Defeating Antibiotic-Resistant Bacteria: Exploring Alternative Therapies for a Post-Antibiotic Era




Abstract
1. Introduction
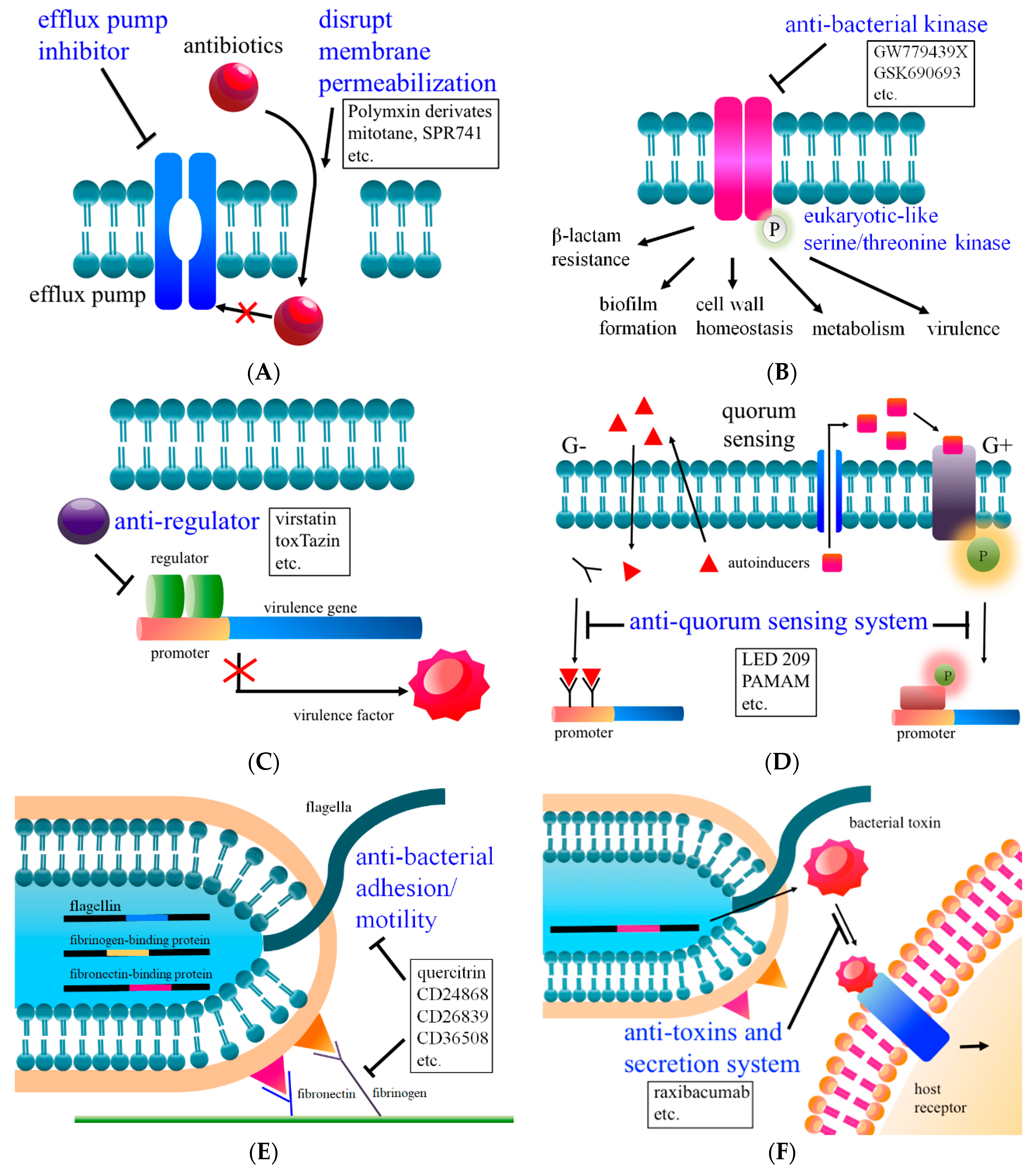
2. Combination Therapy I: Increases Membrane Permeability
3. Combination Therapy II: Reduces Efflux Pump Activity
4. Combination Therapy III: Inhibits Kinase Activity and Intrinsic Antibiotic Resistance
5. Anti-Regulators
6. Anti-Quorum Sensing System
7. Anti-Bacterial Virulence Factors (Adhesion and Motility)
8. Anti-Toxins and Secretion System
9. Engineered Bacteriophage
10. Modulate the Microbiome
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cizman, M.; Plankar Srovin, T. Antibiotic consumption and resistance of gram-negative pathogens (collateral damage). GMS Infect. Dis. 2018, 6, Doc05. [Google Scholar]
- Woolhouse, M.; Waugh, C.; Perry, M.R.; Nair, H. Global disease burden due to antibiotic resistance - state of the evidence. J. Glob. Health 2016, 6, 010306. [Google Scholar] [CrossRef]
- Zhen, X.; Lundborg, C.S.; Sun, X.; Hu, X.; Dong, H. Economic burden of antibiotic resistance in ESKAPE organisms: A systematic review. Antimicrob. Resist. Infect. Control. 2019, 8, 137. [Google Scholar] [CrossRef]
- Rice, L.B. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: No ESKAPE. J. Infect. Dis. 2008, 197, 1079–1081. [Google Scholar] [CrossRef] [PubMed]
- Marturano, J.E.; Lowery, T.J. ESKAPE Pathogens in Bloodstream Infections Are Associated With Higher Cost and Mortality but Can Be Predicted Using Diagnoses Upon Admission. Open Forum Infect. Dis. 2019, 6, ofz503. [Google Scholar] [CrossRef]
- Magiorakos, A.P.; Srinivasan, A.; Carey, R.B.; Carmeli, Y.; Falagas, M.E.; Giske, C.G.; Harbarth, S.; Hindler, J.F.; Kahlmeter, G.; Olsson-Liljequist, B.; et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: An international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 2012, 18, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, V.; Dupont, H.; Glorion, S.; Aupee, M.; Kipnis, E.; Gerard, J.L.; Hanouz, J.L.; Fischer, M.O. Influence of bacterial resistance on mortality in intensive care units: A registry study from 2000 to 2013 (IICU Study). J. Hosp. Infect. 2019, 102, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Naylor, N.R.; Pouwels, K.B.; Hope, R.; Green, N.; Henderson, K.L.; Knight, G.M.; Atun, R.; Robotham, J.V.; Deeny, S.R. The health and cost burden of antibiotic resistant and susceptible Escherichia coli bacteraemia in the English hospital setting: A national retrospective cohort study. PLoS ONE 2019, 14, e0221944. [Google Scholar] [CrossRef] [PubMed]
- Blair, J.M.; Webber, M.A.; Baylay, A.J.; Ogbolu, D.O.; Piddock, L.J. Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 2015, 13, 42–51. [Google Scholar] [CrossRef]
- Lerminiaux, N.A.; Cameron, A.D.S. Horizontal transfer of antibiotic resistance genes in clinical environments. Can. J. Microbiol. 2019, 65, 34–44. [Google Scholar] [CrossRef]
- Munita, J.M.; Arias, C.A. Mechanisms of Antibiotic Resistance. Microbiol. Spectr. 2016, 4, 34. [Google Scholar] [CrossRef] [PubMed]
- Osei Sekyere, J.; Govinden, U.; Bester, L.A.; Essack, S.Y. Colistin and tigecycline resistance in carbapenemase-producing Gram-negative bacteria: Emerging resistance mechanisms and detection methods. J. Appl. Microbiol. 2016, 121, 601–617. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, P.; Zhao, D.; Jiang, Y.; Zhao, F.; Wang, Y.; Li, X.; Du, X.; Yu, Y. Emergence of tigecycline resistance in Escherichia coli co-producing MCR-1 and NDM-5 during tigecycline salvage treatment. Infect. Drug Resist. 2018, 11, 2241–2248. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Dong, N.; Huang, Y.; Zhou, H.; Xie, M.; Chan, E.W.; Hu, Y.; Cai, J.; Chen, S. Evolution of tigecycline- and colistin-resistant CRKP (carbapenem-resistant Klebsiella pneumoniae) in vivo and its persistence in the GI tract. Emerg. Microbes. Infect. 2018, 7, 127. [Google Scholar] [CrossRef]
- Juan, C.H.; Huang, Y.W.; Lin, Y.T.; Yang, T.C.; Wang, F.D. Risk Factors, Outcomes, and Mechanisms of Tigecycline-Nonsusceptible Klebsiella pneumoniae Bacteremia. Antimicrob. Agents Chemother. 2016, 60, 7357–7363. [Google Scholar]
- Rojas, L.J.; Salim, M.; Cober, E.; Richter, S.S.; Perez, F.; Salata, R.A.; Kalayjian, R.C.; Watkins, R.R.; Marshall, S.; Rudin, S.D.; et al. Colistin Resistance in Carbapenem-Resistant Klebsiella pneumoniae: Laboratory Detection and Impact on Mortality. Clin. Infect. Dis. 2017, 64, 711–718. [Google Scholar] [CrossRef]
- Dijkmans, A.C.; Wilms, E.B.; Kamerling, I.M.; Birkhoff, W.; Ortiz-Zacarias, N.V.; van Nieuwkoop, C.; Verbrugh, H.A.; Touw, D.J. Colistin: Revival of an Old Polymyxin Antibiotic. Ther. Drug Monit. 2015, 37, 419–427. [Google Scholar] [CrossRef]
- Moffatt, J.H.; Harper, M.; Boyce, J.D. Mechanisms of Polymyxin Resistance. Adv. Exp. Med. Biol. 2019, 1145, 55–71. [Google Scholar]
- Akiyama, T.; Presedo, J.; Khan, A.A. The tetA gene decreases tigecycline sensitivity of Salmonella enterica isolates. Int. J. Antimicrob. Agents 2013, 42, 133–140. [Google Scholar] [CrossRef]
- Taniguchi, Y.; Maeyama, Y.; Ohsaki, Y.; Hayashi, W.; Osaka, S.; Koide, S.; Tamai, K.; Nagano, Y.; Arakawa, Y.; Nagano, N. Co-resistance to colistin and tigecycline by disrupting mgrB and ramR with IS insertions in a canine Klebsiella pneumoniae ST37 isolate producing SHV-12, DHA-1 and FosA3. Int. J. Antimicrob. Agents 2017, 50, 697–698. [Google Scholar] [CrossRef]
- O’Neill, J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations; Review on Antimicrobial Resistance; Wellcome Trust and HM Government: London, UK, 2016; pp. 1–84. [Google Scholar]
- Coates, A.R.; Halls, G.; Hu, Y. Novel classes of antibiotics or more of the same? Br. J. Pharmacol. 2011, 163, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Gwynn, M.N.; Portnoy, A.; Rittenhouse, S.F.; Payne, D.J. Challenges of antibacterial discovery revisited. Ann. N. Y. Acad. Sci. 2010, 1213, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.B.; Wang, J.; Doi, Y.; Velkov, T.; Bergen, P.J.; Li, J. Novel Polymyxin Combination With Antineoplastic Mitotane Improved the Bacterial Killing Against Polymyxin-Resistant Multidrug-Resistant Gram-Negative Pathogens. Front. Microbiol. 2018, 9, 721. [Google Scholar] [CrossRef] [PubMed]
- Corbett, D.; Wise, A.; Langley, T.; Skinner, K.; Trimby, E.; Birchall, S.; Dorali, A.; Sandiford, S.; Williams, J.; Warn, P.; et al. Potentiation of Antibiotic Activity by a Novel Cationic Peptide: Potency and Spectrum of Activity of SPR741. Antimicrob. Agents Chemother. 2017, 61, e00200-17. [Google Scholar] [CrossRef]
- Zurawski, D.V.; Reinhart, A.A.; Alamneh, Y.A.; Pucci, M.J.; Si, Y.; Abu-Taleb, R.; Shearer, J.P.; Demons, S.T.; Tyner, S.D.; Lister, T. SPR741, an Antibiotic Adjuvant, Potentiates the In Vitro and In Vivo Activity of Rifampin against Clinically Relevant Extensively Drug-Resistant Acinetobacter baumannii. Antimicrob. Agents Chemother. 2017, 61, e01239-17. [Google Scholar] [CrossRef]
- Mendes, R.E.; Rhomberg, P.R.; Lister, T.; Cotroneo, N.; Rubio, A.; Flamm, R.K. Evaluation of Antimicrobial Effects of a New Polymyxin Molecule (SPR741) When Tested in Combination with a Series of beta-Lactam Agents Against a Challenge Set of Gram-Negative Pathogens. Microb. Drug Resist. 2019. [Google Scholar] [CrossRef]
- Stainton, S.M.; Abdelraouf, K.; Utley, L.; Pucci, M.J.; Lister, T.; Nicolau, D.P. Assessment of the In Vivo Activity of SPR741 in Combination with Azithromycin against Multidrug-Resistant Enterobacteriaceae Isolates in the Neutropenic Murine Thigh Infection Model. Antimicrob. Agents Chemother. 2018, 62, e00239-18. [Google Scholar] [CrossRef]
- Sugawara, E.; Kojima, S.; Nikaido, H. Klebsiella pneumoniae Major Porins OmpK35 and OmpK36 Allow More Efficient Diffusion of beta-Lactams than Their Escherichia coli Homologs OmpF and OmpC. J. Bacteriol. 2016, 198, 3200–3208. [Google Scholar] [CrossRef]
- Ye, Y.; Xu, L.; Han, Y.; Chen, Z.; Liu, C.; Ming, L. Mechanism for carbapenem resistance of clinical Enterobacteriaceae isolates. Exp. Ther. Med. 2018, 15, 1143–1149. [Google Scholar] [CrossRef]
- Barrero, M.A.; Pietralonga, P.A.; Schwarz, D.G.; Silva, A., Jr.; Paula, S.O.; Moreira, M.A. Effect of the inhibitors phenylalanine arginyl ss-naphthylamide (PAssN) and 1-(1-naphthylmethyl)-piperazine (NMP) on expression of genes in multidrug efflux systems of Escherichia coli isolates from bovine mastitis. Res. Vet. Sci. 2014, 97, 176–181. [Google Scholar] [CrossRef]
- Lomovskaya, O.; Warren, M.S.; Lee, A.; Galazzo, J.; Fronko, R.; Lee, M.; Blais, J.; Cho, D.; Chamberland, S.; Renau, T.; et al. Identification and characterization of inhibitors of multidrug resistance efflux pumps in Pseudomonas aeruginosa: Novel agents for combination therapy. Antimicrob. Agents Chemother. 2001, 45, 105–116. [Google Scholar] [CrossRef]
- Negi, N.; Prakash, P.; Gupta, M.L.; Mohapatra, T.M. Possible Role of Curcumin as an Efflux Pump Inhibitor in Multi Drug Resistant Clinical Isolates of Pseudomonas aeruginosa. J. Clin. Diagn. Res. 2014, 8, DC04-7. [Google Scholar] [CrossRef]
- Carabajal, M.A.; Asquith, C.R.M.; Laitinen, T.; Tizzard, G.J.; Yim, L.; Rial, A.; Chabalgoity, J.A.; Zuercher, W.J.; Garcia Vescovi, E. Quinazoline-based anti-virulence compounds selectively target Salmonella PhoP/PhoQ signal transduction system. Antimicrob. Agents Chemother. 2019. [Google Scholar] [CrossRef] [PubMed]
- Schaenzer, A.J.; Wlodarchak, N.; Drewry, D.H.; Zuercher, W.J.; Rose, W.E.; Ferrer, C.A.; Sauer, J.D.; Striker, R. GW779439X and Its Pyrazolopyridazine Derivatives Inhibit the Serine/Threonine Kinase Stk1 and Act As Antibiotic Adjuvants against beta-Lactam-Resistant Staphylococcus aureus. ACS Infect. Dis. 2018, 4, 1508–1518. [Google Scholar] [CrossRef] [PubMed]
- Hung, D.T.; Shakhnovich, E.A.; Pierson, E.; Mekalanos, J.J. Small-molecule inhibitor of Vibrio cholerae virulence and intestinal colonization. Science 2005, 310, 670–674. [Google Scholar] [CrossRef] [PubMed]
- Shakhnovich, E.A.; Hung, D.T.; Pierson, E.; Lee, K.; Mekalanos, J.J. Virstatin inhibits dimerization of the transcriptional activator ToxT. Proc. Natl. Acad. Sci. USA 2007, 104, 2372–2377. [Google Scholar] [CrossRef] [PubMed]
- Rasko, D.A.; Moreira, C.G.; Li de, R.; Reading, N.C.; Ritchie, J.M.; Waldor, M.K.; Williams, N.; Taussig, R.; Wei, S.; Roth, M.; et al. Targeting QseC signaling and virulence for antibiotic development. Science 2008, 321, 1078–1080. [Google Scholar] [CrossRef]
- Liu, B.; Chen, F.; Bi, C.; Wang, L.; Zhong, X.; Cai, H.; Deng, X.; Niu, X.; Wang, D. Quercitrin, an inhibitor of Sortase A, interferes with the adhesion of Staphylococcal aureus. Molecules 2015, 20, 6533–6543. [Google Scholar] [CrossRef]
- Wang, J.; Song, M.; Pan, J.; Shen, X.; Liu, W.; Zhang, X.; Li, H.; Deng, X. Quercetin impairs Streptococcus pneumoniae biofilm formation by inhibiting sortase A activity. J. Cell. Mol. Med. 2018, 22, 6228–6237. [Google Scholar] [CrossRef]
- Li, X.Z.; Plesiat, P.; Nikaido, H. The challenge of efflux-mediated antibiotic resistance in Gram-negative bacteria. Clin. Microbiol. Rev. 2015, 28, 337–418. [Google Scholar] [CrossRef]
- Puzari, M.; Chetia, P. RND efflux pump mediated antibiotic resistance in Gram-negative bacteria Escherichia coli and Pseudomonas aeruginosa: A major issue worldwide. World J. Microbiol. Biotechnol. 2017, 33, 24. [Google Scholar] [CrossRef] [PubMed]
- Blanco, P.; Hernando-Amado, S.; Reales-Calderon, J.A.; Corona, F.; Lira, F.; Alcalde-Rico, M.; Bernardini, A.; Sanchez, M.B.; Martinez, J.L. Bacterial Multidrug Efflux Pumps: Much More Than Antibiotic Resistance Determinants. Microorganisms 2016, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- Mao, W.; Warren, M.S.; Black, D.S.; Satou, T.; Murata, T.; Nishino, T.; Gotoh, N.; Lomovskaya, O. On the mechanism of substrate specificity by resistance nodulation division (RND)-type multidrug resistance pumps: The large periplasmic loops of MexD from Pseudomonas aeruginosa are involved in substrate recognition. Mol. Microbiol. 2002, 46, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Yoon, E.J.; Courvalin, P.; Grillot-Courvalin, C. RND-type efflux pumps in multidrug-resistant clinical isolates of Acinetobacter baumannii: Major role for AdeABC overexpression and AdeRS mutations. Antimicrob. Agents Chemother. 2013, 57, 2989–2995. [Google Scholar] [CrossRef] [PubMed]
- Osei Sekyere, J.; Amoako, D.G. Carbonyl Cyanide m-Chlorophenylhydrazine (CCCP) Reverses Resistance to Colistin, but Not to Carbapenems and Tigecycline in Multidrug-Resistant Enterobacteriaceae. Front. Microbiol. 2017, 8, 228. [Google Scholar] [CrossRef] [PubMed]
- Kmeid, J.G.; Youssef, M.M.; Kanafani, Z.A.; Kanj, S.S. Combination therapy for Gram-negative bacteria: What is the evidence? Expert Rev. Anti-Infect. Ther. 2013, 11, 1355–1362. [Google Scholar] [CrossRef]
- Lamers, R.P.; Cavallari, J.F.; Burrows, L.L. The efflux inhibitor phenylalanine-arginine beta-naphthylamide (PAbetaN) permeabilizes the outer membrane of gram-negative bacteria. PLoS ONE 2013, 8, e60666. [Google Scholar] [CrossRef]
- Lee, M.D.; Galazzo, J.L.; Staley, A.L.; Lee, J.C.; Warren, M.S.; Fuernkranz, H.; Chamberland, S.; Lomovskaya, O.; Miller, G.H. Microbial fermentation-derived inhibitors of efflux-pump-mediated drug resistance. Farmaco 2001, 56, 81–85. [Google Scholar] [CrossRef]
- Schillaci, D.; Spano, V.; Parrino, B.; Carbone, A.; Montalbano, A.; Barraja, P.; Diana, P.; Cirrincione, G.; Cascioferro, S. Pharmaceutical Approaches to Target Antibiotic Resistance Mechanisms. J. Med. Chem. 2017, 60, 8268–8297. [Google Scholar] [CrossRef]
- Spengler, G.; Kincses, A.; Gajdacs, M.; Amaral, L. New Roads Leading to Old Destinations: Efflux Pumps as Targets to Reverse Multidrug Resistance in Bacteria. Molecules 2017, 22, 468. [Google Scholar] [CrossRef]
- Liu, Y.; Li, S.; Li, W.; Wang, P.; Ding, P.; Li, L.; Wang, J.; Yang, P.; Wang, Q.; Xu, T.; et al. RstA, a two-component response regulator, plays important roles in multiple virulence-associated processes in enterohemorrhagic Escherichia coli O157:H7. Gut Pathog. 2019, 11, 53. [Google Scholar] [CrossRef] [PubMed]
- Pardo-Este, C.; Hidalgo, A.A.; Aguirre, C.; Briones, A.C.; Cabezas, C.E.; Castro-Severyn, J.; Fuentes, J.A.; Opazo, C.M.; Riedel, C.A.; Otero, C.; et al. The ArcAB two-component regulatory system promotes resistance to reactive oxygen species and systemic infection by Salmonella Typhimurium. PLoS ONE 2018, 13, e0203497. [Google Scholar] [CrossRef] [PubMed]
- Zschiedrich, C.P.; Keidel, V.; Szurmant, H. Molecular Mechanisms of Two-Component Signal Transduction. J. Mol. Biol. 2016, 428, 3752–3775. [Google Scholar] [CrossRef] [PubMed]
- Hoch, J.A. Two-component and phosphorelay signal transduction. Curr. Opin. Microbiol. 2000, 3, 165–170. [Google Scholar] [CrossRef]
- Stock, J.B.; Stock, A.M.; Mottonen, J.M. Signal transduction in bacteria. Nature 1990, 344, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Navarre, W.W.; Halsey, T.A.; Walthers, D.; Frye, J.; McClelland, M.; Potter, J.L.; Kenney, L.J.; Gunn, J.S.; Fang, F.C.; Libby, S.J. Co-regulation of Salmonella enterica genes required for virulence and resistance to antimicrobial peptides by SlyA and PhoP/PhoQ. Mol. Microbiol. 2005, 56, 492–508. [Google Scholar] [CrossRef]
- Munoz-Dorado, J.; Inouye, S.; Inouye, M. A gene encoding a protein serine/threonine kinase is required for normal development of M. xanthus, a gram-negative bacterium. Cell 1991, 67, 995–1006. [Google Scholar] [CrossRef]
- Liu, Q.; Fan, J.; Niu, C.; Wang, D.; Wang, J.; Wang, X.; Villaruz, A.E.; Li, M.; Otto, M.; Gao, Q. The eukaryotic-type serine/threonine protein kinase Stk is required for biofilm formation and virulence in Staphylococcus epidermidis. PLoS ONE 2011, 6, e25380. [Google Scholar] [CrossRef]
- Foulquier, E.; Pompeo, F.; Freton, C.; Cordier, B.; Grangeasse, C.; Galinier, A. PrkC-mediated phosphorylation of overexpressed YvcK protein regulates PBP1 protein localization in Bacillus subtilis mreB mutant cells. J. Biol. Chem. 2014, 289, 23662–23669. [Google Scholar] [CrossRef]
- Leiba, J.; Hartmann, T.; Cluzel, M.E.; Cohen-Gonsaud, M.; Delolme, F.; Bischoff, M.; Molle, V. A novel mode of regulation of the Staphylococcus aureus catabolite control protein A (CcpA) mediated by Stk1 protein phosphorylation. J. Biol. Chem. 2012, 287, 43607–43619. [Google Scholar] [CrossRef]
- Cheung, A.; Duclos, B. Stp1 and Stk1: The Yin and Yang of vancomycin sensitivity and virulence in vancomycin-intermediate Staphylococcus aureus strains. J. Infect. Dis. 2012, 205, 1625–1627. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pensinger, D.A.; Boldon, K.M.; Chen, G.Y.; Vincent, W.J.; Sherman, K.; Xiong, M.; Schaenzer, A.J.; Forster, E.R.; Coers, J.; Striker, R.; et al. The Listeria monocytogenes PASTA Kinase PrkA and Its Substrate YvcK Are Required for Cell Wall Homeostasis, Metabolism, and Virulence. PLoS Pathog. 2016, 12, e1006001. [Google Scholar] [CrossRef] [PubMed]
- Beltramini, A.M.; Mukhopadhyay, C.D.; Pancholi, V. Modulation of cell wall structure and antimicrobial susceptibility by a Staphylococcus aureus eukaryote-like serine/threonine kinase and phosphatase. Infect. Immun. 2009, 77, 1406–1416. [Google Scholar] [CrossRef] [PubMed]
- Pensinger, D.A.; Aliota, M.T.; Schaenzer, A.J.; Boldon, K.M.; Ansari, I.U.; Vincent, W.J.; Knight, B.; Reniere, M.L.; Striker, R.; Sauer, J.D. Selective pharmacologic inhibition of a PASTA kinase increases Listeria monocytogenes susceptibility to beta-lactam antibiotics. Antimicrob. Agents Chemother. 2014, 58, 4486–4494. [Google Scholar] [CrossRef]
- Schaenzer, A.J.; Wlodarchak, N.; Drewry, D.H.; Zuercher, W.J.; Rose, W.E.; Striker, R.; Sauer, J.D. A screen for kinase inhibitors identifies antimicrobial imidazopyridine aminofurazans as specific inhibitors of the Listeria monocytogenes PASTA kinase PrkA. J. Biol. Chem. 2017, 292, 17037–17045. [Google Scholar] [CrossRef]
- Gupta, A.; Pal, S.K.; Pandey, D.; Fakir, N.A.; Rathod, S.; Sinha, D.; SivaKumar, S.; Sinha, P.; Periera, M.; Balgam, S.; et al. PknB remains an essential and a conserved target for drug development in susceptible and MDR strains of M. Tuberculosis. Ann. Clin. Microbiol. Antimicrob. 2017, 16, 56. [Google Scholar] [CrossRef]
- Komiazyk, M.; Palczewska, M.; Pikula, S.; Groves, P. Bacterial type AB enterotoxins--structure, function and mechanism of action. Postepy Biochem. 2015, 61, 430–435. [Google Scholar]
- Childers, B.M.; Klose, K.E. Regulation of virulence in Vibrio cholerae: The ToxR regulon. Future Microbiol. 2007, 2, 335–344. [Google Scholar] [CrossRef]
- Weber, G.G.; Klose, K.E. The complexity of ToxT-dependent transcription in Vibrio cholerae. Indian J. Med. Res. 2011, 133, 201–206. [Google Scholar]
- Anthouard, R.; DiRita, V.J. Small-molecule inhibitors of toxT expression in Vibrio cholerae. mBio 2013, 4, e00403-13. [Google Scholar] [CrossRef]
- Abisado, R.G.; Benomar, S.; Klaus, J.R.; Dandekar, A.A.; Chandler, J.R. Bacterial Quorum Sensing and Microbial Community Interactions. mBio 2018, 9, e02331-17. [Google Scholar] [CrossRef] [PubMed]
- Kaur, A.; Capalash, N.; Sharma, P. Quorum sensing in thermophiles: Prevalence of autoinducer-2 system. BMC Microbiol. 2018, 18, 62. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, X.; Song, C.; Zhang, Y.; Wang, Z.; Liu, Z.; Wei, H.; Yu, J. Autoinducer-2 Facilitates Pseudomonas aeruginosa PAO1 Pathogenicity In Vitro and In Vivo. Front. Microbiol. 2017, 8, 1944. [Google Scholar] [CrossRef] [PubMed]
- Sheng, L.; Lv, Y.; Liu, Q.; Wang, Q.; Zhang, Y. Connecting type VI secretion, quorum sensing, and c-di-GMP production in fish pathogen Vibrio alginolyticus through phosphatase PppA. Vet. Microbiol. 2013, 162, 652–662. [Google Scholar] [CrossRef]
- Rader, B.A.; Campagna, S.R.; Semmelhack, M.F.; Bassler, B.L.; Guillemin, K. The quorum-sensing molecule autoinducer 2 regulates motility and flagellar morphogenesis in Helicobacter pylori. J. Bacteriol. 2007, 189, 6109–6117. [Google Scholar] [CrossRef]
- Slamti, L.; Perchat, S.; Huillet, E.; Lereclus, D. Quorum sensing in Bacillus thuringiensis is required for completion of a full infectious cycle in the insect. Toxins 2014, 6, 2239–2255. [Google Scholar] [CrossRef]
- Ha, C.; Kim, S.K.; Lee, M.N.; Lee, J.H. Quorum sensing-dependent metalloprotease VvpE is important in the virulence of Vibrio vulnificus to invertebrates. Microb. Pathog. 2014, 71, 8–14. [Google Scholar] [CrossRef]
- Clarke, M.B.; Hughes, D.T.; Zhu, C.; Boedeker, E.C.; Sperandio, V. The QseC sensor kinase: A bacterial adrenergic receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 10420–10425. [Google Scholar] [CrossRef]
- Hughes, D.T.; Clarke, M.B.; Yamamoto, K.; Rasko, D.A.; Sperandio, V. The QseC adrenergic signaling cascade in Enterohemorrhagic E. coli (EHEC). PLoS Pathog. 2009, 5, e1000553. [Google Scholar] [CrossRef]
- Merighi, M.; Carroll-Portillo, A.; Septer, A.N.; Bhatiya, A.; Gunn, J.S. Role of Salmonella enterica serovar Typhimurium two-component system PreA/PreB in modulating PmrA-regulated gene transcription. J. Bacteriol. 2006, 188, 141–149. [Google Scholar] [CrossRef]
- Weiss, D.S.; Brotcke, A.; Henry, T.; Margolis, J.J.; Chan, K.; Monack, D.M. In vivo negative selection screen identifies genes required for Francisella virulence. Proc. Natl. Acad. Sci. USA 2007, 104, 6037–6042. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Chen, X.; Mao, X.; Hou, Z.; Zhou, Y.; Bai, H.; Meng, J.; Da, F.; Sang, G.; Wang, Y.; et al. Amino-terminated generation 2 poly(amidoamine) dendrimer as a potential broad-spectrum, nonresistance-inducing antibacterial agent. AAPS J. 2013, 15, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Lopez, A.I.; Reins, R.Y.; McDermott, A.M.; Trautner, B.W.; Cai, C. Antibacterial activity and cytotoxicity of PEGylated poly(amidoamine) dendrimers. Mol Biosyst. 2009, 5, 1148–1156. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.Y.; Mao, X.G.; Li, Z.; Chen, Z.; Zhou, Y.; Hou, Z.; Li, M.K.; Meng, J.R.; Luo, X.X. A potent and selective antimicrobial poly(amidoamine) dendrimer conjugate with LED209 targeting QseC receptor to inhibit the virulence genes of gram negative bacteria. Nanomedicine 2015, 11, 329–339. [Google Scholar] [CrossRef]
- Lee, J.; Zhang, L. The hierarchy quorum sensing network in Pseudomonas aeruginosa. Protein Cell 2015, 6, 26–41. [Google Scholar] [CrossRef]
- Pattnaik, S.S.; Ranganathan, S.; Ampasala, D.R.; Syed, A.; Ameen, F.; Busi, S. Attenuation of quorum sensing regulated virulence and biofilm development in Pseudomonas aeruginosa PAO1 by Diaporthe phaseolorum SSP12. Microb. Pathog. 2018, 118, 177–189. [Google Scholar] [CrossRef]
- Kao, C.Y.; Lin, W.H.; Tseng, C.C.; Wu, A.B.; Wang, M.C.; Wu, J.J. The complex interplay among bacterial motility and virulence factors in different Escherichia coli infections. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 2157–2162. [Google Scholar] [CrossRef]
- Kao, C.Y.; Sheu, B.S.; Sheu, S.M.; Yang, H.B.; Chang, W.L.; Cheng, H.C.; Wu, J.J. Higher motility enhances bacterial density and inflammatory response in dyspeptic patients infected with Helicobacter pylori. Helicobacter 2012, 17, 411–416. [Google Scholar] [CrossRef]
- Cascioferro, S.; Cusimano, M.G.; Schillaci, D. Antiadhesion agents against Gram-positive pathogens. Future Microbiol. 2014, 9, 1209–1220. [Google Scholar] [CrossRef]
- Menard, R.; Schoenhofen, I.C.; Tao, L.; Aubry, A.; Bouchard, P.; Reid, C.W.; Lachance, P.; Twine, S.M.; Fulton, K.M.; Cui, Q.; et al. Small-molecule inhibitors of the pseudaminic acid biosynthetic pathway: Targeting motility as a key bacterial virulence factor. Antimicrob. Agents Chemother. 2014, 58, 7430–7440. [Google Scholar] [CrossRef]
- Kharat, A.S.; Tomasz, A. Inactivation of the srtA gene affects localization of surface proteins and decreases adhesion of Streptococcus pneumoniae to human pharyngeal cells in vitro. Infect. Immun. 2003, 71, 2758–2765. [Google Scholar] [CrossRef]
- Lalioui, L.; Pellegrini, E.; Dramsi, S.; Baptista, M.; Bourgeois, N.; Doucet-Populaire, F.; Rusniok, C.; Zouine, M.; Glaser, P.; Kunst, F.; et al. The SrtA Sortase of Streptococcus agalactiae is required for cell wall anchoring of proteins containing the LPXTG motif, for adhesion to epithelial cells, and for colonization of the mouse intestine. Infect. Immun. 2005, 73, 3342–3350. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Terao, Y.; Ogawa, T.; Takahashi, T.; Hamada, S.; Kawabata, S. Role of Streptococcus sanguinis sortase A in bacterial colonization. Microbes Infect. 2006, 8, 2791–2796. [Google Scholar] [CrossRef] [PubMed]
- Mazmanian, S.K.; Liu, G.; Jensen, E.R.; Lenoy, E.; Schneewind, O. Staphylococcus aureus sortase mutants defective in the display of surface proteins and in the pathogenesis of animal infections. Proc. Natl. Acad. Sci. USA 2000, 97, 5510–5515. [Google Scholar] [CrossRef]
- Kaakoush, N.O.; Castano-Rodriguez, N.; Mitchell, H.M.; Man, S.M. Global Epidemiology of Campylobacter Infection. Clin. Microbiol. Rev. 2015, 28, 687–720. [Google Scholar] [CrossRef]
- Grant, C.C.; Konkel, M.E.; Cieplak, W., Jr.; Tompkins, L.S. Role of flagella in adherence, internalization, and translocation of Campylobacter jejuni in nonpolarized and polarized epithelial cell cultures. Infect. Immun. 1993, 61, 1764–1771. [Google Scholar] [CrossRef]
- Lertsethtakarn, P.; Ottemann, K.M.; Hendrixson, D.R. Motility and chemotaxis in Campylobacter and Helicobacter. Annu. Rev. Microbiol. 2011, 65, 389–410. [Google Scholar] [CrossRef]
- Nachamkin, I.; Yang, X.H.; Stern, N.J. Role of Campylobacter jejuni flagella as colonization factors for three-day-old chicks: Analysis with flagellar mutants. Appl. Environ. Microbiol. 1993, 59, 1269–1273. [Google Scholar] [CrossRef]
- Salah Ud-Din, A.I.M.; Roujeinikova, A. Flagellin glycosylation with pseudaminic acid in Campylobacter and Helicobacter: Prospects for development of novel therapeutics. Cell. Mol. Life Sci. 2018, 75, 1163–1178. [Google Scholar] [CrossRef]
- Martinovic, T.; Andjelkovic, U.; Gajdosik, M.S.; Resetar, D.; Josic, D. Foodborne pathogens and their toxins. J. Proteom. 2016, 147, 226–235. [Google Scholar] [CrossRef]
- Liu, S.; Moayeri, M.; Leppla, S.H. Anthrax lethal and edema toxins in anthrax pathogenesis. Trends Microbiol. 2014, 22, 317–325. [Google Scholar] [CrossRef]
- Moayeri, M.; Leppla, S.H.; Vrentas, C.; Pomerantsev, A.P.; Liu, S. Anthrax Pathogenesis. Annu. Rev. Microbiol. 2015, 69, 185–208. [Google Scholar] [CrossRef]
- Raxibacumab for anthrax. Med. Lett. Drugs Ther. 2013, 55, 27–28.
- Singh, H.; Ratol, S.; Thangaraju, P.; Kumar, S.; Goel, A. Pharmacology and Anti-infective Role of Raxibacumab: A Novel Monoclonal Antibody for the Treatment of Anthrax. West Indian Med. J. 2016, 65, 358–362. [Google Scholar] [PubMed]
- Tsai, C.W.; Morris, S. Approval of Raxibacumab for the Treatment of Inhalation Anthrax Under the US Food and Drug Administration “Animal Rule”. Front. Microbiol. 2015, 6, 1320. [Google Scholar] [CrossRef] [PubMed]
- Kummerfeldt, C.E. Raxibacumab: Potential role in the treatment of inhalational anthrax. Infect. Drug Resist. 2014, 7, 101–109. [Google Scholar] [CrossRef]
- Xu, W.; Ohanjanian, L.; Sun, J.; Cui, X.; Suffredini, D.; Li, Y.; Welsh, J.; Eichacker, P.Q. A systematic review and meta-analysis of preclinical trials testing anti-toxin therapies for B. anthracis infection: A need for more robust study designs and results. PLoS ONE 2017, 12, e0182879. [Google Scholar] [CrossRef]
- Pirazzini, M.; Rossetto, O.; Eleopra, R.; Montecucco, C. Botulinum Neurotoxins: Biology, Pharmacology, and Toxicology. Pharmacol. Rev. 2017, 69, 200–235. [Google Scholar] [CrossRef]
- Smith, T.J.; Hill, K.K.; Raphael, B.H. Historical and current perspectives on Clostridium botulinum diversity. Res. Microbiol. 2015, 166, 290–302. [Google Scholar] [CrossRef]
- Fan, Y.; Garcia-Rodriguez, C.; Lou, J.; Wen, W.; Conrad, F.; Zhai, W.; Smith, T.J.; Smith, L.A.; Marks, J.D. A three monoclonal antibody combination potently neutralizes multiple botulinum neurotoxin serotype F subtypes. PLoS ONE 2017, 12, e0174187. [Google Scholar] [CrossRef]
- Garcia-Rodriguez, C.; Razai, A.; Geren, I.N.; Lou, J.; Conrad, F.; Wen, W.H.; Farr-Jones, S.; Smith, T.J.; Brown, J.L.; Skerry, J.C.; et al. A Three Monoclonal Antibody Combination Potently Neutralizes Multiple Botulinum Neurotoxin Serotype E Subtypes. Toxins 2018, 10, 105. [Google Scholar] [CrossRef] [PubMed]
- Anand, T.; Virmani, N.; Kumar, S.; Kumar Mohanty, A.; Pavulraj, S.; Ch Bera, B.; Vaid, R.K.; Ahlawat, U.; Tripathi, B.N. Phage Therapy for treatment of virulent Klebsiella pneumoniae infection in mouse model. J. Glob. Antimicrob. Resist. 2019. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, M.M.; Frezza, D.; Romano, E.; Marmo, P.; De Angelis, L.H.; Perini, N.; Thaller, M.C.; Di Lallo, G. The lytic bacteriophage vB_EfaH_EF1TV, a new member of the Herelleviridae family, disrupts biofilm produced by Enterococcus faecalis clinical strains. J. Glob. Antimicrob. Resist. 2019. [Google Scholar] [CrossRef]
- Fu, W.; Forster, T.; Mayer, O.; Curtin, J.J.; Lehman, S.M.; Donlan, R.M. Bacteriophage cocktail for the prevention of biofilm formation by Pseudomonas aeruginosa on catheters in an in vitro model system. Antimicrob. Agents Chemother. 2010, 54, 397–404. [Google Scholar] [CrossRef]
- Furfaro, L.L.; Payne, M.S.; Chang, B.J. Bacteriophage Therapy: Clinical Trials and Regulatory Hurdles. Front. Cell. Infect. Microbiol. 2018, 8, 376. [Google Scholar] [CrossRef]
- Rhoads, D.D.; Wolcott, R.D.; Kuskowski, M.A.; Wolcott, B.M.; Ward, L.S.; Sulakvelidze, A. Bacteriophage therapy of venous leg ulcers in humans: Results of a phase I safety trial. J. Wound Care 2009, 18, 237–243. [Google Scholar] [CrossRef]
- Leshkasheli, L.; Kutateladze, M.; Balarjishvili, N.; Bolkvadze, D.; Save, J.; Oechslin, F.; Que, Y.A.; Resch, G. Efficacy of newly isolated and highly potent bacteriophages in a mouse model of extensively drug-resistant Acinetobacter baumannii bacteraemia. J. Glob. Antimicrob. Resist. 2019, 19, 255–261. [Google Scholar] [CrossRef]
- Abedon, S.T.; Kuhl, S.J.; Blasdel, B.G.; Kutter, E.M. Phage treatment of human infections. Bacteriophage 2011, 1, 66–85. [Google Scholar] [CrossRef]
- Dissanayake, U.; Ukhanova, M.; Moye, Z.D.; Sulakvelidze, A.; Mai, V. Bacteriophages Reduce Pathogenic Escherichia coli Counts in Mice Without Distorting Gut Microbiota. Front. Microbiol. 2019, 10, 1984. [Google Scholar] [CrossRef]
- Bruttin, A.; Brussow, H. Human volunteers receiving Escherichia coli phage T4 orally: A safety test of phage therapy. Antimicrob. Agents Chemother. 2005, 49, 2874–2878. [Google Scholar] [CrossRef]
- Sarker, S.A.; McCallin, S.; Barretto, C.; Berger, B.; Pittet, A.C.; Sultana, S.; Krause, L.; Huq, S.; Bibiloni, R.; Bruttin, A.; et al. Oral T4-like phage cocktail application to healthy adult volunteers from Bangladesh. Virology 2012, 434, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.; Hawkins, C.H.; Anggard, E.E.; Harper, D.R. A controlled clinical trial of a therapeutic bacteriophage preparation in chronic otitis due to antibiotic-resistant Pseudomonas aeruginosa; a preliminary report of efficacy. Clin. Otolaryngol. 2009, 34, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Abdelkader, K.; Gerstmans, H.; Saafan, A.; Dishisha, T.; Briers, Y. The Preclinical and Clinical Progress of Bacteriophages and Their Lytic Enzymes: The Parts are Easier than the Whole. Viruses 2019, 11, 96. [Google Scholar] [CrossRef] [PubMed]
- Castillo, D.; Christiansen, R.H.; Dalsgaard, I.; Madsen, L.; Middelboe, M. Bacteriophage resistance mechanisms in the fish pathogen Flavobacterium psychrophilum: Linking genomic mutations to changes in bacterial virulence factors. Appl. Environ. Microbiol. 2015, 81, 1157–1167. [Google Scholar] [CrossRef]
- Goldfarb, T.; Sberro, H.; Weinstock, E.; Cohen, O.; Doron, S.; Charpak-Amikam, Y.; Afik, S.; Ofir, G.; Sorek, R. BREX is a novel phage resistance system widespread in microbial genomes. EMBO J. 2015, 34, 169–183. [Google Scholar] [CrossRef]
- Labrie, S.J.; Samson, J.E.; Moineau, S. Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 2010, 8, 317–327. [Google Scholar] [CrossRef]
- Wang, C.; Nie, T.; Lin, F.; Connerton, I.F.; Lu, Z.; Zhou, S.; Hang, H. Resistance mechanisms adopted by a Salmonella Typhimurium mutant against bacteriophage. Virus Res. 2019, 273, 197759. [Google Scholar] [CrossRef]
- de Jonge, P.A.; Nobrega, F.L.; Brouns, S.J.J.; Dutilh, B.E. Molecular and Evolutionary Determinants of Bacteriophage Host Range. Trends Microbiol. 2019, 27, 51–63. [Google Scholar] [CrossRef]
- Krut, O.; Bekeredjian-Ding, I. Contribution of the Immune Response to Phage Therapy. J. Immunol. 2018, 200, 3037–3044. [Google Scholar] [CrossRef]
- Torres-Barcelo, C. The disparate effects of bacteriophages on antibiotic-resistant bacteria. Emerg. Microbes Infect. 2018, 7, 168. [Google Scholar] [CrossRef]
- Lynch, S.V.; Pedersen, O. The Human Intestinal Microbiome in Health and Disease. N. Engl. J. Med. 2016, 375, 2369–2379. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.T.; Taur, Y.; Walkup, J.T. Gut Microbiota and Autism: Key Concepts and Findings. J. Autism Dev. Disord. 2017, 47, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Weingarden, A.R.; Vaughn, B.P. Intestinal microbiota, fecal microbiota transplantation, and inflammatory bowel disease. Gut Microbes 2017, 8, 238–252. [Google Scholar] [CrossRef] [PubMed]
- Theriot, C.M.; Young, V.B. Interactions between the Gastrointestinal Microbiome and Clostridium difficile. Annu. Rev. Microbiol. 2015, 69, 445–461. [Google Scholar] [CrossRef]
- Chai, J.; Lee, C.H. Management of Primary and Recurrent Clostridium difficile Infection: An Update. Antibiotics 2018, 7, 54. [Google Scholar] [CrossRef]
- Cammarota, G.; Gallo, A.; Bibbo, S. Fecal microbiota transplant for C. difficile infection: Just say yes. Anaerobe 2019, 60, 102109. [Google Scholar] [CrossRef]
- Shogbesan, O.; Poudel, D.R.; Victor, S.; Jehangir, A.; Fadahunsi, O.; Shogbesan, G.; Donato, A. A Systematic Review of the Efficacy and Safety of Fecal Microbiota Transplant for Clostridium difficile Infection in Immunocompromised Patients. Can J. Gastroenterol. Hepatol. 2018, 2018, 1394379. [Google Scholar] [CrossRef]
- Wang, J.W.; Kuo, C.H.; Kuo, F.C.; Wang, Y.K.; Hsu, W.H.; Yu, F.J.; Hu, H.M.; Hsu, P.I.; Wang, J.Y.; Wu, D.C. Fecal microbiota transplantation: Review and update. J. Formos. Med. Assoc. 2019, 118 Suppl. 1, S23–S31. [Google Scholar] [CrossRef]
- Ianiro, G.; Maida, M.; Burisch, J.; Simonelli, C.; Hold, G.; Ventimiglia, M.; Gasbarrini, A.; Cammarota, G. Efficacy of different faecal microbiota transplantation protocols for Clostridium difficile infection: A systematic review and meta-analysis. United Eur. Gastroenterol. J. 2018, 6, 1232–1244. [Google Scholar] [CrossRef]
- Sartor, R.B. Probiotic therapy of intestinal inflammation and infections. Curr. Opin. Gastroenterol. 2005, 21, 44–50. [Google Scholar]
- Van Hoogmoed, C.G.; Geertsema-Doornbusch, G.I.; Teughels, W.; Quirynen, M.; Busscher, H.J.; Van der Mei, H.C. Reduction of periodontal pathogens adhesion by antagonistic strains. Oral. Microbiol. Immunol. 2008, 23, 43–48. [Google Scholar] [CrossRef]
- Ahern, P.P.; Maloy, K.J. Understanding immune-microbiota interactions in the intestine. Immunology 2020, 159, 4–14. [Google Scholar] [CrossRef]
- Arnoriaga-Rodriguez, M.; Fernandez-Real, J.M. Microbiota impacts on chronic inflammation and metabolic syndrome-related cognitive dysfunction. Rev. Endocr. Metab. Disord. 2019, 20, 473–480. [Google Scholar] [CrossRef]
- Thomas, R.M.; Jobin, C. Microbiota in pancreatic health and disease: The next frontier in microbiome research. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 53–64. [Google Scholar] [CrossRef]


{kind=link}
| Compound | Structure and Molecular Weight | Action | Function | Reference |
|---|---|---|---|---|
| Mitotane |  320 g/mol | Unknown | Combine with polymyxin B to target carbapenem-resistant P. aeruginosa, A. baumannii, and K. pneumoniae | [24] |
| SPR741 |  992.1 g/mol | Disrupts the bacterial outer membrane, permeabilizing it to antibiotics | Combine with antibiotics to target antibiotic-resistant E. coli, K. pneumoniae, and A. baumannii | [25,26] |
| Phenylalanyl arginyl β-naphthylamide (PAβN) |  446.5 g/mol | Inhibits Gram-negative efflux pumps | Combine with antibiotics to target fluoroquinolones- resistant P. aeruginosa | [31,32] |
| Curcumin |  368.4 g/mol | Efflux pump inhibitor | Combine with antibiotics target multidrug- resistant P. aeruginosa | [33] |
| Quinazoline compounds-GI261520A |  357.4 g/mol | Inhibits kinase activity of PhoP/PhoQ two-component system | Anti-virulence effect by blocking S. Typhimurium intramacrophage replication | [34] |
| GW779439X |  454.5 g/mol | Inhibits S. aureus PASTA kinase Stk1 activity | Resensitizes methicillin-resistant S. aureus to various β-lactams | [35] |
| Virstatin |  283.28 g/mol | Inhibits dimerization of regulator ToxT of V. cholerae | Reduces the nization of V. cholera in a murine model of infection | [36,37] |
| LED209 |  383.5 g/mol | Anti-E. coli quorum sensing system | Abolishes EHEC A/E lesion formation and decreases expression of shiga toxin | [38] |
| Quercitrin |  448.4 g/mol | Inhibits the enzymatic activity of surface protein sortase A (SrtA) | Reduces attachment of S. aureus to fibronectin-coated or fibrinogen-coated surface; inhibits biofilm formation of S. pneumoniae | [39,40] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.-H.; Hsieh, Y.-H.; Powers, Z.M.; Kao, C.-Y. Defeating Antibiotic-Resistant Bacteria: Exploring Alternative Therapies for a Post-Antibiotic Era. Int. J. Mol. Sci. 2020, 21, 1061. https://doi.org/10.3390/ijms21031061
Wang C-H, Hsieh Y-H, Powers ZM, Kao C-Y. Defeating Antibiotic-Resistant Bacteria: Exploring Alternative Therapies for a Post-Antibiotic Era. International Journal of Molecular Sciences. 2020; 21(3):1061. https://doi.org/10.3390/ijms21031061
Chicago/Turabian StyleWang, Chih-Hung, Yi-Hsien Hsieh, Zachary M. Powers, and Cheng-Yen Kao. 2020. "Defeating Antibiotic-Resistant Bacteria: Exploring Alternative Therapies for a Post-Antibiotic Era" International Journal of Molecular Sciences 21, no. 3: 1061. https://doi.org/10.3390/ijms21031061
APA StyleWang, C.-H., Hsieh, Y.-H., Powers, Z. M., & Kao, C.-Y. (2020). Defeating Antibiotic-Resistant Bacteria: Exploring Alternative Therapies for a Post-Antibiotic Era. International Journal of Molecular Sciences, 21(3), 1061. https://doi.org/10.3390/ijms21031061