Arginase Inhibition Supports Survival and Differentiation of Neuronal Precursors in Adult Alzheimer’s Disease Mice

 ,
, 
 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. 3 × Tg Mice Show a Significantly Reduced DCX-Positive NPC Density, Compared to Wild Type Mice, which Demonstrate a Dissimilar Phenotype, and are Unaffected by Arginase Inhibition
2.2. Norvaline Caused an Escalation of the PSA-NCAM Levels in the Hippocampi of 3 × Tg Mice, as Evidenced by an Increase in Immunopositive Surface Area and Stain Intensity
2.3. Norvaline Rescues Neuronal and Dendritic Loss in 3 × Tg Mice, as Evidenced by MAP2 Staining
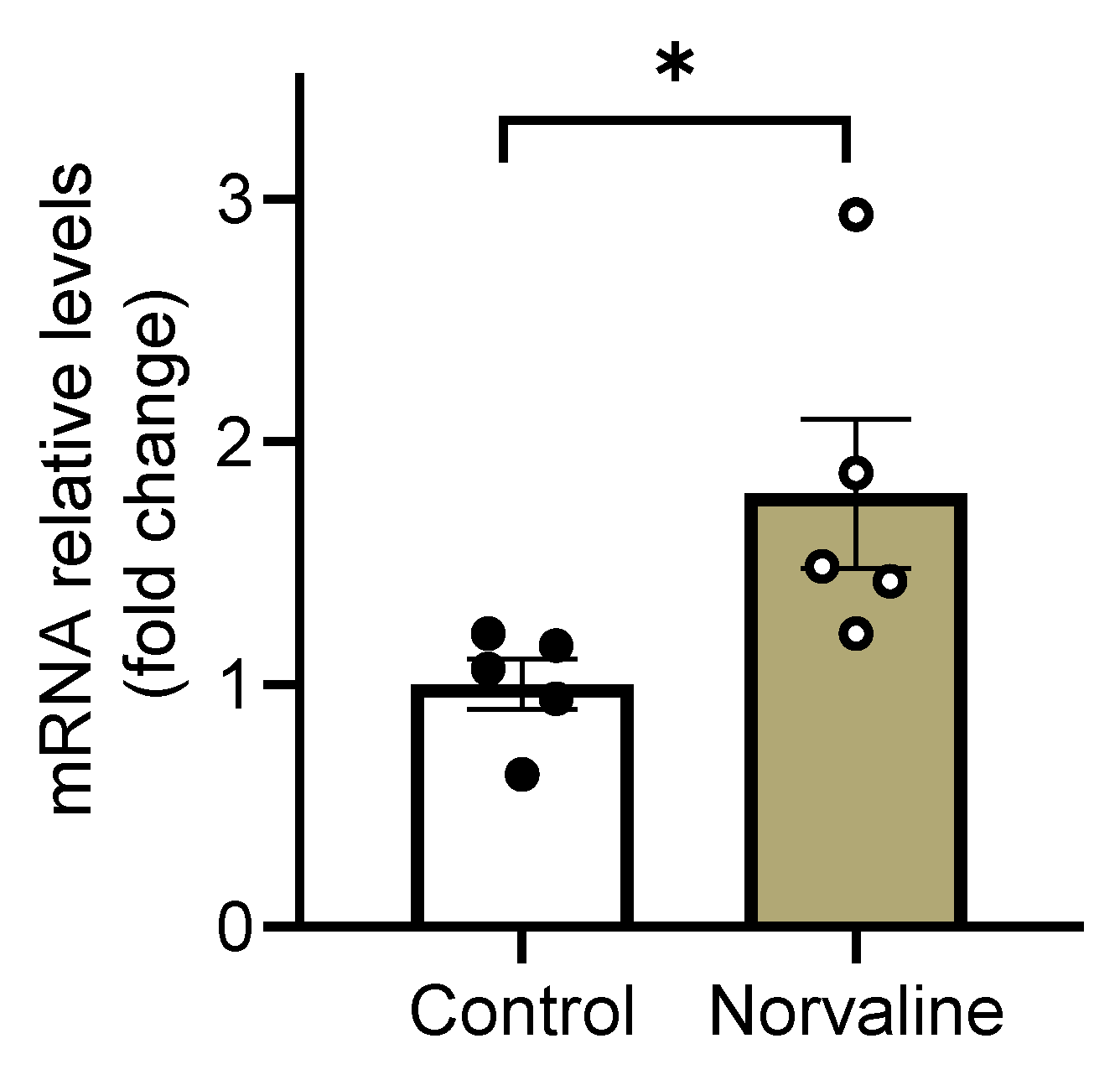
2.4. Norvaline Escalates the Transcription Levels of C-C Motif Chemokine 11
2.5. Arginase Inhibition with Norvaline Increases the Hippocampal Expression Levels of PAX6 Protein
2.6. Norvaline Activates MAPK/ERK Pathway
3. Discussion
4. Materials and Methods
4.1. Animals and Treatment
4.2. Tissue Preparation and Slicing
4.3. Quantitative Immunohistochemistry
4.4. Doublecortin Labeling and Staining
4.5. Polysialylated Neuronal Cell Adhesion Molecule Staining
4.6. Microtubule-Associated Protein 2 Staining
4.7. Imaging and Quantification
4.8. Tissue Sampling, RNA Extraction, Reverse Transcription, and Real-Time Polymerase Chain Reaction
4.9. Western Blotting
4.10. Antibody Microarray
4.11. KiNetscape Analysis and Representation
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| NPCs | neuronal progenitor cells |
| 3 × Tg | triple-transgenic mouse model of Alzheimer’s disease |
| NOS | nitric oxide synthase |
| Aβ | amyloid-beta |
| NFT | neurofibrillary tangles |
| SVZ | subventricular zone |
| APP | amyloid precursor protein |
| SGZ | subgranular zone |
| VEGF | vascular endothelial growth factor |
| BDNF | brain-derived neurotrophic factor |
| CNS | central nervous system |
| NRG | neuregulin |
| RT-PCR | real-time polymerase chain reaction |
| GDNF | glial cell-derived neurotrophic factor |
| RET | REarranged during Transfection |
| WT | wild-type |
| OD | optical density |
| ANOVA | analysis of variance |
| CA | cornus Ammonis |
| PSANCAM | polysialylated neuronal cell adhesion molecule |
| SEM | standard error of the mean |
| NGF | nerve growth factor |
| DCX | doublecortin |
| mRNA | messenger ribonucleic acid |
| PSD-95 | postsynaptic density protein 95 |
| MAP2 | microtubule-associated protein 2 |
| CCL11 | C-C motif chemokine 11 |
| PAX6 | paired box protein 6 |
| MAP | mitogen-activated protein |
| MEK1 | dual specificity mitogen-activated protein kinase kinase 1 |
| ERK | extracellular signal–regulated kinases |
| NF-κB | nuclear factor kappa-B |
| S | Serine |
| Cdk5 | cyclin-dependent protein-serine kinase 5 |
| p38dMAPK | mitogen-activated protein-serine kinase p38 |
| HDAC | histone deacetylase |
References
- Pilz, G.A.; Bottes, S.; Betizeau, M.; Jörg, D.J.; Carta, S.; Simons, B.D.; Helmchen, F.; Jessberger, S. Live imaging of neurogenesis in the adult mouse hippocampus. Science 2018, 359, 658–662. [Google Scholar] [CrossRef] [PubMed]
- Tobin, M.K.; Musaraca, K.; Disouky, A.; Shetti, A.; Bheri, A.; Honer, W.G.; Kim, N.; Dawe, R.J.; Bennett, D.A.; Arfanakis, K.; et al. Human Hippocampal Neurogenesis Persists in Aged Adults and Alzheimer’s Disease Patients. Cell Stem Cell 2019, 24, 974–982.e3. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, J.J.; Jones, V.C.; Tabuchi, M.; Allan, S.M.; Knight, E.M.; LaFerla, F.M.; Oddo, S.; Verkhratsky, A. Impaired adult neurogenesis in the dentate gyrus of a triple transgenic mouse model of Alzheimer’s disease. PLoS ONE 2008, 3, e2935. [Google Scholar] [CrossRef] [PubMed]
- Kempermann, G. The neurogenic reserve hypothesis: What is adult hippocampal neurogenesis good for? Trends Neurosci. 2008, 31, 163–169. [Google Scholar] [CrossRef]
- Choi, S.H.; Bylykbashi, E.; Chatila, Z.K.; Lee, S.W.; Pulli, B.; Clemenson, G.D.; Kim, E.; Rompala, A.; Oram, M.K.; Asselin, C.; et al. Combined adult neurogenesis and BDNF mimic exercise effects on cognition in an Alzheimer’s mouse model. Science 2018, 361. [Google Scholar] [CrossRef]
- Zhang, R.; Zhang, L.; Zhang, Z.; Wang, Y.; Lu, M.; LaPointe, M.; Chopp, M. A nitric oxide donor induces neurogenesis and reduces functional deficits after stroke in rats. Ann. Neurol. 2001, 50, 602–611. [Google Scholar] [CrossRef]
- Lu, D.; Mahmood, A.; Zhang, R.; Li, Y.; Chopp, M. Upregulation of neurogenesis and reduction in functional deficits following administration of DETA/NONOate, a nitric oxide donor, after traumatic brain injury in rats. J. Neurosurg. 2003, 99, 351–361. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Zhang, R.; Katakowski, M.; Gautam, S.C.; Xu, Y.; Lu, M.; Zhang, Z.; Chopp, M. Combination therapy of stroke in rats with a nitric oxide donor and human bone marrow stromal cells enhances angiogenesis and neurogenesis. Brain Res. 2004, 1005, 21–28. [Google Scholar] [CrossRef]
- Reif, A.; Schmitt, A.; Fritzen, S.; Chourbaji, S.; Bartsch, C.; Urani, A.; Wycislo, M.; Mössner, R.; Sommer, C.; Gass, P.; et al. Differential effect of endothelial nitric oxide synthase (NOS-III) on the regulation of adult neurogenesis and behaviour. Eur. J. Neurosci. 2004, 885–895. [Google Scholar] [CrossRef]
- Chen, J.; Zacharek, A.; Zhang, C.; Jiang, H.; Li, Y.; Roberts, C.; Lu, M.; Kapke, A.; Chopp, M. Endothelial nitric oxide synthase regulates brain-derived neurotrophic factor expression and neurogenesis after stroke in mice. J. Neurosci. 2005, 25, 2366–2375. [Google Scholar] [CrossRef]
- Jin, X.; Yu, Z.F.; Chen, F.; Lu, G.X.; Ding, X.Y.; Xie, L.J.; Sun, J.T. Neuronal nitric oxide synthase in neural stem cells induces neuronal fate commitment via the inhibition of histone deacetylase 2. Front. Cell. Neurosci. 2017, 11, 66. [Google Scholar] [CrossRef] [PubMed]
- Kan, M.J.; Lee, J.E.; Wilson, J.G.; Everhart, A.L.; Brown, C.M.; Hoofnagle, A.N.; Jansen, M.; Vitek, M.P.; Gunn, M.D.; Colton, C.A. Arginine Deprivation and Immune Suppression in a Mouse Model of Alzheimer’s Disease. J. Neurosci. 2015, 35, 5969–5982. [Google Scholar] [CrossRef] [PubMed]
- Fonar, G.; Polis, B.; Meirson, T.; Maltsev, A.; Samson, A.O. Intracerebroventricular Administration of L-arginine Improves Spatial Memory Acquisition in Triple Transgenic Mice Via Reduction of Oxidative Stress and Apoptosis. Transl. Neurosci. 2018, 9, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Polis, B.; Samson, A.O. Arginase as a Potential Target in the Treatment of Alzheimer’s Disease. Adv. Alzheimer’s Dis. 2018, 7, 119–140. [Google Scholar] [CrossRef]
- Polis, B.; Srikanth, K.; Gurevich, V.; Gil-Henn, H.; Samson, A. L-Norvaline, a new therapeutic agent against Alzheimer’s disease. Neural Regen. Res. 2019, 14, 1562–1572. [Google Scholar] [CrossRef]
- Polis, B.; Srikanth, K.D.; Elliott, E.; Gil-Henn, H.; Samson, A.O. L-Norvaline Reverses Cognitive Decline and Synaptic Loss in a Murine Model of Alzheimer’s Disease. Neurotherapeutics 2018, 15, 1036–1054. [Google Scholar] [CrossRef]
- Polis, B.; Gurevich, V.; Assa, M.; Samson, A.O. Norvaline Restores the BBB Integrity in a Mouse Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2019, 20, 4616. [Google Scholar] [CrossRef]
- Mei, L.; Nave, K.A. Neuregulin-ERBB signaling in the nervous system and neuropsychiatric diseases. Neuron 2014, 83, 27–49. [Google Scholar] [CrossRef]
- Xu, J.; de Winter, F.; Farrokhi, C.; Rockenstein, E.; Mante, M.; Adame, A.; Cook, J.; Jin, X.; Masliah, E.; Lee, K.F. Neuregulin 1 improves cognitive deficits and neuropathology in an Alzheimer’s disease model. Sci. Rep. 2016, 6, 31692. [Google Scholar] [CrossRef]
- Mahar, I.; Tan, S.; Davoli, M.A.; Dominguez-Lopez, S.; Qiang, C.; Rachalski, A.; Turecki, G.; Mechawar, N. Subchronic peripheral neuregulin-1 increases ventral hippocampal neurogenesis and induces antidepressant-like effects. PLoS ONE 2011, 6, e26610. [Google Scholar] [CrossRef]
- Mahar, I.; Macisaac, A.; Kim, J.J.; Qiang, C.; Davoli, M.A.; Turecki, G.; Mechawar, N. Effects of neuregulin-1 administration on neurogenesis in the adult mouse hippocampus, and characterization of immature neurons along the septotemporal axis. Sci. Rep. 2016, 6, 30467. [Google Scholar] [CrossRef] [PubMed]
- Rosenstein, J.M.; Krum, J.M.; Ruhrberg, C. VEGF in the nervous system. Organogenesis 2010, 6, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Hohman, T.J.; Bell, S.P.; Jefferson, A.L. The Role of Vascular Endothelial Growth Factor in Neurodegeneration and Cognitive Decline: Exploring Interactions with Biomarkers of Alzheimer’s Disease. JAMA Neurol. 2015, 72, 520–529. [Google Scholar] [CrossRef] [PubMed]
- Licht, T.; Rothe, G.; Kreisel, T.; Wolf, B.; Benny, O.; Rooney, A.G.; Ffrench-Constant, C.; Enikolopov, G.; Keshet, E. VEGF preconditioning leads to stem cell remodeling and attenuates age-related decay of adult hippocampal neurogenesis. Proc. Natl. Acad. Sci. USA 2016, 113, E7828–E7836. [Google Scholar] [CrossRef]
- Sun, Y.; Jin, K.; Xie, L.; Childs, J.; Mao, X.O.; Logvinova, A.; Greenberg, D.A. VEGF-induced neuroprotection, neurogenesis, and angiogenesis after focal cerebral ischemia. J. Clin. Invest. 2003, 111, 1843–1851. [Google Scholar] [CrossRef] [PubMed]
- Airaksinen, M.S.; Saarma, M. The GDNF family: Signalling, biological functions and therapeutic value. Nat. Rev. Neurosci. 2002, 3, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Revilla, S.; Suñol, C.; García-Mesa, Y.; Giménez-Llort, L.; Sanfeliu, C.; Cristòfol, R. Physical exercise improves synaptic dysfunction and recovers the loss of survival factors in 3xTg-AD mouse brain. Neuropharmacology 2014, 81, 55–63. [Google Scholar] [CrossRef]
- Revilla, S.; Ursulet, S.; Álvarez-López, M.J.; Castro-Freire, M.; Perpiñá, U.; García-Mesa, Y.; Bortolozzi, A.; Giménez-Llort, L.; Kaliman, P.; Cristòfol, R.; et al. Lenti-GDNF Gene Therapy Protects Against Alzheimer’s Disease-Like Neuropathology in 3xTg-AD Mice and MC65 Cells. CNS Neurosci. Ther. 2014, 20, 961–972. [Google Scholar] [CrossRef]
- Allen, S.J.; Watson, J.J.; Shoemark, D.K.; Barua, N.U.; Patel, N.K. GDNF, NGF and BDNF as therapeutic options for neurodegeneration. Pharmacol. Ther. 2013, 138, 155–175. [Google Scholar] [CrossRef]
- Drinkut, A.; Tillack, K.; Meka, D.P.; Schulz, J.B.; Kügler, S.; Kramer, E.R. Ret is essential to mediate GDNF’s neuroprotective and neuroregenerative effect in a Parkinson disease mouse model. Cell Death Dis. 2016, 7, e2359. [Google Scholar] [CrossRef]
- Boku, S.; Nakagawa, S.; Takamura, N.; Kato, A.; Takebayashi, M.; Hisaoka-Nakashima, K.; Omiya, Y.; Inoue, T.; Kusumi, I. GDNF facilitates differentiation of the adult dentate gyrus-derived neural precursor cells into astrocytes via STAT3. Biochem. Biophys. Res. Commun. 2013, 434, 779–784. [Google Scholar] [CrossRef] [PubMed]
- Paratcha, G.; Ledda, F.; Ibáñez, C.F. The neural cell adhesion molecule NCAM is an alternative signaling receptor for GDNF family ligands. Cell 2003, 113, 867–879. [Google Scholar] [CrossRef]
- Lüthi, A.; Laurent, J.P.; Figurovt, A.; Mullert, D.; Schachnert, M. Hippocampal long-term potentiation and neural cell adhesion molecules L1 and NCAM. Nature 1994, 372, 777–779. [Google Scholar] [CrossRef]
- Iulita, M.F.; Cuello, A.C. Nerve growth factor metabolic dysfunction in Alzheimer’s disease and Down syndrome. Trends Pharmacol. Sci. 2014, 35, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Cheng, S.; Xu, G.; Ma, M.; Zhou, Z.; Liu, D.; Liu, X. Intranasal nerve growth factor enhances striatal neurogenesis in adult rats with focal cerebral ischemia. Drug Deliv. 2011, 18, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Eyjolfsdottir, H.; Eriksdotter, M.; Linderoth, B.; Lind, G.; Juliusson, B.; Kusk, P.; Almkvist, O.; Andreasen, N.; Blennow, K.; Ferreira, D.; et al. Targeted delivery of nerve growth factor to the cholinergic basal forebrain of Alzheimer’s disease patients: Application of a second-generation encapsulated cell biodelivery device. Alzheimer’s Res. Ther. 2016, 8, 30. [Google Scholar] [CrossRef]
- Nguyen, T.L.X.; Kim, C.K.; Cho, J.H.; Lee, K.H.; Ahn, J.Y. Neuroprotection signaling pathway of nerve growth factor and brain-derived neurotrophic factor against staurosporine induced apoptosis in hippocampal H19-7 cells. Exp. Mol. Med. 2010, 42, 583–595. [Google Scholar] [CrossRef]
- Schnell, E.; Long, T.H.; Bensen, A.L.; Washburn, E.K.; Westbrook, G.L. Neuroligin-1 knockdown reduces survival of adult-generated newborn hippocampal neurons. Front. Neurosci. 2014, 8, 71. [Google Scholar] [CrossRef]
- Schnell, E.; Bensen, A.S.L.; Washburn, E.K.; Westbrook, G.L. Neuroligin-1 Overexpression in Newborn Granule Cells In Vivo. PLoS ONE 2012, 7, e48045. [Google Scholar] [CrossRef][Green Version]
- Von Bohlen Und Halbach, O. Immunohistological markers for staging neurogenesis in adult hippocampus. Cell Tissue Res. 2007, 329, 409–420. [Google Scholar] [CrossRef]
- Brown, J.P.; Couillard-Després, S.; Cooper-Kuhn, C.M.; Winkler, J.; Aigner, L.; Kuhn, H.G. Transient Expression of Doublecortin during Adult Neurogenesis. J. Comp. Neurol. 2003. [Google Scholar] [CrossRef] [PubMed]
- Friocourt, G.; Koulakoff, A.; Chafey, P.; Boucher, D.; Fauchereau, F.; Chelly, J.; Francis, F. Doublecortin functions at the extremities of growing neuronal processes. Cereb. Cortex. 2003, 13, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.S.; Shetty, A.K. Efficacy of doublecortin as a marker to analyse the absolute number and dendritic growth of newly generated neurons in the adult dentate gyrus. Eur. J. Neurosci. 2004, 19, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Caceres, A.; Banker, G.A.; Binder, L. Immunocytochemical localization of tubulin and microtubule-associated protein 2 during the development of hippocampal neurons in culture. J. Neurosci. 1986, 6, 714–722. [Google Scholar] [CrossRef]
- Chang, A.; Smith, M.C.; Yin, X.; Fox, R.J.; Staugaitis, S.M.; Trapp, B.D. Neurogenesis in the chronic lesions of multiple sclerosis. Brain 2008, 131 Pt 9, 2366–2375. [Google Scholar] [CrossRef]
- Wang, F.; Baba, N.; Shen, Y.; Yamashita, T.; Tsuru, E.; Tsuda, M.; Maeda, N.; Sagara, Y. CCL11 promotes migration and proliferation of mouse neural progenitor cells. Stem Cell Res. Ther. 2017, 8, 26. [Google Scholar] [CrossRef]
- Nacher, J.; Varea, E.; Blasco-Ibañez, J.M.; Castillo-Gomez, E.; Crespo, C.; Martinez-Guijarro, F.J.; McEwen, B.S. Expression of the transcription factor Pax6 in the adult rat dentate gyrus. J. Neurosci. Res. 2005, 81, 753–761. [Google Scholar] [CrossRef]
- de Jesus, D.F.; Zhang, Z.; Kahraman, S.; Brown, N.K.; Chen, M.; Hu, J.; Gupta, M.K.; He, C.; Kulkarni, R.N. m6A mRNA methylation regulates human β-cell biology in physiological states and in type 2 diabetes. Nat. Metab. 2019, 1, 765–774. [Google Scholar] [CrossRef]
- Raivich, G.; Bohatschek, M.; da Costa, C.; Iwata, O.; Galiano, M.; Hristova, M.; Nateri, A.S.; Makwana, M.; Riera-Sans, L.; Wolfer, D.P.; et al. The AP-1 transcription factor c-Jun is required for efficient axonal regeneration. Neuron 2004, 43, 57–67. [Google Scholar] [CrossRef]
- Gao, B.; Lee, S.M.; Fang, D. The tyrosine kinase c-Abl protects c-Jun from ubiquitination-mediated degradation in T cells. J. Biol. Chem. 2006, 281, 29711–29718. [Google Scholar] [CrossRef]
- Kravtsova-Ivantsiv, Y.; Ciechanover, A. The ubiquitin-proteasome system and activation of NF-κB: Involvement of the ubiquitin ligase KPC1 in p105 processing and tumor suppression. Mol. Cell. Oncol. 2015, 2, e1054552. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, J.; Yao, S.; Li, F.; Xin, L.; Lai, M.; Bracchi-Ricard, V.; Xu, H.; Yen, W.; Meng, W.; et al. Nuclear factor kappa B signaling initiates early differentiation of neural stem cells. Stem Cells. 2012, 30, 510–524. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.; Lahiri, D.K. Cdk5 activity in the brain―multiple paths of regulation. J. Cell Sci. 2014. [Google Scholar] [CrossRef] [PubMed]
- Kanungo, J.; Zheng, Y.L.; Amin, N.D.; Pant, H.C. Targeting Cdk5 activity in neuronal degeneration and regeneration. Cell. Mol. Neurobiol. 2009, 127 Pt 11, 2391–2400. [Google Scholar] [CrossRef]
- Giraldo, E.; Lloret, A.; Fuchsberger, T.; Viña, J. Aβ and tau toxicities in Alzheimer’s are linked via oxidative stress-induced p38 activation: Protective role of vitamin E. Redox Biol. 2014, 2, 873–877. [Google Scholar] [CrossRef]
- Lee, J.K.; Kim, N.J. Recent advances in the inhibition of p38 MAPK as a potential strategy for the treatment of Alzheimer’s disease. Molecules 2017, 22, 1287. [Google Scholar] [CrossRef]
- Huynh, Q.K. Evidence for the phosphorylation of serine259 of histone deacetylase 5 by protein kinase Cδ. Arch. Biochem. Biophys. 2011, 506, 173–180. [Google Scholar] [CrossRef]
- Xu, K.; Dai, X.L.; Huang, H.C.; Jiang, Z.F. Targeting HDACs: A promising therapy for Alzheimer’s disease. Oxid. Med. Cell. Longev. 2011, 2011, 143269. [Google Scholar] [CrossRef]
- Yang, S.S.; Zhang, R.; Wang, G.; Zhang, Y.F. The development prospection of HDAC inhibitors as a potential therapeutic direction in Alzheimer’s disease. Transl. Neurodegener. 2017, 6, 19. [Google Scholar] [CrossRef]
- Grozinger, C.M.; Schreiber, S.L. Regulation of histone deacetylase 4 and 5 and transcriptional activity by 14-3-3-dependent cellular localization. Proc. Natl. Acad. Sci. USA 2000, 97, 7835–7840. [Google Scholar] [CrossRef]
- Mielcarek, M.; Zielonka, D.; Carnemolla, A.; Marcinkowski, J.T.; Guidez, F. HDAC4 as a potential therapeutic target in neurodegenerative diseases: A summary of recent achievements. Front. Cell. Neurosci. 2015, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Kilgore, M.; Miller, C.A.; Fass, D.M.; Hennig, K.M.; Haggarty, S.J.; Sweatt, J.D.; Rumbaugh, G. Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of alzheimer’s disease. Neuropsychopharmacology 2010, 35, 870–880. [Google Scholar] [CrossRef] [PubMed]
- Bull, N.D.; Bartlett, P.F. The adult mouse hippocampal progenitor is neurogenic but not a stem cell. J. Neurosci. 2005, 25, 10815–10821. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Tanzi, R.E. Is Alzheimer’s Disease a Neurogenesis Disorder? Cell Stem Cell 2019, 25, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Taniuchi, N.; Niidome, T.; Goto, Y.; Akaike, A.; Kihara, T.; Sugimoto, H. Decreased proliferation of hippocampal progenitor cells in APPswe/PS1dE9 transgenic mice. Neuroreport 2007, 18, 1801–1805. [Google Scholar] [CrossRef]
- Kempermann, G.; Fabel, K.; Ehninger, D.; Babu, H.; Leal-Galicia, P.; Garthe, A.; Wolf, S.A. Why and how physical activity promotes experience-induced brain plasticity. Front. Neurosci. 2010, 4, 189. [Google Scholar] [CrossRef]
- Pérez-González, R.; Antequera, D.; Vargas, T.; Spuch, C.; Bolós, M.; Carro, E. Leptin induces proliferation of neuronal progenitors and neuroprotection in a mouse model of alzheimer’s disease. J. Alzheimer’s Dis. 2011, 24 (Suppl. 2), 17–25. [Google Scholar] [CrossRef]
- Marlatt, M.W.; Potter, M.C.; Bayer, T.A.; van Praag, H.; Lucassen, P.J. Prolonged running, not fluoxetine treatment, increases neurogenesis, but does not alter neuropathology, in the 3xTg mouse model of alzheimer’s disease. Curr. Top. Behav. Neurosci. 2013, 24 (Suppl. 2), 17–25. [Google Scholar] [CrossRef]
- Chai, G.S.; Wang, Y.Y.; Yasheng, A.; Zhao, P. Beta 2-adrenergic receptor activation enhances neurogenesis in Alzheimer’s disease mice. Neural Regen. Res. 2016, 11, 1617–1624. [Google Scholar] [CrossRef]
- Chong, C.M.; Ai, N.; Ke, M.; Tan, Y.; Huang, Z.; Li, Y.; Lu, J.H.; Ge, W.; Su, H. Roles of Nitric Oxide Synthase Isoforms in Neurogenesis. Mol. Neurobiol. 2018, 55, 2645–2652. [Google Scholar] [CrossRef]
- Mahmoudi, R.; Enant, E.; Delaviz, H.; Rad, P.; Roozbehi, A.; Barmak, M.J.; Azizi, A. The effects of l-arginine on the hippocampus of male rat fetuses under maternal stress. Basic Clin. Neurosci. 2016, 7, 5–11. [Google Scholar] [PubMed]
- Seki, T. Hippocampal adult neurogenesis occurs in a microenvironment provided by PSA-NCAM-expressing immature neurons. J. Neurosci. Res. 2002, 69, 772–783. [Google Scholar] [CrossRef] [PubMed]
- Casadesus, G.; Shukitt-Hale, B.; Stellwagen, H.M.; Smith, M.A.; Rabin, B.M.; Joseph, J.A. Hippocampal neurogenesis and PSA-NCAM expression following exposure to 56Fe particles mimics that seen during aging in rats. Exp. Gerontol. 2005, 40, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Vutskits, L.; Gascon, E.; Kiss, J.Z. Removal of PSA from NCAM affects the survival of magnocellular vasopressin- and oxytocin-producing neurons in organotypic cultures of the paraventricular nucleus. Eur. J. Neurosci. 2003, 17, 2119–2126. [Google Scholar] [CrossRef] [PubMed]
- Gascon, E.; Vutskits, L.; Jenny, B.; Durbec, P.; Kiss, J.Z. PSA-NCAM in postnatally generated immature neurons of the olfactory bulb: A crucial role in regulating p75 expression and cell survival. Development 2007, 134, 1181–1190. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gascon, E.; Vutskits, L.; Kiss, J.Z. The role of PSA-NCAM in adult neurogenesis. Adv. Exp. Med. Biol. 2010, 663, 127–136. [Google Scholar] [CrossRef]
- Gray, S.C.; Kinghorn, K.J.; Woodling, N.S. Shifting equilibriums in Alzheimer’s disease: The complex roles of microglia in neuroinflammation, neuronal survival and neurogenesis. Neural Regen. Res. 2019, 15, 1208–1219. [Google Scholar] [CrossRef]
- Lipton, S.A.; Rezaie, T.; Nutter, A.; Lopez, K.M.; Parker, J.; Kosaka, K.; Satoh, T.; McKercher, S.R.; Masliah, E.; Nakanishi, N. Therapeutic advantage of pro-electrophilic drugs to activate the Nrf2/ARE pathway in Alzheimer’s disease models. Cell Death Dis. 2016, 7, e2499. [Google Scholar] [CrossRef]
- Venero, C.; Herrero, A.I.; Touyarot, K.; Cambon, K.; López-Fernández, M.A.; Berezin, V.; Bock, E.; Sandi, C. Hippocampal up-regulation of NCAM expression and polysialylation plays a key role on spatial memory. Eur. J. Neurosci. 2006, 23, 1585–1595. [Google Scholar] [CrossRef]
- Fifre, A.; Sponne, I.; Koziel, V.; Kriem, B.; Potin, F.T.Y.; Bihain, B.E.; Olivier, J.L.; Oster, T.; Pillot, T. Microtubule-associated protein MAP1A, MAP1B, and MAP2 proteolysis during soluble amyloid β-peptide-induced neuronal apoptosis: Synergistic involvement of calpain and caspase-3. J. Biol. Chem. 2006, 281, 229–240. [Google Scholar] [CrossRef]
- Adlard, P.A.; Vickers, J.C. Morphologically distinct plaque types differentially affect dendritic structure and organisation in the early and late states of Alzheimer’s disease. Acta Neuropathol. 2002, 103, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Lin, L.; Liu, Z.; Ji, F.; Shao, W.; Wang, M.; Liu, L.; Li, S.; Li, F.; Bu, X. Potential therapeutic effects of curcumin: Relationship to microtubule-associated proteins 2 in Aβ1-42 insult. Brain Res. 2010, 1361, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Bittner, T.; Fuhrmann, M.; Burgold, S.; Ochs, S.M.; Hoffmann, N.; Mitteregger, G.; Kretzschmar, H.; Laferla, F.M.; Herms, J. Multiple events lead to dendritic spine loss in triple transgenic Alzheimer’s disease mice. PLoS ONE 2010, 5, e15477. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Robbins, D.; Frost, J.; Dang, A.; Lange-Carter, C.; Cobb, M.H. MEKK1 phosphorylates MEK1 and MEK2 but does not cause activation of mitogen-activated protein kinase. Proc. Natl. Acad. Sci. USA 1995, 92, 6808–6812. [Google Scholar] [CrossRef]
- Morrison, R.S.; Kornblum, H.I.; Leslie, F.M.; Bradshaw, R.A. Trophic stimulation of cultured neurons from neonatal rat brain by epidermal growth factor. Science 1987, 238, 72–75. [Google Scholar] [CrossRef]
- Li, X.; Newbern, J.M.; Wu, Y.; Morgan-Smith, M.; Zhong, J.; Charron, J.; Snider, W.D. MEK Is a Key Regulator of Gliogenesis in the Developing. Brain. Neuron. 2012, 75, 1035–1050. [Google Scholar] [CrossRef]
- Guix, F.X.; Uribesalgo, I.; Coma, M.; Muñoz, F.J. The physiology and pathophysiology of nitric oxide in the brain. Prog. Neurobiol. 2005, 76, 126–152. [Google Scholar] [CrossRef]
- Carpentier, P.A.; Palmer, T.D. Immune Influence on Adult Neural Stem Cell Regulation and Function. Neuron 2009, 64, 79–92. [Google Scholar] [CrossRef]
- Iosif, R.E.; Ekdahl, C.T.; Ahlenius, H.; Pronk, C.J.H.; Bonde, S.; Kokaia, Z.; Jacobsen, S.E.W.; Lindvall, O. Tumor necrosis factor receptor 1 is a negative regulator of progenitor proliferation in adult hippocampal neurogenesis. J. Neurosci. 2006, 26, 9703–9712. [Google Scholar] [CrossRef]
- Adzemovic, M.Z.; Öckinger, J.; Zeitelhofer, M.; Hochmeister, S.; Beyeen, A.D.; Paulson, A.; Gillett, A.; Hedreul, M.T.; Covacu, R.; Lassmann, H.; et al. Expression of Ccl11 associates with immune response modulation and protection against neuroinflammation in rats. PLoS ONE 2012, 7, e39794. [Google Scholar] [CrossRef]
- Osumi, N.; Shinohara, H.; Numayama-Tsuruta, K.; Maekawa, M. Concise Review: Pax6 Transcription Factor Contributes to both Embryonic and Adult Neurogenesis as a Multifunctional Regulator. Stem Cells 2008, 26, 1663–1672. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, M.; Takashima, N.; Arai, Y.; Nomura, T.; Inokuchi, K.; Yuasi, S.; Osumi, N. Pax6 is required for production and maintenance of progenitor cells in postnatal hippocampal neurogenesis. Genes Cells 2005, 10, 1001–1014. [Google Scholar] [CrossRef] [PubMed]
- Maurya, S.K.; Mishra, R. Pax6 binds to promoter sequence elements associated with immunological surveillance and energy homeostasis in brain of aging mice. Ann. Neurosci. 2017, 24, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, R.; Mishra, R. Aging-associated modulation in the expression of pax6 in mouse brain. Cell. Mol. Neurobiol. 2012, 32, 209–218. [Google Scholar] [CrossRef]
- Srivastava, K.; Tripathi, R.; Mishra, R. Age-dependent alterations in expression and co-localization of Pax6 and Ras-GAP in brain of aging mice. J. Chem. Neuroanat. 2018, 92, 25–34. [Google Scholar] [CrossRef]
- Hunter, A.; Downs, E. The inhibition of arginase by amino acids. J. Biol. Chem. 1945, 157, 427–446. [Google Scholar]
- Sparapani, M.; Dall’Olio, R.; Gandolfi, O.; Ciani, E.; Contestabile, A. Neurotoxicity of Polyamines and Pharmacological Neuroprotection in Cultures of Rat Cerebellar Granule Cells. Exp. Neurol. 1997, 148, 157–166. [Google Scholar] [CrossRef]
- Stewart, T.M.; Dunston, T.T.; Woster, P.M.; Casero, R.A. Polyamine catabolism and oxidative damage. J. Biol. Chem. 2018, 293, 18736–18745. [Google Scholar] [CrossRef]
- Polis, B.; Samson, A. A New Perspective on Alzheimer’s Disease as a Brain Expression of a Complex Metabolic Disorder. In Alzheimer’s Disease, 1st ed.; Wisniewski, T., Ed.; Codon Publication: Brisbane, Australia, 2019; pp. 1–22. [Google Scholar] [CrossRef][Green Version]
- Poluha, W.; Schonhoff, C.M.; Harrington, K.S.; Lachyankar, M.B.; Crosbie, N.E.; Bulseco, D.A.; Ross, A.H. A novel, nerve growth factor-activated pathway involving nitric oxide, p53, and p21(WAF1) regulates neuronal differentiation of PC12 cells. J. Biol. Chem. 1997, 272, 24002–24007. [Google Scholar] [CrossRef]
- Laube, G.; Bernstein, H.G. Agmatine: Multifunctional arginine metabolite and magic bullet in clinical neuroscience? Biochem. J. 2017, 474, 2619–2640. [Google Scholar] [CrossRef]
- Li, Y.F.; Chen, H.X.; Liu, Y.; Zhang, Y.Z.; Liu, Y.Q.; Li, J. Agmatine increases proliferation of cultured hippocampal progenitor cells and hippocampal neurogenesis in chronically stressed mice. Acta Pharmacol. Sin. 2006, 27, 1395–1400. [Google Scholar] [CrossRef]
- Kuo, J.R.; Lo, C.J.; Chang, C.P.; Lin, K.C.; Lin, M.T.; Chio, C.C. Agmatine-promoted angiogenesis, neurogenesis, and inhibition of gliosis-reduced traumatic brain injury in rats. J. Trauma. 2011, 71, E87–E93. [Google Scholar] [CrossRef] [PubMed]
- Song, H.W.; Kumar, B.K.; Kim, S.H.; Jeon, Y.H.; Lee, Y.A.; Lee, W.T.; Park, K.A.; Lee, J.E. Agmatine enhances neurogenesis by increasing ERK1/2 expression, and suppresses astrogenesis by decreasing BMP 2,4 and SMAD 1,5,8 expression in subventricular zone neural stem cells. Life Sci. 2011, 89, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s Disease with plaques and tangles: Intracellular Aβ and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef]
- Albus, U. Guide for the Care and Use of Laboratory Animals (8th edn). Lab. Anim. 2012. [Google Scholar] [CrossRef]
- Bonfanti, L. PSA-NCAM in mammalian structural plasticity and neurogenesis. Prog. Neurobiol. 2006, 80, 129–164. [Google Scholar] [CrossRef]
- Seki, T.; Arai, Y. The persistent expression of a highly polysialylated NCAM in the dentate gyrus of the adult rat. Neurosci. Res. 1991, 12, 503–513. [Google Scholar] [CrossRef]
- Dehmelt, L.; Halpain, S. Protein family review The MAP2/Tau family of microtubule-associated proteins. Genome Biol. 2004, 6, 204. Available online: http://genomebiology.com/2004/6/1/204 (accessed on 10 September 2019). [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Cheadle, C.; Vawter, M.P.; Freed, W.J.; Becker, K.G. Analysis of microarray data using Z score transformation. J. Mol. Diagn. 2003, 5, 73–81. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Polis, B.; Srikanth, K.D.; Gurevich, V.; Bloch, N.; Gil-Henn, H.; Samson, A.O. Arginase Inhibition Supports Survival and Differentiation of Neuronal Precursors in Adult Alzheimer’s Disease Mice. Int. J. Mol. Sci. 2020, 21, 1133. https://doi.org/10.3390/ijms21031133
Polis B, Srikanth KD, Gurevich V, Bloch N, Gil-Henn H, Samson AO. Arginase Inhibition Supports Survival and Differentiation of Neuronal Precursors in Adult Alzheimer’s Disease Mice. International Journal of Molecular Sciences. 2020; 21(3):1133. https://doi.org/10.3390/ijms21031133
Chicago/Turabian StylePolis, Baruh, Kolluru D. Srikanth, Vyacheslav Gurevich, Naamah Bloch, Hava Gil-Henn, and Abraham O. Samson. 2020. "Arginase Inhibition Supports Survival and Differentiation of Neuronal Precursors in Adult Alzheimer’s Disease Mice" International Journal of Molecular Sciences 21, no. 3: 1133. https://doi.org/10.3390/ijms21031133
APA StylePolis, B., Srikanth, K. D., Gurevich, V., Bloch, N., Gil-Henn, H., & Samson, A. O. (2020). Arginase Inhibition Supports Survival and Differentiation of Neuronal Precursors in Adult Alzheimer’s Disease Mice. International Journal of Molecular Sciences, 21(3), 1133. https://doi.org/10.3390/ijms21031133