, K.; Zheng, Y. Cdc42 in oncogenic transformation, invasion, and tumorigenesis. Cell. Signal. 2011, 23, 1415–1423. [Google Scholar] [CrossRef]
- Elsaraj, S.M.; Bhullar, R.P. Regulation of platelet Rac1 and Cdc42 activation through interaction with calmodulin. Biochim. Biophys. Acta 2008, 1783, 770–778. [Google Scholar] [CrossRef]
- Vidal-Quadras, M.; Gelabert-Baldrich, M.; Soriano-Castell, D.; Lladó, A.; Rentero, C.; Calvo, M.; Pol, A.; Enrich, C.; Tebar, F. Rac1 and calmodulin interactions modulate dynamics of ARF6-dependent endocytosis. Traffic 2011, 12, 1879–1896. [Google Scholar] [CrossRef]
- Fivaz, M.; Meyer, T. Reversible intracellular translocation of K-Ras but not H-Ras in hippocampal neurons regulated by Ca2+/calmodulin. J. Cell Biol. 2005, 170, 429–441. [Google Scholar] [CrossRef]
- Lopez-Alcalá, C.; Alvarez-Moya, B.; Villalonga, P.; Calvo, M.; Bachs, O.; Agell, N. Identification of essential interacting elements in K-Ras/calmodulin binding and its role in K-Ras localization. J. Biol. Chem. 2008, 283, 10621–10631. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Planchon, S.M.; Wolfman, J.C.; Wolfman, A. Growth factor-dependent AKT activation and cell migration requires the function of c-K(B)-Ras versus other cellular Ras isoforms. J. Biol. Chem. 2006, 281, 29730–29738. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Manser, E. Myotonic dystrophy kinase-related Cdc42-binding kinases (MRCK), the ROCK-like effectors of Cdc42 and Rac1. Small GTPases 2015, 6, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Fukata, M. Identification of IQGAP as a putative target for the small GTPases, Cdc42 and Rac1. J. Biol. Chem. 1996, 271, 23363–23367. [Google Scholar]
- Erickson, J.W.; Cerione, R.A.; Hart, M.J. Identification of an actin cytoskeletal complex that includes IQGAP and the Cdc42 GTPase. J. Biol. Chem. 1997, 272, 24443–24447. [Google Scholar] [CrossRef] [PubMed]
- Suetsugu, S.; Miki, H.; Takenawa, T. Identification of two human WAVE/SCAR homologues as general actin regulatory molecules which associate with the Arp2/3 complex. Biochem. Biophys. Res. Commun. 1999, 260, 296–302. [Google Scholar] [CrossRef]
- Takenawa, T.; Miki, H. WASP and WAVE family proteins: Key molecules for rapid rearrangement of cortical actin filaments and cell movement. J. Cell Sci. 2001, 114, 1801–1809. [Google Scholar]
- Tebar, F.; Enrich, C.; Rentero, C.; Grewal, T. GTPases Rac1 and Ras signaling from endosomes. Prog. Mol. Subcell. Biol. 2018, 57, 65–105. [Google Scholar]
- Krauthammer, M.; Kong, Y.; Ha, B.H.; Evans, P.; Bacchiocchi, A.; McCusker, J.P.; Cheng, E.; Davis, M.J.; Goh, G.; Choi, M.; et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat. Genet. 2012, 44, 1006–1014. [Google Scholar] [CrossRef]
- Feltham, J.L.; Dötsch, V.; Raza, S.; Manor, D.; Cerione, R.A.; Sutcliffe, M.J.; Wagner, G.; Oswald, R.E. Definition of the switch surface in the solution structure of Cdc42Hs. Biochemistry 1997, 36, 8755–8766. [Google Scholar] [CrossRef]
- Mills, E.; Truong, K. Ca2+-mediated synthetic biosystems offer protein design versatility, signal specificity, and pathway rewiring. Chem. Biol. 2011, 18, 1611–1619. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.; Pham, E.; Nagaraj, S.; Truong, K. Engineered networks of synthetic and natural proteins to control cell migration. ACS Synth. Biol. 2012, 1, 211–220. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, R.S.; Casanova, J.E. The BRAG/IQSec family of Arf GEFs. Small GTPases 2016, 7, 257–264. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Welch, D.R.; Hurst, D.R. Defining the hallmarks of metastasis. Cancer Res. 2019. [Google Scholar] [CrossRef]
- Pearson, G.W. Control of invasion by epithelial-to-mesenchymal transition programs during metastasis. J. Clin. Med. 2019, 8, 646. [Google Scholar] [CrossRef]
- Roy, A.; Ye, J.; Deng, F.; Wang, Q.J. Protein kinase D signaling in cancer: A friend or foe? Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 283–294. [Google Scholar] [CrossRef]
- Ungefroren, H.; Witte, D.; Lehnert, H. The role of small GTPases of the Rho/Rac family in TGF-β-induced EMT and cell motility in cancer. Dev. Dyn. 2018, 247, 451–461. [Google Scholar] [CrossRef]
- Sanz-Moreno, V.; Gadéa, G.; Ahn, J.; Paterson, H.; Marra, P.; Pinner, S.; Sahai, E.; Marshall, C.J. Rac Activation and inactivation control plasticity of tumor cell movement. Cell 2008, 135, 510–523. [Google Scholar] [CrossRef]
- Joyce, J.A.; Pollard, J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer 2009, 9, 239–252. [Google Scholar] [CrossRef]
- Doglioni, G.; Parik, S.; Fendt, S.-M. Interactions in the (pre)metastatic niche support metastasis formation. Front. Oncol. 2019, 9, 219. [Google Scholar] [CrossRef]
- Wortzel, I.; Dror, S.; Kenific, C.M.; Lyden, D. Exosome-mediated metastasis: Communication from a distance. Dev. Cell 2019, 49, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Martínez, A.; Sew, W.Q.G.; Molano-Fernández, M.; Carretero-Junquera, M.; Herranz, H. Mechanisms of oncogenic cell competition–Paths of victory. Semin. Cancer Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Jackson, T.R.; Davidson, L.A. On the role of mechanics in driving mesenchymal-to-epithelial transitions. Semin. Cell Dev. Biol. 2017, 67, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Destaing, O.; Block, M.R.; Planus, E.; Albiges-Rizo, C. Invadosome regulation by adhesion signaling. Curr. Opin. Cell Biol. 2011, 23, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Foxall, E.; Pipili, A.; Jones, G.E.; Wells, C.M. Significance of kinase activity in the dynamic invadosome. Eur. J. Cell Biol. 2016, 95, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Di Martino, J.; Henriet, E.; Ezzoukhry, Z.; Goetz, J.G.; Moreau, V.; Saltel, F. The microenvironment controls invadosome plasticity. J. Cell Sci. 2016, 129, 1759–1768. [Google Scholar] [CrossRef]
- Zeng, Q.; Michael, I.P.; Zhang, P.; Saghafinia, S.; Knott, G.; Jiao, W.; McCabe, B.D.; Galván, J.A.; Robinson, H.P.C.; Zlobec, I.; et al. Synaptic proximity enables NMDAR signalling to promote brain metastasis. Nature 2019, 573, 526–531. [Google Scholar] [CrossRef]
- Venkataramani, V.; Tanev, D.I.; Strahle, C.; Studier-Fischer, A.; Fankhauser, L.; Kessler, T.; Körber, C.; Kardorff, M.; Ratliff, M.; Xie, R.; et al. Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature 2019, 573, 532–538. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Morishita, W.; Geraghty, A.C.; Silverbush, D.; Gillespie, S.M.; Arzt, M.; Tam, L.T.; Espenel, C.; Ponnuswami, A.; Ni, L.; et al. Electrical and synaptic integration of glioma into neural circuits. Nature 2019, 573, 539–545. [Google Scholar] [CrossRef]
- Barria, A. Dangerous liaisons as tumour cells form synapses with neurons. Nature 2019, 573, 499–501. [Google Scholar] [CrossRef]
- Affara, N.I.; Robertson, F.M. Vascular endothelial growth factor as a survival factor in tumor-associated angiogenesis. In Vivo 2004, 18, 525–542. [Google Scholar] [PubMed]
- Gerber, P.A.; Hippe, A.; Buhren, B.A.; Müller, A.; Homey, B. Chemokines in tumor-associated angiogenesis. Biol. Chem. 2009, 390, 1213–1223. [Google Scholar] [CrossRef] [PubMed]
- Attalla, H.; Mäkelä, T.P.; Adlercreutz, H.; Andersson, L.C. 2-Methoxyestradiol arrests cells in mitosis without depolymerizing tubulin. Biochem. Biophys. Res. Commun. 1996, 228, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, M.; Takeoka, M.; Oguchi, M.; Koganehira, Y.; Murata, H.; Ehara, T.; Tozuka, M.; Saida, T.; Taniguchi, S. Calponin h1 suppresses tumor growth of Src-induced transformed 3Y1 cells in association with a decrease in angiogenesis. Jpn. J. Cancer Res. 2002, 93, 935–943. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Wang, Y.; Meng, C.; Fang, N. FMRP regulates endothelial cell proliferation and angiogenesis via the miR-181a-CaM-CaMKII pathway. Cell Biol. Int. 2018, 42, 1432–1444. [Google Scholar] [CrossRef] [PubMed]
- Brzozowski, J.S.; Skelding, K.A. The multi-functional calcium/calmodulin stimulated protein kinase (CaMK) family: emerging targets for anti-cancer therapeutic intervention. Pharmaceuticals 2019, 12, 8. [Google Scholar] [CrossRef] [PubMed]
- Umemura, M.; Baljinnyam, E.; Feske, S.; de Lorenzo, M.S.; Xie, L.H.; Feng, X.; Oda, K.; Makino, A.; Fujita, T.; Yokoyama, U.; et al. Store-operated Ca2+ entry (SOCE) regulates melanoma proliferation and cell migration. PLoS ONE 2014, 9, e89292. [Google Scholar] [CrossRef]
- Hodeify, R.; Yu, F.; Courjaret, R.; Nader, N.; Dib, M.; Sun, L.; Adap, E.; Hubrack, S.; Machaca, K. Regulation and role of store-operated Ca2+ entry in cellular proliferation. In Calcium Entry Channels in Non-Excitable Cells; Kozak, J.A., Putney, J.W., Jr., Eds.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2018; pp. 215–240. [Google Scholar]
- Davare, M.A.; Saneyoshi, T.; Soderling, T.R. Calmodulin-kinases regulate basal and estrogen stimulated medulloblastoma migration via Rac1. J. Neurooncol. 2011, 104, 65–82. [Google Scholar] [CrossRef]
- Kim, E.-K.; Park, J.-M.; Lim, S.; Choi, J.W.; Kim, H.S.; Seok, H.; Seo, J.K.; Oh, K.; Lee, N.-S.; Kim, K.T.; et al. Activation of AMP-activated protein kinase is essential for lysophosphatidic acid-induced cell migration in ovarian cancer cells. J. Biol. Chem. 2011, 286, 24036–24045. [Google Scholar] [CrossRef]
- Frigo, D.E.; Howe, M.K.; Wittmann, B.M.; Brunner, A.M.; Cushman, I.; Wang, Q.; Brown, M.; Means, A.R.; McDonnell, D.P. CaM kinase kinase-β-mediated activation of the growth regulatory kinase AMPK is required for androgen-dependent migration of prostate cancer cells. Cancer Res. 2011, 71, 528–537. [Google Scholar] [CrossRef]
- Popovics, P.; Frigo, D.E.; Schally, A.V.; Rick, F.G. Targeting the 5′-AMP-activated protein kinase and related metabolic pathways for the treatment of prostate cancer. Expert Opin. Ther. Targets 2015, 19, 617–632. [Google Scholar] [CrossRef] [PubMed]
- Dadwal, U.C.; Chang, E.S.; Sankar, U. Androgen receptor-CaMKK2 axis in prostate cancer and bone microenvironment. Front. Endocrinol. 2018, 9, 335. [Google Scholar] [CrossRef] [PubMed]
- Bergamaschi, A.; Kim, Y.H.; Kwei, K.A.; La Choi, Y.; Bocanegra, M.; Langerød, A.; Han, W.; Noh, N.-Y.; Huntsman, D.G.; Jeffrey, S.S.; et al. CAMK1D amplification implicated in epithelial–mesenchymal transition in basal-like breast cancer. Mol. Oncol. 2008, 2, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Cai, Y.; Li, Y.; Li, Y.; Hu, N.; Ma, S.; Hu, S.; Zhu, P.; Wang, W.; Zhou, H. Yap promotes hepatocellular carcinoma metastasis and mobilization via governing cofilin/F-actin/lamellipodium axis by regulation of JNK/Bnip3/SERCA/CaMKII pathways. Redox Biol. 2018, 14, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, E.K.; Koval, O.M.; Noble, P.; Broadhurst, K.; Allamargot, C.; Wu, M.; Strack, S.; Thiel, W.H.; Grumbach, I.M. CaMKII (Ca2+/calmodulin-dependent kinase II) in mitochondria of smooth muscle cells controls mitochondrial mobility, migration, and neointima formation. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1333–1345. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Cheng, C.J.; Lin, S.C.; Lee, Y.C.; Frigo, D.E.; Yu-Lee, L.Y.; Gallick, G.E.; Titus, M.A.; Nutt, L.K.; Lin, S.H. Organelle-derived acetyl-CoA promotes prostate cancer cell survival, migration, and metastasis via activation of calmodulin ksinase II. Cancer Res. 2018, 78, 2490–2502. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; An, P.; Quan, X.J.; Zhang, J.; Zhou, Z.Y.; Zou, L.P.; Luo, H.S. Ca2+/calmodulin-dependent protein kinase II regulates colon cancer proliferation and migration via ERK1/2 and p38 pathways. World J. Gastroenterol. 2017, 23, 6111–6118. [Google Scholar] [CrossRef]
- Tian, S.; Hu, J.; Tao, K.; Wang, J.; Chu, Y.; Li, J.; Liu, Z.; Ding, X.; Xu, L.; Li, Q.; et al. Secreted AGR2 promotes invasion of colorectal cancer cells via Wnt11-mediated non-canonical Wnt signaling. Exp. Cell Res. 2018, 364, 198–207. [Google Scholar] [CrossRef]
- Chi, M.; Evans, H.; Gilchrist, J.; Mayhew, J.; Hoffman, A.; Pearsall, E.A.; Jankowski, H.; Brzozowski, J.S.; Skelding, K.A. Phosphorylation of calcium/calmodulin-stimulated protein kinase II at T286 enhances invasion and migration of human breast cancer cells. Sci. Rep. 2016, 6, 33132. [Google Scholar] [CrossRef]
- Liu, Z.; Han, G.; Cao, Y.; Wang, Y.; Gong, H. Calcium/calmodulindependent protein kinase II enhances metastasis of human gastric cancer by upregulating nuclear factor-κB and Akt-mediated matrix metalloproteinase-9 production. Mol. Med. Rep. 2014, 10, 2459–2464. [Google Scholar] [CrossRef]
- Choi, H.S.; Kim, D.-A.; Chung, H.; Park, I.H.; Kim, B.H.; Oh, E.-S.; Kang, D.-H. Screening of breast cancer stem cell inhibitors using a protein kinase inhibitor library. Cancer Cell Int. 2017, 17, 25. [Google Scholar] [CrossRef] [PubMed]
- Daft, P.G.; Yuan, K.; Warram, J.M.; Klein, M.J.; Siegal, G.P.; Zayzafoon, M. α-CaMKII plays a critical role in determining the aggressive behavior of human osteosarcoma. Mol. Cancer Res. 2013, 11, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Russo, E.; Salzano, M.; De Falco, V.; Mian, C.; Barollo, S.; Secondo, A.; Bifulco, M.; Vitale, M. Calcium/calmodulin-dependent protein kinase II and its endogenous inhibitor in medullary thyroid Cancer. Clin. Cancer Res. 2014, 20, 1513–1520. [Google Scholar] [CrossRef] [PubMed]
- Asghar, M.Y.; Magnusson, M.; Kemppainen, K.; Sukumaran, P.; Lof, C.; Pulli, I.; Kalhori, V.; Tornquist, K. Transient receptor potential canonical 1 (TRPC1) channels as regulators of sphingolipid and VEGF receptor expression: Implications for thyroid cancer cell migration and proliferation. J. Biol. Chem. 2015, 290, 16116–16131. [Google Scholar] [CrossRef]
- Cheng, L.; Wu, Q.; Guryanova, O.A.; Huang, Z.; Huang, Q.; Rich, J.N.; Bao, S. Elevated invasive potential of glioblastoma stem cells. Biochem. Biophys. Res. Commun. 2011, 406, 643–648. [Google Scholar] [CrossRef]
- Li, T.; Yi, L.; Hai, L.; Ma, H.; Tao, Z.; Zhang, C.; Abeysekera, I.R.; Zhao, K.; Yang, Y.; Wang, W.; et al. The interactome and spatial redistribution feature of Ca2+ receptor protein calmodulin reveals a novel role in invadopodia-mediated invasion. Cell Death Dis. 2018, 9, 292. [Google Scholar] [CrossRef]
- Bourguignon, L.Y.; Gunja-Smith, Z.; Iida, N.; Zhu, H.; Young, L.; Muller, W.J.; Cardiff, R. CD44v3,8-10 is involved in cytoskeleton-mediated tumor cell migration and matrix metalloproteinase (MMP-9) association in metastatic breast cancer cells. J. Cell. Physiol. 1998, 176, 206–215. [Google Scholar] [CrossRef]
- Shin, H.J.; Lee, S.; Jung, H.J. A curcumin derivative hydrazinobenzoylcurcumin suppresses stem-like features of glioblastoma cells by targeting Ca2+/calmodulin-dependent protein kinase II. J. Cell Biochem. 2019, 120, 6741–6752. [Google Scholar] [CrossRef]
- Wang, X. Structural studies of GDNF family ligands with their receptors—Insights into ligand recognition and activation of receptor tyrosine kinase RET. Biochim. Biophys. Acta 2013, 1834, 2205–2212. [Google Scholar] [CrossRef]
- Chen, C.-C.; Sureshbabul, M.; Chen, H.-W.; Lin, Y.-S.; Lee, J.-Y.; Hong, Q.-S.; Yang, Y.-C.; Yu, S.-L. Curcumin suppresses metastasis via Sp-1, FAK inhibition, and E-cadherin upregulation in colorectal cancer. Biochim. Biophys. Acta 2013, 2013, 1–17. [Google Scholar] [CrossRef]
- Cuddapah, V.A.; Sontheimer, H. Molecular interaction and functional regulation of ClC-3 by Ca2+/calmodulin-dependent protein kinase II (CaMKII) in human malignant glioma. J. Biol. Chem. 2010, 285, 11188–11196. [Google Scholar] [CrossRef] [PubMed]
- Cuddapah, V.A.; Turner, K.L.; Seifert, S.; Sontheimer, H. Bradykinin-induced chemotaxis of human gliomas requires the activation of KCa3.1 and ClC-3. J. Neurosci. 2013, 33, 1427–1440. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Zhao, D.; Li, Y.L.; Sun, Y.; Lei, X.H.; Zhang, J.N.; Wu, M.M.; Li, R.Y.; Zhao, Z.F.; Zhang, Z.R.; et al. Regulation of ASIC1 by Ca2+/calmodulin-dependent protein kinase II in human glioblastoma multiforme. Oncol. Rep. 2013, 30, 2852–2858. [Google Scholar] [CrossRef] [PubMed]
- Ganapathi, S.B.; Wei, S.G.; Zaremba, A.; Lamb, F.S.; Shears, S.B. Functional regulation of ClC-3 in the migration of vascular smooth muscle cells. Hypertension 2013, 61, 174–179. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ishida, A.; Sueyoshi, N.; Kameshita, I. Functions and dysfunctions of Ca2+/calmodulin-dependent protein kinase phosphatase (CaMKP/PPM1F) and CaMKP-N/PPM1E. Arch. Biochem. Biophys. 2018, 640, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Weng, T.; Cheruba, E.; Guo, T.; Chan, H.; Sze, S.K.; Koh, C.G. Phosphatase POPX2 exhibits dual regulatory functions in cancer metastasis. J. Proteome Res. 2017, 16, 698–711. [Google Scholar] [CrossRef]
- Luo, G.; Chao, Y.L.; Tang, B.; Li, B.S.; Xiao, Y.F.; Xie, R.; Wang, S.M.; Wu, Y.Y.; Dong, H.; Liu, X.D.; et al. miR-149 represses metastasis of hepatocellular carcinoma by targeting actin-regulatory proteins PPM1F. Oncotarget 2015, 6, 37808–37823. [Google Scholar] [CrossRef]
- Geering, B. Death-associated protein kinase 2: Regulator of apoptosis, autophagy and inflammation. Int. J. Biochem. Cell Biol. 2015, 65, 151–154. [Google Scholar] [CrossRef]
- Lin, Y.; Hupp, T.R.; Stevens, C. Death-associated protein kinase (DAPK) and signal transduction: Additional roles beyond cell death. FEBS J. 2010, 277, 48–57. [Google Scholar] [CrossRef]
- Inbal, B.; Cohen, O.; Polak-Charcon, S.; Kopolovic, J.; Vadai, E.; Eisenbach, L.; Kimchi, A. DAP kinase links the control of apoptosis to metastasis. Nature 1997, 390, 180–184. [Google Scholar] [CrossRef]
- Kuo, J.-C.; Wang, W.-J.; Yao, C.-C.; Wu, P.-R.; Chen, R.-H. The tumor suppressor DAPK inhibits cell motility by blocking the integrin-mediated polarity pathway. J. Cell Biol. 2006, 172, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Simpson, D.J.; Clayton, R.N.; Farrell, W.E. Preferential loss of death associated protein kinase expression in invasive pituitary tumours is associated with either CpG island methylation or homozygous deletion. Oncogene 2002, 21, 1217–1224. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-F.; Kong, F.-M.; Xu, Z.; Yu, S.-P.; Sun, F.-B.; Zhang, C.-S.; Huang, Q.-X.; Zhou, X.-T.; Song, Z.-W. Promoter hypermethylation of death-associated protein kinase gene in cholangiocarcinoma. Hepatobiliary Pancreat. Dis. Int. 2007, 6, 407–411. [Google Scholar] [PubMed]
- Gade, P.; Singh, A.K.; Roy, S.K.; Reddy, S.P.; Kalvakolanu, D.V. Down-regulation of the transcriptional mediator subunit Med1 contributes to the loss of expression of metastasis-associated DAPK1 in human cancers and cancer cells. Int. J. Cancer 2009, 125, 1566–1574. [Google Scholar] [CrossRef]
- Wang, W.-J.; Kuo, J.-C.; Ku, W.; Lee, Y.-R.; Lin, F.-C.; Chang, Y.-L.; Lin, Y.-M.; Chen, C.-H.; Huang, Y.-P.; Chiang, M.-J.; et al. The tumor suppressor DAPK is reciprocally regulated by tyrosine kinase Src and phosphatase LAR. Mol. Cell 2007, 27, 701–716. [Google Scholar] [CrossRef]
- LaConte, L.; Mukherjee, K. Structural constraints and functional divergences in CASK evolution. Biochem. Soc. Trans. 2013, 41, 1017–1022. [Google Scholar] [CrossRef]
- Hsueh, Y.-P. The role of the MAGUK protein CASK in neural development and synaptic function. Curr. Med. Chem. 2006, 13, 1915–1927. [Google Scholar] [CrossRef]
- Wei, J.-L.; Fu, Z.-X.; Fang, M.; Zhou, Q.-Y.; Zhao, Q.-N.; Guo, J.-B.; Lü, W.-D.; Wang, H. High expression of CASK correlates with progression and poor prognosis of colorectal cancer. Tumor Biol. 2014, 35, 9185–9194. [Google Scholar] [CrossRef]
- Zhou, X.; Xu, G.; Yin, C.; Jin, W.; Zhang, G. Down-regulation of miR-203 induced by Helicobacter pylori infection promotes the proliferation and invasion of gastric cancer by targeting CASK. Oncotarget 2014, 5, 11631–11640. [Google Scholar] [CrossRef]
- Bayraktar, R.; Pichler, M.; Kanlikilicer, P.; Ivan, C.; Bayraktar, E.; Kahraman, N.; Aslan, B.; Oguztuzun, S.; Ulasli, M.; Arslan, A.; et al. MicroRNA 603 acts as a tumor suppressor and inhibits triple-negative breast cancer tumorigenesis by targeting elongation factor 2 kinase. Oncotarget 2017, 8, 11641–11658. [Google Scholar] [CrossRef]
- Ashour, A.A.; Gurbuz, N.; Alpay, S.N.; Abdel-Aziz, A.A.; Mansour, A.M.; Huo, L.; Ozpolat, B. Elongation factor-2 kinase regulates TGFβ1 integrin/Src/uPAR pathway and epithelial-mesenchymal transition mediating pancreatic cancer cells invasion. J. Cell Mol. Med. 2014, 18, 2235–2251. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Hou, Y.; Yin, X.; Su, J.; Zhao, Z.; Ye, X.; Zhou, X.; Zhou, L.; Wang, Z. Rottlerin inhibits cell growth and invasion via down-regulation of Cdc20 in glioma cells. Oncotarget 2016, 7, 69770–69782. [Google Scholar] [CrossRef] [PubMed]
- Mujica, A.O.; Brauksiepe, B.; Saaler-Reinhardt, S.; Reuss, S.; Schmidt, E.R. Differential expression pattern of the novel serine/threonine kinase, STK33, in mice and men. FEBS J. 2005, 272, 4884–4898. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.D.; Ma, S.P.; Liu, F.; Chen, Y.Z. Role of serine/threonine kinase 33 methylation in colorectal cancer and its clinical significance. Oncol. Lett. 2018, 15, 2153–2160. [Google Scholar] [CrossRef] [PubMed]
- Quang, C.T.; Leboucher, S.; Passaro, D.; Fuhrmann, L.; Nourieh, M.; Vincent-Salomon, A.; Ghysdael, J. The calcineurin/NFAT pathway is activated in diagnostic breast cancer cases and is essential to survival and metastasis of mammary cancer cells. Cell Death Dis. 2015, 6, e1658. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.-L.; Zhao, S.-H.; Wang, Z.; Qiu, B.; Li, B.-Z.; Zhou, F.; Tan, X.-G.; He, J. Expression and unique functions of four nuclear factor of activated T cells isoforms in non-small cell lung cancer. Chin. J. Cancer 2011, 30, 62–68. [Google Scholar] [CrossRef]
- Hojo, M.; Morimoto, T.; Maluccio, M.; Asano, T.; Morimoto, K.; Lagman, M.; Shimbo, T.; Suthanthiran, M. Cyclosporine induces cancer progression by a cell-autonomous mechanism. Nature 1999, 397, 530–534. [Google Scholar] [CrossRef]
- Sliwa, M.; Markovic, D.; Gabrusiewicz, K.; Synowitz, M.; Glass, R.; Zawadzka, M.; Wesolowska, A.; Kettenmann, H.; Kaminska, B. The invasion promoting effect of microglia on glioblastoma cells is inhibited by cyclosporin A. Brain 2007, 130, 476–489. [Google Scholar] [CrossRef]
- Yiu, G.K.; Toker, A. NFAT induces breast cancer cell invasion by promoting the induction of cyclooxygenase-2. J. Biol. Chem. 2006, 281, 12210–12217. [Google Scholar] [CrossRef]
- Juhász, T.; Matta, C.; Veress, G.; Nagy, G.; Szíjgyártó, Z.; Molnár, Z.; Fodor, J.; Zákány, R.; Gergely, P. Inhibition of calcineurin by cyclosporine A exerts multiple effects on human melanoma cell lines HT168 and WM35. Int. J. Oncol. 2009, 34, 995–1003. [Google Scholar]
- Liu, Y.; Zhang, Y.; Min, J.; Liu, L.-L.; Ma, N.-Q.; Feng, Y.-M.; Liu, N.; Wang, P.-Z.; Huang, D.-D.; Zhuang, Y.; et al. Calcineurin promotes proliferation, migration, and invasion of small cell lung cancer. Tumor Biol. 2010, 31, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.Q.; Liu, L.L.; Min, J.; Wang, J.W.; Jiang, W.F.; Liu, Y.; Feng, Y.G.; Su, H.C.; Feng, Y.M.; Zhang, H.L. The effect of down regulation of calcineurin Aα by lentiviral vector-mediated RNAi on the biological behavior of small-cell lung cancer and its bone metastasis. Clin. Exp. Metastasis 2011, 28, 765–778. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-L.; Wang, Y.; Tong, L.; Wei, Q. Overexpression of calcineurin B subunit (CnB) enhances the oncogenic potential of HEK293 cells. Cancer Sci. 2008, 99, 1100–1108. [Google Scholar] [CrossRef] [PubMed]
- Stetler-Stevenson, W.G. The role of matrix metalloproteinases in tumor invasion, metastasis, and angiogenesis. Surg. Oncol. Clin. N. Am. 2001, 10, 383–392. [Google Scholar] [CrossRef]
- Stetler-Stevenson, W.G.; Yu, A.E. Proteases in invasion: Matrix metalloproteinases. Semin. Cancer Biol. 2001, 11, 143–152. [Google Scholar] [CrossRef]
- Yip, C.; Foidart, P.; Noël, A.; Sounni, N.E. MT4-MMP: The GPI-anchored membrane-type matrix metalloprotease with multiple functions in diseases. Int. J. Mol. Sci. 2019, 20, 354. [Google Scholar] [CrossRef]
- Matter, H.; Schudok, M. Recent advances in the design of matrix metalloprotease inhibitors. Curr. Opin. Drug Discov. Dev. 2004, 7, 513–535. [Google Scholar]
- Tsai, Y.F.; Tseng, L.M.; Hsu, C.Y.; Yang, M.H.; Chiu, J.H.; Shyr, Y.M. Brain-derived neurotrophic factor (BDNF)-TrKB signaling modulates cancer-endothelial cells interaction and affects the outcomes of triple negative breast cancer. PLoS ONE 2017, 12, e0178173. [Google Scholar] [CrossRef]
- Karp, C.M.; Shukla, M.N.; Buckley, D.J.; Buckley, A.R. HRPAP20: A novel calmodulin-binding protein that increases breast cancer cell invasion. Oncogene 2007, 26, 1780–1788. [Google Scholar] [CrossRef][Green Version]
- Zhao, H.; Zhang, L.; Zhang, Y.; Zhao, L.; Wan, Q.; Wang, B.; Bu, X.; Wan, M.; Shen, C. Calmodulin promotes matrix metalloproteinase 9 production and cell migration by inhibiting the ubiquitination and degradation of TBC1D3 oncoprotein in human breast cancer cells. Oncotarget 2017, 8, 36383–36398. [Google Scholar] [CrossRef][Green Version]
- Nagano, O.; Murakami, D.; Hartmann, D.; de Strooper, B.; Saftig, P.; Iwatsubo, T.; Nakajima, M.; Shinohara, M.; Saya, H. Cell-matrix interaction via CD44 is independently regulated by different metalloproteinases activated in response to extracellular Ca2+ influx and PKC activation. J. Cell Biol. 2004, 165, 893–902. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Han, C.; Wang, C.; Hu, G.; Luo, C.; Gan, X.; Zhang, F.; Lu, Y.; Ding, X. ADAM10 promotes pituitary adenoma cell migration by regulating cleavage of CD44 and L1. J. Mol. Endocrinol. 2012, 49, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Sánchez-Torres, J.; Del Carpio, A.F.; Nogales-González, A.; Molina-Ortiz, P.; Moreno, M.J.; Török, K.; Villalobo, A. The adaptor Grb7 is a novel calmodulin-binding protein: Functional implications of the interaction of calmodulin with Grb7. Oncogene 2005, 24, 4206–4219. [Google Scholar] [CrossRef] [PubMed][Green Version]
- García-Palmero, I.; Pompas-Veganzones, N.; Villalobo, E.; Gioria, S.; Haiech, J.; Villalobo, A. The adaptors Grb10 and Grb14 are calmodulin-binding proteins. FEBS Lett. 2017, 591, 1176–1186. [Google Scholar] [CrossRef]
- Daly, R.J. The Grb7 family of signalling proteins. Cell. Signal. 1998, 10, 613–618. [Google Scholar] [CrossRef]
- Lucas-Fernandez, E.; Garcia-Palmero, I.; Villalobo, A. Genomic organization and control of the Grb7 gene family. Curr. Genom. 2008, 9, 60–68. [Google Scholar] [CrossRef][Green Version]
- García-Palmero, I.; Villalobo, A. Deletion of the calmodulin-binding domain of Grb7 impairs cell attachment to the extracellular matrix and migration. Biochem. Biophys. Res. Commun. 2013, 436, 271–277. [Google Scholar] [CrossRef]
- García-Palmero, I.; López-Larrubia, P.; Cerdán, S.; Villalobo, A. Nuclear magnetic resonance imaging of tumour growth and neovasculature performance in vivo reveals Grb7 as a novel antiangiogenic target. NMR Biomed. 2013, 26, 1059–1069. [Google Scholar] [CrossRef]
- Pelikan-Conchaudron, A.; le Clainche, C.; Didry, D.; Carlier, M.F. The IQGAP1 protein is a calmodulin-regulated barbed end capper of actin filaments: Possible implications in its function in cell migration. J. Biol. Chem. 2011, 286, 35119–35128. [Google Scholar] [CrossRef]
- Jang, D.-J.; Ban, B.; Lee, J.-A. Characterization of novel calmodulin binding domains within IQ motifs of IQGAP1. Mol. Cells 2011, 32, 511–518. [Google Scholar] [CrossRef]
- Mataraza, J.M.; Briggs, M.W.; Li, Z.; Entwistle, A.; Ridley, A.J.; Sacks, D.B. IQGAP1 promotes cell motility and invasion. J. Biol. Chem. 2003, 278, 41237–41245. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Sacks, D.B. Elucidation of the interaction of calmodulin with the IQ motifs of IQGAP1. J. Biol. Chem. 2003, 278, 4347–4352. [Google Scholar] [CrossRef] [PubMed]
- Briggs, M.W.; Sacks, D.B. IQGAP1 as signal integrator: Ca2+, calmodulin, Cdc42 and the cytoskeleton. FEBS Lett. 2003, 542, 7–11. [Google Scholar] [CrossRef]
- Brill, S.; Li, S.; Lyman, C.W.; Church, D.M.; Wasmuth, J.J.; Weissbach, L.; Bernards, A.; Snijders, A.J. The Ras GTPase-activating-protein-related human protein IQGAP2 harbors a potential actin binding domain and interacts with calmodulin and Rho family GTPases. Mol. Cell. Biol. 1996, 16, 4869–4878. [Google Scholar] [CrossRef]
- White, C.D.; Brown, M.D.; Sacks, D.B. IQGAPs in cancer: A family of scaffold proteins underlying tumorigenesis. FEBS Lett. 2009, 583, 1817–1824. [Google Scholar] [CrossRef]
- Foroutannejad, S.; Rohner, N.; Reimer, M.; Kwon, G.; Schober, J.M. A novel role for IQGAP1 protein in cell motility through cell retraction. Biochem. Biophys. Res. Commun. 2014, 448, 39–44. [Google Scholar] [CrossRef][Green Version]
- Mataraza, J.M.; Li, Z.; Jeong, H.-W.; Brown, M.D.; Sacks, D.B. Multiple proteins mediate IQGAP1-stimulated cell migration. Cell. Signal. 2007, 19, 1857–1865. [Google Scholar] [CrossRef]
- Hu, W.; Wang, Z.; Zhang, S.; Lu, X.; Wu, J.; Yu, K.; Ji, A.; Lu, W.; Wang, Z.; Wu, J.; et al. IQGAP1 promotes pancreatic cancer progression and epithelial-mesenchymal transition (EMT) through Wnt/β-catenin signaling. Sci. Rep. 2019, 9, 7539. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Gelman, I.H. Suppression of tumor and metastasis progression through the scaffolding functions of SSeCKS/Gravin/AKAP12. Cancer Metastasis Rev. 2012, 31, 493–500. [Google Scholar] [CrossRef]
- Amith, S.R.; Wilkinson, J.M.; Fliegel, L. Na+/H+ exchanger NHE1 regulation modulates metastatic potential and epithelial-mesenchymal transition of triple-negative breast cancer cells. Oncotarget 2016, 7, 21091–21113. [Google Scholar] [CrossRef] [PubMed]
- Amith, S.R.; Wilkinson, J.M.; Fliegel, L. KR-33028, a potent inhibitor of the Na+/H+ exchanger NHE1, suppresses metastatic potential of triple-negative breast cancer cells. Biochem. Pharmacol. 2016, 118, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Ji, B.; Wen, G.; Yang, Y.; Jin, H.; Liu, X.; Xie, R.; Song, W.; Song, P.; Dong, H.; et al. Na+/H+ exchanger 1, Na+/Ca2+ exchanger 1 and calmodulin complex regulates interleukin 6-mediated cellular behavior of human hepatocellular carcinoma. Carcinogenesis 2016, 37, 290–300. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Olschewski, D.N.; Hofschröer, V.; Nielsen, N.; Seidler, D.G.; Schwab, A.; Stock, C. The angiotensin II type 1 receptor antagonist Losartan affects NHE1-dependent melanoma cell behavior. Cell. Physiol. Biochem. 2018, 45, 2560–2576. [Google Scholar] [CrossRef]
- Gandalovičová, A.; Rosel, D.; Fernandes, M.; Veselý, P.; Heneberg, P.; Čermák, V.; Petruželka, L.; Kumar, S.; Sanz-Moreno, V.; Brábek, J. Migrastatics—Anti-metastatic and anti-invasion drugs: promises and challenges. Trends Cancer 2017, 3, 391–406. [Google Scholar] [CrossRef]
- Wang, H.; Gao, X.; Yang, J.J.; Liu, Z.R. Interaction between p68 RNA helicase and Ca2+-calmodulin promotes cell migration and metastasis. Nat. Commun. 2013, 4, 1354. [Google Scholar] [CrossRef][Green Version]
- Hamada, T.; Souda, M.; Yoshimura, T.; Sasaguri, S.; Hatanaka, K.; Tasaki, T.; Yoshioka, T.; Ohi, Y.; Yamada, S.; Tsutsui, M.; et al. Anti-apoptotic effects of PCP4/PEP19 in human breast cancer cell lines: A novel oncotarget. Oncotarget 2014, 5, 6076–6086. [Google Scholar] [CrossRef]
- Yoshimura, T.; Hamada, T.; Hijioka, H.; Souda, M.; Hatanaka, K.; Yoshioka, T.; Yamada, S.; Tsutsui, M.; Umekita, Y.; Nakamura, N.; et al. PCP4/PEP19 promotes migration, invasion and adhesion in human breast cancer MCF-7 and T47D cells. Oncotarget 2016, 7, 49065–49074. [Google Scholar] [CrossRef]
- Hendershott, M.C.; Vale, R.D. Regulation of microtubule minus-end dynamics by CAMSAPs and patronin. Proc. Natl. Acad. Sci. USA 2014, 111, 5860–5865. [Google Scholar] [CrossRef]
- Akhmanova, A.; Steinmetz, M.O. Control of microtubule organization and dynamics: Two ends in the limelight. Nat. Rev. Mol. Cell Biol. 2015, 16, 711–726. [Google Scholar] [CrossRef]
- Sun, X.; Wang, Z.-M.; Song, Y.; Tai, X.-H.; Ji, W.-Y.; Gu, H. MicroRNA-126 modulates the tumor microenvironment by targeting calmodulin-regulated spectrin-associated protein 1 (Camsap 1). Int. J. Oncol. 2014, 44, 1678–1684. [Google Scholar] [CrossRef] [PubMed]
- Pongrakhananon, V.; Wattanathamsan, O.; Takeichi, M.; Chetprayoon, P.; Chanvorachote, P. Loss of CAMSAP3 promotes EMT via the modification of microtubule–Akt machinery. J. Cell Sci. 2018, 131, jcs216168. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Jiang, P.; Du, W.; Wu, Z.; Li, C.; Qiao, M.; Yang, X.; Wu, M. Siva1 suppresses epithelial–mesenchymal transition and metastasis of tumor cells by inhibiting stathmin and stabilizing microtubules. Proc. Natl. Acad. Sci. USA 2011, 108, 12851–12856. [Google Scholar] [CrossRef] [PubMed]
- Panina, S.; Stephan, A.; La Cour, J.M.; Jacobsen, K.; Kallerup, L.K.; Bumbuleviciute, R.; Knudsen, K.V.K.; Sánchez-González, P.; Villalobo, A.; Olesen, U.H.; et al. Significance of calcium binding, tyrosine phosphorylation, and lysine trimethylation for the essential function of calmodulin in vertebrate cells analyzed in a novel gene replacement system. J. Biol. Chem. 2012, 287, 18173–18181. [Google Scholar] [CrossRef] [PubMed]
- Kellar, A.; Egan, C.; Morris, D. Preclinical murine models for lung cancer: Clinical Trial Applications. BioMed Res. Int. 2015, 2015, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Grigorian, M.S.; Tulchinsky, E.M.; Zain, S.; Ebralidze, A.K.; Kramerov, D.A.; Kriajevska, M.V.; Georgiev, G.P.; Lukanidin, E.M. The mts1 gene and control of tumor metastasis. Gene 1993, 135, 229–238. [Google Scholar] [CrossRef]
- Golden, A.; Benedict, M.; Shearn, A.; Kimura, N.; Leone, A.; Liotta, L.A.; Steeg, P.S. Nucleoside diphosphate kinases, nm23, and tumor metastasis: Possible biochemical mechanisms. Adv. Nutr. Cancer 1992, 63, 345–358. [Google Scholar]
- Lacombe, M.-L.; Milon, L.; Munier, A.; Mehus, J.G.; Lambeth, D.O. The human Nm23/nucleoside diphosphate kinases. J. Bioenerg. Biomembr. 2000, 32, 247–258. [Google Scholar] [CrossRef]
- Wright, N.T.; Prosser, B.L.; Varney, K.M.; Zimmer, D.B.; Schneider, M.F.; Weber, D.J. S100A1 and calmodulin compete for the same binding site on ryanodine receptor. J. Biol. Chem. 2008, 283, 26676–26683. [Google Scholar] [CrossRef]
- Prosser, B.L.; Wright, N.T.; Hernandez-Ochoa, E.O.; Varney, K.M.; Liu, Y.; Olojo, R.O.; Zimmer, D.B.; Weber, D.J.; Schneider, M.F. S100A1 binds to the calmodulin-binding site of ryanodine receptor and modulates skeletal muscle excitation-contraction coupling. J. Biol. Chem. 2008, 283, 5046–5057. [Google Scholar] [CrossRef]
- Wafer, L.N.; Tzul, F.O.; Pandharipande, P.P.; McCallum, S.A.; Makhatadze, G.I. Structural and thermodynamic characterization of the recognition of the S100-binding peptides TRTK12 and p53 by calmodulin. Protein Sci. 2014, 23, 1247–1261. [Google Scholar] [CrossRef] [PubMed]
- Patel-King, R.S.; Gorbatyuk, O.; Takebe, S.; King, S.M. Flagellar radial spokes contain a Ca2+-stimulated nucleoside diphosphate kinase. Mol. Biol. Cell 2004, 15, 3891–3902. [Google Scholar] [CrossRef] [PubMed]
- Lukas, T.J.; Marshak, D.R.; Watterson, D.M. Drug protein interactions: Isolation and characterization of covalent adducts of phenoxybenzamine and calmodulin. Biochemistry 1985, 24, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.B.; Jiang, L.; Ding, M.H.; Chen, Z.H.; Bao, Y.; Chen, Y.; Sun, W.; Zhang, C.R.; Hu, H.K.; Cai, Z.; et al. Anti-tumor activity of phenoxybenzamine hydrochloride on malignant glioma cells. Tumor Biol. 2016, 37, 2901–2908. [Google Scholar] [CrossRef]
- Mac Neil, S.; Wagner, M.; Rennie, I.G. Investigation of the role of signal transduction in attachment of ocular melanoma cells to matrix proteins: Inhibition of attachment by calmodulin antagonists including tamoxifen. Clin. Exp. Metastasis 1994, 12, 375–384. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, Y.; Hedman, A.C.; Ames, J.B.; Sacks, D.B. Calmodulin lobes facilitate dimerization and activation of estrogen receptor-α. J. Biol. Chem. 2017, 292, 4614–4622. [Google Scholar] [CrossRef]
- Leclercq, G. Calcium-induced activation of estrogen receptor-α—New insight. Steroids 2012, 77, 924–927. [Google Scholar] [CrossRef]
- Menéndez-Menéndez, J.; Martínez-Campa, C. Melatonin: An anti-tumor agent in hormone-dependent cancers. Int. J. Endocrinol. 2018, 2018, 1–20. [Google Scholar] [CrossRef]
- Greenberg, D.A.; Carpenter, C.L.; Messing, R.O. Calcium channel antagonist properties of the antineoplastic antiestrogen tamoxifen in the PC12 neurosecretory cell line. Cancer Res. 1987, 47, 70–74. [Google Scholar]
- Li, H.; Panina, S.; Kaur, A.; Ruano, M.J.; Sánchez-González, P.; la Cour, J.M.; Stephan, A.; Olesen, U.H.; Berchtold, M.W.; Villalobo, A. Regulation of the ligand-dependent activation of the epidermal growth factor receptor by calmodulin. J. Biol. Chem. 2012, 287, 3273–3281. [Google Scholar] [CrossRef]
- Goetz, S.C.; Anderson, K.V. The primary cilium: A signalling centre during vertebrate development. Nat. Rev. Genet. 2010, 11, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Wheway, G.; Nazlamova, L.; Hancock, J.T. Signaling through the primary cilium. Front. Cell Dev. Biol. 2018, 6, 8. [Google Scholar] [CrossRef] [PubMed]
- Christensen, S.T.; Pedersen, S.F.; Satir, P.; Veland, I.R.; Schneider, L. Chapter 10 The primary cilium coordinates signaling pathways in cell cycle control and migration during development and tissue repair. Curr. Top. Dev. Biol. 2008, 85, 261–301. [Google Scholar] [PubMed]
- Reiter, J.F.; Leroux, M.R. Genes and molecular pathways underpinning ciliopathies. Nat. Rev. Mol. Cell Biol. 2017, 18, 533–547. [Google Scholar] [CrossRef] [PubMed]
- Phua, S.C.; Lin, Y.-C.; Inoue, T. An intelligent nano-antenna: Primary cilium harnesses TRP channels to decode polymodal stimuli. Cell Calcium 2015, 58, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Phua, S.C.; Derose, R.; Chiba, S.; Narita, K.; Kalugin, P.N.; Katada, T.; Kontani, K.; Takeda, S.; Inoue, T. Genetically encoded calcium indicator illuminates calcium dynamics in primary cilia. Nat. Methods 2013, 10, 1105–1107. [Google Scholar] [CrossRef] [PubMed]
- Delling, M.; Indzhykulian, A.A.; Liu, X.; Li, Y.; Xie, T.; Corey, D.P.; Clapham, D.E.; Liu, Y. Primary cilia are not calcium-responsive mechanosensors. Nature 2016, 531, 656–660. [Google Scholar] [CrossRef]
- Delling, M.; DeCaen, P.G.; Doerner, J.F.; Febvay, S.; Clapham, D.E. Primary cilia are specialized calcium signalling organelles. Nature 2013, 504, 311–314. [Google Scholar] [CrossRef]
- DeCaen, P.G.; Delling, M.; Vien, T.N.; Clapham, D.E. Direct recording and molecular identification of the calcium channel of primary cilia. Nature 2013, 504, 315–318. [Google Scholar] [CrossRef]
- Otto, E.A.; Loeys, B.; Khanna, H.; Hellemans, J.; Sudbrak, R.; Fan, S.; Muerb, U.; O’Toole, J.F.; Helou, J.; Attanasio, M.; et al. Nephrocystin-5, a ciliary IQ domain protein, is mutated in Senior-Loken syndrome and interacts with RPGR and calmodulin. Nat. Genet. 2005, 37, 282–288. [Google Scholar] [CrossRef]
- Badone, B.; Ronchi, C.; Kotta, M.-C.; Sala, L.; Ghidoni, A.; Crotti, L.; Zaza, A. Calmodulinopathy: Functional effects of CALM mutations and their relationship with clinical phenotypes. Front. Cardiovasc. Med. 2018, 5, 176. [Google Scholar] [CrossRef] [PubMed]
- Villalobo, A.; García-Palmero, I.; Stateva, S.R.; Jellali, K. Targeting the calmodulin-regulated ErbB/Grb7 signaling axis in cancer therapy. J. Pharm. Pharm. Sci. 2013, 16, 177–189. [Google Scholar] [CrossRef] [PubMed]




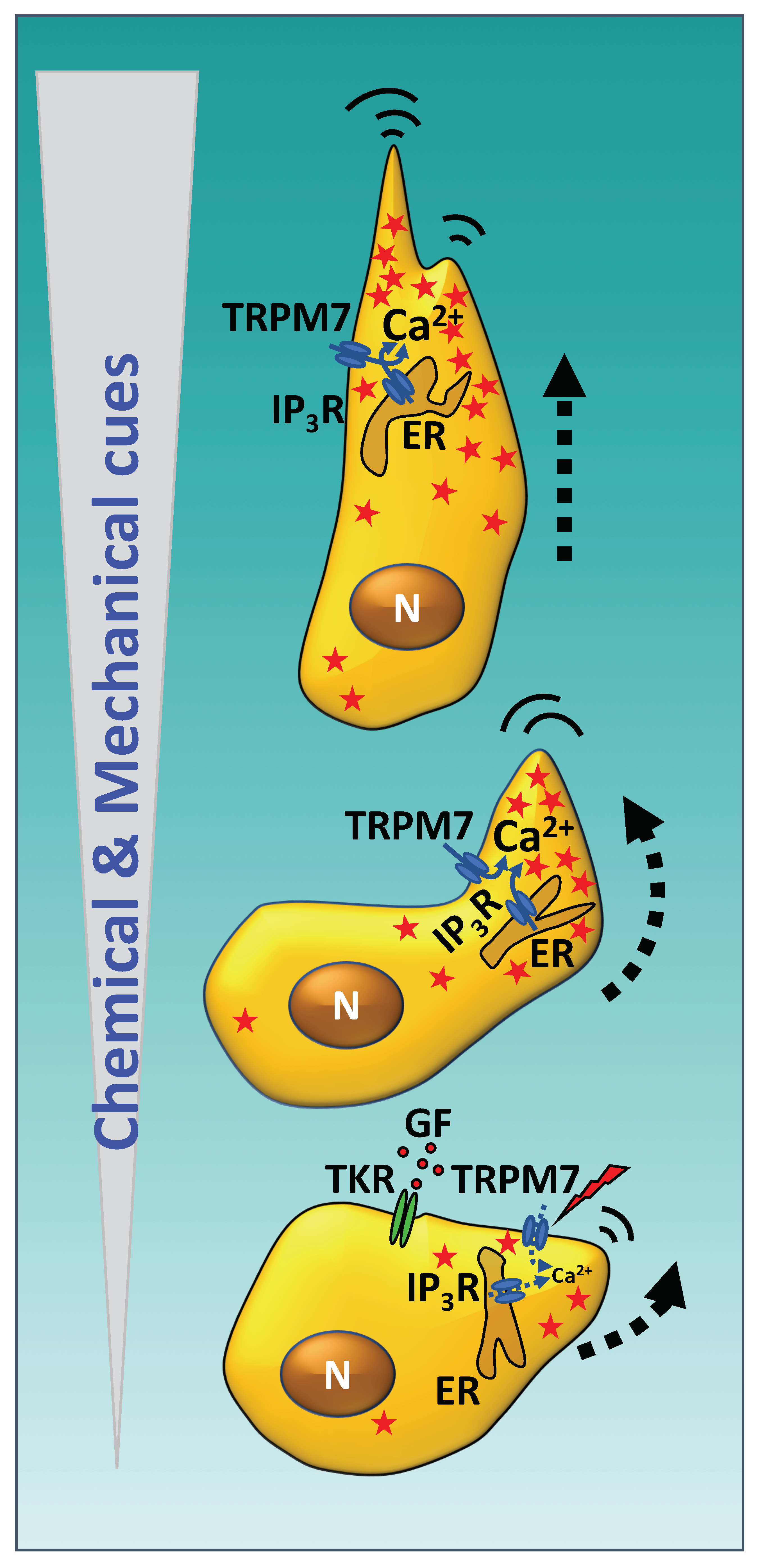{kind=link}
{kind=link}
{kind=link}
| CaM Antagonist | Cell Type (Species) | Effects | Mechanisms | Ref. |
|---|---|---|---|---|
| Calmidazolium (R24571) | Cerebellar granule cells (mice) Gastric epithelial cells (rabbit) | Inhibits cell migration | Reduces Ca2+ transients | [50,61] |
| Chlorpromazine | WIRL liver epithelial cells (rat) | Inhibits cell spreading and decreases F-actin levels | Alters microfilaments function; inhibits actin polymerization | [45] |
| Fluphenazine | Fibroblasts (human) (1) Fibroblasts (rat) | Inhibits cell migration and contractility; inhibits wound contraction and re-epithelization in vivo | Inhibits MLCK | [62,63] |
| Trifluoperazine | 3T3 fibroblasts (mouse) SV40 transformed 3T3 fibroblasts (mouse) WIRL liver epithelial cells (rat) SV40 transformed WIRL cells (rat) NRK-LA23 kidney cells (rat) NIL-2 fibroblasts (hamster) Fibroblasts (human) MRC-5 diploid fibroblasts (human) Aortic smooth muscle cells (rat) Neutrophils (human) (2) ECV304 umbilical vein endothelial cells (human) Corneal epithelial cells (rat) Monocytes (human) (3) | Inhibits cell migration, cell spreading, cell adhesion; decreases F-actin levels; inhibits ATP-induced cell contraction and prevents MLC phosphorylation | Alters microfilaments function;inhibits actin polymerization and aggregation; inhibits MLCK and actomyosin function | [45,46,48,55,64,65] |
| W-7 | CFSC-8B myofibroblast-like hepatic stellate cells (rat) Bronchial epithelial cells (human) ECV304 umbilical vein endothelial cells (human) Corneal epithelial cells (rat) Gastric mucosal cells (rabbit) Neutrophils (human) Myofibroblasts (rat) Vascular smooth muscle cells (rat) Fibroblasts (rat) | Inhibits PDGF-mediated cell migration; inhibits VIP-, IL-8-, CT-1-induced chemotaxis; inhibits wound contraction and re-epithelization in vivo | Inhibits CaMK-II, MLCK and actomyosin function | [49,51,55,63,64,66,67,68,69] |
| CaM Antagonist | Cell Type (Species) | Effects | Mechanisms | Ref. |
|---|---|---|---|---|
| Calmidazolium (R24571) | Colo201 colon cancer cells (human) | Inhibits cell adhesion | Inhibits calcineurin | [70] |
| CBP501 | A549 NSLC cells (human) H1299 NSLC cells (human) Lewis lung carcinoma cells (mouse) (1) | Inhibits EGF-dependent cell migration and invasiveness; inhibits EMT; inhibits metastasis; inhibits tumor spheroid formation | Inhibits CaM binding to K-Ras; inhibits Akt and ERK1/2 activation; inhibits EGF-induced transcription factor Zeb1 and vimentin expression; inhibits tumor-promoting IL-6 and TNFα production by tumor-promoting macrophages | [71,72] |
| Chlorpromazine | MOLT-4 lymphoma T cells (human) | Inhibits invasiveness | Inhibits actin polymerization; inhibits CaMK-II | [47,73] |
| Diphenylbutylpiperidine | MOLT-4 lymphoma T cells (human) | Inhibits invasiveness | Inhibits CaMK-II | [73] |
| Flunarizine | B16 melanoma cells (mouse) K1735-M2 melanoma cells (mouse) M5076 macrophage-like sarcoma cells (mouse) | Inhibits cell migration and cell invasiveness | [53,54] | |
| J8 | A375-SM melanoma cells (human) Choroidal melanoma cells (human) | Inhibits cell invasiveness | [74,75] | |
| Ophiobolin A | HeLa cervix carcinoma cells (human) PC3 prostate carcinoma cells (human) | Inhibits cell migration and enhances Ca2+ leak from ER | Prevents CaM binding to Sec61 Ca2+ leak channel and increases SOCE | [59] |
| Prochlorperazine | MOLT-4 lymphoma T cells (human) | Decreases F-actin | Inhibits actin polymerization | [47] |
| Squalamine | 13762 mammary carcinoma (rat) Lewis lung carcinoma (mouse) MV-522 lung carcinoma (human) Calu-6 lung adenocarcinoma (human) H460 large-cell lung carcinoma (human) NCI-H23 non-small cell lung carcinoma (human) | Synergistic anti-metastatic action in combination with different anti-tumor agents | Disrupts F-actin stress fibers | [76] |
| Tamoxifen | C-1300 neuroblastoma cells (mice) A375-SM melanoma cells (human) Uveal melanoma cells (human) Choroidal melanoma cells (human) B16 melanoma cells (mouse) MCF7 breast carcinoma cells (human) | Inhibits cell attachment; inhibits invasiveness; inhibits bone marrow metastasis | [74,75,77,78] | |
| Trifluoperazine | HeLa cervix carcinoma cells (human) PC3 prostate carcinoma cells (human) TA3 breast carcinoma cells (mouse) MB6A lymphosarcoma cells (mouse) HCT116 colon carcinoma cells (human) | Inhibits cell migration, invasiveness, EMT and enhances Ca2+ leak from ER | Prevents CaM binding to Sec61 Ca2+ leak channel; increases SOCE; inhibits adhesion to hepatocytes and decreases expression of N-cadherin and the transcription factors Snail and Slug; inhibits CaMK-II | [59,79,80] |
| Triflupromazine | MOLT-4 lymphoma T cells (human) | Decreases F-actin | Inhibits actin polymerization | [47] |
| W-7 | HeLa cervix carcinoma cells (human) Lewis lung carcinoma cells (mouse) HT-1080 fibrosarcoma cells (human) MOLT-4 lymphoma T cells (human) CHRF-28-11 megakaryoblastic leukemia cells (human) Colo201 colon cancer cells (human) B16 melanoma cells (mouse) SK-MEL118 melanoma cells (human) | Inhibits cell migration, cell adhesion, cell invasiveness, and metastasis in vivo | Decreases PMA-induced MMP-9 secretion; inhibits EGF- and thrombin-dependent Rac1 activation; inhibits calcineurin; inhibits TPA-induced integrin-α2/β1 expression; inhibits MTS1 and NM23 metastasis-associated genes | [70,73,81,82,83,84,85,86,87] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villalobo, A.; Berchtold, M.W. The Role of Calmodulin in Tumor Cell Migration, Invasiveness, and Metastasis. Int. J. Mol. Sci. 2020, 21, 765. https://doi.org/10.3390/ijms21030765
Villalobo A, Berchtold MW. The Role of Calmodulin in Tumor Cell Migration, Invasiveness, and Metastasis. International Journal of Molecular Sciences. 2020; 21(3):765. https://doi.org/10.3390/ijms21030765
Chicago/Turabian StyleVillalobo, Antonio, and Martin W. Berchtold. 2020. "The Role of Calmodulin in Tumor Cell Migration, Invasiveness, and Metastasis" International Journal of Molecular Sciences 21, no. 3: 765. https://doi.org/10.3390/ijms21030765
APA StyleVillalobo, A., & Berchtold, M. W. (2020). The Role of Calmodulin in Tumor Cell Migration, Invasiveness, and Metastasis. International Journal of Molecular Sciences, 21(3), 765. https://doi.org/10.3390/ijms21030765