BDNF as a Promising Therapeutic Agent in Parkinson’s Disease

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
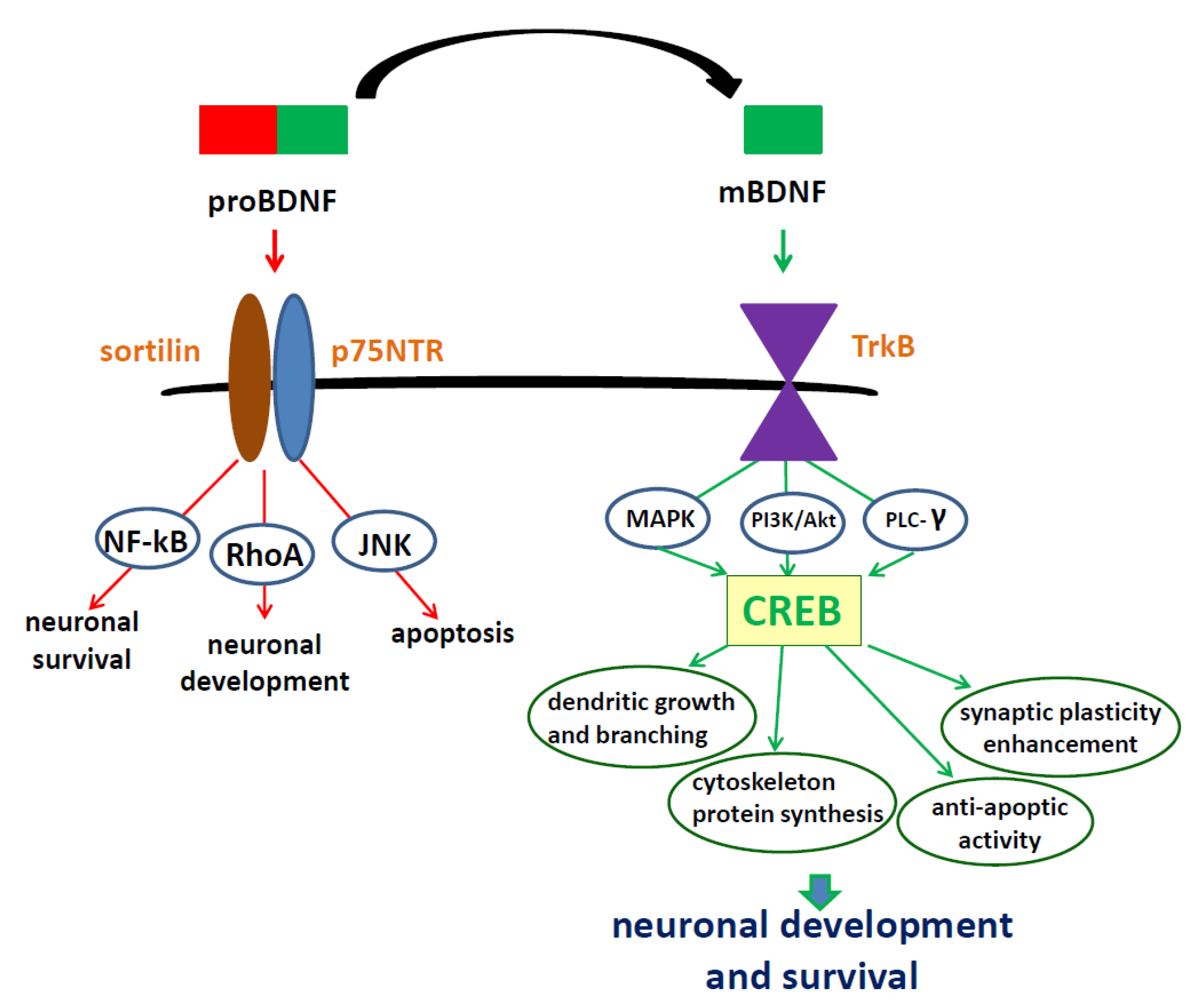
2. Brain-Derived Neurotrophic Factor—Regulation of Synthesis, Activation of Specific Receptors, Location and Function in the Nervous System
3. Role of BDNF in Neurodegeneration and Neuroregeneration
4. BDNF as a Promising Compound in the Therapy of Parkinson’s Disease
4.1. Upregulation of BDNF Signaling through Direct Injection and Gene Therapy
4.1.1. Study in PD Animal Models
4.1.2. Study in Humans
4.2. Stimulation of Endogenous BDNF Level by Physical Effort
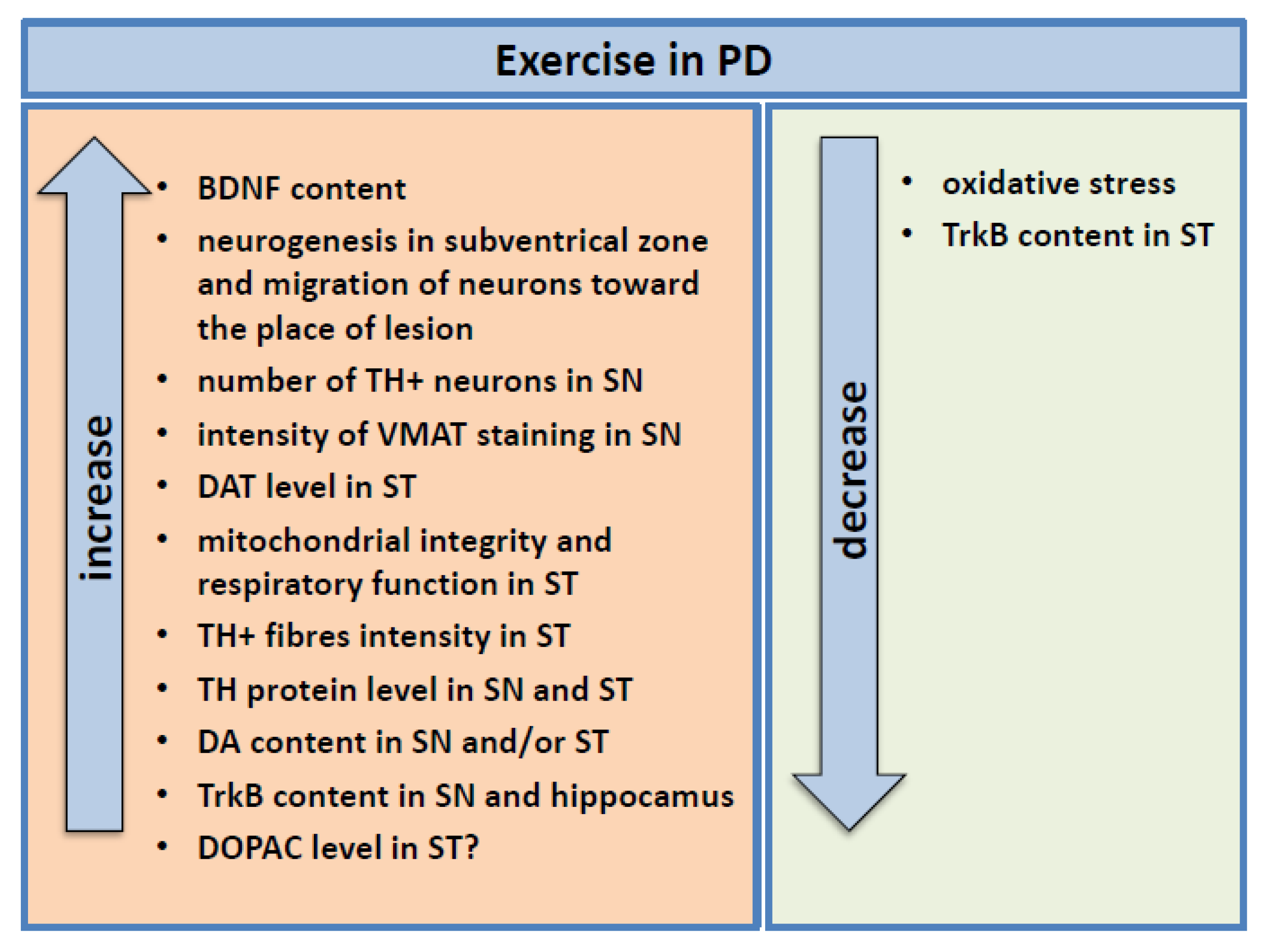
4.2.1. Study in PD Animal Models
4.2.2. Study in Humans
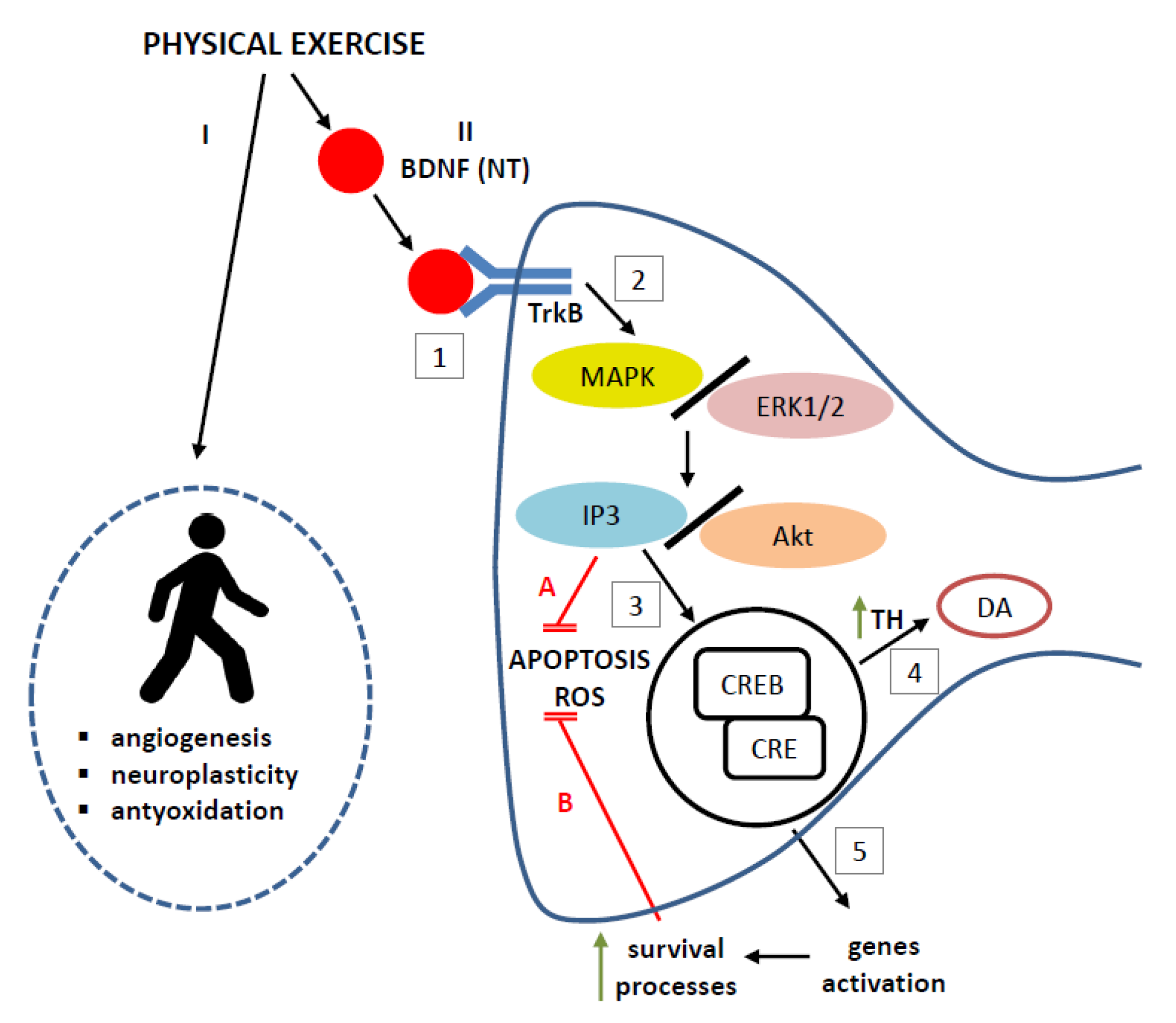
5. Possible Mechanisms Underlying the Protective Effect of BNDF Induced by Exercise
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 5-HT | 5-hydroxytryptamine, serotonine |
| 6-OHDA | 6-hydroxydopamine |
| AAV2 | adeno-associated virus serotype 2 vector |
| AD | Alzheimer’s disease |
| Akt | Akt enzyme, also known as protein kinase B |
| ALS | amyotrophic lateral sclerosis |
| ASN | alpha-synuclein |
| BBB | blood-brain-barrier |
| BDNF | brain-derived neurotrophic factor |
| CaMKII | calmodulin-dependent protein kinase II |
| cAMP | cyclicAMP |
| CNS | central nervous system |
| CREB | cAMP-response element-binding protein |
| CSF | cerebrospinal fluid |
| DA | dopamine |
| DAG | diacylglycerol |
| DAT | dopamine transporter |
| DOPAC | 3,4-dihydroxyphenylacetic acid |
| ERK | extracellular signal-regulated kinases |
| GDNF | glial cell-derived neurotrophic factor |
| GSK-3β | glycogen synthase kinase 3 beta |
| HVA | homovanillic acid |
| IGF-1 | insulin-like growth factor-1 |
| IL-1β | interleukin 1 beta |
| IL-6 | interleukin 6 |
| IP3 | inositol trisphosphate |
| JNK | c-Jun N-terminal kinases |
| L-DOPA | levodopa |
| LTP | long-term potentiation |
| MAPK | mitogen-activated protein kinase |
| mBDNF | mature BDNF |
| miRNAs | microRNAs |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| NF-κB | nuclear factor kappa B |
| NGF | nerve growth factor |
| NMDA-R | N-methyl-D-aspartate receptor |
| NO | nitric oxide |
| NRTN | neurturin |
| NT-3 | neurotrophin-3 |
| NT-4 | neurotrophin-4 |
| p75NTR | p75 neurotrophin receptor |
| PD | Parkinson’s disease |
| PET | positron emission tomography |
| PGC1α | peroxisome proliferator-activated receptor gamma coactivator 1-alpha |
| PI3K | phosphatidylinositol-3 kinase |
| PKA | protein kinase A |
| PLCγ | phospholipase Cγ |
| RhoA | Ras homolog family member A |
| ROS | reactive oxygen species |
| r-metHuBDNF | recombinant methionyl human BDNF |
| SN | substantia nigra |
| ST | striatum |
| TH | tyrosine hydroxylase |
| TrkB | tropomyosin receptor kinase B |
| VEGF | vascular endothelial growth factor |
| VMAT2 | vesicular monoamine transporter 2 |
References
- Conner, J.M.; Lauterborn, J.C.; Yan, Q.; Gall, C.M.; Varon, S. Distribution of brain-derived neurotrophic factor (BDNF) protein and mRNA in the normal adult rat CNS: Evidence for anterograde axonal transport. J. Neurosci. 1997, 17, 2295–2313. [Google Scholar] [CrossRef] [PubMed]
- Kerschensteiner, M.; Gallmeier, E.; Behrens, L.; Leal, V.V.; Misgeld, T.; Klinkert, W.E.F.; Kolbeck, R.; Hoppe, E.; Oropeza-Wekerle, R.-L.; Bartke, I.; et al. Activated Human T Cells, B Cells, and Monocytes Produce Brain-derived Neurotrophic Factor In Vitro and in Inflammatory Brain Lesions: A Neuroprotective Role of Inflammation? J. Exp. Med. 1999, 189, 865–870. [Google Scholar] [CrossRef] [PubMed]
- Nakahashi, T.; Fujimura, H.; Altar, C.A.; Li, J.; Kambayashi, J.I.; Tandon, N.N.; Sun, B. Vascular endothelial cells synthesize and secrete brain-derived neurotrophic factor. FEBS Lett. 2000, 470, 113–117. [Google Scholar] [CrossRef]
- Donovan, M.J.; Miranda, R.C.; Kraemer, R.; McCaffrey, T.A.; Tessarollo, L.; Mahadeo, D.; Sharif, S.; Kaplan, D.R.; Tsoulfas, P.; Parada, L.; et al. Neurotrophin and neurotrophin receptors in vascular smooth muscle cells: Regulation of expression in response to injury. Am. J. Pathol. 1995, 147, 309–324. [Google Scholar]
- Yarrow, J.F.; White, L.J.; McCoy, S.C.; Borst, S.E. Training augments resistance exercise induced elevation of circulating brain derived neurotrophic factor (BDNF). Neurosci. Lett. 2010, 479, 161–165. [Google Scholar] [CrossRef]
- Murer, M.; Yan, Q.; Raisman-Vozari, R. Brain-derived neurotrophic factor in the control human brain, and in Alzheimer’s disease and Parkinson’s disease. Prog. Neurobiol. 2001, 63, 71–124. [Google Scholar] [CrossRef]
- Tapia-Arancibia, L.; Aliaga, E.; Silhol, M.; Arancibia, S. New insights into brain BDNF function in normal aging and Alzheimer disease. Brain Res. Rev. 2008, 59, 201–220. [Google Scholar] [CrossRef]
- Lee, J.; Duan, W.; Mattson, M.P. Evidence that brain-derived neurotrophic factor is required for basal neurogenesis and mediates, in part, the enhancement of neurogenesis by dietary restriction in the hippocampus of adult mice. J. Neurochem. 2002, 82, 1367–1375. [Google Scholar] [CrossRef]
- Bath, K.G.; Lee, F.S. Neurotrophic factor control of adult SVZ neurogenesis. Dev. Neurobiol. 2010, 70, 339–349. [Google Scholar] [CrossRef]
- Lu, B.; Nagappan, G.; Lu, Y. BDNF and Synaptic Plasticity, Cognitive Function, and Dysfunction. In Neurotrophic Factors. Handbook of Experimental Pharmacology; Lewin, G., Carter, B., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 223–250. ISBN 978-3-642-45105-8. [Google Scholar]
- Zuccato, C.; Cattaneo, E. Brain-derived neurotrophic factor in neurodegenerative diseases. Nat. Rev. Neurol. 2009, 5, 311–322. [Google Scholar] [CrossRef]
- Autry, A.E.; Monteggia, L.M. Brain-Derived Neurotrophic Factor and Neuropsychiatric Disorders. Pharmacol. Rev. 2012, 64, 238–258. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.J.; Watson, J.J.; Shoemark, D.K.; Barua, N.U.; Patel, N.K. GDNF, NGF and BDNF as therapeutic options for neurodegeneration. Pharmacol. Ther. 2013, 138, 155–175. [Google Scholar] [CrossRef] [PubMed]
- Kowiański, P.; Lietzau, G.; Czuba, E.; Waśkow, M.; Steliga, A.; Moryś, J. BDNF: A Key Factor with Multipotent Impact on Brain Signaling and Synaptic Plasticity. Cell. Mol. Neurobiol. 2018, 38, 579–593. [Google Scholar] [CrossRef] [PubMed]
- Aid, T.; Kazantseva, A.; Piirsoo, M.; Palm, K.; Timmusk, T. Mouse and ratBDNF gene structure and expression revisited. J. Neurosci. Res. 2007, 85, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.-J.; Yang, M.; Sun, Y.; Zhou, X.-F. Synthesis, Trafficking and Release of BDNF. In Handbook of Neurotoxicity; Springer: New York, NY, USA, 2014; pp. 1955–1971. [Google Scholar]
- Yang, J.; Siao, C.J.; Nagappan, G.; Marinic, T.; Jing, D.; McGrath, K.; Chen, Z.Y.; Mark, W.; Tessarollo, L.; Lee, F.S.; et al. Neuronal release of proBDNF. Nat. Neurosci. 2009, 12, 113–115. [Google Scholar] [CrossRef]
- Rauskolb, S.; Zagrebelsky, M.; Dreznjak, A.; Deogracias, R.; Matsumoto, T.; Wiese, S.; Erne, B.; Sendtner, M.; Schaeren-Wiemers, N.; Korte, M.; et al. Global deprivation of brain-derived neurotrophic factor in the CNS reveals an area-specific requirement for dendritic growth. J. Neurosci. 2010, 30, 1739–1749. [Google Scholar] [CrossRef]
- Yeh, C.-M.; Huang, C.-C.; Hsu, K.-S. Prenatal stress alters hippocampal synaptic plasticity in young rat offspring through preventing the proteolytic conversion of pro-brain-derived neurotrophic factor (BDNF) to mature BDNF. J. Physiol. 2012, 590, 991–1010. [Google Scholar] [CrossRef]
- Je, H.S.; Yang, F.; Ji, Y.; Nagappan, G.; Hempstead, B.L.; Lu, B. Role of pro-brain-derived neurotrophic factor (proBDNF) to mature BDNF conversion in activity-dependent competition at developing neuromuscular synapses. Proc. Natl. Acad. Sci. USA 2012, 109, 15924–15929. [Google Scholar] [CrossRef]
- Castrén, E.; Antila, H. Neuronal plasticity and neurotrophic factors in drug responses. Mol. Psychiatry 2017, 22, 1085–1095. [Google Scholar] [CrossRef]
- Mellios, N.; Huang, H.-S.; Grigorenko, A.; Rogaev, E.; Akbarian, S. A set of differentially expressed miRNAs, including miR-30a-5p, act as post-transcriptional inhibitors of BDNF in prefrontal cortex. Hum. Mol. Genet. 2008, 17, 3030–3042. [Google Scholar] [CrossRef]
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of post-transcriptional regulation by microRNAs: Are the answers in sight? Nat. Rev. Genet. 2008, 9, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Shi, J. Regulatory networks between neurotrophins and miRNAs in brain diseases and cancers. Acta Pharmacol. Sin. 2015, 36, 149–157. [Google Scholar] [CrossRef]
- Caputo, V.; Sinibaldi, L.; Fiorentino, A.; Parisi, C.; Catalanotto, C.; Pasini, A.; Cogoni, C.; Pizzuti, A. Brain Derived Neurotrophic Factor (BDNF) Expression Is Regulated by MicroRNAs miR-26a and miR-26b Allele-Specific Binding. PLoS ONE 2011, 6, e28656. [Google Scholar] [CrossRef] [PubMed]
- Tu, Z.; Li, Y.; Dai, Y.; Li, L.; Lv, G.; Chen, I.; Wang, B. MiR-140/BDNF axis regulates normal human astrocyte proliferation and LPS-induced IL-6 and TNF-α secretion. Biomed. Pharmacother. 2017, 91, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Wu, S.; Li, Z.; Zhou, J. MicroRNA-211/BDNF axis regulates LPS-induced proliferation of normal human astrocyte through PI3K/AKT pathway. Biosci. Rep. 2017, 37, BSR20170755. [Google Scholar] [CrossRef] [PubMed]
- Thomas, K.T.; Gross, C.; Bassell, G.J. MicroRNAs Sculpt Neuronal Communication in a Tight Balance That Is Lost in Neurological Disease. Front. Mol. Neurosci. 2018, 11, 455. [Google Scholar] [CrossRef]
- Li, Y.-J.; Xu, M.; Gao, Z.-H.; Wang, Y.-Q.; Yue, Z.; Zhang, Y.-X.; Li, X.-X.; Zhang, C.; Xie, S.-Y.; Wang, P.-Y. Alterations of Serum Levels of BDNF-Related miRNAs in Patients with Depression. PLoS ONE 2013, 8, e63648. [Google Scholar] [CrossRef]
- Zheng, P.; Bin, H.; Chen, W. Inhibition of microRNA-103a inhibits the activation of astrocytes in hippocampus tissues and improves the pathological injury of neurons of epilepsy rats by regulating BDNF. Cancer Cell Int. 2019, 19, 109. [Google Scholar] [CrossRef]
- Cagni, F.C.; das Campêlo, C.L.C.; Coimbra, D.G.; Barbosa, M.R.; Júnior, L.G.O.; Neto, A.B.S.; Ribeiro, A.M.; Júnior, C.O.G.; Gomes de Andrade, T.; Silva, R.H. Association of BDNF Val66MET Polymorphism With Parkinson’s Disease and Depression and Anxiety Symptoms. J. Neuropsychiatry Clin. Neurosci. 2017, 29, 142–147. [Google Scholar] [CrossRef]
- Lemos, J.R.; Alves, C.R.; de Souza, S.B.C.; Marsiglia, J.D.C.; Silva, M.S.M.; Pereira, A.C.; Teixeira, A.L.; Vieira, E.L.M.; Krieger, J.E.; Negrão, C.E.; et al. Peripheral vascular reactivity and serum BDNF responses to aerobic training are impaired by the BDNF Val66Met polymorphism. Physiol. Genom. 2016, 48, 116–123. [Google Scholar] [CrossRef]
- Tsai, S.-J. Critical Issues in BDNF Val66Met Genetic Studies of Neuropsychiatric Disorders. Front. Mol. Neurosci. 2018, 11, 156. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, D.R.; Miller, F.D. Neurotrophin signal transduction in the nervous system. Curr. Opin. Neurobiol. 2000, 10, 381–391. [Google Scholar] [CrossRef]
- Borodinova, A.A.; Salozhin, S.V. Differences in the Biological Functions of BDNF and proBDNF in the Central Nervous System. Neurosci. Behav. Physiol. 2017, 47, 251–265. [Google Scholar] [CrossRef]
- Miranda, M.; Morici, J.F.; Zanoni, M.B.; Bekinschtein, P. Brain-Derived Neurotrophic Factor: A Key Molecule for Memory in the Healthy and the Pathological Brain. Front. Cell. Neurosci. 2019, 13, 363. [Google Scholar] [CrossRef] [PubMed]
- Minichiello, L. TrkB signalling pathways in LTP and learning. Nat. Rev. Neurosci. 2009, 10, 850–860. [Google Scholar] [CrossRef]
- Liu, X.; Obiany, O.; Chan, C.B.; Huang, J.; Xue, S.; Yang, J.J.; Zeng, F.; Goodman, M.; Ye, K. Biochemical and biophysical investigation of the brain-derived neurotrophic factor mimetic 7,8-dihydroxyflavone in the binding and activation of the trkb receptor. J. Biol. Chem. 2014, 289, 27571–27584. [Google Scholar] [CrossRef]
- Reichardt, L.F. Neurotrophin-regulated signalling pathways. Philos. Trans. R. Soc. B Biol. Sci. 2006, 361, 1545–1564. [Google Scholar] [CrossRef]
- Leal, G.; Bramham, C.R.; Duarte, C.B. BDNF and Hippocampal Synaptic Plasticity. Vitam. Horm. 2017, 104, 153–195. [Google Scholar]
- Gorski, J.A.; Zeiler, S.R.; Tamowski, S.; Jones, K.R. Brain-derived neurotrophic factor is required for the maintenance of cortical dendrites. J. Neurosci. 2003, 23, 6856–6865. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.; Fernandez, J.R.; Zegarek, G.F.; Lo, S.B.; Firestein, B.L. BDNF-Promoted Increases in Proximal Dendrites Occur via CREB-Dependent Transcriptional Regulation of Cypin. J. Neurosci. 2011, 31, 9735–9745. [Google Scholar] [CrossRef]
- Orefice, L.L.; Waterhouse, E.G.; Partridge, J.G.; Lalchandani, R.R.; Vicini, S.; Xu, B. Distinct Roles for Somatically and Dendritically Synthesized Brain-Derived Neurotrophic Factor in Morphogenesis of Dendritic Spines. J. Neurosci. 2013, 33, 11618–11632. [Google Scholar] [CrossRef] [PubMed]
- Yoshii, A.; Constantine-Paton, M. Postsynaptic localization of PSD-95 is regulated by all three pathways downstream of TrkB signaling. Front. Synaptic Neurosci. 2014, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Wu, Y.; Wang, Y.; Zhu, J.; Chu, H.; Kong, L.; Yin, L.; Ma, H. Brain-Derived Neurotrophic Factor Increases Synaptic Protein Levels via the MAPK/Erk Signaling Pathway and Nrf2/Trx Axis Following the Transplantation of Neural Stem Cells in a Rat Model of Traumatic Brain Injury. Neurochem. Res. 2017, 42, 3073–3083. [Google Scholar] [CrossRef] [PubMed]
- Opazo, P.; Watabe, A.M.; Grant, S.G.N.; O’Dell, T.J. Phosphatidylinositol 3-Kinase Regulates the Induction of Long-Term Potentiation through Extracellular Signal-Related Kinase-Independent Mechanisms. J. Neurosci. 2003, 23, 3679–3688. [Google Scholar] [CrossRef]
- Kay, J.C.; Xia, C.-M.; Liu, M.; Shen, S.; Yu, S.J.; Chung, C.; Qiao, L.-Y. Endogenous PI3K/Akt and NMDAR act independently in the regulation of CREB activity in lumbosacral spinal cord in cystitis. Exp. Neurol. 2013, 250, 366–375. [Google Scholar] [CrossRef]
- Petersén, Å.; Larsen, K.E.; Behr, G.G.; Romero, N.; Przedborski, S.; Brundin, P.; Sulzer, D. Brain-derived neurotrophic factor inhibits apoptosis and dopamine-induced free radical production in striatal neurons but does not prevent cell death. Brain Res. Bull. 2001, 56, 331–335. [Google Scholar] [CrossRef]
- Patel, A.V.; Krimm, R.F. BDNF is required for the survival of differentiated geniculate ganglion neurons. Dev. Biol. 2010, 340, 419–429. [Google Scholar] [CrossRef][Green Version]
- Chen, A.; Xiong, L.-J.; Tong, Y.; Mao, M. Neuroprotective effect of brain-derived neurotrophic factor mediated by autophagy through the PI3K/Akt/mTOR pathway. Mol. Med. Rep. 2013, 8, 1011–1016. [Google Scholar] [CrossRef]
- Wu, C.-H.; Chen, C.-C.; Hung, T.-H.; Chuang, Y.-C.; Chao, M.; Shyue, S.-K.; Chen, S.-F. Activation of TrkB/Akt signaling by a TrkB receptor agonist improves long-term histological and functional outcomes in experimental intracerebral hemorrhage. J. Biomed. Sci. 2019, 26, 53. [Google Scholar] [CrossRef]
- Awad, B.I.; Carmody, M.A.; Steinmetz, M.P. Potential role of growth factors in the management of spinal cord injury. World Neurosurg. 2015, 83, 120–131. [Google Scholar] [CrossRef]
- Sampaio, T.; Savall, A.; Gutierrez, M.Z.; Pinton, S. Neurotrophic factors in Alzheimer’s and Parkinson’s diseases: Implications for pathogenesis and therapy. Neural Regen. Res. 2017, 12, 549–557. [Google Scholar] [PubMed]
- Pramanik, S.; Sulistio, Y.A.; Heese, K. Neurotrophin Signaling and Stem Cells-Implications for Neurodegenerative Diseases and Stem Cell Therapy. Mol. Neurobiol. 2017, 54, 7401–7459. [Google Scholar] [CrossRef]
- Zoladz, J.A.; Pilc, A. The effect of physical activity on the brain derived neurotrophic factor: From animal to human studies. J. Physiol. Pharmacol. 2010, 61, 533–541. [Google Scholar] [PubMed]
- Song, D.; Diao, J.; Yang, Y.; Chen, Y. MicroRNA-382 inhibits cell proliferation and invasion of retinoblastoma by targeting BDNF-mediated PI3K/AKT signalling pathway. Mol. Med. Rep. 2017, 16, 6428–6436. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.-M.; Lee, B.D.; Sok, D.-E.; Ma, J.Y.; Kim, M.R. Neuroprotective action of N-acetyl serotonin in oxidative stress-induced apoptosis through the activation of both TrkB/CREB/BDNF pathway and Akt/Nrf2/Antioxidant enzyme in neuronal cells. Redox Biol. 2017, 11, 592–599. [Google Scholar] [CrossRef]
- Porritt, M.J.; Batchelor, P.E.; Howells, D. Inhibiting BDNF expression by antisense oligonucleotide infusion causes loss of nigral dopaminergic neurons. Exp. Neurol. 2005, 192, 226–234. [Google Scholar] [CrossRef]
- Scalzo, P.; Kümmer, A.; Bretas, T.L.; Cardoso, F.; Teixeira, A.L. Serum levels of brain-derived neurotrophic factor correlate with motor impairment in Parkinson’s disease. J. Neurol. 2010, 257, 540–545. [Google Scholar] [CrossRef]
- Grah, M.; Mihanovic, M.; Ruljancic, N.; Restek-Petrovic, B.; Molnar, S.; Jelavic, S. Brain-derived neurotrophic factor as a suicide factor in mental disorders. Acta Neuropsychiatr. 2014, 26, 356–363. [Google Scholar] [CrossRef]
- Ventriglia, M.; Zanardini, R.; Bonomini, C.; Zanetti, O.; Volpe, D.; Pasqualetti, P.; Gennarelli, M.; Bocchio-Chiavetto, L. Serum Brain-Derived Neurotrophic Factor Levels in Different Neurological Diseases. Biomed Res. Int. 2013, 2013, 901082. [Google Scholar] [CrossRef]
- Howells, D.W.; Porritt, M.J.; Wong, J.Y.F.; Batchelor, P.E.; Kalnins, R.; Hughes, A.J.; Donnan, G.A. Reduced BDNF mRNA expression in the Parkinson’s disease substantia nigra. Exp. Neurol. 2000, 166, 127–135. [Google Scholar] [CrossRef]
- Lin, J.-G.; Chen, C.-J.; Yang, H.-B.; Chen, Y.-H.; Hung, S.-Y. Electroacupuncture Promotes Recovery of Motor Function and Reduces Dopaminergic Neuron Degeneration in Rodent Models of Parkinson’s disease. Int. J. Mol. Sci. 2017, 18, 1846. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, H.; Zhang, B.-S.; Soares, J.C.; Zhang, X.Y. Low BDNF is associated with cognitive impairments in patients with Parkinson’s disease. Parkinsonism Relat. Disord. 2016, 29, 66–71. [Google Scholar] [CrossRef]
- Huang, Y.; Yun, W.; Zhang, M.; Luo, W.; Zhou, X. Serum concentration and clinical significance of brain-derived neurotrophic factor in patients with Parkinson’s disease or essential tremor. J. Int. Med. Res. 2018, 46, 1477–1485. [Google Scholar] [CrossRef] [PubMed]
- Siuda, J.; Patalong-Ogiewa, M.; Żmuda, W.; Targosz-Gajniak, M.; Niewiadomska, E.; Matuszek, I.; Jędrzejowska-Szypułka, H.; Rudzińska-Bar, M. Cognitive impairment and BDNF serum levels. Neurol. Neurochir. Pol. 2017, 51, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Leyhe, T.; Eschweiler, G.W.; Stransky, E.; Gasser, T.; Annas, P.; Basun, H.; Laske, C. Increase of BDNF Serum Concentration in Lithium Treated Patients with Early Alzheimer’s Disease. J. Alzheimer’s Dis. 2009, 16, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Song, J.-H.; Yu, J.-T.; Tan, L. Brain-Derived Neurotrophic Factor in Alzheimer’s disease: Risk, Mechanisms, and Therapy. Mol. Neurobiol. 2015, 52, 1477–1493. [Google Scholar] [CrossRef]
- Kang, S.S.; Zhang, Z.; Liu, X.; Manfredsson, F.P.; Benskey, M.J.; Cao, X.; Xu, J.; Sun, Y.E.; Ye, K. TrkB neurotrophic activities are blocked by α-synuclein, triggering dopaminergic cell death in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, 10773–10778. [Google Scholar] [CrossRef]
- Fang, F.; Yang, W.; Florio, J.B.; Rockenstein, E.; Spencer, B.; Orain, X.M.; Dong, S.X.; Li, H.; Chen, X.; Sung, K.; et al. Synuclein impairs trafficking and signaling of BDNF in a mouse model of Parkinson’s disease. Sci. Rep. 2017, 7, 3868. [Google Scholar] [CrossRef]
- Katila, N.; Bhurtel, S.; Shadfar, S.; Srivastav, S.; Neupane, S.; Ojha, U.; Jeong, G.-S.; Choi, D.-Y. Metformin lowers α-synuclein phosphorylation and upregulates neurotrophic factor in the MPTP mouse model of Parkinson’s disease. Neuropharmacology 2017, 125, 396–407. [Google Scholar] [CrossRef]
- Baquet, Z.C.; Bickford, P.C.; Jones, K.R. Brain-Derived Neurotrophic Factor Is Required for the Establishment of the Proper Number of Dopaminergic Neurons in the Substantia Nigra Pars Compacta. J. Neurosci. 2005, 25, 6251–6259. [Google Scholar] [CrossRef]
- Baydyuk, M.; Nguyen, M.T.; Xu, B. Chronic deprivation of TrkB signaling leads to selective late-onset nigrostriatal dopaminergic degeneration. Exp. Neurol. 2011, 228, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Ziebell, M.; Khalid, U.; Klein, A.B.; Aznar, S.; Thomsen, G.; Jensen, P.; Knudsen, G.M. Striatal dopamine transporter binding correlates with serum BDNF levels in patients with striatal dopaminergic neurodegeneration. Neurobiol. Aging 2012, 33, 428.e1–428.e5. [Google Scholar] [CrossRef] [PubMed]
- Gerecke, K.M.; Jiao, Y.; Pani, A.; Pagala, V.; Smeyne, R.J. Exercise protects against MPTP-induced neurotoxicity in mice. Brain Res. 2010, 1341, 72–83. [Google Scholar] [CrossRef]
- Baker, S.A.; Stanford, L.E.; Brown, R.E.; Hagg, T. Maturation but not survival of dopaminergic nigrostriatal neurons is affected in developing and aging BDNF-deficient mice. Brain Res. 2005, 1039, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Pałasz, E.; Bąk, A.; Gąsiorowska, A.; Niewiadomska, G. The role of trophic factors and inflammatory processes in physical activity-induced neuroprotection in Parkinson’s disease. Postepy Hig. Med. Doswiadczalnej (Online) 2017, 71, 713–726. [Google Scholar] [CrossRef] [PubMed]
- Drinkut, A.; Tillack, K.; Meka, D.P.; Schulz, J.B.; Kügler, S.; Kramer, E.R. Ret is essential to mediate GDNF’s neuroprotective and neuroregenerative effect in a Parkinson disease mouse model. Cell Death Dis. 2016, 7, e2359. [Google Scholar] [CrossRef]
- Numakawa, T.; Suzuki, S.; Kumamaru, E.; Adachi, N.; Richards, M.; Kunugi, H. BDNF function and intracellular signaling in neurons. Histol. Histopathol. 2010, 25, 237–258. [Google Scholar] [PubMed]
- Kaur, R.; Mehan, S.; Singh, S. Understanding multifactorial architecture of Parkinson’s disease: Pathophysiology to management. Neurol. Sci. 2019, 40, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Andero, R.; Choi, D.C.; Ressler, K.J. BDNF-TrkB receptor regulation of distributed adult neural plasticity, memory formation, and psychiatric disorders. Prog. Mol. Biol. Transl. Sci. 2014, 122, 169–192. [Google Scholar]
- Park, H.; Kang, S.; Nam, E.; Suh, Y.-H.; Chang, K.-A. The Protective Effects of PSM-04 Against Beta Amyloid-Induced Neurotoxicity in Primary Cortical Neurons and an Animal Model of Alzheimer’s Disease. Front. Pharmacol. 2019, 10, 2. [Google Scholar] [CrossRef]
- Linnarsson, S.; Björklund, A.; Ernfors, P. Learning deficit in BDNF mutant mice. Eur. J. Neurosci. 1997, 9, 2581–2587. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Iacovitti, L. Multiple signaling pathways direct the initiation of tyrosine hydroxylase gene expression in cultured brain neurons. Mol. Brain Res. 1997, 50, 1–8. [Google Scholar] [CrossRef]
- Parain, K.; Murer, M.; Yan, Q.; Faucheux, B.; Agid, Y.; Hirsch, E.; Raisman-Vozari, R. Reduced expression of brain derived neurotrophic factor protein in Parkinson’s disease substantia nigra. Neuroreport 1999, 10, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Guillin, O.; Diaz, J.; Carroll, P.; Griffon, N.; Schwartz, J.-C.; Sokoloff, P. BDNF controls dopamine D3 receptor expression and triggers behavioural sensitization. Nature 2001, 411, 86–89. [Google Scholar] [CrossRef]
- Nagatsu, T.; Sawada, M. Biochemistry of postmortem brains in Parkinson’s disease: Historical overview and future prospects. J. Neural Transm. Suppl. 2007, 72, 113–120. [Google Scholar]
- Hung, H.C.; Lee, E.H.Y. The mesolimbic dopaminergic pathway is more resistant than the nigrostriatal dopaminergic pathway to MPTP and MPP+ toxicity: Role of BDNF gene expression. Mol. Brain Res. 1996, 41, 16–26. [Google Scholar] [CrossRef]
- Tsukahara, T.; Takeda, M.; Shimohama, S.; Ohara, O.; Hashimoto, N. Effects of Brain-derived Neurotrophic Factor on 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinsonism in Monkeys. Neurosurgery 1995, 37, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.L.; Lewis, M.H.; Muzyczka, N.; Meyer, E.M. Prevention of 6-hydroxydopamine-induced rotational behavior by BDNF somatic gene transfer. Brain Res. 1999, 847, 314–320. [Google Scholar] [CrossRef]
- Sun, M.; Kong, L.; Wang, X.; Lu, X.; Gao, Q.; Geller, A.I. Comparison of the capability of GDNF, BDNF, or both, to protect nigrostriatal neurons in a rat model of Parkinson’s disease. Brain Res. 2005, 1052, 119–129. [Google Scholar] [CrossRef]
- Kim, S.R.; Kareva, T.; Yarygina, O.; Kholodilov, N.; Burke, R.E. AAV transduction of dopamine neurons with constitutively active rheb protects from neurodegeneration and mediates axon regrowth. Mol. Ther. 2012, 20, 275–286. [Google Scholar] [CrossRef]
- Nam, J.H.; Leem, E.; Jeon, M.T.; Jeong, K.H.; Park, J.W.; Jung, U.J.; Kholodilov, N.; Burke, R.E.; Jin, B.K.; Kim, S.R. Induction of GDNF and BDNF by hRheb(S16H) Transduction of SNpc Neurons: Neuroprotective Mechanisms of hRheb(S16H) in a Model of Parkinson’s Disease. Mol. Neurobiol. 2015, 51, 487–499. [Google Scholar] [CrossRef]
- Tronci, E.; Napolitano, F.; Muñoz, A.; Fidalgo, C.; Rossi, F.; Björklund, A.; Usiello, A.; Carta, M. BDNF over-expression induces striatal serotonin fiber sprouting and increases the susceptibility to L-DOPA-induced dyskinesia in 6-OHDA-lesioned rats. Exp. Neurol. 2017, 297, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Chan, N.G.; Bannon, M.J.; Orozco-Barrios, C.E.; Escobedo, L.; Zamudio, S.; De La Cruz, F.; Gongora-Alfaro, J.L.; Armendáriz-Borunda, J.; Reyes-Corona, D.; Espadas-Alvarez, A.J.; et al. Neurotensin-polyplex-mediated brain-derived neurotrophic factor gene delivery into nigral dopamine neurons prevents nigrostriatal degeneration in a rat model of early Parkinson’s disease. J. Biomed. Sci. 2015, 22. [Google Scholar] [CrossRef] [PubMed]
- Razgado-Hernandez, L.F.; Espadas-Alvarez, A.J.; Reyna-Velazquez, P.; Sierra-Sanchez, A.; Anaya-Martinez, V.; Jimenez-Estrada, I.; Bannon, M.J.; Martinez-Fong, D.; Aceves-Ruiz, J. The transfection of BDNF to dopamine neurons potentiates the effect of dopamine D3 receptor agonist recovering the striatal innervation, dendritic spines and motor behavior in an aged rat model of Parkinson’s disease. PLoS ONE 2015, 10, e0117391. [Google Scholar] [CrossRef] [PubMed]
- Lucidi-Phillipi, C.A.; Gage, F.H.; Shults, C.W.; Jones, K.R.; Reichardt, L.F.; Kang, U.J. Brain-derived neurotrophic factor-transduced fibroblasts: Production of BDNF and effects of grafting to the adult rat brain. J. Comp. Neurol. 1995, 354, 361–376. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, Y.; Lin, Q.; Collier, T.J.; Frim, D.M.; Breakefield, X.O.; Bohn, M.C. Astrocytes retrovirally transduced with BDNF elicit behavioral improvement in a rat model of Parkinson’s disease. Brain Res. 1995, 691, 25–36. [Google Scholar] [CrossRef]
- Frim, D.M.; Uhler, T.A.; Galpern, W.R.; Beal, M.F.; Breakefield, X.O.; Isacson, O. Implanted fibroblasts genetically engineered to produce brain-derived neurotrophic factor prevent 1-methyl-4-phenylpyridinium toxicity to dopaminergic neurons in the rat. Proc. Natl. Acad. Sci. USA 1994, 91, 5104–5108. [Google Scholar] [CrossRef]
- Galpern, W.R.; Frim, D.M.; Tatter, S.B.; Altar, C.A.; Beal, M.F.; Isacson, O. Cell-mediated delivery of brain-derived neurotrophic factor enhances dopamine levels in an MPP+ rat model of substantia nigra degeneration. Cell Transplant. 1996, 5, 225–232. [Google Scholar] [CrossRef]
- Levivier, M.; Przedborski, S.; Bencsics, C.; Kang, U.J. Intrastriatal implantation of fibroblasts genetically engineered to produce brain-derived neurotrophic factor prevents degeneration of dopaminergic neurons in a rat model of Parkinson’s disease. J. Neurosci. 1995, 15, 7810–7820. [Google Scholar] [CrossRef]
- Zhu, G.; Li, J.; He, L.; Wang, X.; Hong, X. MPTP-induced changes in hippocampal synaptic plasticity and memory are prevented by memantine through the BDNF-TrkB pathway. Br. J. Pharmacol. 2015, 172, 2354–2368. [Google Scholar] [CrossRef]
- Kinoshita, K.I.; Muroi, Y.; Unno, T.; Ishii, T. Rolipram improves facilitation of contextual fear extinction in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced mouse model of Parkinson’s disease. J. Pharmacol. Sci. 2017, 134, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Nagahara, A.H.; Tuszynski, M.H. Potential therapeutic uses of BDNF in neurological and psychiatric disorders. Nat. Rev. Drug Discov. 2011, 10, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Bradley, W.G. A controlled trial of recombinant methionyl human BDNF in ALS. Neurology 1999, 52, 1427–1433. [Google Scholar]
- Ochs, G.; Penn, R.D.; York, M.; Giess, R.; Beck, M.; Tonn, J.; Haigh, J.; Malta, E.; Traub, M.; Sendtner, M.; et al. A phase I/II trial of recombinant methionyl human brain derived neurotrophic factor administered by intrathecal infusion to patients with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2000, 1, 201–206. [Google Scholar] [CrossRef]
- Gill, S.S.; Patel, N.K.; Hotton, G.R.; O’Sullivan, K.; McCarter, R.; Bunnage, M.; Brooks, D.J.; Svendsen, C.N.; Heywood, P. Direct brain infusion of glial cell line–derived neurotrophic factor in Parkinson disease. Nat. Med. 2003, 9, 589–595. [Google Scholar] [CrossRef]
- Slevin, J.T.; Gash, D.M.; Smith, C.D.; Gerhardt, G.A.; Kryscio, R.; Chebrolu, H.; Walton, A.; Wagner, R.; Young, A.B. Unilateral intraputamenal glial cell line–derived neurotrophic factor in patients with Parkinson disease: Response to 1 year of treatment and 1 year of withdrawal. J. Neurosurg. 2007, 106, 614–620. [Google Scholar] [CrossRef]
- Kordower, J.H.; Palfi, S.; Chen, E.-Y.; Ma, S.Y.; Sendera, T.; Cochran, E.J.; Mufson, E.J.; Penn, R.; Goetz, C.G.; Comella, C.D. Clinicopathological findings following intraventricular glial-derived neurotrophic factor treatment in a patient with Parkinson’s disease. Ann. Neurol. 1999, 46, 419–424. [Google Scholar] [CrossRef]
- Whone, A.; Luz, M.; Boca, M.; Woolley, M.; Mooney, L.; Dharia, S.; Broadfoot, J.; Cronin, D.; Schroers, C.; Barua, N.U.; et al. Randomized trial of intermittent intraputamenal glial cell line-derived neurotrophic factor in Parkinson’s disease. Brain 2019, 142, 512–525. [Google Scholar] [CrossRef]
- Géral, C.; Angelova, A.; Lesieur, S. From Molecular to Nanotechnology Strategies for Delivery of Neurotrophins: Emphasis on Brain-Derived Neurotrophic Factor (BDNF). Pharmaceutics 2013, 5, 127–167. [Google Scholar] [CrossRef]
- Zhao, H.; Alam, A.; San, C.-Y.; Eguchi, S.; Chen, Q.; Lian, Q.; Ma, D. Molecular mechanisms of brain-derived neurotrophic factor in neuro-protection: Recent developments. Brain Res. 2017, 1665, 1–21. [Google Scholar] [CrossRef]
- Denyer, R.; Douglas, M.R. Gene Therapy for Parkinson’s disease. Parkinsons Dis. 2012, 2012, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Bunker, D.L.J. Delivery Techniques in Gene Therapy: A Brief Overview. J. Phys. Chem. Biophys. 2014, 4. [Google Scholar] [CrossRef]
- Bjorklund, T.; Kordower, J.H. Gene therapy for Parkinson’s disease. Mov. Disord. 2010, 25, S161–S173. [Google Scholar] [CrossRef] [PubMed]
- Bemelmans, A.-P.; Horellou, P.; Pradier, L.; Brunet, I.; Colin, P.; Mallet, J. Brain-Derived Neurotrophic Factor-Mediated Protection of Striatal Neurons in an Excitotoxic Rat Model of Huntington’s Disease, as Demonstrated by Adenoviral Gene Transfer. Hum. Gene Ther. 1999, 10, 2987–2997. [Google Scholar] [CrossRef] [PubMed]
- Kells, A.P.; Fong, D.M.; Dragunow, M.; During, M.J.; Young, D.; Connor, B. AAV-Mediated gene delivery of BDNF or GDNF is neuroprotective in a model of huntington disease. Mol. Ther. 2004, 9, 682–688. [Google Scholar] [CrossRef] [PubMed]
- Paradiso, B.; Marconi, P.; Zucchini, S.; Berto, E.; Binaschi, A.; Bozac, A.; Buzzi, A.; Mazzuferi, M.; Magri, E.; Mora, G.N.; et al. Localized delivery of fibroblast growth factor–2 and brain-derived neurotrophic factor reduces spontaneous seizures in an epilepsy model. Proc. Natl. Acad. Sci. USA 2009, 106, 7191–7196. [Google Scholar] [CrossRef]
- Nagahara, A.H.; Merrill, D.A.; Coppola, G.; Tsukada, S.; Schroeder, B.E.; Shaked, G.M.; Wang, L.; Blesch, A.; Kim, A.; Conner, J.M.; et al. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat. Med. 2009, 15, 331–337. [Google Scholar] [CrossRef]
- Majláth, Z.; Török, N.; Toldi, J.; Vécsei, L. Promising therapeutic agents for the treatment of Parkinson’s disease. Expert Opin. Biol. Ther. 2016, 16, 787–799. [Google Scholar] [CrossRef][Green Version]
- Kirik, D.; Cederfjäll, E.; Halliday, G. Petersén Gene therapy for Parkinson’s disease: Disease modification by GDNF family of ligands. Neurobiol. Dis. 2017, 97, 179–188. [Google Scholar] [CrossRef]
- Valles, F.; Fiandaca, M.S.; Eberling, J.L.; Starr, P.A.; Larson, P.S.; Christine, C.W.; Forsayeth, J.; Richardson, R.M.; Su, X.; Aminoff, M.J.; et al. Qualitative imaging of adeno-associated virus serotype 2-human aromatic L-amino acid decarboxylase gene therapy in a phase i study for the treatment of parkinson disease. Neurosurgery 2010, 67, 1377–1385. [Google Scholar] [CrossRef]
- Marks, W.J.; Ostrem, J.L.; Verhagen, L.; Starr, P.A.; Larson, P.S.; Bakay, R.A.; Taylor, R.; Cahn-Weiner, D.A.; Stoessl, A.J.; Olanow, C.W.; et al. Safety and tolerability of intraputaminal delivery of CERE-120 (adeno-associated virus serotype 2–neurturin) to patients with idiopathic Parkinson’s disease: An open-label, phase I trial. Lancet Neurol. 2008, 7, 400–408. [Google Scholar] [CrossRef]
- Marks, W.J.; Bartus, R.T.; Siffert, J.; Davis, C.S.; Lozano, A.; Boulis, N.; Vitek, J.; Stacy, M.; Turner, D.; Verhagen, L.; et al. Gene delivery of AAV2-neurturin for Parkinson’s disease: A double-blind, randomised, controlled trial. Lancet Neurol. 2010, 9, 1164–1172. [Google Scholar] [CrossRef]
- Palasz, E.; Niewiadomski, W.; Gasiorowska, A.; Wysocka, A.; Stepniewska, A.; Niewiadomska, G. Exercise-Induced Neuroprotection and Recovery of Motor Function in Animal Models of Parkinson’s Disease. Front. Neurol. 2019, 10, 1143. [Google Scholar] [CrossRef] [PubMed]
- Lau, Y.-S.; Patki, G.; Das-Panja, K.; Le, W.-D.; Ahmad, S.O. Neuroprotective effects and mechanisms of exercise in a chronic mouse model of Parkinson’s disease with moderate neurodegeneration. Eur. J. Neurosci. 2011, 33, 1264–1274. [Google Scholar] [CrossRef] [PubMed]
- Tajiri, N.; Yasuhara, T.; Shingo, T.; Kondo, A.; Yuan, W.; Kadota, T.; Wang, F.; Baba, T.; Tayra, J.T.; Morimoto, T.; et al. Exercise exerts neuroprotective effects on Parkinson’s disease model of rats. Brain Res. 2010, 1310, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Real, C.C.; Ferreira, A.F.B.; Chaves-Kirsten, G.P.; Torrão, A.S.; Pires, R.S.; Britto, L.R.G. BDNF receptor blockade hinders the beneficial effects of exercise in a rat model of Parkinson’s disease. Neuroscience 2013, 237, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-Y.; Wang, T.-F.; Yu, L.; Jen, C.J.; Chuang, J.-I.; Wu, F.-S.; Wu, C.-W.; Kuo, Y.-M. Running exercise protects the substantia nigra dopaminergic neurons against inflammation-induced degeneration via the activation of BDNF signaling pathway. Brain Behav. Immun. 2011, 25, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; He, L.X.; Huang, S.N.; Gong, L.J.; Li, L.; Lv, Y.Y.; Qian, Z.M. Protection of dopamine neurons by vibration training and up-regulation of brain-derived neurotrophic factor in a MPTP mouse model of Parkinson’s disease. Physiol. Res. 2014, 63, 649–657. [Google Scholar] [PubMed]
- Tuon, T.; Valvassori, S.S.; Dal Pont, G.C.; Paganini, C.S.; Pozzi, B.G.; Luciano, T.F.; Souza, P.S.; Quevedo, J.; Souza, C.T.; Pinho, R.A. Physical training prevents depressive symptoms and a decrease in brain-derived neurotrophic factor in Parkinson’s disease. Brain Res. Bull. 2014, 108, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Fredriksson, A.; Stigsdotter, I.M.; Hurtig, A.; Ewalds-Kvist, B.; Archer, T. Running wheel activity restores MPTP-induced functional deficits. J. Neural Transm. 2011, 118, 407–420. [Google Scholar] [CrossRef]
- Da Costa, R.O.; Gadelha-Filho, C.V.J.; da Costa, A.E.M.; Feitosa, M.L.; de Araújo, D.P.; de Lucena, J.D.; de Aquino, P.E.A.; Lima, F.A.V.; Neves, K.R.T.; de Barros Viana, G.S. The Treadmill Exercise Protects against Dopaminergic Neuron Loss and Brain Oxidative Stress in Parkinsonian Rats. Oxidative Med. Cell. Longev. 2017, 2017, 2138169. [Google Scholar] [CrossRef] [PubMed]
- Choe, M.-A.; Koo, B.-S.; An, G.J.; Jeon, S. Effects of Treadmill Exercise on the Recovery of Dopaminergic Neuron Loss and Muscle Atrophy in the 6-OHDA Lesioned Parkinson’s Disease Rat Model. Korean J. Physiol. Pharmacol. 2012, 16, 305–312. [Google Scholar] [CrossRef]
- Elsworth, J.D.; Roth, R.H. Dopamine synthesis, uptake, metabolism, and receptors: Relevance to gene therapy of Parkinson’s disease. Exp. Neurol. 1997, 144, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, D.S.; Holmes, C.; Bentho, O.; Sato, T.; Moak, J.; Sharabi, Y.; Imrich, R.; Conant, S.; Eldadah, B.A. Biomarkers to detect central dopamine deficiency and distinguish Parkinson disease from multiple system atrophy. Park. Relat. Disord. 2008, 14, 600–607. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Ebina, J.; Kawabe, K.; Iwasaki, Y. Dopamine transporter imaging in parkinson disease: Progressive changes and therapeutic modification after anti-parkinsonian medications. Intern. Med. 2019, 58, 1665–1672. [Google Scholar] [CrossRef]
- Chen, G.; Bower, K.A.; Ma, C.; Fang, S.; Thiele, C.J.; Luo, J. Glycogen synthase kinase 3β (GSK3β) mediates 6-hydroxydopamine-induced neuronal death. FASEB J. 2004, 18, 1162–1164. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yang, Y.; Ying, C.; Li, W.; Ruan, H.; Zhu, X.; You, Y.; Han, Y.; Chen, R.; Wang, Y.; et al. Inhibition of glycogen synthase kinase-3β protects dopaminergic neurons from MPTP toxicity. Neuropharmacology 2007, 52, 1678–1684. [Google Scholar] [CrossRef]
- Xie, C.; Lin, J.-Y.; Wang, M.-H.; Zhang, Y.; Zhang, S.; Wang, X.-J.; Liu, Z.-G. Inhibition of Glycogen Synthase Kinase-3β (GSK-3β) as potent therapeutic strategy to ameliorates L-dopa-induced dyskinesia in 6-OHDA parkinsonian rats. Sci. Rep. 2016, 6, 23527. [Google Scholar] [CrossRef]
- Petit-Paitel, A.; Brau, F.; Cazareth, J.; Chabry, J. Involvment of Cytosolic and Mitochondrial GSK-3β in Mitochondrial Dysfunction and Neuronal Cell Death of MPTP/MPP+-Treated Neurons. PLoS ONE 2009, 4, e5491. [Google Scholar] [CrossRef]
- Pérez-Sen, R.; Ortega, F.; Morente, V.; Delicado, E.G.; Miras-Portugal, M.T. P2X7, NMDA and BDNF receptors converge on GSK3 phosphorylation and cooperate to promote survival in cerebellar granule neurons. Cell. Mol. Life Sci. 2010, 67, 1723–1733. [Google Scholar]
- Gerecke, K.M.; Jiao, Y.; Pagala, V.; Smeyne, R.J. Exercise Does Not Protect against MPTP-Induced Neurotoxicity in BDNF Happloinsufficent Mice. PLoS ONE 2012, 7, e43250. [Google Scholar] [CrossRef]
- Paillard, T.; Rolland, Y.; de Souto Barreto, P. Protective Effects of Physical Exercise in Alzheimer’s disease and Parkinson’s disease: A Narrative Review. J. Clin. Neurol. 2015, 11, 212–219. [Google Scholar] [CrossRef]
- Seifert, T.; Brassard, P.; Wissenberg, M.; Rasmussen, P.; Nordby, P.; Stallknecht, B.; Adser, H.; Jakobsen, A.H.; Pilegaard, H.; Nielsen, H.B.; et al. Endurance training enhances BDNF release from the human brain. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R372–R377. [Google Scholar] [CrossRef]
- Conradsson, D.; Löfgren, N.; Nero, H.; Hagströmer, M.; Ståhle, A.; Lökk, J.; Franzén, E. The Effects of Highly Challenging Balance Training in Elderly with Parkinson’s disease: A Randomized Controlled Trial. Neurorehabilit. Neural Repair 2015, 29, 827–836. [Google Scholar] [CrossRef]
- Uhrbrand, A.; Stenager, E.; Pedersen, M.S.; Dalgas, U. Parkinson’s disease and intensive exercise therapy—A systematic review and meta-analysis of randomized controlled trials. J. Neurol. Sci. 2015, 353, 9–19. [Google Scholar] [CrossRef]
- Lamotte, G.; Rafferty, M.R.; Prodoehl, J.; Kohrt, W.M.; Comella, C.L.; Simuni, T.; Corcos, D.M. Effects of Endurance Exercise Training on The Motor and Non-Motor Features of Parkinson’s Disease: A Review. J. Parkinsons Dis. 2015, 5, 21–41. [Google Scholar] [CrossRef] [PubMed]
- Santos, S.M.; da Silva, R.A.; Terra, M.B.; Almeida, I.A.; de Melo, L.B.; Ferraz, H.B. Balance versus resistance training on postural control in patients with Parkinson’s disease: A randomized controlled trial. Eur. J. Phys. Rehabil. Med. 2017, 53, 173–183. [Google Scholar] [PubMed]
- O’Callaghan, A.; Harvey, M.; Houghton, D.; Gray, W.K.; Weston, K.L.; Oates, L.L.; Romano, B.; Walker, R.W. Comparing the influence of exercise intensity on brain-derived neurotrophic factor serum levels in people with Parkinson’s disease: A pilot study. Aging Clin. Exp. Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, B.; Barbieri, F.A.; Arthuso, F.Z.; Silva, F.A.; Moretto, G.F.; Imaizumi, L.F.I.; Ngomane, A.Y.; Guimarães, G.V.; Ciolac, E.G. High-Intensity Interval Versus Moderate-Intensity Continuous Training in Individuals With Parkinson’s Disease: Hemodynamic and Functional Adaptation. J. Phys. Act. Health 2019, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Fiorelli, C.M.; Ciolac, E.G.; Simieli, L.; Silva, F.A.; Fernandes, B.; Christofoletti, G.; Barbieri, F.A. Differential Acute Effect of High-Intensity Interval or Continuous Moderate Exercise on Cognition in Individuals With Parkinson’s Disease. J. Phys. Act. Health 2019, 16, 157–164. [Google Scholar] [CrossRef]
- Marusiak, J.; Zeligowska, E.; Mencel, J.; Kisiel-Sajewicz, K.; Majerczak, J.; Zoladz, J.A.; Jaskólski, A.; Jaskólska, A. Interval training-induced alleviation of rigidity and hypertonia in patients with Parkinson’s disease is accompanied by increased basal serum brain-derived neurotrophic factor: A repeated-measures, case series pilot study. J. Rehabil. Med. 2015, 47, 372–375. [Google Scholar] [CrossRef]
- Santana-Sosa, E.; Barriopedro, M.; López-Mojares, L.; Pérez, M.; Lucia, A. Exercise Training is Beneficial for Alzheimer’s Patients. Int. J. Sports Med. 2008, 29, 845–850. [Google Scholar] [CrossRef] [PubMed]
- Brienesse, L.A.; Emerson, M.N. Effects of Resistance Training for People with Parkinson’s disease: A Systematic Review. J. Am. Med. Dir. Assoc. 2013, 14, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Bressel, E.; Wing, J.E.; Miller, A.I.; Dolny, D.G. High-Intensity Interval Training on an Aquatic Treadmill in Adults with Osteoarthritis. J. Strength Cond. Res. 2014, 28, 2088–2096. [Google Scholar] [CrossRef] [PubMed]
- Skriver, K.; Roig, M.; Lundbye-Jensen, J.; Pingel, J.; Helge, J.W.; Kiens, B.; Nielsen, J.B. Acute exercise improves motor memory: Exploring potential biomarkers. Neurobiol. Learn. Mem. 2014, 116, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Filus, J.; Rybakowski, J. Serum BDNF levels and intensity of depressive symptoms. Neuropsychiatr. Neuropsychol. 2010, 5, 155–162. [Google Scholar]
- Piotrowicz, Z.; Czuba, M.; Langfort, J.; Chalimoniuk, M. Alterations in serum BDNF and catecholamines during exercise to volitional exhaustion—The influence of normobaric hypoxia and endurance training. Folia Neuropathol. 2017, 55, 173. [Google Scholar]
- Knaepen, K.; Goekint, M.; Heyman, E.M.; Meeusen, R. Neuroplasticity—Exercise-Induced Response of Peripheral Brain-Derived Neurotrophic Factor. Sports Med. 2010, 40, 765–801. [Google Scholar] [CrossRef]
- Sakuma, K.; Yamaguchi, A. The Recent Understanding of the Neurotrophin’s Role in Skeletal Muscle Adaptation. J. Biomed. Biotechnol. 2011, 2011, 201696. [Google Scholar] [CrossRef]
- Ahlskog, J.E. Does vigorous exercise have a neuroprotective effect in Parkinson disease? Neurology 2011, 77, 288–294. [Google Scholar] [CrossRef]
- Liu, P.Z.; Nusslock, R. Exercise-Mediated Neurogenesis in the Hippocampus via BDNF. Front. Neurosci. 2018, 12, 52. [Google Scholar] [CrossRef] [PubMed]
- Schmolesky, M.T.; Webb, D.L.; Hansen, R.A. The effects of aerobic exercise intensity and duration on levels of brain-derived neurotrophic factor in healthy men. J. Sports Sci. Med. 2013, 12, 502–511. [Google Scholar] [PubMed]
- Zhang, J.; Sokal, I.; Peskind, E.R.; Quinn, J.F.; Jankovic, J.; Kenney, C.; Chung, K.A.; Millard, S.P.; Nutt, J.G.; Montine, T.J. CSF Multianalyte Profile Distinguishes Alzheimer and Parkinson Diseases. Am. J. Clin. Pathol. 2008, 129, 526–529. [Google Scholar] [CrossRef]
- Salehi, Z.; Mashayekhi, F. Brain-derived neurotrophic factor concentrations in the cerebrospinal fluid of patients with Parkinson’s disease. J. Clin. Neurosci. 2009, 16, 90–93. [Google Scholar] [CrossRef]
- Hirsch, M.A.; van Wegen, E.E.H.; Newman, M.A.; Heyn, P.C. Exercise-induced increase in brain-derived neurotrophic factor in human Parkinson’s disease: A systematic review and meta-analysis. Transl. Neurodegener. 2018, 7. [Google Scholar] [CrossRef]
- Matthews, V.B.; Åström, M.-B.; Chan, M.H.S.; Bruce, C.R.; Krabbe, K.S.; Prelovsek, O.; Åkerström, T.; Yfanti, C.; Broholm, C.; Mortensen, O.H.; et al. Brain-derived neurotrophic factor is produced by skeletal muscle cells in response to contraction and enhances fat oxidation via activation of AMP-activated protein kinase. Diabetologia 2009, 52, 1409–1418. [Google Scholar] [CrossRef]
- Pratesi, A. Skeletal muscle: An endocrine organ. Clin. Cases Miner. Bone Metab. 2013, 10, 11–14. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, B.; Fei, A. BDNF contributes to the skeletal muscle anti-atrophic effect of exercise training through AMPK-PGC1α signaling in heart failure mice. Arch. Med. Sci. 2019, 15, 214–222. [Google Scholar] [CrossRef]
- Pareja-Galeano, H.; Alis, R.; Sanchis-Gomar, F.; Cabo, H.; Cortell-Ballester, J.; Gomez-Cabrera, M.C.; Lucia, A.; Viña, J. Methodological considerations to determine the effect of exercise on brain-derived neurotrophic factor levels. Clin. Biochem. 2015, 48, 162–166. [Google Scholar] [CrossRef]
- Kallies, G.; Rapp, M.A.; Fydrich, T.; Fehm, L.; Tschorn, M.; Terán, C.; Schwefel, M.; Pietrek, A.; Henze, R.; Hellweg, R.; et al. Serum brain-derived neurotrophic factor (BDNF) at rest and after acute aerobic exercise in major depressive disorder. Psychoneuroendocrinology 2019, 102, 212–215. [Google Scholar] [CrossRef]
- Walsh, J.J.; Tschakovsky, M.E. Exercise and circulating BDNF: Mechanisms of release and implications for the design of exercise interventions. Appl. Physiol. Nutr. Metab. 2018, 43, 1095–1104. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, P.; Brassard, P.; Adser, H.; Pedersen, M.V.; Leick, L.; Hart, E.; Secher, N.H.; Pedersen, B.K.; Pilegaard, H. Evidence for a release of brain-derived neurotrophic factor from the brain during exercise. Exp. Physiol. 2009, 94, 1062–1069. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.B.; Williamson, R.; Santini, M.A.; Clemmensen, C.; Ettrup, A.; Rios, M.; Knudsen, G.M.; Aznar, S. Blood BDNF concentrations reflect brain-tissue BDNF levels across species. Int. J. Neuropsychopharmacol. 2011, 14, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, G.; Lira, C.M.; Johansson, J.; Wisén, A.; Wohlfart, B.; Ekman, R.; Westrin, Å. The acute response of plasma brain-derived neurotrophic factor as a result of exercise in major depressive disorder. Psychiatry Res. 2009, 169, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Rojas Vega, S.; Strüder, H.K.; Vera Wahrmann, B.; Schmidt, A.; Bloch, W.; Hollmann, W. Acute BDNF and cortisol response to low intensity exercise and following ramp incremental exercise to exhaustion in humans. Brain Res. 2006, 1121, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Zoladz, J.A.; Majerczak, J.; Zeligowska, E.; Mencel, J.; Jaskolski, A.; Jaskolska, A.; Marusiak, J. Moderate-intensity interval training increases serum brain-derived neurotrophic factor level and decreases inflammation in Parkinson’s disease patients. J. Physiol. Pharmacol. 2014, 65, 441–448. [Google Scholar]
- Palasz, E.; Niewiadomski, W.; Gasiorowska, A.; Mietelska-Porowska, A.; Niewiadomska, G. Neuroplasticity and Neuroprotective Effect of Treadmill Training in the Chronic Mouse Model of Parkinson’s Disease. Neural Plast. 2019, 2019, 8215017. [Google Scholar] [CrossRef]
- Fontanesi, C.; Kvint, S.; Frazzitta, G.; Bera, R.; Ferrazzoli, D.; Di Rocco, A.; Rebholz, H.; Friedman, E.; Pezzoli, G.; Quartarone, A.; et al. Intensive Rehabilitation Enhances Lymphocyte BDNF-TrkB Signaling in Patients With Parkinson’s Disease. Neurorehabilit. Neural Repair 2016, 30, 411–418. [Google Scholar] [CrossRef]
- Da Silva, P.G.C.; Domingues, D.D.; De Carvalho, L.A.; Allodi, S.; Correa, C.L. Neurotrophic factors in Parkinson’s disease are regulated by exercise: Evidence-based practice. J. Neurol. Sci. 2016, 363, 5–15. [Google Scholar] [CrossRef]
- Gómez-Pinilla, F.; Ying, Z.; Opazo, P.; Roy, R.R.; Edgerton, V.R. Differential regulation by exercise of BDNF and NT-3 in rat spinal cord and skeletal muscle. Eur. J. Neurosci. 2001, 13, 1078–1084. [Google Scholar] [CrossRef]
- Fahimi, A.; Baktir, M.A.; Moghadam, S.; Mojabi, F.S.; Sumanth, K.; McNerney, M.W.; Ponnusamy, R.; Salehi, A. Physical exercise induces structural alterations in the hippocampal astrocytes: Exploring the role of BDNF-TrkB signaling. Brain Struct. Funct. 2017, 222, 1797–1808. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.-S.; Shin, M.-S.; Song, W.; Jun, T.-W.; Lim, B.-V.; Kim, Y.-P.; Kim, C.-J. Treadmill exercise alleviates short-term memory impairment in 6-hydroxydopamine-induced Parkinson’s rats. J. Exerc. Rehabil. 2013, 9, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Sung, Y.-H. Effects of treadmill exercise on hippocampal neurogenesis in an MPTP/probenecid-induced Parkinson’s disease mouse model. J. Phys. Ther. Sci. 2015, 27, 3203–3206. [Google Scholar] [CrossRef]
- Zheng, W.-H.; Quirion, R. Comparative signaling pathways of insulin-like growth factor-1 and brain-derived neurotrophic factor in hippocampal neurons and the role of the PI3 kinase pathway in cell survival. J. Neurochem. 2004, 89, 844–852. [Google Scholar] [CrossRef] [PubMed]
- Wrann, C.D.; White, J.P.; Salogiannnis, J.; Laznik-Bogoslavski, D.; Wu, J.; Ma, D.; Lin, J.D.; Greenberg, M.E.; Spiegelman, B.M. Exercise Induces Hippocampal BDNF through a PGC-1α/FNDC5 Pathway. Cell Metab. 2013, 18, 649–659. [Google Scholar] [CrossRef]
- Jodeiri Farshbaf, M.; Ghaedi, K.; Megraw, T.L.; Curtiss, J.; Shirani Faradonbeh, M.; Vaziri, P.; Nasr-Esfahani, M.H. Does PGC1α/FNDC5/BDNF Elicit the Beneficial Effects of Exercise on Neurodegenerative Disorders? Neuromol. Med. 2016, 18, 1–15. [Google Scholar] [CrossRef]
- Pyrzak, B.; Demkow, U.; Kucharska, A.M. Brown Adipose Tissue and Browning Agents: Irisin and FGF21 in the Development of Obesity in Children and Adolescents. Adv. Exp. Med. Biol. 2015, 866, 25–34. [Google Scholar]
- Natalicchio, A.; Marrano, N.; Biondi, G.; Dipaola, L.; Spagnuolo, R.; Cignarelli, A.; Perrini, S.; Laviola, L.; Giorgino, F. Irisin Increases the Expression of Anorexigenic and Neurotrophic Genes in Mouse Brain. Diabetes Metab. Res. Rev. 2019, 19, e3238. [Google Scholar] [CrossRef]
- Guo, Z.; Du, X.; Iacovitti, L. Regulation of tyrosine hydroxylase gene expression during transdifferentiation of striatal neurons: Changes in transcription factors binding the AP-1 site. J. Neurosci. 1998, 18, 8163–8174. [Google Scholar] [CrossRef]
- Nagamoto-Combs, K.; Piech, K.M.; Best, J.A.; Sun, B.; Tank, A.W. Tyrosine Hydroxylase Gene Promoter Activity Is Regulated by Both Cyclic AMP-responsive Element and AP1 Sites following Calcium Influx. J. Biol. Chem. 1997, 272, 6051–6058. [Google Scholar] [CrossRef]
- He, X.; Yang, S.; Zhang, R.; Hou, L.; Xu, J.; Hu, Y.; Xu, R.; Wang, H.; Zhang, Y. Smilagenin Protects Dopaminergic Neurons in Chronic MPTP/Probenecid—Lesioned Parkinson’s Disease Models. Front. Cell. Neurosci. 2019, 13, 18. [Google Scholar] [CrossRef] [PubMed]
- Rangasamy, S.B.; Dasarathi, S.; Pahan, P.; Jana, M.; Pahan, K. Low-Dose Aspirin Upregulates Tyrosine Hydroxylase and Increases Dopamine Production in Dopaminergic Neurons: Implications for Parkinson’s disease. J. Neuroimmune Pharmacol. 2019, 14, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Vaynman, S.; Ying, Z.; Gomez-Pinilla, F. Interplay between brain-derived neurotrophic factor and signal transduction modulators in the regulation of the effects of exercise on synaptic-plasticity. Neuroscience 2003, 122, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Rabie, M.A.; Abd El Fattah, M.A.; Nassar, N.N.; El-Abhar, H.S.; Abdallah, D.M. Angiotensin 1–7 ameliorates 6-hydroxydopamine lesions in hemiparkinsonian rats through activation of MAS receptor/PI3K/Akt/BDNF pathway and inhibition of angiotensin II type-1 receptor/NF-κB axis. Biochem. Pharmacol. 2018, 151, 126–134. [Google Scholar] [CrossRef] [PubMed]
- McMorris, T.; Collard, K.; Corbett, J.; Dicks, M.; Swain, J.P. A test of the catecholamines hypothesis for an acute exercise-cognition interaction. Pharmacol. Biochem. Behav. 2008, 89, 106–115. [Google Scholar] [CrossRef] [PubMed]
- McMorris, T. Developing the catecholamines hypothesis for the acute exercise-cognition interaction in humans: Lessons from animal studies. Physiol. Behav. 2016, 165, 291–299. [Google Scholar] [CrossRef]
- Ma, Q. Beneficial effects of moderate voluntary physical exercise and its biological mechanisms on brain health. Neurosci. Bull. 2008, 24, 265–270. [Google Scholar] [CrossRef]
- Lopez-Alvarez, V.M.; Puigdomenech, M.; Navarro, X.; Cobianchi, S. Monoaminergic descending pathways contribute to modulation of neuropathic pain by increasing-intensity treadmill exercise after peripheral nerve injury. Exp. Neurol. 2018, 299, 42–55. [Google Scholar] [CrossRef]
- Huat, T.J.; Khan, A.A.; Pati, S.; Mustafa, Z.; Abdullah, J.M.; Jaafar, H. IGF-1 enhances cell proliferation and survival during early differentiation of mesenchymal stem cells to neural progenitor-like cells. BMC Neurosci. 2014, 15. [Google Scholar] [CrossRef]
- Maass, A.; Düzel, S.; Brigadski, T.; Goerke, M.; Becke, A.; Sobieray, U.; Neumann, K.; Lövdén, M.; Lindenberger, U.; Bäckman, L.; et al. Relationships of peripheral IGF-1, VEGF and BDNF levels to exercise-related changes in memory, hippocampal perfusion and volumes in older adults. Neuroimage 2016, 131, 142–154. [Google Scholar] [CrossRef]
- Cobianchi, S.; Arbat-Plana, A.; Lopez-Alvarez, V.M.; Navarro, X. Neuroprotective Effects of Exercise Treatments after Injury: The Dual Role of Neurotrophic Factors. Curr. Neuropharmacol. 2017, 15, 495–518. [Google Scholar] [CrossRef] [PubMed]
- Yuskaitis, C.J.; Jope, R.S. Glycogen synthase kinase-3 regulates microglial migration, inflammation, and inflammation-induced neurotoxicity. Cell. Signal. 2009, 21, 264–273. [Google Scholar] [CrossRef]
- Green, H.F.; Nolan, Y.M. GSK-3 mediates the release of IL-1β, TNF-α and IL-10 from cortical glia. Neurochem. Int. 2012, 61, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Ru, J.; Ma, W.; Gao, Y.; Liang, Z.; Liu, J.; Guo, J.; Li, L. BDNF promotes the growth of human neurons through crosstalk with the Wnt/β-catenin signaling pathway via GSK-3β. Neuropeptides 2015, 54, 35–46. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palasz, E.; Wysocka, A.; Gasiorowska, A.; Chalimoniuk, M.; Niewiadomski, W.; Niewiadomska, G. BDNF as a Promising Therapeutic Agent in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 1170. https://doi.org/10.3390/ijms21031170
Palasz E, Wysocka A, Gasiorowska A, Chalimoniuk M, Niewiadomski W, Niewiadomska G. BDNF as a Promising Therapeutic Agent in Parkinson’s Disease. International Journal of Molecular Sciences. 2020; 21(3):1170. https://doi.org/10.3390/ijms21031170
Chicago/Turabian StylePalasz, Ewelina, Adrianna Wysocka, Anna Gasiorowska, Malgorzata Chalimoniuk, Wiktor Niewiadomski, and Grazyna Niewiadomska. 2020. "BDNF as a Promising Therapeutic Agent in Parkinson’s Disease" International Journal of Molecular Sciences 21, no. 3: 1170. https://doi.org/10.3390/ijms21031170
APA StylePalasz, E., Wysocka, A., Gasiorowska, A., Chalimoniuk, M., Niewiadomski, W., & Niewiadomska, G. (2020). BDNF as a Promising Therapeutic Agent in Parkinson’s Disease. International Journal of Molecular Sciences, 21(3), 1170. https://doi.org/10.3390/ijms21031170