High Cell Selectivity and Bactericidal Mechanism of Symmetric Peptides Centered on d-Pro–Gly Pairs

Abstract
1. Introduction
2. Results
2.1. Peptide Design and Characteristics
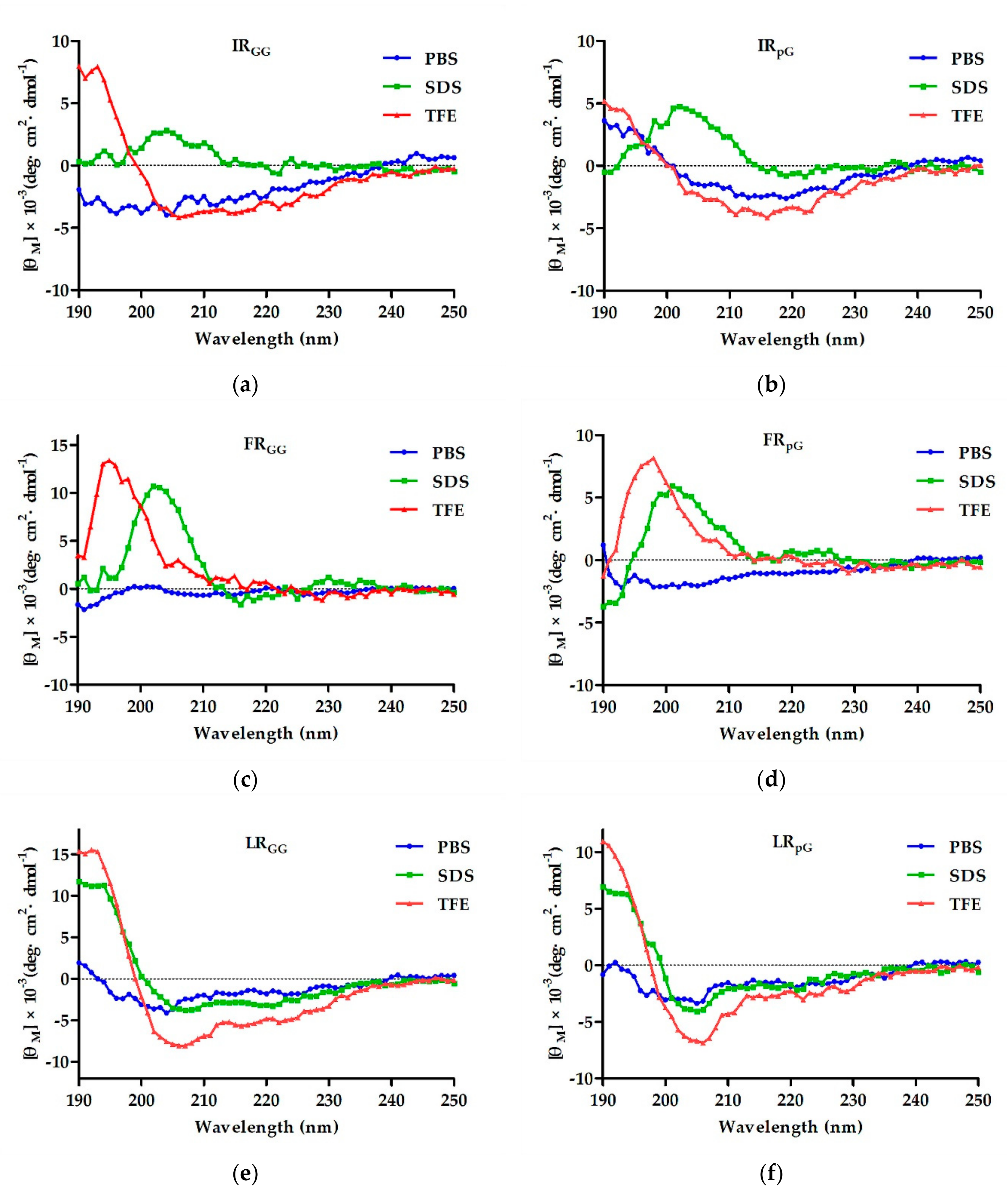
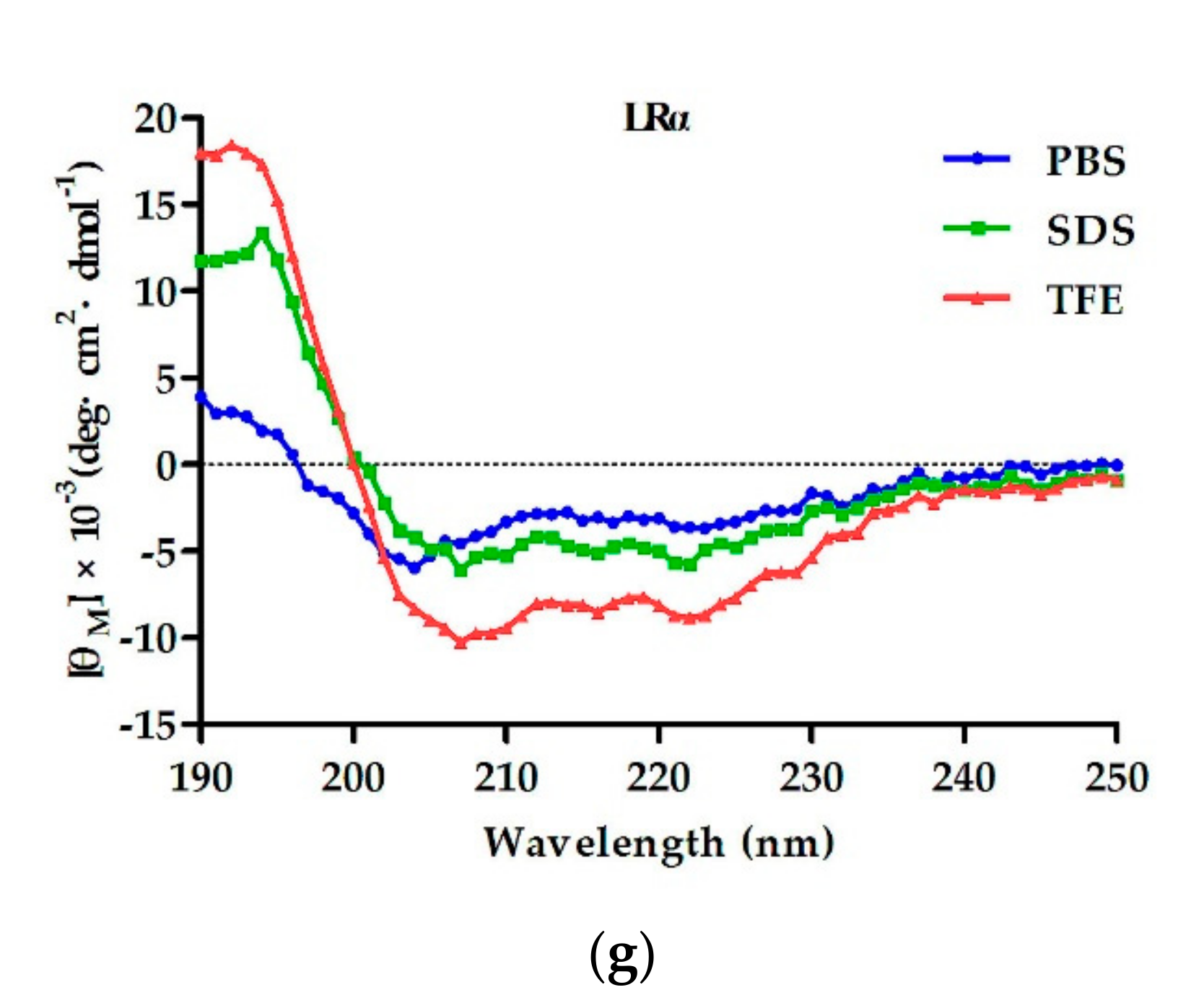
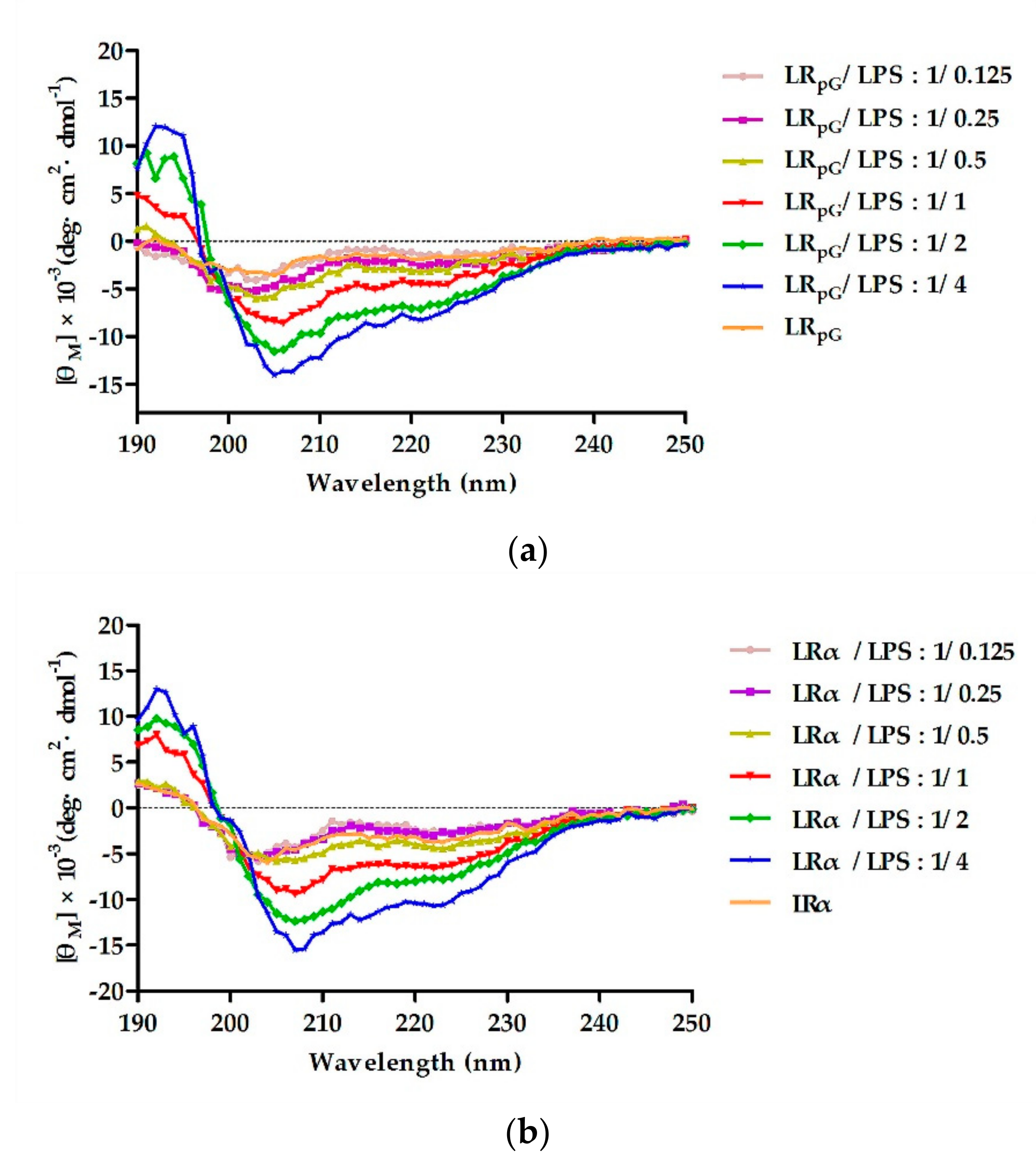
2.2. Circular Dichroism (CD) Spectroscopy
2.3. Antimicrobial Activity
2.4. Biocompatibility Assays
2.5. Condition Sensitivity Assays
2.6. Additive Effect of AMPs and Conventional Antibiotics
2.7. Antimicrobial Mechanism Study
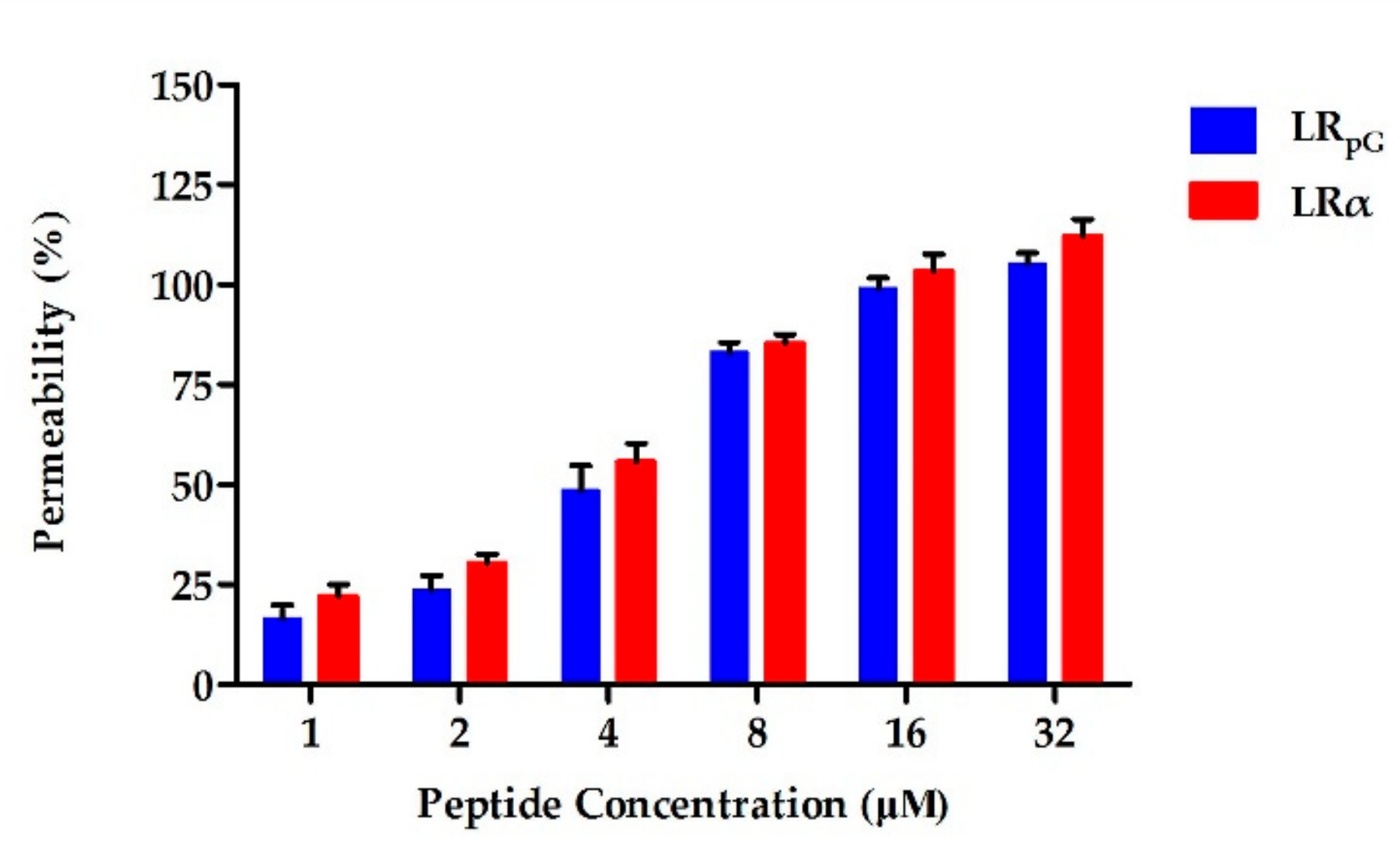
2.7.1. Outer Membrane Permeability Assay
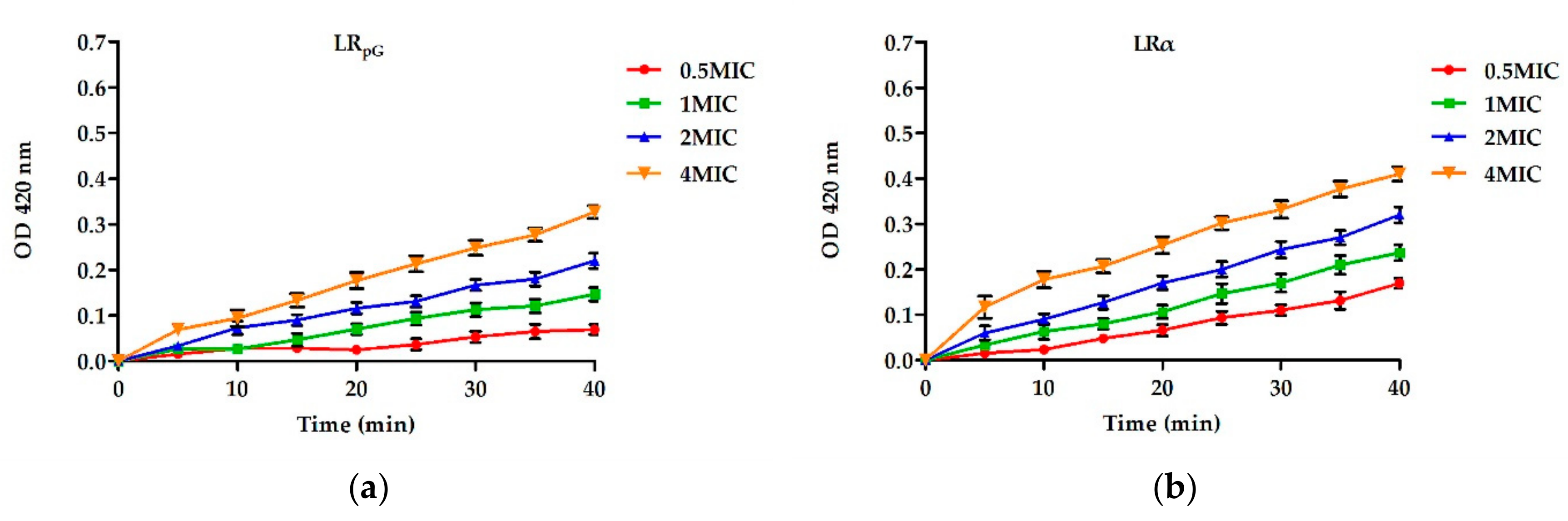
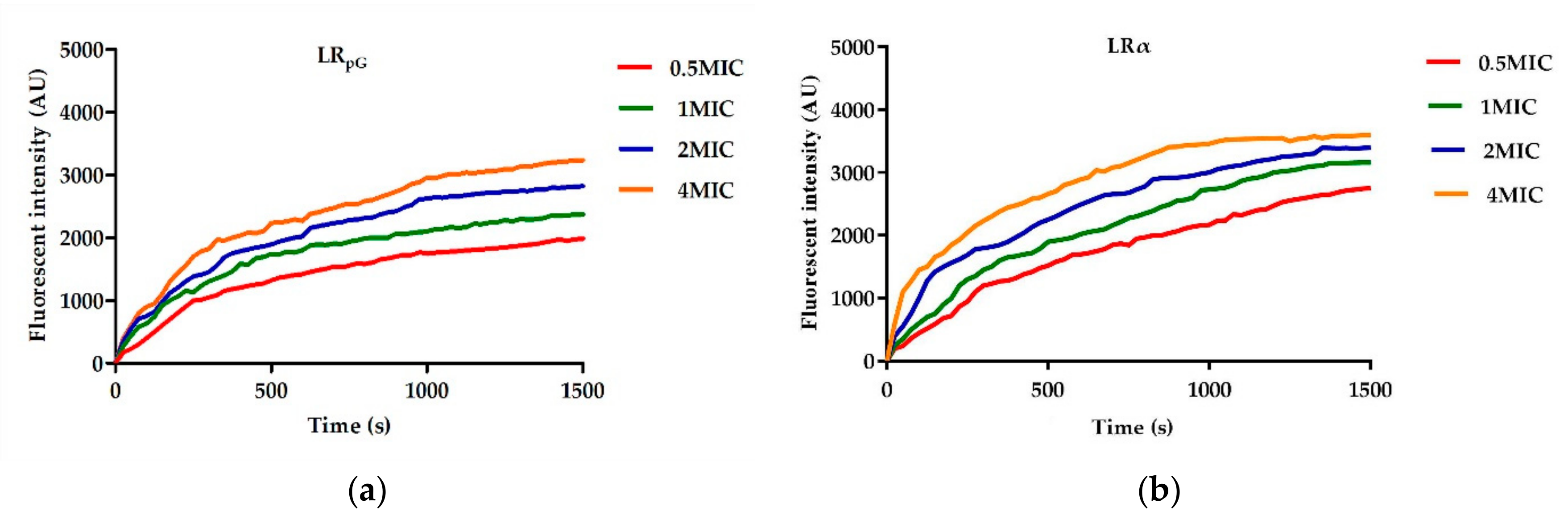
2.7.2. Inner Membrane Permeability Assay
2.7.3. Cytoplasmic Membrane Depolarization
2.7.4. Scanning Electron Microscopy (SEM)
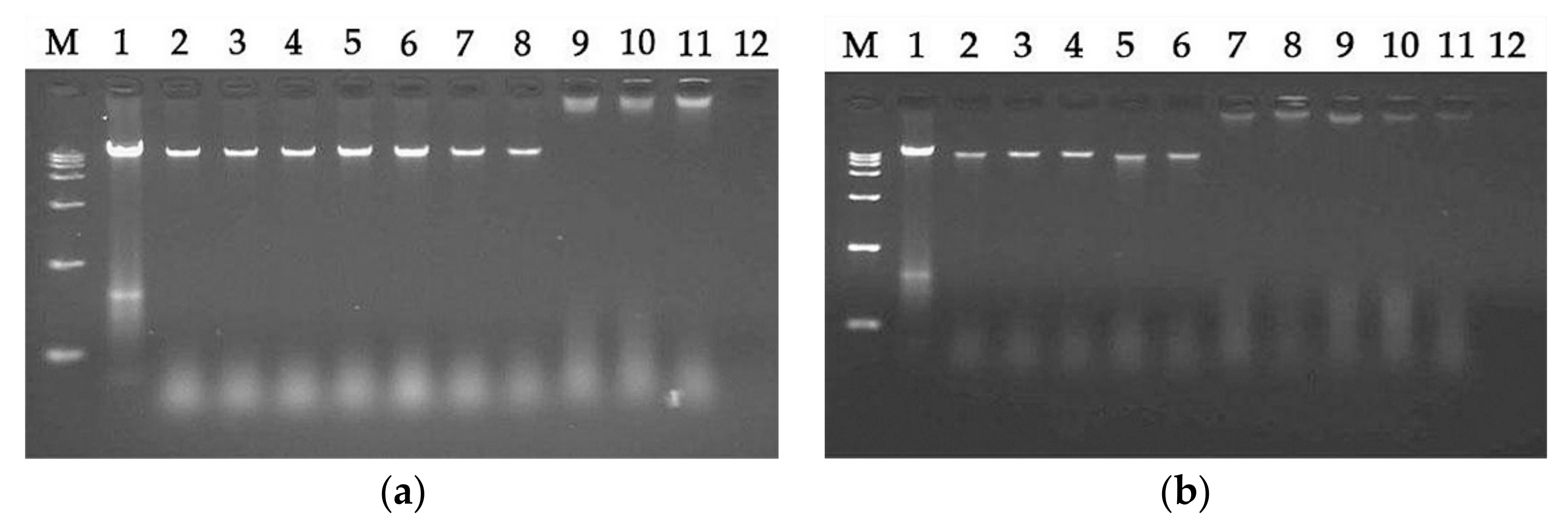
2.7.5. DNA Binding Assay
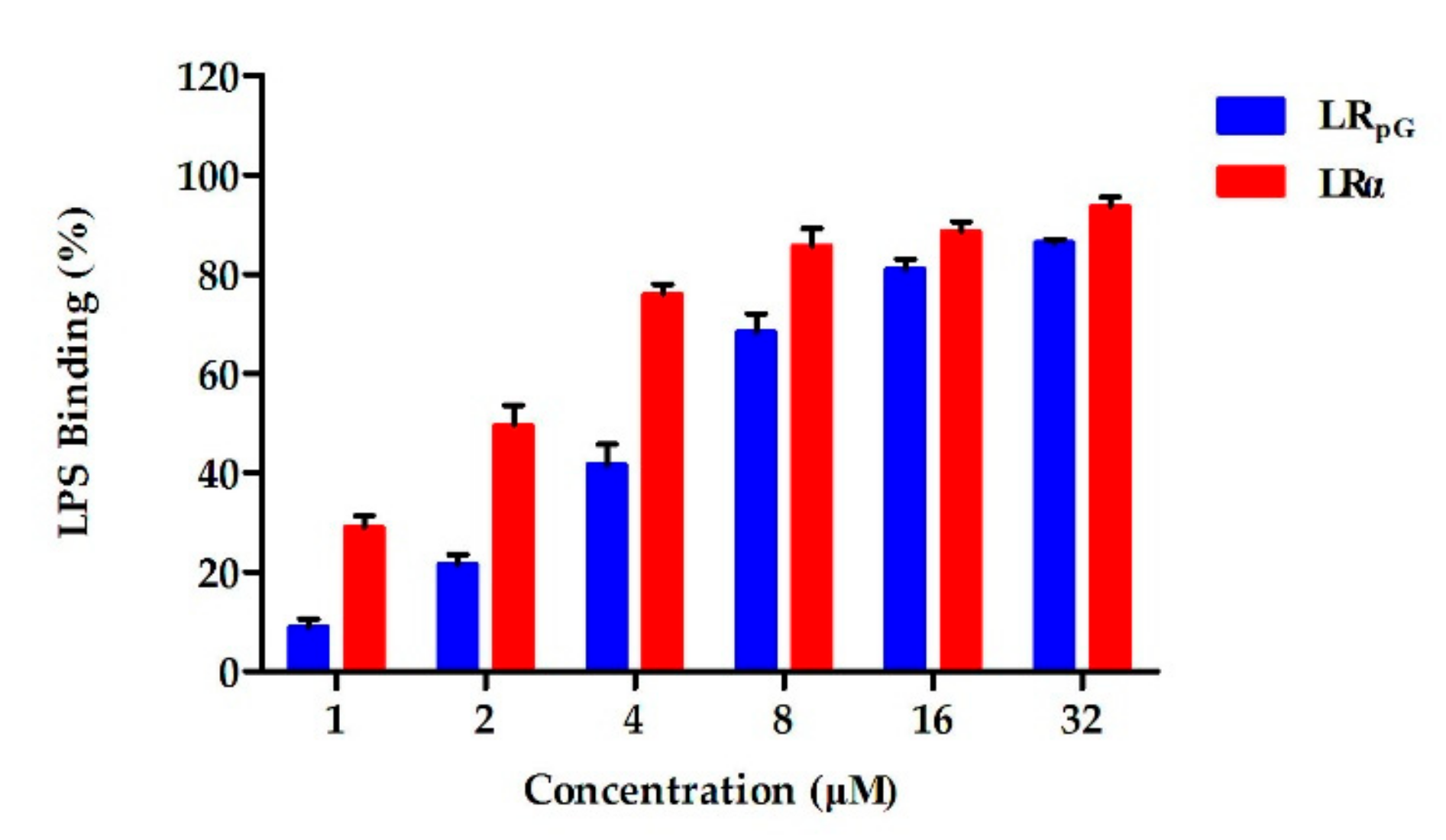
2.7.6. Lipopolysaccharide (LPS) Binding Assay
2.7.7. Limulus Amoebocyte Lysate (LAL) Assay
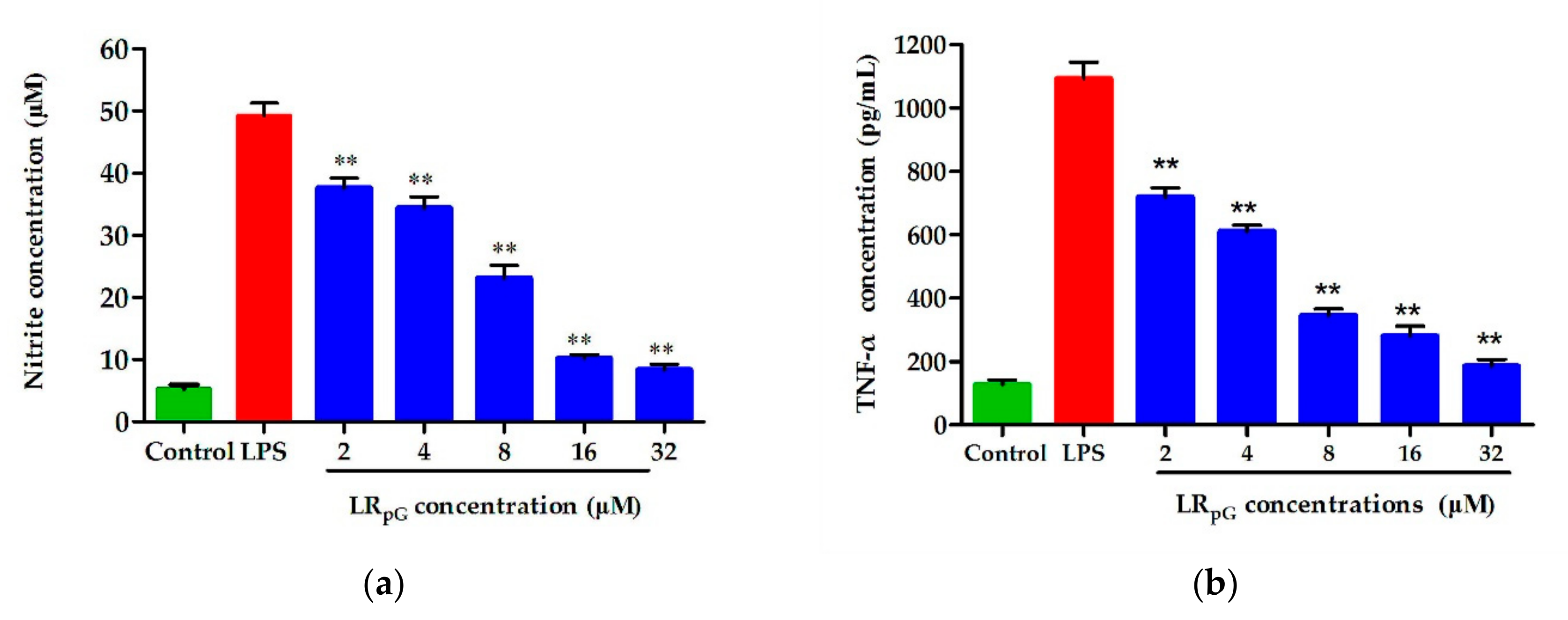
2.7.8. Endotoxin Neutralization Assay
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Synthesis and Sequence Analysis of Peptides
4.3. CD Spectroscopy
4.4. MIC Measurements
4.5. Biocompatibility Assays
4.6. Condition Sensitivity Assays
4.7. Synergy with Conventional Antibiotics
4.8. Outer Membrane Permeability Assay
4.9. Inner Membrane Permeability Assay
4.10. Cytoplasmic Membrane Depolarization Assay
4.11. Scanning Electron Microscopy
4.12. DNA Binding Assay
4.13. LPS Binding Assay
4.14. LAL Assay
4.15. Endotoxin Neutralization Assay
4.16. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AMPs | antimicrobial peptides |
| CD | circular dichroism |
| MIC | minimal inhibitory concentration |
| TI | theoretical index |
| hRBCs | human red blood cells |
| E. coli | Escherichia coli |
| FICI | fractional inhibitory concentration index |
| SEM | scanning electron microscopy |
| LPS | lipopolysaccharide |
References
- Van Boeckel, T.P.; Brower, C.; Gilbert, M.; Grenfell, B.T.; Levin, S.A.; Robinson, T.P.; Teillant, A.; Laxminarayan, R. Global trends in antimicrobial use in food animals. Proc. Natl. Acad. Sci. USA 2015, 112, 5649–5654. [Google Scholar] [CrossRef]
- Wang, J.J.; Dou, X.J.; Song, J.; Lyu, Y.F.; Zhu, X.; Xu, L.; Li, W.Z.; Shan, A.S. Antimicrobial peptides: Promising alternatives in the post feeding antibiotic era. Med. Res. Rev. 2019, 39, 831–859. [Google Scholar] [CrossRef] [PubMed]
- Tellez, G.A.; Zapata, J.A.; Toro, L.J.; Henao, D.C.; Bedoya, J.P.; Rivera, J.D.; Trujillo, J.Y.; Rivas-Santiago, B.; Hoyos, R.O.; Castano, J.C. Identification, characterization, immunolocalization, and biological activity of lucilin peptide. Acta Trop. 2018, 185, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.X.; Chen, Y.C.; Yang, C.; Zeng, P.; Xu, H.; Pan, F.; Lu, J.R. High selective performance of designed antibacterial and anticancer peptide amphiphiles. ACS Appl. Mater. Interfaces 2015, 7, 17346–17355. [Google Scholar] [CrossRef] [PubMed]
- Omardien, S.; Brul, S.; Zaat, S.A. Antimicrobial activity of cationic antimicrobial peptides against gram-positives: Current progress made in understanding the mode of action and the response of bacteria. Front. Cell Dev. Biol. 2016, 4, 111. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Chen, T.W.; Pan, Z.M.; Sun, X.B.; Yin, X.; He, M.; Xiao, S.Y.; Liang, H.J. Theoretical insights into the interactions between star-shaped antimicrobial polypeptides and bacterial membranes. Langmuir. 2018, 34, 13438–13448. [Google Scholar] [CrossRef]
- Melo, M.N.; Ferre, R.; Castanho, M.A. Antimicrobial peptides: Linking partition, activity and high membrane-bound concentrations. Nat. Rev. Microbiol. 2009, 7, 245–250. [Google Scholar] [CrossRef]
- Ong, Z.Y.; Wiradharma, N.; Yang, Y.Y. Strategies employed in the design and optimization of synthetic antimicrobial peptide amphiphiles with enhanced therapeutic potentials. Adv. Drug Deliv. Rev. 2014, 78, 28–45. [Google Scholar] [CrossRef]
- Tripathi, A.K.; Kumari, T.; Tandon, A.; Sayeed, M.; Afshan, T.; Kathuria, M.; Shukla, P.K.; Mitra, K.; Ghosh, J.K. Selective phenylalanine to proline substitution for improved antimicrobial and anticancer activities of peptides designed on phenylalanine heptad repeat. Acta Biomater. 2017, 57, 170–186. [Google Scholar] [CrossRef]
- Ahmad, A.; Azmi, S.; Srivastava, R.M.; Srivastava, S.; Pandey, B.K.; Saxena, R.; Bajpai, V.K.; Ghosh, J.K. Design of nontoxic analogues of cathelicidin-derived bovine antimicrobial peptide BMAP-27: The role of leucine as well as phenylalanine zipper sequences in determining its toxicity. Biochemistry 2009, 48, 10905–10917. [Google Scholar] [CrossRef]
- Asthana, N.; Yadav, S.P.; Ghosh, J.K. Dissection of antibacterial and toxic activity of melittin: A leucine zipper motif plays a crucial role in determining its hemolytic activity but not antibacterial activity. J. Biol. Chem. 2004, 279, 55042–55050. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Tripathi, A.K.; Kathuria, M.; Shree, S.; Tripathi, J.K.; Purshottam, R.K.; Ramachandran, R.; Mitra, K.; Ghosh, J.K. Single Amino acid substitutions at specific positions of the heptad repeat sequence of piscidin-1 yielded novel analogs that show low cytotoxicity and in vitro and in vivo antiendotoxin activity. Antimicrob. Agents Chemother. 2016, 60, 3687–3699. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R.M.; Srivastava, S.M.; Singh, M.; Bajpai, V.K.; Ghosh, J.K. Consequences of alteration in leucine zipper sequence of melittin in its neutralization of lipopolysaccharide-induced proinflammatory response in macrophage cells and interaction with lipopolysaccharide. J. Biol. Chem. 2012, 287, 1980–1995. [Google Scholar] [CrossRef] [PubMed]
- Dong, N.; Ma, Q.Q.; Shan, A.S.; Lv, Y.F.; Hu, W.N.; Gu, Y.; Li, Y.Z. Strand length-dependent antimicrobial activity and membrane-active mechanism of arginine- and valine-rich β-hairpin-like antimicrobial peptides. Antimicrob. Agents Chemother. 2012, 56, 2994–3003. [Google Scholar] [CrossRef]
- Chou, S.L.; Shao, C.X.; Wang, J.J.; Shan, A.S.; Xu, L.; Dong, N.; Li, Z.Y. Short, multiple-stranded β-hairpin peptides have antimicrobial potency with high selectivity and salt resistance. Acta Biomater. 2016, 30, 78–93. [Google Scholar] [CrossRef]
- Dong, N.; Chou, S.L.; Li, J.W.; Xue, C.Y.; Li, X.R.; Chen, B.J.; Shan, A.S. Short symmetric-end antimicrobial peptides centered on β-turn amino acids unit improve selectivity and stability. Front. Microbiol. 2018, 27, 2832. [Google Scholar] [CrossRef]
- Wang, J.J.; Chou, S.L.; Xu, L.; Zhu, X.; Dong, N.; Shan, A.S.; Chen, Z. High specific selectivity and membrane-active mechanism of the synthetic centrosymmetric α-helical peptides with gly-gly pairs. Sci. Rep. 2015, 5, 15963. [Google Scholar] [CrossRef]
- Chou, S.L.; Wang, J.J.; Shang, L.; Akhtar, M.U.; Wang, Z.H.; Shi, B.M.; Feng, X.J.; Shan, A.S. Short, symmetric-helical peptides have narrow-spectrum activity with low resistance potential and high selectivity. Biomater. Sci. 2019, 7, 2394–2409. [Google Scholar] [CrossRef]
- Ma, Z.; Yang, J.; Han, J.Z.; Gao, L.; Liu, H.X.; Lu, Z.X.; Zhao, H.Z.; Bie, X.M. Insights into the antimicrobial activity and cytotoxicity of engineered α-helical peptide amphiphiles. J. Med. Chem. 2016, 59, 10946–10962. [Google Scholar] [CrossRef]
- Shao, C.X.; Tian, H.T.; Wang, T.Y.; Wang, Z.H.; Chou, S.L.; Shan, A.S.; Chen, B.J. Central β-turn increases the cell selectivity of imperfectly amphipathic α-helical peptides. Acta Biomater. 2018, 69, 243–255. [Google Scholar] [CrossRef]
- Dong, N.; Wang, C.S.; Zhang, T.T.; Zhang, L.; Xue, C.Y.; Feng, X.J.; Bi, C.P.; Shan, A.S. Bioactivity and bactericidal mechanism of histidine-rich β-hairpin peptide against Gram-negative bacteria. Int. J. Mol. Sci. 2019, 20, 3954. [Google Scholar] [CrossRef] [PubMed]
- Shao, C.X.; Li, W.Z.; Tan, P.; Shan, A.S.; Dou, X.J.; Ma, D.Y.; Liu, C.Y. Symmetrical modification of minimized dermaseptins to extend the spectrum of antimicrobials with endotoxin neutralization potency. Int. J. Mol. Sci. 2019, 20, 1417. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Kizhakkedathu, J.N.; Straus, S.K. Antimicrobial peptides: Diversity, mechanism of action and strategies to improve the activity and biocompatibility in vivo. Biomolecules 2018, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Zelezetsky, I.; Tossi, A. Alpha-helical antimicrobial peptides-using a sequence template to guide structure-activity relationship studies. Biochim. Biophys. Acta 2006, 1758, 1436–1449. [Google Scholar] [CrossRef]
- Takahashi, D.; Shukla, S.K.; Prakash, O.; Zhang, G. Structural determinants of host defense peptides for antimicrobial activity and target cell selectivity. Biochimie 2010, 92, 1236–1241. [Google Scholar] [CrossRef]
- Kumar, A.; Mahajan, M.; Awasthi, B.; Tandon, A.; Harioudh, M.K.; Shree, S.; Singh, P.; Shukla, P.K.; Ramachandran, R.; Mitra, K.; et al. Piscidin-1-analogs with double L- and D-lysine residues exhibited different conformations in lipopolysaccharide but comparable anti-endotoxin activities. Sci. Rep. 2017, 7, 39925. [Google Scholar] [CrossRef]
- Lee, J.K.; Park, S.C.; Hahm, K.S.; Park, Y. A helix-PXXP-helix peptide with antibacterial activity without cytotoxicity against MDRPA-infected mice. Biomaterials 2014, 35, 1025–1039. [Google Scholar] [CrossRef]
- Zhu, X.; Zhang, L.C.; Wang, J.; Ma, Z.; Xu, W.; Li, J.P.; Shan, A.S. Characterization of antimicrobial activity and mechanisms of low amphipathic peptides with different α-helical propensity. Acta Biomater. 2015, 18, 155–167. [Google Scholar] [CrossRef]
- Wang, J.J.; Chou, S.L.; Yang, Z.Y.; Yang, Y.; Wang, Z.H.; Song, J.; Dou, X.J.; Shan, A.S. Combating drug-resistant fungi with novel imperfectly amphipathic palindromic peptides. J. Med. Chem. 2018, 61, 3889–3907. [Google Scholar] [CrossRef]
- Park, I.Y.; Cho, J.H.; Kim, K.S.; Kim, Y.B.; Kim, M.S.; Kim, S.C. Helix stability confers salt resistance upon helical antimicrobial peptides. J. Biol. Chem. 2004, 279, 13896–13901. [Google Scholar] [CrossRef]
- Jin, Y.; Hammer, J.; Pate, M.; Zhang, Y.; Zhu, F.; Zmuda, E.; Blazy, J. Antimicrobial activities and structures of two linear cationic peptide families with various amphipathic β-sheet and α-helical potentials. Antimicrob. Agents Chemother. 2005, 49, 4957–4964. [Google Scholar] [CrossRef] [PubMed]
- Arouri, A.; Dathe, M.; Blume, A. The helical propensity of KLA amphipathic peptides enhances their binding to gel-state lipid membranes. Biophys. Chem. 2013, 180, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.Q.; Dong, N.; Shan, A.S.; Lv, Y.F.; Li, Y.Z.; Chen, Z.H.; Cheng, B.J.; Li, Z.Y. Biochemical property and membrane-peptide interactions of de novo antimicrobial peptides designed by helix-forming units. Amino Acids 2012, 43, 2527–2536. [Google Scholar] [CrossRef]
- Khara, J.S.; Wang, Y.; Ke, X.Y.; Liu, S.; Newton, S.M.; Langford, P.R.; Yang, Y.Y.; Ee, P.L. Anti-mycobacterial activities of synthetic cationic α-helical peptides and their synergism with rifampicin. Biomaterials 2014, 35, 2032–2038. [Google Scholar] [CrossRef]
- Liu, Y.F.; Xia, X.; Xu, L.; Wang, Y.Z. Design of hybrid β-hairpin peptides with enhanced cell specificity and potent anti-inflammatory activity. Biomaterials 2013, 34, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.; Haney, E.F.; Gill, E.E. The immunology of host defence peptides: Beyond antimicrobial activity. Nat. Rev. Immunol. 2016, 16, 321–334. [Google Scholar] [CrossRef]
- Cohen, J.; Vincent, J.L.; Adhikari, N.K.; Machado, F.R.; Angus, D.C.; Calandra, T.; Jaton, K.; Giulieri, S.; Delaloye, J.; Opal, S.; et al. Sepsis: A roadmap for future research. Lancet Infect. Dis. 2015, 15, 581–614. [Google Scholar] [CrossRef]
- Zong, X.; Song, D.; Wang, T.; Xia, X.; Hu, W.; Han, F.; Wang, Y. LFP-20, a porcine lactoferrin peptide, ameliorates LPS-induced inflammation via the MyD88/NF-κB and MyD88/MAPK signaling pathways. Dev. Comp. Immunol. 2015, 52, 123–131. [Google Scholar] [CrossRef]
- Khara, J.S.; Obuobi, S.; Wang, Y.; Hamilton, M.S.; Robertson, B.D.; Newton, S.M.; Yang, Y.Y.; Langford, P.R.; Ee, P.L.R. Disruption of drug-resistant biofilms using, de novo, designed short α-helical antimicrobial peptides with idealized facial amphiphilicity. Acta Biomater. 2017, 57, 103–114. [Google Scholar] [CrossRef]
- Wang, J.J.; Song, J.; Yang, Z.Y.; He, S.Q.; Yang, Y.; Feng, X.J.; Dou, X.J.; Shan, A.S. Antimicrobial peptides with high proteolytic resistance for combating gram-negative bacteria. J. Med. Chem. 2019, 62, 2286–2304. [Google Scholar] [CrossRef]
- Dong, N.; Zhu, X.; Chou, S.; Shan, A.S.; Li, W.Z.; Jiang, J.G. Antimicrobial potency and selectivity of simplified symmetricend peptides. Biomaterials 2014, 27, 8028–8039. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.T.; Ma, Q.Q.; Zhu, D.D.; Shao, C.X.; Lv, Y.F.; Shan, A.S. The C-terminal sequences of porcine thrombin are active as antimicrobial peptides. Chem. Biol. Drug Des. 2016, 88, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Dong, W.B.; Sun, L.; Ma, L.J.; Shang, D.J. Insights into the membrane interaction mechanism and antibacterial properties of chensinin-1b. Biomaterials 2015, 37, 299–311. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptides | Sequence | Theoretical MW | Measured MW 1 | Net Charge | H 2 | µHre 3 |
|---|---|---|---|---|---|---|
| IRGG | IIRIIRRGGRRIIRII-NH2 | 1974.52 | 1973.5 | 6 | 0.521 | 0.770 |
| IRpG | IIRIIRRp 4 GRRIIRII-NH2 | 2014.58 | 2013.8 | 6 | 0.566 | 0.800 |
| FRGG | FFRFFRRGGRRFFRFF-NH2 | 2246.66 | 2245.68 | 6 | 0.516 | 0.767 |
| FRpG | FFRFFRRp 4 GRRFFRFF-NH2 | 2286.72 | 2285.74 | 6 | 0.561 | 0.797 |
| LRGG | LLRLLRRGGRRLLRLL-NH2 | 1974.52 | 1973.51 | 6 | 0.471 | 0.743 |
| LRpG | LLRLLRRp 4 GRRLLRLL-NH2 | 2014.58 | 2013.58 | 6 | 0.516 | 0.773 |
| LRα | GLRLLRRLLRRLLRLp 4 -NH2 | 2014.58 | 2013.58 | 6 | 0.516 | 0.735 |
| Bacterial Species | IRGG | IRpG | FRGG | FRpG | LRGG | LRpG | LRα | Melittin | |
|---|---|---|---|---|---|---|---|---|---|
| MIC (µM) | |||||||||
| Gram (−) | |||||||||
| Escherichia coli ATCC25922 | 8 | 4 | 8 | 8 | 2 | 2 | 2 | 1 | |
| E. coli K88 | 16 | 16 | 16 | 8 | 4 | 2 | 4 | 2 | |
| Salmonella Pullorum NCTC5776 | 32 | 16 | 16 | 16 | 4 | 4 | 2 | 2 | |
| Klebsiella pneumonia CMCC46117 | 32 | 32 | 16 | 16 | 8 | 4 | 4 | 4 | |
| Pseudomonas aeruginosa ATCC27853 | 32 | 32 | 32 | 32 | 8 | 8 | 4 | 2 | |
| Gram (+) | |||||||||
| Staphylococcus aureus ATCC25923 | >128 | >128 | 32 | 16 | 16 | 16 | 4 | 2 | |
| S. aureus ATCC29213 | >128 | >128 | 16 | 16 | 16 | 16 | 8 | 1 | |
| S. aureus ATCC43300 | >128 | >128 | 32 | 32 | 32 | 16 | 8 | 2 | |
| Enterococcus faecalis ATCC29212 | >128 | >128 | 32 | 32 | 32 | 32 | 16 | 1 | |
| GM 1 (µM) | |||||||||
| Gram (−) | 21.1 | 16 | 16 | 13.9 | 4.6 | 3.5 | 3 | 2 | |
| Gram (+) | 256 | 256 | 26.9 | 22.6 | 22.6 | 19.0 | 8 | 1.4 | |
| MHC10 2 (µM) | 512 | 512 | 128 | 128 | 256 | 256 | 4 | 0.25 | |
| TI 3 | |||||||||
| TI (−) | 24.2 | 32 | 8 | 9.2 | 55.6 | 73.1 | 1.3 | 0.125 | |
| TI (+) | 2 | 2 | 4.8 | 5.7 | 11.2 | 13.5 | 0.5 | 0.178 | |
| Peptides | Control 1 | Physiological Salts 2 | |||||
|---|---|---|---|---|---|---|---|
| NaCl | KCl | NH4Cl | MgCl2 | ZnCl2 | FeCl3 | ||
| IRGG | 8 | 64 | 8 | 8 | 16 | 16 | 8 |
| IRpG | 4 | 32 | 4 | 4 | 8 | 4 | 4 |
| FRGG | 8 | 64 | 8 | 8 | 16 | 16 | 8 |
| FRpG | 8 | 32 | 8 | 8 | 8 | 8 | 8 |
| LRGG | 2 | 8 | 2 | 2 | 4 | 2 | 2 |
| LRpG | 2 | 8 | 2 | 2 | 2 | 2 | 2 |
| LRα | 2 | 8 | 2 | 2 | 2 | 2 | 2 |
| Melittin | 1 | 4 | 1 | 1 | 1 | 1 | 1 |
| Peptides | Control (pH 7) | Temperature | pH | |||||
|---|---|---|---|---|---|---|---|---|
| 0 °C | 37 °C | 100 °C | pH 4 | pH 6 | pH 8 | pH 10 | ||
| IRGG | 8 | 8 | 8 | 8 | 16 | 8 | 8 | 16 |
| IRpG | 4 | 4 | 4 | 4 | 16 | 4 | 8 | 16 |
| FRGG | 8 | 8 | 8 | 8 | 32 | 8 | 8 | 32 |
| FRpG | 8 | 8 | 8 | 8 | 16 | 8 | 8 | 32 |
| LRGG | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 4 |
| LRpG | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 4 |
| LRα | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 4 |
| Melittin | 1 | 1 | 1 | 1 | 2 | 1 | 1 | 2 |
| Peptide | Streptomycin 2 | Ciprofloxacin 2 | Chloramphenicol 2 | Cefotaxime 2 |
|---|---|---|---|---|
| LRpG | 0.5 | 0.625 | 0.75 | 1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jia, B.; Wang, Y.; Zhang, Y.; Wang, Z.; Wang, X.; Muhammad, I.; Kong, L.; Pei, Z.; Ma, H.; Jiang, X. High Cell Selectivity and Bactericidal Mechanism of Symmetric Peptides Centered on d-Pro–Gly Pairs. Int. J. Mol. Sci. 2020, 21, 1140. https://doi.org/10.3390/ijms21031140
Jia B, Wang Y, Zhang Y, Wang Z, Wang X, Muhammad I, Kong L, Pei Z, Ma H, Jiang X. High Cell Selectivity and Bactericidal Mechanism of Symmetric Peptides Centered on d-Pro–Gly Pairs. International Journal of Molecular Sciences. 2020; 21(3):1140. https://doi.org/10.3390/ijms21031140
Chicago/Turabian StyleJia, Boyan, Yiming Wang, Ying Zhang, Zi Wang, Xue Wang, Inam Muhammad, Lingcong Kong, Zhihua Pei, Hongxia Ma, and Xiuyun Jiang. 2020. "High Cell Selectivity and Bactericidal Mechanism of Symmetric Peptides Centered on d-Pro–Gly Pairs" International Journal of Molecular Sciences 21, no. 3: 1140. https://doi.org/10.3390/ijms21031140
APA StyleJia, B., Wang, Y., Zhang, Y., Wang, Z., Wang, X., Muhammad, I., Kong, L., Pei, Z., Ma, H., & Jiang, X. (2020). High Cell Selectivity and Bactericidal Mechanism of Symmetric Peptides Centered on d-Pro–Gly Pairs. International Journal of Molecular Sciences, 21(3), 1140. https://doi.org/10.3390/ijms21031140