Abstract
Nevoid basal cell carcinoma syndrome (NBCCS), also named Gorlin syndrome, is a rare multisystem genetic disorder characterized by marked predisposition to basal cell carcinomas (BCCs), childhood medulloblastomas, maxillary keratocysts, celebral calcifications, in addition to various skeletal and soft tissue developmental abnormalities. Mutations in the tumor suppressor gene PATCHED1 (PTCH1) have been found to be associated in the majority of NBCCS cases. PATCH1 somatic mutations and loss of heterozygosity are also very frequent in sporadic BCCs. Unlike non-syndromic patients, NBCCS patients develop multiple BCCs in sun-protected skin area starting from early adulthood. Recent studies suggest that dermo/epidermal interaction could be implicated in BCC predisposition. According to this idea, NBCCS fibroblasts, sharing with keratinocytes the same PTCH1 germline mutation and consequent constitutive activation of the Hh pathway, display features of carcinoma-associated fibroblasts (CAF). This phenotypic traits include the overexpression of growth factors, specific microRNAs profile, modification of extracellular matrix and basement membrane composition, increased cytokines and pro-angiogenic factors secretion, and a complex alteration of the Wnt/β-catenin pathway. Here, we review studies about the involvement of dermal fibroblasts in BCC predisposition of Gorlin syndrome patients. Further, we matched the emerged NBCCS fibroblast profile to those of CAF to compare the impact of cell autonomous “pre-activated state” due to PTCH1 mutations to those of skin tumor stroma.
1. Introduction
Nevoid basal cell carcinoma syndrome (NBCCS OMIM #109400), also known as Gorlin–Goltz syndrome or Gorlin syndrome (GS), is a rare genetic alteration with an estimated prevalence of around 1 in 100,000 on average [1,2] and of 1 in 256,000 in Italy [3]. It is most frequently produced by a mutation in the patched homologue 1 (PTCH1) tumor suppressor gene, a component of the Hedgehog (Hh) pathway, and it is inherited in an autosomal dominant manner, though about 20–30% of cases present a de novo pathogenic variant. This syndrome shows a high penetrance and variable expressiveness. It is characterized by jaw keratocysts, palmar and/or plantar pits and calcification of celebral falx, malformations of the ribs, macrocephaly, generalized overgrowth and a predisposition to tumors, specifically multiple basocellular carcinomas, medulloblastoma, meningioma and benign ovarian cysts and cardiac fibromas [4,5,6,7,8,9,10,11,12]. Recently, the model of patient-derived induced pluripotent stem cells (iPSC) clarified some of the molecular mechanisms underlying skeletal abnormalities [13]. Clinical diagnosis of Gorlin syndrome requires either two major criteria (multiple basal cell carcinomas (BCCs) (≥5 in a lifetime) or a BCC before 30 years, lamellar calcification of the falx cerebri, jaw keratocysts, palmar or plantar pits, first-degree relative with NBCCS) or one major and two minor ones (lympho-mesenteric or pleural cysts, macrocephaly, cleft lip/palate, vertebral/rib anomalies, preaxial or postaxial polydactyly, ovarian/cardiac fibromas, medulloblastoma, ocular anomalies). Clinical features of NBCCS arise in the first, second, and third decade [2,9]. Although patients with NBCCS can often be diagnosed on the basis of clinical data, genetic tests are useful in many situations. Genetic analysis can be performed to confirm the diagnosis in patients lacking sufficient clinical diagnostic criteria and for prenatal and pre-implantation diagnostic purposes if a defined mutation has been identified in a proband family member. Because patients with NBCCS are predisposed to neoplastic disease, patients who are at risk for the syndrome are encouraged to undergo regular surveillance.
1.1. Clinical and Histopathological Features of Nevoid Basal Cell Carcinoma Syndrome-Basal Cell Carcinomas (NBCCS-BCCs)
Life expectancy in NBCCS patients is not significantly different from the general population [2]. Early diagnosis is important due to complications of NBCCS (most notably medulloblastoma) during childhood and susceptibility to multiple neoplasms in early age. The patients affected by Gorlin syndrome often show frequent skin lesions including multiple nevi, epidermal sebaceous and dermoid cysts, small palmo-plantar depression (pits), hair skin pathes, and numerous basocellular carcinomas (BCCs) distributed randomly over the entire face or body including sun-unexposed area [14,15]. The risk of developing a second BCC is 10 times the risk of unaffected individuals and the number of BCCs may vary from few to thousands [16]. The treatment of choice for multiple BCCs is surgical eradication. Consequently, the aesthetic outcome of surgical treatment of multiple skin tumors represents one of the major problem for these patients. However, depending on BCCs location, number and size, medical therapies used for sporadic BCC can be considered. Combined approaches including surgery supplemented by cryotherapy, laser, photodynamic therapy or topical treatments also are used to preserve normal tissue and prevent disfigurement. Radiation therapy must be avoided as it can cause disease relapse and invasion of BCC years later [17]. Interferon injected directly into tumor lesion demonstrated complete resolution of BCC in some patients [17]. Use of oral retinoids has also been suggested [18]. Recently, several small molecules inhibitors of the Hh pathway, impeding ligand-mediated signal transmission, demonstrated efficacy in the clinical practice. Among these, oral administration of Vismodegib and Sonidegib reduce the development of BCC, rendering the surgical resections less challenging for these patients [19,20,21]. However, side effects related to these systemic therapies limit their long-term use in clinical practice [22]. Although BCCs from syndromic individuals are histopathologically indistinguishable from sporadic BCCs [17,23], the corresponding molecular phenotypes do not fully overlap. NBCCS-BCCs present lower mutation load and UV-induced mutagenesis compared with sporadic BCCs, suggesting that strict sun protection is effective in this genetically predisposed population [24]. Reduced mutational load could explain the relatively indolent clinical course NBCCS-BCCs and the responsiveness to target therapy. In fact, it has been observed that NBCCS patients lack intrinsic resistance to Hh target therapy, such as Vismodegib and Sonidegib [25,26]. However, it is unclear if the scarcity of drug resistance to Vismodegib in Gorlin syndrome is an intrinsic disease trait or if it refers to the fact that in these studies, locally advanced and metastatic sporadic basal cell carcinomas were compared to a more heterogeneous group of NBCCS-BCCs [22,27,28]. As demonstrated for sporadic BCC, exposure to the sun promotes the development of BCCs in patients with NBCCS [29]. The lower frequency of BCCs in the African-American syndromic patients compared with Caucasian populations, presumably due to the protective effect of melanin, confirm that the sun may exacerbate the development of BCCs in NBCCS [30]. However, since NBCCS-BCCs commonly develop also in sun-protected skin area, the role of sun exposure is still debated. Also, data emerging from mouse models are not conclusive since patch knockout mice develop more BCCs than wild mice when exposed to ultraviolet (UV) but about one-third of skin tumors develop in absence of exposure to external genotoxic stress [29] Ptch1-deficent mice develop several basaloid hyperproliferation but these cancer precursors progress to nodular and infiltrative BCCs only in X-ray irradiated areas [31]. Also, in vitro studies showed some incongruity. NBCCS fibroblasts display hypersensitivity to UVB, evidenced by accumulation of oxidative damage (8-hydroxydeoxyguanosine and pyrimidine dimers) and reduced cell survival [32,33]. By contrast, Brellier and collaborators reported normal nucleotide excision repair and survival capacity in UVB-treated NBCCS keratinocytes and fibroblasts [34].
1.2. Genetic Aspects
The majority of NBCCS cases can be associated with heterozygous germline mutations in PTCH1 (Chr9q22.32), the human homolog of the Drosophila gene, patched [35], and is transmitted in an autosomal dominant way with high penetrance and variable expressivity [36]. Males and females are equally affected. Most germline mutations in NBCCS lead to a premature termination of the PATCHED protein [37,38]. PTCH1 gene is composed of 23 exons and codifies a transmembrane glycoprotein (PTCH1) composed of 1447 amino acids and 12 domains [7,8,36]. Since the discovery of the PTCH1 gene as being responsible for Gorlin syndrome in 1996 [4], more than 200 sequence mutations have been reported to date including nonsense, missense, and splicing variants, as well as insertions and deletions [38]. Nearly 20–30% of the syndrome cases present as new mutations [4,6,7,22] and no clustering of mutations has been identified [39]. Nevertheless, missense mutations do not occur with the same frequency as frameshift or nonsense mutations [19]. Data in the literature reported that, despite exhaustive molecular analysis, PTCH1 mutation could not be determined in some patients, probably due to large deletions or the existence of mutations outside the regions analyzed, such as in introns, intron-exon junctions, or regulatory elements. Other rare causative genes have also been identified in Gorlin syndrome including PTCH2 and SUFU encoding for patched 2 and suppressor of fused homolog proteins, respectively [40,41,42]. In this hereditary setting, the genotype–phenotype correlation is not constantly present: family members sharing the same PTCH1 germline mutation have a variable phenotype [43]. The missing relationship between the genotype and the phenotype suggests the existence of a very complex variability of this syndrome, possibly originated by the interaction of genetic background and environmental factors [2,44,45]. However, it has been proposed that additional simultaneous mutation may contribute to phenotype variation [46].
The developmental defects associated with NBCCS are mostly due to haploinsufficiency. Instead, as reported for other tumor suppressor genes, tumors develop in the presence of two genetic alterations. The first genetic defect is inheritance of a mutation and the second hit is inactivation of the normal homologue by environmental mutagenesis or random genetic rearrangement that definitely confer ligand-independent activation of the Hh pathway, cellular growth, genetic instability and, potentially, tumor development [47]. This loss has also been observed in basocellular carcinomas independently of the syndrome, in the meduloblastoma, in odontogenic keratocysts and trichoblastomas [6,48]. In sporadic BCCs, mutations of one homologue have been described at high frequency (67%) [49] and loss of heterozygosity (LOH) by nondisjunction, deletion, or mitotic recombination in the chromosomal region surrounding PATCH1 gene has also frequently been found (93%) [50,51]. Interestingly, <50% of the PTCH1 mutations in sporadic BCCs have the typical UVB signature [52]. Sporadic BCCs, which do not have mutations in this gene, carry gain-of-function mutations in the Smoothened (SMO) gene, another member of the Hh pathway [53,54]. Thus, inactivation of PTCH1 or oncogenic activation of SMO occurs in almost all BCCs. Therefore, deregulation of Hh signaling is considered the pivotal aberration in all BCCs (whether in syndromic and non-syndromic patients). Patched may function as a “gatekeeper gene” for BCC formation; i.e., a precursor cell gains a survival advantage with inactivation of a gatekeeper gene, and inactivation would be necessary before clonal expansion and accumulation of multiple genetic events preceding BCC formation [55].
1.3. Hedgehog Signaling
The Hh pathway is one of several major signaling pathways that demonstrated a connection between embryogenesis, regeneration, and cancer, highlighting common signaling networks in these processes [12,36]. Hh signaling also participates in control of adult stem cell proliferation, tissue homeostasis maintenance, and regeneration [56,57]. Hh signaling is turned on locally as a physiological response to wound, ischemic stroke, or myocardial infarction [58,59]. The hedgehog pathway appears to be evolutionary conserved from Drosophila to vertebrates [60,61]. This raises the important question of how long-term use of Hh inhibitors can disturb normal physiology. Despite overall similarity, many vertebrate-specific components have been identified [62]. In contrast to most signaling pathways, in which the ligand activates its receptor, in Hh signaling, the ligand inhibits the PTCH receptor. In the canonical pathway, binding of Sonic Hedgehog (SHH), the most widely expressed hedgehog protein to its receptor, PTCH, releases inhibition on SMO, which transduces the signal to the cytoplasm [63,64,65,66], ultimately leading to the activation of the glioma-associated oncogene homolog zinc finger transcription factors (Gli1, -2 and -3) [67,68,69] (Figure 1). Among them, Gli1 exists only as its activated form, while Gli2 and Gli3 have both activator and repressor forms. Gli1 and Gli2 increase the expression of key regulators of G1/S and G2/M phase progression of the cell cycle, thereby promoting the transition from a quiescent to the proliferative state. The pathway normally is regulated by the spatially and temporally restricted expression of Shh, and two other less expressed homologues, Desert hedgehog (DHH), and Indian hedgehog (IHH). In the absence of functional PTCH, smoothened may be constitutively active, independent of hedgehog control, and may induce overexpression of several target genes including Wnts, CyclinD1 and D2, N-myc, Snail, the anti-apoptotic factor Bcl2 as well as feedback genes of Hh signaling (PATCH1 and 2, Gli1 and huntingtin interacting protein-1, Hip1) [70,71]. The Hh signaling pathway is in conjunction with other important pathways, such as epidermal growth factors (EGF/EGFR), transforming growth factor-β (TGF-β), and Wnt/β-catenin signaling [72,73]. Gli1 is also implicated in replicative immortality achieved by human telomerase reverse transcription transcriptase (hTERT) protein expression regulation [74].
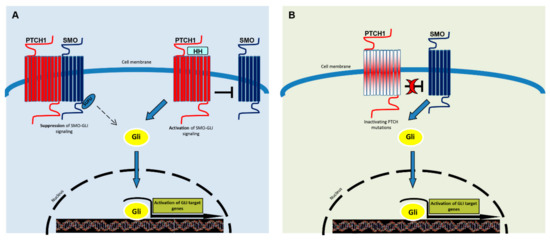
Figure 1.
A simplified representation of the Hedgehog signaling pathway. SMO is the key signal transducer of the Hh pathway. (A) In the absence of Hedgehog (Shh, Dhh, Ihh) ligands, PTCH1 inhibits SMO signaling. Gli molecules are processed with the help of SuFu molecules into repressor forms, which disable the Hh signaling pathway. The binding of Hh promotes SMO conformational change, leading to activation of the GLI transcription factors (the activators Gli1 and -2 and the repressor Gli3). Activated GLI accumulates in the nucleus and controls the transcription of hedgehog target genes. (B) Mutated PTCH1 does not inhibit SMO and the pathway results activated, as in the presence of ligands.
2. Hh Signaling in Tumor Microenvironment
In addition to cancer cells, tumors exhibit a co-operational and evolving coexistence of a number of cell types comprehending immune cells, endothelial cells, pericytes, adipocytes, and fibroblasts. These cells, of which fibroblasts constitute a major part, are embedded in the extracellular matrix composed of a multitude of scaffold proteins (polysaccharides, fibrous proteins, and proteoglycans), growth factors, chemokines and cytokines, that are released by various cell types. These autocrine/paracrine acting factors have a crucial role in the survival and progression of tumors [75]. Evidence has been documented that alteration in the microenvironment plays an important role in both BCC and squamous cell carcinoma (SCC) [76,77,78]. In the skin, accumulation of senescent fibroblasts mostly due to photo-aging alters the surrounding tissue into a neoplasia-promoting environment [78]. BCC-associated CAF expression profile is mainly linked to extracellular matrix metabolism such as MMPs, metallopeptidase, lysol oxidases, 4-hydroxylase, fibronectin, fibronectin, proteoglycans and factor involved in epithelial to mesenchymal transition [78]. Moreover, in tissue surrounding BCC abundant collagen XI, which is not normally present in skin, argues for a pro-invasive phenotype. BCC peritumoral stroma also is a source of cytokines and chemokines implicated in local immunosuppression. In BCC lesion, CAF expressed an increased level of two WNT pathway proteins, WISP1 and Lef1 [78]. Although several reports have described a cell-autonomous autocrine role for Hh signaling in tumors, recent data support an additional paracrine model of Hh-mediated tumorigenesis, in which neoplastic cells secrete Hh ligand to activate tumor-promoting hedgehog target genes in adjacent stroma [79,80]. Even though in NBCCS patients the activation of the Hh pathway originates in a different context, specifically by an intrinsic ligand-independent mechanism, the resulting stroma may support cancer development similarly to tumor stroma. Hence, NBCCS represent a useful model not only to evaluate keratinocytes during BCC onset but also for studying BCC/microenvironment molecular cross-talk.
2.1. Hh Signaling in Cancer-Associated Fibroblasts
Previous studies on various human cancers suggested that Hh signaling effects tumourigenesis by controlling cell proliferation, angiogenesis, and epithelial-mesenchymal transition (EMT) in an autocrine–juxtacrine manner [81]. However, tumor microenvironment may significantly influence cancer evolution, both in cancer cells hedgehog-dependent and -independent [82]. In the process of tumor formation, quiescent fibroblasts change to adapted reactive stromal elements phenotypically closely resemble myofibroblasts with enlarged spindle-shape and elongated projections, expressing α-smooth muscle actin. Collectively, the desmoplastic reaction secondary to neoplasm generates scar tissue similar to the wound healing process [83,84]. Thus, tumors are often referred to as “wounds that never heal” [82]. Cancer-associated fibroblasts (CAF) express specific markers such as fibroblast activation protein (FAP), fibroblast-specific protein 1 (FSP1), neuronglial antigen-2 (NG2), vimentin, tenascin (TNC), and display an increased secretion of various extracellular matrix (ECM) proteins (i.e., collagens I, III, IV), proteoglycans (i.e., fibronectin, laminin), chemokines (e.g., CXCL and CCL), cytokines and other tumor-promoting factors which affect vascularisation (i.e., platelet-derived growth factor (PDGF), vascular endothelial growth factor (VEGF)), stromal-derived factor-1 (SDF-1), matrix metalloproteinase (MMPs), and tissue inhibitors of MMPs (TIMPs), capable of promoting tumor cell invasiveness and survival. Intensive tumor-stroma bidirectional cross-talk is responsible for fibroblast secretome switch onto CAF secretome [85,86,87]. Cancer stimulates stromal cells to also produce a huge number of growth factors that support neoplastic cell proliferation (i.e., TGF-β, epidermal growth factor (EGF), hepatocyte growth factor (HGF) or fibroblasts growth factor (FGF), and Wnts [88,89]. Recent studies have shown that Hh signaling activation, driven by ligand expression in carcinoma cells, can modulate the stromal cells to create a favorable environment for faster cancer progression [90,91].
In mice models, increased Hh activity in tumor fibroblasts results in overexpression of VEGF which increases endothelial cell proliferation and vascular density in tumor [92]. Furthermore, Hh signaling exerts a critical role in tumor-associated macrophages (TAMs) immunosuppression. In myeloid cells, sonic hedgehog secreted by tumor cells induces functional M2 polarization with consequent suppression of CD8 + T cell recruitment via reduction of CXCL9 and CXCL10 [93,94]. Wei and coauthors recently demonstrated that SHH ligands increase PTCH1 and Gli1 expression leading to Hh signaling pathway activation in human CAF but not in normal fibroblasts. In the same study, Hh signaling stimulation augmented CAF proliferation and confirmed lymphangiogenesis in a xenograft model [95]. The Hh pathway also supports tumor cell invasion by stimulating cell motility and MMPs expression and by directly targeting genes implicated in EMT. In addition to paracrine factors, a tumor-stromal direct contact seems to be necessary for Hh signaling-dependent mobilization of non-small cell lung cancer cells [96]. One important point is the fact that in in situ skin tumor, cancer cells and stromal components can communicate through the basement membrane by diffusible factors while exclusively in invasive tumor, cancer and stromal cells are in direct contact and establish a complex cross-talk capable of altering fibroblast phenotype. Therefore, alterations in the composition of basement membrane represents an early microenviroment modification for skin cancer progression. In general, indirect CAF-cancer cell interaction is responsible for tumor growth promotion by the paracrine release of pro-mitogenic factors, whilst direct CAF-cancer contact mediates cell migration and invasion [88]. NBCCS fibroblasts, at least during BCC initiation, are in a situation similar to those of in situ non-invasive BCC, whereas CAF are more likely in proximity or in direct cell–cell contact with tumor cells. This unique scenario does not find similarity in non-syndromic skin carcinoma and could, in part, explain the prevalent indolent phenotype of NBCCS-BCCs.
Moreover, the Hh pathway supports cancer genomic instability and inflammation [94]. Another important issue regarding the role of fibroblasts in cancer evolution is the frequency of somatic genetic mutations in CAFs. Manifold studies demonstrated that high percentage of CAFs undergo genetic alterations, including loss of heterozygosity or mutation of tumor suppressor genes (i.e., PTEN and p53) [97]. This is a relevant point for cutaneous tumor since skin is constantly exposed to extrinsic stressor including ultraviolet radiation. Until now, somatic mutations in the Hh pathway have not been investigated in sporadic skin cancer.
2.2. Dermal Fibroblasts from Gorlin Syndrome Have Phenotypic Traits Reminiscent BCC Cancer Associated Fibroblasts
The first evidence that NBCCS fibroblasts are prone to acquire CAF phenotype arose from a report by Majmudar and co-workers in 1993. In this study, all the fibroblast cell cultures isolated from skin adjacent to BCC tumors of NBCCS patients demonstrated overexpression of MMP3, whereas in corresponding nonsyndromic fibroblast cultures, abnormal MMP3 expression was restricted to 25% of BCCs. More interestingly, the abnormal expression of MMP3 has been found extended to uninvolved skin fibroblasts, demonstrating the CAF-prone phenotype in GS patients [98]. Next, this observation was extended to other CAF markers, spanning from molecules involved in tissue organization such as MMP1, collagen type 11 alpha 1 (COL11A1), matrix gla protein (MGP), and TNC to chemokines as CXCL12, and other bioactive factors governing cell proliferation and survival as keratinocyte growth factors (KGF), angiopoietin-related protein 2 and 4 (ANGPTL2/4). Among the Wnt pathway regulator factors, secreted frizzled-related protein-1 and 2 (SFRP2/2), Dickkopf 1 (DKK1), and WNT inhibitory factor 1 (WIF1), WNT1, inducible signaling pathway protein 2 (WISP2) have been found upregulated, while DKK3 was downregulated. Activation of the WNT/β-catenin signaling pathway has been increasingly appreciated in CAF biology [99,100] especially in the skin background [101]. The expression level of proteins involved in extracellular matrix remodeling, such as MMP1, MMP3, TNC as well as KGF and stroma cell-derived factor 1 alpha, has been associated with the NBCCS-BCCs aggressive clinical phenotype since patients presenting a high number of large BCCs show a higher level of expression compared to non-aggressive clinical phenotype [102]. By contrast, the difference in the protein profile among distinct genetic groups (missense and nonsense) failed to demonstrated definitive data [103,104]. The idea that fibroblasts carrying heterozygous PTCH1 mutations present a phenotype overlapping with those of skin CAF is also supported by the adhesion properties and intracellular cytoskeleton organization since α-SMA and vinculin are highly expressed and organized than in healthy fibroblasts [104]. Functional dermo-epidermal interaction in 3D organic cultures evidenced reduced epidermal thickness and dysregulation of keratinocyte differentiation proteins when healthy keratinocytes were maintained in the presence of NBCCS fibroblasts [105]. In the same experimental system, diffusible factors produced by NBCCS fibroblasts strongly stimulate the expression of p53 in an organotypic in vitro model containing normal keratinocytes, whereas carcinoma cells carrying mutate p53 acquired an invasive phenotype in the presence of NBCCS dermis [106]. The possible contribute of syndromic dermal cells in photodynamic therapy success has also been highlighted demonstrating that GS fibroblasts are predisposed to accumulate UV-dependent oxidative stress and consequence cell death [106]. In addition to an abnormal response to UV, NBCCS fibroblasts display increased oxidative perturbation following radiation associated with a marked deficiency in aldehyde dehydrogenase 1A1, one of the rate-limiting enzymes in retinoic acid synthesis [105]. Recently, additional evidence arose from microRNAs profiling in fibroblasts derived from Gorlin syndrome patients. Two miRNAs, has-miR-196a-5p and has-miR-4485, involved in mitochondrial function and tumorigenesis [107], were found to be downregulated and upregulated, respectively, [108] prospecting innovative miRNA-based therapies.
Even if the overall description of NBCCS fibroblasts in the literature strongly argues for a cancer associated phenotype, it is important to take into consideration that CAFs are reported to be a heterogeneous cell population that include both tumor-promoting and tumor-suppressing phenotypes expressing partially overlapping markers [68]. The provocative possible involvement of syndromic fibroblasts in the indolent phenotype presented by NBCCS-BCCs has not been investigated yet. Moreover, CAFs progressively co-evolve with the disease, whereas NBCCS fibroblasts are genetically determined to assume cancer microenvironment features. Co-evolution of CAFs and neoplastic cells includes intrinsic and extrinsic aging since sporadic BCCs are particularly prevalent in the elderly. Senescent phenotype is not necessary involved in NBCCS predisposition to BCC since hyperproliferation frequently develops early in life and in sun-unexposed body area. Lastly, in tumor, the stroma contains activated fibroblasts that are also increased in number, while no alteration in the number of this type of cells has been reported in NBCCS skin. The insight of all these interesting points needs to be clarified in future investigations.
3. Conclusions
Although many aspects of the Drosophila Hh pathway are conserved in vertebrates, mechanistic differences between the two species have begun to emerge, disabling some important notions previously acquired. Thus, patients with GS provide an important human model of hyperactivity in hedgehog signaling. On the other hand, the similarity of syndromic fibroblasts to CAFs complemented our knowledge about the predisposition of these patients to develop a huge number of BCCs in early life and in sun-unexposed body areas. In line with the idea that fibroblasts may be key players in cancer-prone hereditary diseases, abnormal dermal fibroblast activation and extracellular matrix remodeling has been described as a common hallmark of genodermatoses (recessive dystrophic epidermolysis bullosa, Kindler syndrome and xeroderma pigmentosum) [109]. It is incontrovertible that the accumulation of senescent cells in the skin provides a better milieu and stromal support for tumor cells. According to this notion, it will be of great importance to understand if NBCCS genetic defects are linked to the accumulation of senescent fibroblasts and limited regenerative potential independent of sun explosion and chronological age. Future research will clarify whether regenerative therapies are useful for Gorlin syndrome patients. Moreover, the characterization of NBCCS-derived fibroblasts and correlation of molecular data with clinical data may help to address the question of their role in therapy efficacy and resistance as well as provide a useful model to evaluate new therapeutic approaches of BCC. In particular, the effect of promising treatment, such as target therapies on the NBCCS dermal microenvironment, needs to be investigated.
Author Contributions
B.B. planned the Review and prepared the manuscript. F.P. and S.C. reviewed contributed to the conception of the work. A.C., V.S., L.E. contributed analysing clinical data. M.P. coordinated all the activities. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by public funds from the Italian Ministry of Health, Ricerca Corrente 2018–2020.
Conflicts of Interest
Authors have no conflict of interest to disclose.
Abbreviations
| NBCCS | Nevoid basal cell carcinoma syndrome |
| BCC | Basal cell carcinoma |
| PTCH | protein patched homolog |
| Hh | Hedgehog |
| CAF | Carcinoma associated fibroblast |
| iPCS | induced pluripotent stem cells |
| UV | ultraviolet |
| UVB | ultraviolet B |
| SUFU | suppressor of fused homolog |
| LOH | loss of heterozygosity |
| SMO | Smoothened gene |
| SHH | Sonic Hedgehog |
| Gli1, 2, 3 | glioma-associated oncogene 1,2,3 |
| DHH | Desert hedgehog |
| IHH | Indian hedgehog |
| Wnt | wingless signaling pathway |
| N-myc | neuroblastoma oncogene |
| Bcl2 | B-cell lymphoma 2 |
| Hip1 | Huntingtin interacting protein-1 |
| EGF | epidermal growth factor |
| EGFR | epidermal growth factor receptor |
| SCC | Squamous cell carcinoma |
| TGF-β | transforming growth factor beta |
| hTert | human telomerase reverse transcription transcriptase |
| EMT | epithelial-mesenchymal transition |
| FAP | fibroblast activation protein |
| FSP1 | fibroblast-specific protein 1 |
| GS | Gorlyn Syndrome |
| NG2 | neuroglial antigen-2 |
| TNC | tenascin |
| ECM | extracellular matrix |
| CXCL | chemokines |
| CCL | cytokines |
| PDGF | platelet- derived growth factor |
| VEGF | vascular endothelial growth factor |
| SDF-1 | stromal derived factor-1 |
| MMP | matrix metalloproteinase |
| TIMP | tissue inhibitors of MMP |
| HGF | hepatocyte growth factor |
| FGF | fibroblasts growth factor |
| TAMS | tumor-associated macrophages |
| CXCL9 | chemokine ligand 9 |
| CXCL10 | chemokine ligand 10 |
| PTEN | phosphatase and tensin homolog |
| COLL11A1 | collagen type 11 α1 |
| MGP | Matrix gla protein |
| TNC | tenascin |
| KGF | keratinocytes growth factor |
| Angptl2/4 | angiopoietin-related protein 2 and 4 |
| SFRP | secreted frizzled-related protein |
| DKK1 | Dickkopf1 |
| WIF1 | Wnt inhibitory factor 1 |
| WISP2 | Wnt1-inducible-signaling pathway protein 2 |
| Wnt1 | wingless-type MMTV integration site family, member 1 |
| α-Sma | α smooth muscle actin |
| miRNA | small non-coding RNA molecule |
References
- Evans, D.; Howard, E.; Giblin, C.; Clancy, T.; Spencer, H.; Huson, S.; Lalloo, F.; Evans, G. Birth incidence and prevalence of tumor-prone syndromes: Estimates from a UK family genetic register service. Am. J. Med Genet. Part A 2010, 152, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.G.; Ladusans, E.J.; Rimmer, S.; Burnell, L.D.; Thakker, N.; Farndon, P.A.; Evans, G. Complications of the naevoid basal cell carcinoma syndrome: Results of a population based study. J. Med. Genet. 1993, 30, 460–464. [Google Scholar] [CrossRef] [PubMed]
- Muzio, L.L.; Nocini, P.F.; Savoia, A.; Consolo, U.; Procaccini, M.; Zelante, L.; Pannone, G.; Bucci, P.; Dolci, M.; Bambini, F.; et al. Nevoid basal cell carcinoma syndrome. Clinical findings in 37 Italian affected individuals. Clin. Genet. 1999, 55, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.-M.; Zhao, H.-S.; Sun, L.-S.; Li, T.-J. PTCH mutations in sporadic and Gorlin-syndrome-related odontogenic keratocysts. J. Dent. Res. 2006, 85, 859–863. [Google Scholar] [CrossRef]
- Yang, X.; Pfeiffer, R.M.; Goldstein, A.M. Influence of glutathione-Stransferase (GSTM1, GSTP1, GSTT1) and cytochrome p450 (CYP1A1, CYP2D6) polymorphims on numbers of basal cell carcinomas (BCCs) in families with the naevoid basal cell carcinoma syndrome. J. Med. Genet. 2006, 43, e16. [Google Scholar] [CrossRef]
- Veenstra-Knol, H.E.; Scheewe, J.H.; van der Vlist, G.J.; van Doorn, M.E.; Ausems, M.G. Early recognition of basal cell naevus syndrome. Eur. J. Pediatr. 2005, 164, 126–130. [Google Scholar] [CrossRef]
- Marsh, A.; Wicking, C.; Wainwright, B.; Chenevix-Trench, G. DHPLC analysis of patients with Nevoid Basal Cell Carcinoma Syndrome reveals novel PTCH missense mutations in the sterol-sensing domain. Hum. Mutat. 2005, 26, 283. [Google Scholar] [CrossRef]
- Pastorino, L.; Cusano, R.; Nasti, S.; Faravelli, F.; Forzano, F.; Baldó, C.; Barile, M.; Gliori, S.; Muggianu, M.; Ghigliotti, G.; et al. Molecular characterization of Italian nevoid basal cell carcinoma syndrome patients. Hum. Mutat. 2005, 25, 322–323. [Google Scholar] [CrossRef]
- Gorlin, R.J. Nevoid basal cell carcinoma (Gorlin) syndrome: Unanswered issues. J. Lab. Clin. Med. 1999, 134, 551–552. [Google Scholar] [CrossRef]
- Kimonis, V.E.; Goldstein, A.M.; Pastakia, B.; Yang, M.L.; Kase, R.; DiGiovanna, J.J.; Bale, A.E.; Bale, S.J. Clinical manifestations in 105 persons with nevoid basal cell carcinoma syndrome. Am. J. Med. Genet. 1997, 69, 299–308. [Google Scholar] [CrossRef]
- Muzio, L.L.; Staibano, S.; Pannone, G.; Bucci, P.; Nocini, P.; Bucci, E.; De Rosa, G. Expression of cell cycle and apoptosis-related proteins in sporadic odontogenic keratocysts and odontogenic keratocysts associated with the nevoid basal cell carcinoma syndrome. J. Dent. Res. 1999, 78, 1345–1353. [Google Scholar] [CrossRef] [PubMed]
- Gorlin, R.J. Nevoid Basal Cell Carcinoma Syndrome. Dermatol. Clin. 1995, 13, 113–125. [Google Scholar] [CrossRef]
- Hasegawa, D.; Ochiai-Shino, H.; Onodera, S.; Nakamura, T.; Saito, A.; Onda, T.; Watanabe, K.; Nishimura, K.; Ohtaka, M.; Nakanishi, M.; et al. Gorlin syndrome-derived induced pluripotent stem cells are hypersensitive to hedgehog-mediated osteogenic induction. PLoS ONE 2017, 12, e0186879. [Google Scholar] [CrossRef] [PubMed]
- Wilson, L.C.; Bernhard, B.; Maas, S.M.; Ajayi-Obe, E. Patched mutations and hairy skin patches: A new sign in Gorlin syndrome. Am. J. Med. Genet. Part A 2006, 140, 2625–2630. [Google Scholar] [CrossRef] [PubMed]
- Muzio, L.L. Nevoid basal cell carcinoma syndrome (Gorlin syndrome). Orphanet J. Rare Dis. 2008, 3, 32. [Google Scholar] [CrossRef]
- Marcil, I.; Stern, R.S. Risk of developing a subsequent non melanoma skin cancer in patients with a history of skin cancer: A critical review of the literature and meta-analysis. Arch. Dermatol. 2000, 136, 1525–1530. [Google Scholar] [CrossRef]
- Sanchez-Conejo-Mir, J.; Camacho, F. Nevoid Basal Cell Carcinoma Syndrome: Combined Etretinate and Surgical Treatment. J. Dermatol. Surg. Oncol. 1989, 15, 868–871. [Google Scholar] [CrossRef]
- So, P.-L.; Fujimoto, M.A.; Epstein, E.H. Pharmacologic retinoid signaling and physiologic retinoic acid receptor signaling inhibit basal cell carcinoma tumorigenesis. Mol. Cancer Ther. 2008, 7, 1275–1284. [Google Scholar] [CrossRef]
- Tang, T.; Tang, J.Y.; Li, N.; Reich, M.; Callahan, C.A.; Fu, L.; Yauch, R.L.; Wong, F.; Kotkow, K.; Chang, K.S.; et al. Targeting Superficial or Nodular Basal Cell Carcinoma with Topically Formulated Small Molecule Inhibitor of Smoothened. Clin. Cancer Res. 2011, 17, 3378–3387. [Google Scholar] [CrossRef]
- Tang, J.Y.; Mackay-Wiggan, J.M.; Aszterbaum, M.; Yauch, R.L.; Lindgren, J.; Chang, K.; Coppola, C.; Chanana, A.M.; Marji, J.; Bickers, D.R.; et al. Inhibiting the hedgehog pathway in patients with the basal-cell nevus syndrome. New Engl. J. Med. 2012, 366, 2180–2188. [Google Scholar] [CrossRef]
- Chang, A.L.S.; Arron, S.T.; Migden, M.R.; Solomon, J.A.; Yoo, S.; Day, B.-M.; McKenna, E.F.; Sekulic, A. Safety and efficacy of vismodegib in patients with basal cell carcinoma nevus syndrome: Pooled analysis of two trials. Orphanet J. Rare Dis. 2016, 11, 120. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.Y.; Ally, M.S.; Chanana, A.M.; Mackay-Wiggan, J.M.; Aszterbaum, M.; Lindgren, J.A.; Ulerio, G.; Rezaee, M.R.; Gildengorin, G.; Marji, J.; et al. Inhibition of the hedgehog pathway in patients with basal-cell nevus syndrome: Final results from the multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2016, 17, 1720–1731. [Google Scholar] [CrossRef]
- Clendenning, W.E.; Block, J.B.; Radde, I.G. Basal cell nevus syndrome. Arch. Dermatol. 1964, 90, 38–53. [Google Scholar] [CrossRef] [PubMed]
- Chiang, A.; Jaju, P.D.; Batra, P.; Rezaee, M.; Epstein, E.H.; Tang, J.Y.; Sarin, K.Y. Genomic Stability in Syndromic Basal Cell Carcinoma. J. Investig. Dermatol. 2018, 138, 1044–1051. [Google Scholar] [CrossRef] [PubMed]
- Basset-Séguin, N.; Sharpe, H.J.; De Sauvage, F.J. Efficacy of Hedgehog Pathway Inhibitors in Basal Cell Carcinoma. Mol. Cancer Ther. 2015, 14, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, J.B.; Macdonald, B.; Golitz, L.E.; LoRusso, P.; Sekulic, A. Cutaneous adverse effects of targeted therapies: Part II: Inhibitors of intracellular molecular signaling pathways. J. Am. Acad. Dermatol. 2015, 72, 221–236. [Google Scholar] [CrossRef]
- Sekulic, A.; for the Erivance BCC Investigators; Migden, M.R.; Basset-Seguin, N.; Garbe, C.; Gesierich, A.; Lao, C.D.; Miller, C.; Mortier, L.; Murrell, D.F.; et al. Long-term safety and efficacy of vismodegib in patients with advanced basal cell carcinoma: Final update of the pivotal Erivance BCC study. BMC Cancer 2017, 17, 332. [Google Scholar] [CrossRef]
- Goldstein, A.M.; Bale, S.J.; Peck, G.L.; DiGiovanna, J.J. Sun exposure and basal cell carcinomas in the nevoid basal cell carcinoma syndrome. J. Am. Acad. Dermatol. 1993, 29, 34–41. [Google Scholar] [CrossRef]
- Aszterbaum, M.; Beech, J.; Epstein, E.H. Ultraviolet radiation mutagenesis of hedgehog pathway genes in basal cell carcinomas. J. Investig. Dermatol. Symp. Proc. 1999, 4, 41–45. [Google Scholar] [CrossRef]
- Goldstein, A.M.; Pastakia, B.; DiGiovanna, J.J.; Poliak, S.; Santucci, S.; Kase, R.; Bale, A.E.; Bale, S.J. Clinical findings in two African-American families with the nevoid basal cell carcinoma syndrome (NBCC). Am. J. Med Genet. 1994, 50, 272–281. [Google Scholar] [CrossRef]
- Mancuso, M.; Pazzaglia, S.; Tanori, M.; Hahn, H.; Merola, P.; Rebessi, S.; Atkinson, M.J.; Di Majo, V.; Covelli, V.; Saran, A. Basal cell carcinoma and its development: Insights from radiation-induced tumors in Ptch1-deficient mice. Cancer Res. 2004, 64, 934–941. [Google Scholar] [CrossRef] [PubMed]
- Applegate, L.A.; Goldberg, L.H.; Ley, R.D.; Ananthaswamy, H.N. Hypersensitivity of skin fibroblasts from basal cell nevus syndrome patients to killing by ultraviolet B but not by ultraviolet C radiation. Cancer Res. 1990, 50, 637–641. [Google Scholar] [PubMed]
- Nishigori, C.; Arima, Y.; Matsumura, Y.; Matsui, M.; Miyachi, Y. Impaired removal of 8-hydroxydeoxyguanosine induced by UVB radiation in naevoid basal cell carcinoma syndrome cells. Br. J. Dermatol. 2005, 153, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Brellier, F.; Valin, A.; Gorry, P.; Avril, M.-F.; Magnaldo, T.; Chevallier-Lagente, O. Ultraviolet responses of Gorlin syndrome primary skin cells. Br. J. Dermatol. 2008, 159, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Hahn, H.; Wicking, C.; Zaphiropoulos, P.G.; Gailani, M.R.; Shanley, S.; Chidambaram, A.; Vořechovský, I.; Holmberg, E.; Undèn, A.B.; Gillies, S.; et al. Mutations of the Human Homolog of Drosophila patched in the Nevoid Basal Cell Carcinoma Syndrome. Cell 1996, 85, 841–851. [Google Scholar] [CrossRef]
- Boutet, N.; Bignon, Y.-J.; Drouin-Garraud, V.; Sarda, P.; Longy, M.; Lacombe, D.; Gorry, P. Spectrum of PTCH1 Mutations in French Patients with Gorlin Syndrome. J. Investig. Dermatol. 2003, 121, 478–481. [Google Scholar] [CrossRef]
- Wicking, C.; Shanley, S.; Smyth, I.; Gillies, S.; Negus, K.; Graham, S.; Suthers, G.; Haites, N.; Edwards, M.; Wainwright, B.; et al. Most germ-line mutations in the nevoid basal cell carcinoma syndrome lead to a premature termination of the PATCHED protein, and no genotype-phenotype correlations are evident. Am. J. Hum. Genet. 1997, 60, 21–26. [Google Scholar]
- Savino, M.; D’Apolito, M.; Formica, V.; Baorda, F.; Mari, F.; Renieri, A.; Carabba, E.; Tarantino, E.; Andreucci, E.; Belli, S.; et al. Spectrum ofPTCH mutations in Italian nevoid basal cell-carcinoma syndrome patients: Identification of thirteen novel alleles. Hum. Mutat. 2004, 24, 441. [Google Scholar] [CrossRef]
- Lindström, E.; Shimokawa, T.; Toftgard, R.; Zaphiropoulos, P.G. PTCH mutations: Distribution and analyses. Hum. Mutat. 2006, 27, 215–219. [Google Scholar] [CrossRef]
- Fujii, K.; Ohashi, H.; Suzuki, M.; Hatsuse, H.; Shiohama, T.; Uchikawa, H.; Miyashita, T. Frameshift mutation in the PTCH2 gene can cause nevoid basal cell carcinoma syndrome. Fam. Cancer 2013, 12, 611–614. [Google Scholar] [CrossRef]
- Fan, Z.; Li, J.; Du, J.; Zhang, H.; Shen, Y.; Wang, C.-Y.; Wang, S. A missense mutation in PTCH2 underlies dominantly inherited NBCCS in a Chinese family. J. Med. Genet. 2008, 45, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Pastorino, L.; Ghiorzo, P.; Nasti, S.; Battistuzzi, L.; Cusano, R.; Marzocchi, C.; Garrè, M.L.; Clementi, M.; Scarrà, G.B. Identification of a SUFU germline mutation in a family with Gorlin syndrome. Am. J. Med. Genet. A 2009, 149, 1539–1543. [Google Scholar] [CrossRef] [PubMed]
- Ogden, T.; Higgins, S.; Elbaum, D.; Wysong, A. The relevance of a suppressor of fused (SUFU) mutation in the diagnosis and treatment of Gorlin syndrome. JAAD Case Rep. 2018, 4, 196–199. [Google Scholar] [CrossRef] [PubMed]
- Shimkets, R.; Gailani, M.R.; Siu, V.M.; Yang-Feng, T.; Pressman, C.L.; Levanat, S.; Goldstein, A.; Dean, M.; Bale, A.E. Molecular analysis of chromosome 9q deletions in two Gorlin syndrome patients. Am. J. Hum. Genet. 1996, 59, 417–422. [Google Scholar] [PubMed]
- Castori, M.; Morrone, A.; Kanitakis, J.; Grammatico, P. Genetic skin diseases predisposing to basal cell carcinoma. Eur. J. Dermatol. EJD 2012, 22, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Onodera, S.; Saito, A.; Hasegawa, D.; Morita, N.; Watanabe, K.; Nomura, T.; Shibahara, T.; Ohba, S.; Yamaguchi, A.; Azuma, T. Multi-layered mutation in hedgehog-related genes in Gorlin syndrome may affect the phenotype. PLoS ONE 2017, 12, e0184702. [Google Scholar] [CrossRef] [PubMed]
- Ragge, N.K.; Salt, A.; Collin, J.R.O.; Michalski, A.; Farndon, P.A. Gorlin syndrome: The PTCH gene links ocular developmental defects and tumour formation. Br. J. Ophthalmol. 2005, 89, 988–991. [Google Scholar] [CrossRef]
- Reifenberger, J.; Wolter, M.; Knobbe, C.B.; Köhler, B.; Schönicke, A.; Scharwächter, C.; Kumar, K.; Blaschke, B.; Ruzicka, T.; Reifenberger, G. Somatic mutations in the PTCH, SMOH, SUFUH and TP53 genes in sporadic basal cell carcinomas. Br. J. Dermatol. 2005, 152, 43–51. [Google Scholar] [CrossRef]
- Unden, A.B.; Holmberg, E.; Lundh-Rozell, B.; Stähle-Bäckdahl, M.; Zaphiropoulos, P.G.; Toftgård, R.; Vorechovsky, I. Mutations in the human homologue of Drosophila patched (PTCH) in basal cell carcinomas and the Gorlin syndrome: Different in vivo mechanisms of PTCH inactivation. Cancer Res. 1996, 56, 4562–4565. [Google Scholar]
- Quinn, A.G.; Campbell, C.; Healy, E.; Rees, J.L. Chromosome 9 Allele Loss Occurs in both Basal and Squamous Cell Carcinomas of the Skin. J. Investig. Dermatol. 1994, 102, 300–303. [Google Scholar] [CrossRef][Green Version]
- Gailani, M.R.; Ståhle-Bäckdahl, M.; Leffell, D.J.; Glyn, M.; Zaphiropoulos, P.G.; Undèn, A.B.; Dean, M.; Brash, D.E.; Bale, A.E.; Toftgard, R.; et al. The role of the human homologue of Drosophila patched in sporadic basal cell carcinomas. Nat. Genet. 1996, 14, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Crowson, A.N. Basal cell carcinoma: Biology, morphology and clinical implications. Mod. Pathol. 2006, 19, S127–S147. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Murone, M.; Luoh, S.-M.; Ryan, A.; Gu, Q.; Zhang, C.; Bonifas, J.M.; Lam, C.-W.; Hynes, M.; Goddard, A.; et al. Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature 1998, 391, 90–92. [Google Scholar] [CrossRef] [PubMed]
- Sidransky, D. Is human patched the gatekeeper of common skin cancers? Nat. Genet. 1996, 14, 7–8. [Google Scholar] [CrossRef]
- Hooper, J.E.; Scott, M.P. Communicating with Hedgehogs. Nat. Rev. Mol. Cell Boil. 2005, 6, 306–317. [Google Scholar] [CrossRef]
- Currie, K.W.; Molinaro, A.M.; Pearson, B.J. Neuronal sources of hedgehog modulate neurogenesis in the adult planarian brain. Elife 2016, 5, e19735. [Google Scholar] [CrossRef]
- Kan, C.; Chen, L.; Hu, Y.; Ding, N.; Li, Y.; McGuire, T.L.; Lu, H.; Kessler, J.A.; Kan, L. Gli1-labeled adult mesenchymal stem/progenitor cells and hedgehog signaling contribute to endochondral heterotopic ossification. Bone 2018, 109, 71–79. [Google Scholar] [CrossRef]
- Petrova, R.; Joyner, A.L. Roles for Hedgehog signaling in adult organ homeostasis and repair. Development 2014, 141, 3445–3457. [Google Scholar] [CrossRef]
- Shen, X.; Peng, Y.; Li, H. The Injury-Related Activation of Hedgehog Signaling Pathway Modulates the Repair-Associated Inflammation in Liver Fibrosis. Front. Immunol. 2017, 8, 8. [Google Scholar] [CrossRef]
- Goodrich, L.V.; Scott, M.P.; Johnson, R.L.; Milenković, L.; McMahon, J.A. Conservation of the hedgehog/patched signaling pathway from flies to mice: Induction of a mouse patched gene by Hedgehog. Genes Dev. 1996, 10, 301–312. [Google Scholar] [CrossRef]
- Marigo, V.; Scott, M.P.; Johnson, R.L.; Goodrich, L.V.; Tabin, C.J. Conservation in hedgehog signaling: Induction of a chicken patched homolog by Sonic hedgehog in the developing limb. Development 1996, 122, 1225–1233. [Google Scholar] [PubMed]
- Cohen, M.M. The hedgehog signaling network. Am. J. Med. Genet. 2003, 123, 5–28. [Google Scholar] [CrossRef] [PubMed]
- Heuvel, M.V.D.; Ingham, P.W. smoothened encodes a receptor-like serpentine protein required for hedgehog signalling. Nature 1996, 382, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Matise, M.P.; Joyner, A.L. Gli genes in development and cancer. Oncogene 1999, 18, 7852–7859. [Google Scholar] [CrossRef]
- Méthot, N.; Basler, K. An absolute requirement for Cubitus interruptus in Hedgehog signaling. Development 2001, 128, 733–742. [Google Scholar]
- Kasper, M.; Regl, G.; Frischauf, A.-M.; Aberger, F. GLI transcription factors: Mediators of oncogenic Hedgehog signalling. Eur. J. Cancer 2006, 42, 437–445. [Google Scholar] [CrossRef]
- Regl, G.; Kasper, M.; Schnidar, H.; Eichberger, T.; Neill, G.W.; Philpott, M.P.; Esterbauer, H.; Hauser-Kronberger, C.; Frischauf, A.-M.; Aberger, F. Activation of the BCL2 Promoter in Response to Hedgehog/GLI Signal Transduction Is Predominantly Mediated by GLI2. Cancer Res. 2004, 64, 7724–7731. [Google Scholar] [CrossRef]
- Bigelow, R.L.; Chari, N.S.; Unden, A.B.; Spurgers, K.B.; Lee, S.; Roop, D.R.; Toftgard, R.; McDonnell, T.J. Transcriptional regulation of bcl-2 mediated by the Sonic hedgehog signaling pathway through Gli-1. J. Biol. Chem. 2004, 279, 1197–1205. [Google Scholar] [CrossRef]
- Ingham, P.W.; Taylor, A.M.; Nakano, Y. Role of the Drosophila patched gene in positional signalling. Nature 1991, 353, 184–187. [Google Scholar] [CrossRef]
- Ingham, P.W.; Hidalgo, A. Regulation of wingless transcription in the Drosophila embryo. Development 1993, 117, 283–291. [Google Scholar]
- Huang, T.X.; Guan, X.Y.; Fu, L. Wnt, Notch, and TGF-β Pathways Impinge on Hedgehog Signaling Complexity: An Open Window on Cancer. Am. J. Cancer Res. 2019, 9, 1889–1904. [Google Scholar] [PubMed]
- Noubissi, F.K.; Yedjou, C.G.; Spiegelman, V.S.; Tchounwou, P.B. Cross-Talk between Wnt and Hh Signaling Pathways in the Pathology of Basal Cell Carcinoma. Int. J. Environ. Res. Public Heal. 2018, 15, 1442. [Google Scholar] [CrossRef] [PubMed]
- Schnidar, H.; Eberl, M.; Klingler, S.; Mangelberger, R.; Kasper, M.; Hauser-Kronberger, C.; Regl, G.; Kroismayr, R.; Moriggl, R.; Sibilia, M.; et al. Epidermal growth factor receptor signaling synergizes with Hedgehog/GLI in oncogenic transformation via activation of the MEK/ERK/JUN pathway. Cancer Res. 2009, 69, 1284–1292. [Google Scholar] [CrossRef] [PubMed]
- Mazumdar, T.; Sandhu, R.; Qadan, M.; DeVecchio, J.; Magloire, V.; Agyeman, A.; Li, B.; Houghton, J.A. Hedgehog Signaling Regulates Telomerase Reverse Transcriptase in Human Cancer Cells. PLoS ONE 2013, 8, e75253. [Google Scholar] [CrossRef] [PubMed]
- Alphonso, A.; Alahari, S.K. Stromal Cells and Integrins: Conforming to the Needs of the Tumor Microenvironment. Neoplasia 2009, 11, 1264–1271. [Google Scholar] [CrossRef]
- Nissinen, L.; Farshchian, M.; Riihilä, P.; Kähäri, V.-M. New perspectives on role of tumor microenvironment in progression of cutaneous squamous cell carcinoma. Cell Tissue Res. 2016, 365, 691–702. [Google Scholar] [CrossRef]
- Lewis, D.A.; Travers, J.B.; Spandau, D.F. A New Paradigm for the Role of Aging in the Development of Skin Cancer. J. Invest. Dermatol. 2009, 129, 787–791. [Google Scholar] [CrossRef]
- Omland, S.H.; Wettergren, E.E.; Mollerup, S.; Asplund, M.; Mourier, T.; Hansen, A.J.; Gniadecki, R.; Robert, R. Cancer associated fibroblasts (CAFs) are activated in cutaneous basal cell carcinoma and in the peritumoural skin. BMC Cancer 2017, 17, 675. [Google Scholar] [CrossRef]
- Micke, P.; Kappert, K.; Ohshima, M.; Sundquist, C.; Scheidl, S.; Lindahl, P.; Heldin, C.-H.; Botling, J.; Pontén, F.; Östman, A.; et al. In Situ Identification of Genes Regulated Specifically in Fibroblasts of Human Basal Cell Carcinoma. J. Investig. Dermatol. 2007, 127, 1516–1523. [Google Scholar] [CrossRef]
- Mao, Y.; Keller, E.T.; Garfield, D.H.; Shen, K.; Wang, J. Stromal cells in tumor microenvironment and breast cancer. Cancer Metastasis Rev. 2013, 32, 303–315. [Google Scholar] [CrossRef]
- Cirri, P.; Chiarugi, P. Cancer-associated-fibroblasts and tumour cells: A diabolic liaison driving cancer progression. Cancer Metastasis Rev. 2012, 31, 195–208. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; Kalluri, R. A peek into cancer-associated fibroblasts: Origins, functions and translational impact. Dis. Model. Mech. 2018, 11, dmm029447. [Google Scholar] [CrossRef] [PubMed]
- Moinfar, F.; Man, Y.G.; Arnould, L.; Bratthauer, G.L.; Ratschek, M.; Tavassoli, F.A. Concurrent and independent genetic alterations in the stromal and epithelial cells of mammary carcinoma: Implications for tumorigenesis. Cancer Res. 2000, 60, 2562–2566. [Google Scholar] [PubMed]
- Ohlund, D.; Elyada, E.; Tuveson, D. Fibroblast heterogeneity in the cancer wound. J. Exp. Med. 2014, 211, 1503–1523. [Google Scholar] [CrossRef]
- Yauch, R.L.; Gould, S.E.; Scales, S.J.; Tang, T.; Tian, H.; Ahn, C.P.; Marshall, D.; Fu, L.; Januario, T.; Kallop, D.; et al. A paracrine requirement for hedgehog signalling in cancer. Nature 2008, 455, 406–410. [Google Scholar] [CrossRef]
- Nurmik, M.; Ullmann, P.; Rodriguez, F.; Haan, S.; Letellier, E. In search of definitions: Cancer-associated fibroblasts and their markers. Int. J. Cancer 2019, 146, 895–905. [Google Scholar] [CrossRef]
- Gieniec, K.A.; Butler, L.M.; Worthley, D.L.; Woods, S.L. Cancer-associated fibroblasts-heroes or villains? Br. J. Cancer 2019, 121, 293–302. [Google Scholar] [CrossRef]
- Liu, T.; Zhou, L.; Li, D.; Andl, T.; Zhang, Y. Cancer-Associated Fibroblasts Build and Secure the Tumor Microenvironment. Front. Cell Dev. Boil. 2019, 7, 60. [Google Scholar] [CrossRef]
- O’Toole, S.A.; Machalek, D.A.; Shearer, R.F.; Millar, E.K.; Nair, R.; Schofield, P.; McLeod, D.; Cooper, C.L.; McNeil, C.M.; McFarland, A.; et al. Hedgehog Overexpression Is Associated with Stromal Interactions and Predicts for Poor Outcome in Breast Cancer. Cancer Res. 2011, 71, 4002–4014. [Google Scholar] [CrossRef]
- Gascard, P.; Tlsty, T.D. Carcinoma-associated fibroblasts: Orchestrating the composition of malignancy. Genome Res. 2016, 30, 1002–1019. [Google Scholar] [CrossRef]
- Tian, H.; Callahan, C.A.; DuPree, K.J.; Darbonne, W.C.; Ahn, C.P.; Scales, S.J.; De Sauvage, F.J. Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 4254–4259. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Tang, T.; Eastham-Anderson, J.; Dunlap, D.; Alicke, B.; Nannini, M.; Gould, S.; Yauch, R.; Modrusan, Z.; DuPree, K.J.; et al. Canonical hedgehog signaling augments tumor angiogenesis by induction of VEGF-A in stromal perivascular cells. Proc. Natl. Acad. Sci. USA 2011, 108, 9589–9594. [Google Scholar] [CrossRef] [PubMed]
- Petty, J.P.; Li, A.; Wang, X.; Dai, R.; Heyman, B.; Hsu, D.; Huang, X.; Yang, Y. Hedgehog signaling promotes tumor-associated macrophage polarization to suppress intratumoral CD8+ T cell recruitment. J. Clin. Investig. 2019, 129, 5151–5162. [Google Scholar] [CrossRef] [PubMed]
- Merchant, J.L.; Ding, L. Hedgehog Signaling Links Chronic Inflammation to Gastric Cancer Precursor Lesions. Cell. Mol. Gastroenterol. Hepatol. 2017, 3, 201–210. [Google Scholar] [CrossRef]
- Wei, R.; Lv, M.; Li, F.; Cheng, T.; Zhang, Z.; Jiang, G.; Zhou, Y.; Gao, R.; Wei, X.; Lou, J.; et al. Human CAFs promote lymphangiogenesis in ovarian cancer via the Hh-VEGF-C signaling axis. Oncotarget 2017, 8, 67315–67328. [Google Scholar] [CrossRef]
- Choe, C.; Shin, Y.-S.; Kim, S.-H.; Jeon, M.-J.; Choi, S.-J.; Lee, J.; Kim, J. Tumor-stromal interactions with direct cell contacts enhance motility of non-small cell lung cancer cells through the hedgehog signaling pathway. Anticancer. Res. 2013, 33, 3715–3723. [Google Scholar]
- Kurose, K.; Gilley, K.; Matsumoto, S.; Watson, P.H.; Zhou, X.-P.; Eng, C. Frequent somatic mutations in PTEN and TP53 are mutually exclusive in the stroma of breast carcinomas. Nat. Genet. 2002, 32, 355–357. [Google Scholar] [CrossRef]
- Majmudar, G.; Nelson, B.R.; Jensen, T.C.; Johnson, T.M. Increased expression of matrix metalloproteinase-3 (stromelysin-1) in cultured fibroblasts and basal cell carcinomas of nevoid basal cell carcinoma syndrome. Mol. Carcinog. 1994, 11, 29–33. [Google Scholar] [CrossRef]
- Shao, H.; Cai, L.; Grichnik, J.M.; Livingstone, A.S.; Velazquez, O.C.; Liu, Z.-J. Activation of Notch1 signaling in stromal fibroblasts inhibits melanoma growth by upregulating WISP-1. Oncogene 2011, 30, 4316–4326. [Google Scholar] [CrossRef]
- Zhou, L.; Yang, K.; Wickett, R.R.; Kadekaro, A.L.; Zhang, Y. Targeted deactivation of cancer-associated fibroblasts by β-catenin ablation suppresses melanoma growth. Tumor Boil. 2016, 37, 14235–14248. [Google Scholar] [CrossRef]
- Nitzki, F.; Zibat, A.; König, S.; Wijgerde, M.; Rosenberger, A.; Brembeck, F.H.; Carstens, P.-O.; Frommhold, A.; Uhmann, A.; Klingler, S.; et al. Tumor Stroma–Derived Wnt5a Induces Differentiation of Basal Cell Carcinoma ofPtch-Mutant Mice via CaMKII. Cancer Res. 2010, 70, 2739–2748. [Google Scholar] [CrossRef] [PubMed]
- Ponti, G.; Bertazzoni, G.; Pastorino, L.; Monari, E.; Cuoghi, A.; Bergamini, S.; Bellei, E.; Benassi, L.; Azzoni, P.; Petrachi, T.; et al. Proteomic analysis of PTCH1+/- fibroblast lysate and conditioned culture media isolated from the skin of healthy subjects and nevoid basal cell carcinoma syndrome patients. Biomed. Res. Int. 2013, 2013, 794028. [Google Scholar] [CrossRef] [PubMed]
- Valin, A.; Barnay-Verdier, S.; Robert, T.; Ripoche, H.; Brellier, F.; Chevallier-Lagente, O.; Avril, M.F.; Magnaldo, T. PTCH1 +/- dermal fibroblasts isolated from healthy skin of Gorlin syndrome patients exhibit features of carcinoma associated fibroblasts. PLoS ONE 2009, 4, e4818. [Google Scholar] [CrossRef] [PubMed]
- Zamarrón, A.; García, M.; Del Río, M.; Larcher, F.; Juarranz, Á. Effects of photodynamic therapy on dermal fibroblasts from xeroderma pigmentosum and Gorlin-Goltz syndrome patients. Oncotarget 2017, 8, 77385–77399. [Google Scholar] [CrossRef]
- Wright, A.T.; Magnaldo, T.; Sontag, R.L.; Anderson, L.N.; Sadler, N.C.; Piehowski, P.D.; Gache, Y.; Weber, T.J. Deficient expression of aldehyde dehydrogenase 1A1 is consistent with increased sensitivity of Gorlin syndrome patients to radiation carcinogenesis. Mol. Carcinog. 2015, 54, 473–484. [Google Scholar] [CrossRef]
- Gache, Y.; Brellier, F.; Rouanet, S.; Al-Qaraghuli, S.; Goncalves-Maia, M.; Burty-Valin, E.; Barnay, S.; Scarzello, S.; Ruat, M.; Sevenet, N.; et al. Basal Cell Carcinoma in Gorlin’s Patients: A Matter of Fibroblasts-Led Protumoral Microenvironment? PLoS ONE 2015, 10, e0145369. [Google Scholar] [CrossRef]
- Shiohama, T.; Fujii, K.; Miyashita, T.; Takatani, T.; Ikehara, H.; Uchikawa, H.; Motojima, T.; Uchida, T.; Shimojo, N. MicroRNAs profiling in fibroblasts derived from patients with Gorlin syndrome. J. Hum. Genet. 2019, 64, 757–765. [Google Scholar] [CrossRef]
- Sripada, L.; Singh, K.; Lipatova, A.V.; Prajapati, P.; Tomar, D.; Bhatelia, K.; Roy, M.; Godbole, M.M.; Chumakov, P.M. hsa-miR-4485 regulates mitochondrial functions and inhibits the tumorigenicity of breast cancer cells. J. Mol. Med. 2017, 95, 641–651. [Google Scholar] [CrossRef]
- Chacón-Solano, E.; León, C.; Díaz, F.; García-García, F.; García, M.; Escámez, M.; Guerrero-Aspizua, S.; Conti, C.; Mencía, Á.; Martínez-Santamaría, L.; et al. Fibroblast activation and abnormal extracellular matrix remodelling as common hallmarks in three cancer-prone genodermatoses. Br. J. Dermatol. 2019, 181, 512–522. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).