New Carbapenemase Inhibitors: Clearing the Way for the β-Lactams



Abstract
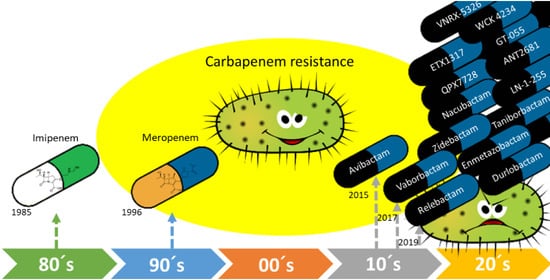
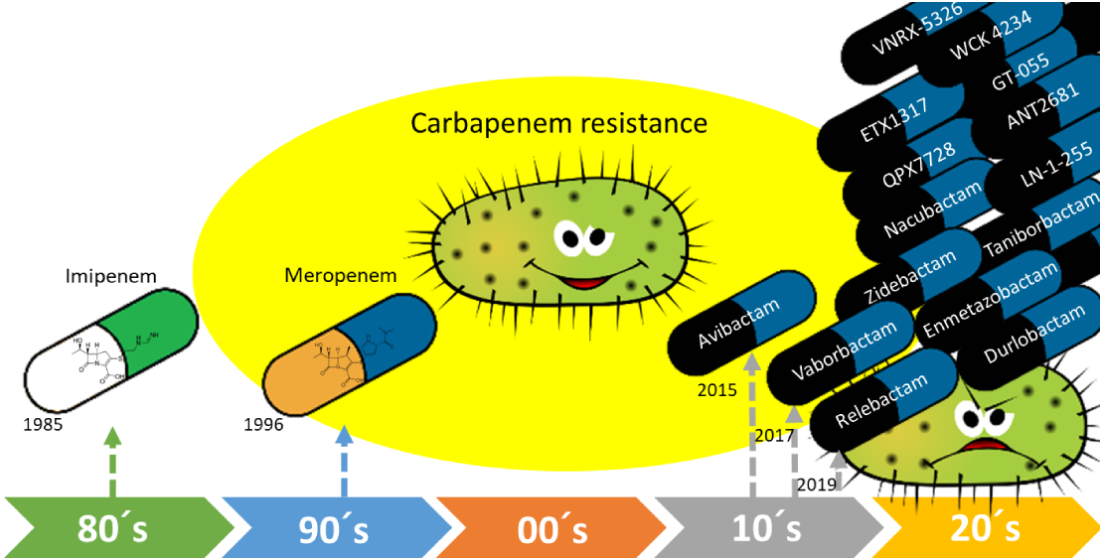
1. Introduction
2. Carbapenemase Inhibitors Recently Approved for Therapeutic Use
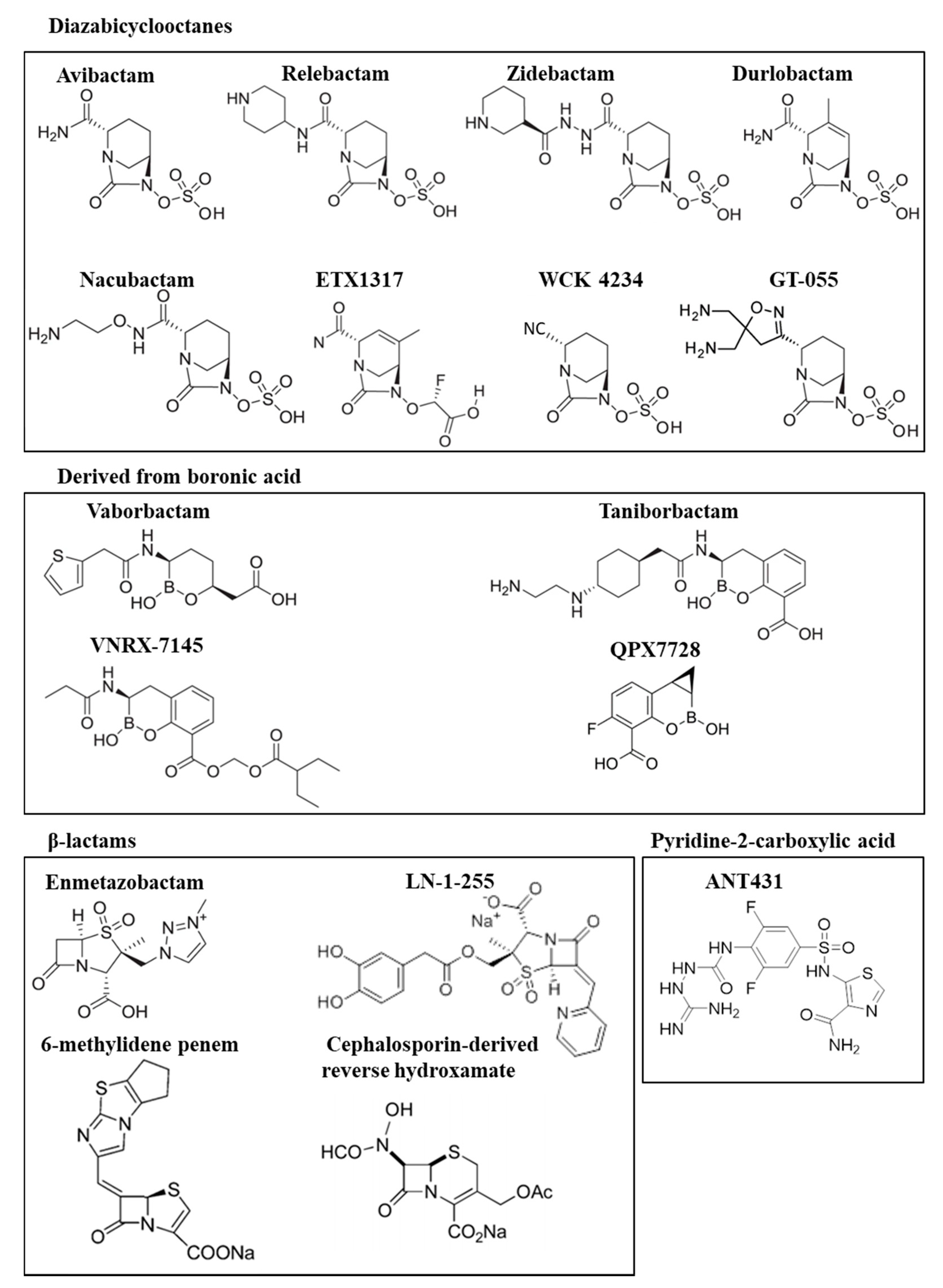
2.1. Diazabicyclooctanes
2.1.1. Avibactam (Ceftazidime/Avibactam)
2.1.2. Relebactam (Imipenem/Relebactam)
2.2. Boronic Acid Derivatives
2.3. Vaborbactam (Meropenem/Vaborbactam)
2.4. Emerging Broad-Spectrum Resistance to Recently Approved β-Lactam/β-Lactamase Inhibitor Combinations Active against Carbapenemase-Producing/Carbapenem-Resistant Gram-Negative Pathogens
3. New Carbapenemase Inhibitors in Development
3.1. Diazabicyclooctane Derived Inhibitors
3.1.1. Avibactam (Aztreonam/Avibactam)
3.1.2. Zidebactam (Cefepime/Zidebactam)
3.1.3. Durlobactam (Sulbactam/Durlobactam)
3.1.4. Nacubactam (Meropenem/Nacubactam)
3.1.5. ETX1317 (Cefpodoxime/ETX1317)
3.1.6. WCK 4234 (Meropenem/WCK 4234)
3.1.7. GT-055 (GT-1/GT-055)
3.2. Boronic Acid Derived Inhibitors
3.2.1. Taniborbactam (Cefepime/Taniborbactam)
3.2.2. VNRX-5236 (Ceftibuten/VNRX-7145)
3.2.3. QPX7728 (Meropenem/QPX7728)
3.3. β-Lactam-Derived Inhibitors. Penicillin Sulfones
3.3.1. Enmetazobactam (Cefepime/Enmetazobactam)
3.3.2. LN-1-255 (Imipenem or Meropenem/LN-1-255)
3.4. Other Promising MBL Inhibitors
4. Major Challenges in the Development of New Carbapenemase Inhibitors
5. Final Considerations
Author Contributions
Funding
Conflicts of Interest
References
- Bush, K.; Bradford, P.A. β-Lactams and β-Lactamase Inhibitors: An Overview. Cold Spring Harb. Perspect. Med. 2016, 6, a025247. [Google Scholar] [CrossRef] [PubMed]
- Tooke, C.L.; Hinchliffe, P.; Bragginton, E.C.; Colenso, C.K.; Hirvonen, V.H.A.; Takebayashi, Y.; Spencer, J. β-Lactamases and β-Lactamase Inhibitors in the 21st Century. J. Mol. Biol. 2019, 431, 3472–3500. [Google Scholar] [CrossRef] [PubMed]
- Kohanski, M.A.; Dwyer, D.J.; Collins, J.J. How antibiotics kill bacteria: From targets to networks. Nat. Rev. Microbiol. 2010, 8, 423–435. [Google Scholar] [CrossRef] [PubMed]
- El-Gamal, M.I.; Brahim, I.; Hisham, N.; Aladdin, R.; Mohammed, H.; Bahaaeldin, A. Recent updates of carbapenem antibiotics. Eur. J. Med. Chem. 2017, 131, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Blair, J.M.A.; Webber, M.A.; Baylay, A.J.; Ogbolu, D.O.; Piddock, L.J.V. Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 2015, 13, 42–51. [Google Scholar] [CrossRef]
- World Health Organization. Global Priority List of Antibiotic-Resistant Bacteria to Guide Research, Discovery, and Development of New Antibiotics. Available online: https://www.who.int/news-room/detail/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed (accessed on 1 September 2020).
- Nordmann, P.; Poirel, L. Epidemiology and Diagnostics of Carbapenem Resistance in Gram-negative Bacteria. Clin. Infect. Dis. 2019, 69, S521–S528. [Google Scholar] [CrossRef]
- Queenan, A.M.; Bush, K. Carbapenemases: The versatile beta-lactamases. Clin. Microbiol. Rev. 2007, 20, 440–458. [Google Scholar] [CrossRef]
- Jeon, J.H.; Lee, J.H.; Lee, J.J.; Park, K.S.; Karim, A.M.; Lee, C.-R.; Jeong, B.C.; Lee, S.H. Structural basis for carbapenem-hydrolyzing mechanisms of carbapenemases conferring antibiotic resistance. Int. J. Mol. Sci. 2015, 16, 9654–9692. [Google Scholar] [CrossRef]
- Naas, T.; Dortet, L.; Iorga, B.I. Structural and Functional Aspects of Class A Carbapenemases. Curr. Drug Targets 2016, 17, 1006–1028. [Google Scholar] [CrossRef]
- Boyd, S.E.; Livermore, D.M.; Hooper, D.C.; Hope, W.W. Metallo-β-Lactamases: Structure, Function, Epidemiology, Treatment Options, and the Development Pipeline. Antimicrob. Agents Chemother. 2020, 64, e00397-20. [Google Scholar] [CrossRef]
- Poirel, L.; Naas, T.; Nordmann, P. Diversity, epidemiology, and genetics of class D beta-lactamases. Antimicrob. Agents Chemother. 2010, 54, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Evans, B.A.; Amyes, S.G.B. OXA β-lactamases. Clin. Microbiol. Rev. 2014, 27, 241–263. [Google Scholar] [CrossRef] [PubMed]
- Drawz, S.M.; Papp-Wallace, K.M.; Bonomo, R.A. New β-lactamase inhibitors: A therapeutic renaissance in an MDR world. Antimicrob. Agents Chemother. 2014, 58, 1835–1846. [Google Scholar] [CrossRef] [PubMed]
- Fernández, L.; Hancock, R.E.W. Adaptive and mutational resistance: Role of porins and efflux pumps in drug resistance. Clin. Microbiol. Rev. 2012, 25, 661–681. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Luo, Y.-F.; Williams, B.J.; Blackwell, T.S.; Xie, C.-M. Structure and function of OprD protein in Pseudomonas aeruginosa: From antibiotic resistance to novel therapies. Int. J. Med. Microbiol. 2012, 302, 63–68. [Google Scholar] [CrossRef]
- Ayoub Moubareck, C.; Hammoudi Halat, D. Insights into Acinetobacter baumannii: A Review of Microbiological, Virulence, and Resistance Traits in a Threatening Nosocomial Pathogen. Antibiotics 2020, 9, 119. [Google Scholar] [CrossRef]
- Moyá, B.; Beceiro, A.; Cabot, G.; Juan, C.; Zamorano, L.; Alberti, S.; Oliver, A. Pan-β-lactam resistance development in Pseudomonas aeruginosa clinical strains: Molecular mechanisms, penicillin-binding protein profiles, and binding affinities. Antimicrob. Agents Chemother. 2012, 56, 4771–4778. [Google Scholar] [CrossRef]
- Yamachika, S.; Sugihara, C.; Kamai, Y.; Yamashita, M. Correlation between penicillin-binding protein 2 mutations and carbapenem resistance in Escherichia coli. J. Med. Microbiol. 2013, 62, 429–436. [Google Scholar] [CrossRef]
- Drawz, S.M.; Bonomo, R.A. Three decades of beta-lactamase inhibitors. Clin. Microbiol. Rev. 2010, 23, 160–201. [Google Scholar] [CrossRef]
- Shahid, M.; Sobia, F.; Singh, A.; Malik, A.; Khan, H.M.; Jonas, D.; Hawkey, P.M. Beta-lactams and beta-lactamase-inhibitors in current- or potential-clinical practice: A comprehensive update. Crit. Rev. Microbiol. 2009, 35, 81–108. [Google Scholar] [CrossRef]
- Pérez-Llarena, F.J.; Bou, G. Beta-lactamase inhibitors: The story so far. Curr. Med. Chem. 2009, 16, 3740–3765. [Google Scholar] [CrossRef] [PubMed]
- Toussaint, K.A.; Gallagher, J.C. β-lactam/β-lactamase inhibitor combinations: From then to now. Ann. Pharmacother. 2015, 49, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Olsen, I. New promising β-lactamase inhibitors for clinical use. Eur. J. Clin. Microbiol. Infect. Dis. 2015, 34, 1303–1308. [Google Scholar] [CrossRef] [PubMed]
- Tehrani, K.H.M.E.; Martin, N.I. β-lactam/β-lactamase inhibitor combinations: An update. MedChemComm 2018, 9, 1439–1456. [Google Scholar] [CrossRef] [PubMed]
- González-Bello, C.; Rodríguez, D.; Pernas, M.; Rodríguez, Á.; Colchón, E. β-Lactamase Inhibitors to Restore the Efficacy of Antibiotics against Superbugs. J. Med. Chem. 2020, 63, 1859–1881. [Google Scholar] [CrossRef]
- Arca-Suárez, J.; Fraile-Ribot, P.; Vázquez-Ucha, J.C.; Cabot, G.; Martínez-Guitián, M.; Lence, E.; González-Bello, C.; Beceiro, A.; Rodríguez-Iglesias, M.; Galán-Sánchez, F.; et al. Challenging Antimicrobial Susceptibility and Evolution of Resistance (OXA-681) during Treatment of a Long-Term Nosocomial Infection Caused by a Pseudomonas aeruginosa ST175 Clone. Antimicrob. Agents Chemother. 2019, 63, e01110-19. [Google Scholar] [CrossRef]
- Papp-Wallace, K.M. The latest advances in β-lactam/β-lactamase inhibitor combinations for the treatment of Gram-negative bacterial infections. Expert Opin. Pharmacother. 2019, 20, 2169–2184. [Google Scholar] [CrossRef]
- Somboro, A.M.; Osei Sekyere, J.; Amoako, D.G.; Essack, S.Y.; Bester, L.A. Diversity and Proliferation of Metallo-β-Lactamases: A Clarion Call for Clinically Effective Metallo-β-Lactamase Inhibitors. Appl. Environ. Microbiol. 2018, 84, e00698-18. [Google Scholar] [CrossRef]
- Durand-Réville, T.F.; Guler, S.; Comita-Prevoir, J.; Chen, B.; Bifulco, N.; Huynh, H.; Lahiri, S.; Shapiro, A.B.; McLeod, S.M.; Carter, N.M.; et al. ETX2514 is a broad-spectrum β-lactamase inhibitor for the treatment of drug-resistant Gram-negative bacteria including Acinetobacter baumannii. Nat. Microbiol. 2017, 2, 17104. [Google Scholar] [CrossRef]
- Tsivkovski, R.; Totrov, M.; Lomovskaya, O. Biochemical Characterization of QPX7728, a New Ultrabroad-Spectrum Beta-Lactamase Inhibitor of Serine and Metallo-Beta-Lactamases. Antimicrob. Agents Chemother. 2020, 64, e00130-20. [Google Scholar] [CrossRef]
- Liu, B.; Trout, R.E.L.; Chu, G.-H.; McGarry, D.; Jackson, R.W.; Hamrick, J.C.; Daigle, D.M.; Cusick, S.M.; Pozzi, C.; De Luca, F.; et al. Discovery of Taniborbactam (VNRX-5133): A Broad-Spectrum Serine- and Metallo-β-lactamase Inhibitor for Carbapenem-Resistant Bacterial Infections. J. Med. Chem. 2020, 63, 2789–2801. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Ucha, J.C.; Maneiro, M.; Martínez-Guitián, M.; Buynak, J.; Bethel, C.R.; Bonomo, R.A.; Bou, G.; Poza, M.; González-Bello, C.; Beceiro, A. Activity of the β-Lactamase Inhibitor LN-1-255 against Carbapenem-Hydrolyzing Class D β-Lactamases from Acinetobacter baumannii. Antimicrob. Agents Chemother. 2017, 61, e01172-17. [Google Scholar] [CrossRef]
- Papp-Wallace, K.M.; Nguyen, N.Q.; Jacobs, M.R.; Bethel, C.R.; Barnes, M.D.; Kumar, V.; Bajaksouzian, S.; Rudin, S.D.; Rather, P.N.; Bhavsar, S.; et al. Strategic approaches to overcome resistance against Gram-negative pathogens using β-lactamase inhibitors and β-lactam enhancers: Activity of three novel diazabicyclooctanes WCK 5153, zidebactam (WCK 5107), and WCK 4234. J. Med. Chem. 2018, 61, 4067–4086. [Google Scholar] [CrossRef] [PubMed]
- Hecker, S.J.; Reddy, K.R.; Lomovskaya, O.; Griffith, D.C.; Rubio-Aparicio, D.; Nelson, K.; Tsivkovski, R.; Sun, D.; Sabet, M.; Tarazi, Z.; et al. Discovery of Cyclic Boronic Acid QPX7728, an Ultrabroad-Spectrum Inhibitor of Serine and Metallo-β-lactamases. J. Med. Chem. 2020, 63, 7491–7507. [Google Scholar] [CrossRef]
- Tooke, C.L.; Hinchliffe, P.; Lang, P.A.; Mulholland, A.J.; Brem, J.; Schofield, C.J.; Spencer, J. Molecular Basis of Class A β-Lactamase Inhibition by Relebactam. Antimicrob. Agents Chemother. 2019, 63, e00564-19. [Google Scholar] [CrossRef] [PubMed]
- Moya, B.; Barcelo, I.M.; Bhagwat, S.; Patel, M.; Bou, G.; Papp-Wallace, K.M.; Bonomo, R.A.; Oliver, A. WCK 5107 (Zidebactam) and WCK 5153 Are Novel Inhibitors of PBP2 Showing Potent “β-Lactam Enhancer” Activity against Pseudomonas aeruginosa, Including Multidrug-Resistant Metallo-β-Lactamase-Producing High-Risk Clones. Antimicrob. Agents Chemother. 2017, 61, e02529-16. [Google Scholar] [CrossRef] [PubMed]
- Morinaka, A.; Tsutsumi, Y.; Yamada, M.; Suzuki, K.; Watanabe, T.; Abe, T.; Furuuchi, T.; Inamura, S.; Sakamaki, Y.; Mitsuhashi, N.; et al. OP0595, a new diazabicyclooctane: Mode of action as a serine β-lactamase inhibitor, antibiotic and β-lactam “enhancer”. J. Antimicrob. Chemother. 2015, 70, 2779–2786. [Google Scholar] [CrossRef]
- Barnes, M.D.; Taracila, M.A.; Good, C.E.; Bajaksouzian, S.; Rojas, L.J.; van Duin, D.; Kreiswirth, B.N.; Jacobs, M.R.; Haldimann, A.; Papp-Wallace, K.M.; et al. Nacubactam Enhances Meropenem Activity against Carbapenem-Resistant Klebsiella pneumoniae Producing KPC. Antimicrob. Agents Chemother. 2019, 63, e00432-19. [Google Scholar] [CrossRef]
- Miller, A.A.; Shapiro, A.B.; McLeod, S.M.; Carter, N.M.; Moussa, S.H.; Tommasi, R.; Mueller, J.P. In Vitro Characterization of ETX1317, a Broad-Spectrum β-Lactamase Inhibitor that Restores and Enhances β-Lactam Activity against Multi-Drug-Resistant Enterobacteriales, Including Carbapenem-Resistant Strains. ACS Infect. Dis. 2020, 6, 1389–1397. [Google Scholar] [CrossRef]
- Hamrick, J.C.; Docquier, J.-D.; Uehara, T.; Myers, C.L.; Six, D.A.; Chatwin, C.L.; John, K.J.; Vernacchio, S.F.; Cusick, S.M.; Trout, R.E.L.; et al. VNRX-5133 (Taniborbactam), a Broad-Spectrum Inhibitor of Serine- and Metallo-β-Lactamases, Restores Activity of Cefepime in Enterobacterales and Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2020, 64, e01963-19. [Google Scholar] [CrossRef]
- Krajnc, A.; Brem, J.; Hinchliffe, P.; Calvopiña, K.; Panduwawala, T.D.; Lang, P.A.; Kamps, J.J.A.G.; Tyrrell, J.M.; Widlake, E.; Saward, B.G.; et al. Bicyclic Boronate VNRX-5133 Inhibits Metallo- and Serine-β-Lactamases. J. Med. Chem. 2019, 62, 8544–8556. [Google Scholar] [CrossRef] [PubMed]
- Myers, C.L.; Daigle, D.M.; Burns, C.J.; Pevear, D.C. Ceftibuten/VNRX-7145, an orally bioavailable β-lactam/β-lactamase inhibitor combination active against serine-β-lactamase-producing Enterobacteriaceae. In Proceedings of the ECCMID 2019, Amsterdam, The Netherlands, 13–16 April 2019. [Google Scholar]
- Papp-Wallace, K.M.; Bethel, C.R.; Caillon, J.; Barnes, M.D.; Potel, G.; Bajaksouzian, S.; Rutter, J.D.; Reghal, A.; Shapiro, S.; Taracila, M.A.; et al. Beyond Piperacillin-Tazobactam: Cefepime and AAI101 as a Potent β-Lactam-β-Lactamase Inhibitor Combination. Antimicrob. Agents Chemother. 2019, 63, e00105-19. [Google Scholar] [CrossRef] [PubMed]
- Vallejo, J.A.; Martínez-Guitián, M.; Vázquez-Ucha, J.C.; González-Bello, C.; Poza, M.; Buynak, J.D.; Bethel, C.R.; Bonomo, R.A.; Bou, G.; Beceiro, A. LN-1-255, a penicillanic acid sulfone able to inhibit the class D carbapenemase OXA-48. J. Antimicrob. Chemother. 2016, 71, 2171–2180. [Google Scholar] [CrossRef] [PubMed]
- Davies, D.T.; Leiris, S.; Sprynski, N.; Castandet, J.; Lozano, C.; Bousquet, J.; Zalacain, M.; Vasa, S.; Dasari, P.K.; Pattipati, R.; et al. ANT2681: SAR Studies Leading to the Identification of a Metallo-β-lactamase Inhibitor with Potential for Clinical Use in Combination with Meropenem for the Treatment of Infections Caused by NDM-Producing Enterobacteriaceae. ACS Infect. Dis. 2020, 6, 2419–2430. [Google Scholar] [CrossRef]
- Coleman, K. Diazabicyclooctanes (DBOs): A potent new class of non-β-lactam β-lactamase inhibitors. Curr. Opin. Microbiol. 2011, 14, 550–555. [Google Scholar] [CrossRef]
- Shlaes, D.M. New β-lactam-β-lactamase inhibitor combinations in clinical development. Ann. N. Y. Acad. Sci. 2013, 1277, 105–114. [Google Scholar] [CrossRef]
- Hirsch, E.B.; Ledesma, K.R.; Chang, K.-T.; Schwartz, M.S.; Motyl, M.R.; Tam, V.H. In Vitro activity of MK-7655, a novel β-lactamase inhibitor, in combination with imipenem against carbapenem-resistant Gram-negative bacteria. Antimicrob. Agents Chemother. 2012, 56, 3753–3757. [Google Scholar] [CrossRef]
- Livermore, D.M.; Mushtaq, S.; Warner, M.; Vickers, A.; Woodford, N. In Vitro activity of cefepime/zidebactam (WCK 5222) against Gram-negative bacteria. J. Antimicrob. Chemother. 2017, 72, 1373–1385. [Google Scholar] [CrossRef]
- Livermore, D.M.; Meunier, D.; Hopkins, K.L.; Doumith, M.; Hill, R.; Pike, R.; Staves, P.; Woodford, N. Activity of ceftazidime/avibactam against problem Enterobacteriaceae and Pseudomonas aeruginosa in the UK, 2015–2016. J. Antimicrob. Chemother. 2018, 73, 648–657. [Google Scholar] [CrossRef]
- Ehmann, D.E.; Jahic, H.; Ross, P.L.; Gu, R.-F.; Hu, J.; Durand-Réville, T.F.; Lahiri, S.; Thresher, J.; Livchak, S.; Gao, N.; et al. Kinetics of avibactam inhibition against Class A, C, and D β-lactamases. J. Biol. Chem. 2013, 288, 27960–27971. [Google Scholar] [CrossRef]
- Ehmann, D.E.; Jahić, H.; Ross, P.L.; Gu, R.-F.; Hu, J.; Kern, G.; Walkup, G.K.; Fisher, S.L. Avibactam is a covalent, reversible, non-β-lactam β-lactamase inhibitor. Proc. Natl. Acad. Sci. USA 2012, 109, 11663–11668. [Google Scholar] [CrossRef] [PubMed]
- Barnes, M.D.; Winkler, M.L.; Taracila, M.A.; Page, M.G.; Desarbre, E.; Kreiswirth, B.N.; Shields, R.K.; Nguyen, M.-H.; Clancy, C.; Spellberg, B.; et al. Klebsiella pneumoniae Carbapenemase-2 (KPC-2), Substitutions at Ambler Position Asp179, and Resistance to Ceftazidime-Avibactam: Unique Antibiotic-Resistant Phenotypes Emerge from β-Lactamase Protein Engineering. MBio 2017, 8, e00528-17. [Google Scholar] [CrossRef] [PubMed]
- Shields, R.K.; Nguyen, M.H.; Chen, L.; Press, E.G.; Potoski, B.A.; Marini, R.V.; Doi, Y.; Kreiswirth, B.N.; Clancy, C.J. Ceftazidime-Avibactam Is Superior to Other Treatment Regimens against Carbapenem-Resistant Klebsiella pneumoniae Bacteremia. Antimicrob. Agents Chemother. 2017, 61, e00883-17. [Google Scholar] [CrossRef] [PubMed]
- Sader, H.S.; Castanheira, M.; Shortridge, D.; Mendes, R.E.; Flamm, R.K. Antimicrobial Activity of Ceftazidime-Avibactam Tested against Multidrug-Resistant Enterobacteriaceae and Pseudomonas aeruginosa Isolates from U.S. Medical Centers, 2013 to 2016. Antimicrob. Agents Chemother. 2017, 61, e01045-17. [Google Scholar] [CrossRef] [PubMed]
- Torrens, G.; Cabot, G.; Ocampo-Sosa, A.A.; Conejo, M.C.; Zamorano, L.; Navarro, F.; Pascual, Á.; Martínez-Martínez, L.; Oliver, A. Activity of Ceftazidime-Avibactam against Clinical and Isogenic Laboratory Pseudomonas aeruginosa Isolates Expressing Combinations of Most Relevant β-Lactam Resistance Mechanisms. Antimicrob. Agents Chemother. 2016, 60, 6407–6410. [Google Scholar] [CrossRef]
- Van Duin, D.; Bonomo, R.A. Ceftazidime/Avibactam and Ceftolozane/Tazobactam: Second-generation β-Lactam/β-Lactamase Inhibitor Combinations. Clin. Infect. Dis. 2016, 63, 234–241. [Google Scholar] [CrossRef]
- Blizzard, T.A.; Chen, H.; Kim, S.; Wu, J.; Bodner, R.; Gude, C.; Imbriglio, J.; Young, K.; Park, Y.-W.; Ogawa, A.; et al. Discovery of MK-7655, a β-lactamase inhibitor for combination with Primaxin®. Bioorg. Med. Chem. Lett. 2014, 24, 780–785. [Google Scholar] [CrossRef]
- Barnes, M.D.; Bethel, C.R.; Alsop, J.; Becka, S.A.; Rutter, J.D.; Papp-Wallace, K.M.; Bonomo, R.A. Inactivation of the Pseudomonas-Derived Cephalosporinase-3 (PDC-3) by Relebactam. Antimicrob. Agents Chemother. 2018, 62, e02406-17. [Google Scholar] [CrossRef]
- Papp-Wallace, K.M.; Barnes, M.D.; Alsop, J.; Taracila, M.A.; Bethel, C.R.; Becka, S.A.; van Duin, D.; Kreiswirth, B.N.; Kaye, K.S.; Bonomo, R.A. Relebactam Is a Potent Inhibitor of the KPC-2 β-Lactamase and Restores Imipenem Susceptibility in KPC-Producing Enterobacteriaceae. Antimicrob. Agents Chemother. 2018, 62, e00174-18. [Google Scholar] [CrossRef]
- Reyes, S.; Abdelraouf, K.; Nicolau, D.P. In vivo activity of human-simulated regimens of imipenem alone and in combination with relebactam against Pseudomonas aeruginosa in the murine thigh infection model. J. Antimicrob. Chemother. 2020, 75, 2197–2205. [Google Scholar] [CrossRef]
- Motsch, J.; Murta de Oliveira, C.; Stus, V.; Köksal, I.; Lyulko, O.; Boucher, H.W.; Kaye, K.S.; File, T.M.; Brown, M.L.; Khan, I.; et al. RESTORE-IMI 1: A Multicenter, Randomized, Double-blind Trial Comparing Efficacy and Safety of Imipenem/Relebactam vs Colistin Plus Imipenem in Patients With Imipenem-nonsusceptible Bacterial Infections. Clin. Infect. Dis. 2020, 70, 1799–1808. [Google Scholar] [CrossRef] [PubMed]
- Titov, I.; Wunderink, R.G.; Roquilly, A.; Rodríguez Gonzalez, D.; David-Wang, A.; Boucher, H.W.; Kaye, K.S.; Losada, M.C.; Du, J.; Tipping, R.; et al. A Randomized, Double-blind, Multicenter Trial Comparing Efficacy and Safety of Imipenem/Cilastatin/Relebactam Versus Piperacillin/Tazobactam in Adults With Hospital-acquired or Ventilator-associated Bacterial Pneumonia (RESTORE-IMI 2 Study). Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, E.J.C.; Citron, D.M.; Tyrrell, K.L.; Leoncio, E.; Merriam, C.V. Comparative In Vitro Activities of Relebactam, Imipenem, the Combination of the Two, and Six Comparator Antimicrobial Agents against 432 Strains of Anaerobic Organisms, Including Imipenem-Resistant Strains. Antimicrob. Agents Chemother. 2018, 62, e01992-17. [Google Scholar] [CrossRef]
- Karlowsky, J.A.; Lob, S.H.; Kazmierczak, K.M.; Young, K.; Motyl, M.R.; Sahm, D.F. In Vitro activity of imipenem/relebactam against Enterobacteriaceae and Pseudomonas aeruginosa isolated from intraabdominal and urinary tract infection samples: SMART Surveillance United States 2015-2017. J. Glob. Antimicrob. Resist. 2020, 21, 223–228. [Google Scholar] [CrossRef]
- Karlowsky, J.A.; Lob, S.H.; Raddatz, J.; DePestel, D.D.; Young, K.; Motyl, M.R.; Sahm, D.F. In Vitro Activity of Imipenem/Relebactam and Ceftolozane/Tazobactam against Clinical Isolates of Gram-Negative Bacilli with Difficult-to-Treat Resistance and Multidrug-Resistant Phenotypes—SMART United States 2015-2017. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Gomez-Simmonds, A.; Stump, S.; Giddins, M.J.; Annavajhala, M.K.; Uhlemann, A.-C. Clonal Background, Resistance Gene Profile, and Porin Gene Mutations Modulate In Vitro Susceptibility to Imipenem-Relebactam in Diverse Enterobacteriaceae. Antimicrob. Agents Chemother. 2018, 62, e00573-18. [Google Scholar] [CrossRef]
- Arca-Suárez, J.; Lasarte-Monterrubio, C.; Rodiño-Janeiro, B.-K.; Cabot, G.; Vázquez-Ucha, J.C.; Rodríguez-Iglesias, M.; Galán-Sánchez, F.; Beceiro, A.; González-Bello, C.; Oliver, A.; et al. Molecular mechanisms driving the in vivo development of OXA-10-mediated resistance to ceftolozane/tazobactam and ceftazidime/avibactam during treatment of XDR Pseudomonas aeruginosa infections. J. Antimicrob. Chemother. 2020. [Google Scholar] [CrossRef] [PubMed]
- Mushtaq, S.; Meunier, D.; Vickers, A.; Woodford, N.; Livermore, D.M. Activity of imipenem/relebactam against Pseudomonas aeruginosa producing ESBLs and carbapenemases. J. Antimicrob. Chemother. 2020. [Google Scholar] [CrossRef]
- Fraile-Ribot, P.A.; Zamorano, L.; Orellana, R.; Del Barrio-Tofiño, E.; Sánchez-Diener, I.; Cortes-Lara, S.; López-Causapé, C.; Cabot, G.; Bou, G.; Martínez-Martínez, L.; et al. Activity of Imipenem-Relebactam against a Large Collection of Pseudomonas aeruginosa Clinical Isolates and Isogenic β-Lactam-Resistant Mutants. Antimicrob. Agents Chemother. 2020, 64, e02165-19. [Google Scholar] [CrossRef] [PubMed]
- Lang, P.A.; Parkova, A.; Leissing, T.M.; Calvopiña, K.; Cain, R.; Krajnc, A.; Panduwawala, T.D.; Philippe, J.; Fishwick, C.W.G.; Trapencieris, P.; et al. Bicyclic Boronates as Potent Inhibitors of ampC, the Class C β-Lactamase from Escherichia coli. Biomolecules 2020, 10, 899. [Google Scholar] [CrossRef]
- Cendron, L.; Quotadamo, A.; Maso, L.; Bellio, P.; Montanari, M.; Celenza, G.; Venturelli, A.; Costi, M.P.; Tondi, D. X-ray Crystallography Deciphers the Activity of Broad-Spectrum Boronic Acid β-Lactamase Inhibitors. ACS Med. Chem. Lett. 2019, 10, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Lang, P.A.; Leissing, T.M.; Page, M.G.P.; Schofield, C.J.; Brem, J. Structural Investigations of the Inhibition of Escherichia coli AmpC β-Lactamase by Diazabicyclooctanes. Antimicrob. Agents Chemother. 2020. [Google Scholar] [CrossRef] [PubMed]
- Krajnc, A.; Lang, P.A.; Panduwawala, T.D.; Brem, J.; Schofield, C.J. Will Morphing Boron-Based Inhibitors Beat the β-Lactamases? Curr. Opin. Chem. Biol. 2019, 50, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Cahill, S.T.; Tyrrell, J.M.; Navratilova, I.H.; Calvopiña, K.; Robinson, S.W.; Lohans, C.T.; McDonough, M.A.; Cain, R.; Fishwick, C.W.G.; Avison, M.B.; et al. Studies on the inhibition of AmpC and other β-lactamases by cyclic boronates. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 742–748. [Google Scholar] [CrossRef] [PubMed]
- Lomovskaya, O.; Sun, D.; Rubio-Aparicio, D.; Nelson, K.; Tsivkovski, R.; Griffith, D.C.; Dudley, M.N. Vaborbactam: Spectrum of Beta-Lactamase Inhibition and Impact of Resistance Mechanisms on Activity in Enterobacteriaceaez. Antimicrob. Agents Chemother. 2017, 61, e01443-17. [Google Scholar] [CrossRef]
- Kinn, P.M.; Chen, D.J.; Gihring, T.M.; Schulz, L.T.; Fox, B.C.; McCreary, E.K.; Lepak, A.J. In Vitro evaluation of meropenem-vaborbactam against clinical CRE isolates at a tertiary care center with low KPC-mediated carbapenem resistance. Diagn. Microbiol. Infect. Dis. 2019, 93, 258–260. [Google Scholar] [CrossRef]
- Tsivkovski, R.; Lomovskayaa, O. Biochemical Activity of Vaborbactam. Antimicrob. Agents Chemother. 2020, 64, e01935-19. [Google Scholar] [CrossRef]
- Kaye, K.S.; Bhowmick, T.; Metallidis, S.; Bleasdale, S.C.; Sagan, O.S.; Stus, V.; Vazquez, J.; Zaitsev, V.; Bidair, M.; Chorvat, E.; et al. Effect of Meropenem-Vaborbactam vs Piperacillin-Tazobactam on Clinical Cure or Improvement and Microbial Eradication in Complicated Urinary Tract Infection: The TANGO I Randomized Clinical Trial. JAMA 2018, 319, 788–799. [Google Scholar] [CrossRef]
- Wunderink, R.G.; Giamarellos-Bourboulis, E.J.; Rahav, G.; Mathers, A.J.; Bassetti, M.; Vazquez, J.; Cornely, O.A.; Solomkin, J.; Bhowmick, T.; Bishara, J.; et al. Effect and Safety of Meropenem-Vaborbactam versus Best-Available Therapy in Patients with Carbapenem-Resistant Enterobacteriaceae Infections: The TANGO II Randomized Clinical Trial. Infect. Dis. Ther. 2018, 7, 439–455. [Google Scholar] [CrossRef]
- Zhou, M.; Yang, Q.; Lomovskaya, O.; Sun, D.; Kudinha, T.; Xu, Z.; Zhang, G.; Chen, X.; Xu, Y. In Vitro activity of meropenem combined with vaborbactam against KPC-producing Enterobacteriaceae in China. J. Antimicrob. Chemother. 2018, 73, 2789–2796. [Google Scholar] [CrossRef]
- Hackel, M.A.; Lomovskaya, O.; Dudley, M.N.; Karlowsky, J.A.; Sahm, D.F. In Vitro Activity of Meropenem-Vaborbactam against Clinical Isolates of KPC-Positive Enterobacteriaceae. Antimicrob. Agents Chemother. 2018, 62, e01904-17. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.R.; Kline, E.G.; Jones, C.E.; Morder, K.T.; Mettus, R.T.; Doi, Y.; Nguyen, M.H.; Clancy, C.J.; Shields, R.K. Effects of KPC Variant and Porin Genotype on the In Vitro Activity of Meropenem-Vaborbactam against Carbapenem-Resistant Enterobacteriaceae. Antimicrob. Agents Chemother. 2019, 63, e02048-18. [Google Scholar] [CrossRef] [PubMed]
- Yahav, D.; Giske, C.G.; Grāmatniece, A.; Abodakpi, H.; Tam, V.H.; Leibovici, L. New β-Lactam-β-Lactamase Inhibitor Combinations. Clin. Microbiol. Rev. 2020, 34, e00115-20. [Google Scholar] [CrossRef] [PubMed]
- Humphries, R.M.; Yang, S.; Hemarajata, P.; Ward, K.W.; Hindler, J.A.; Miller, S.A.; Gregson, A. First Report of Ceftazidime-Avibactam Resistance in a KPC-3-Expressing Klebsiella pneumoniae Isolate. Antimicrob. Agents Chemother. 2015, 59, 6605–6607. [Google Scholar] [CrossRef] [PubMed]
- Fraile-Ribot, P.A.; Cabot, G.; Mulet, X.; Periañez, L.; Luisa Martín-Pena, M.; Juan, C.; Pérez, J.L.; Oliver, A. Mechanisms leading to in vivo ceftolozane/tazobactam resistance development during the treatment of infections caused by MDR Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2018, 73, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Oueslati, S.; Iorga, B.I.; Tlili, L.; Exilie, C.; Zavala, A.; Dortet, L.; Jousset, A.B.; Bernabeu, S.; Bonnin, R.A.; Naas, T. Unravelling ceftazidime/avibactam resistance of KPC-28, a KPC-2 variant lacking carbapenemase activity. J. Antimicrob. Chemother. 2019, 74, 2239–2246. [Google Scholar] [CrossRef]
- Mueller, L.; Masseron, A.; Prod’Hom, G.; Galperine, T.; Greub, G.; Poirel, L.; Nordmann, P. Phenotypic, biochemical and genetic analysis of KPC-41, a KPC-3 variant conferring resistance to ceftazidime-avibactam and exhibiting reduced carbapenemase activity. Antimicrob. Agents Chemother. 2019, 63, e01111-19. [Google Scholar] [CrossRef]
- Hobson, C.A.; Bonacorsi, S.; Jacquier, H.; Choudhury, A.; Magnan, M.; Cointe, A.; Bercot, B.; Tenaillon, O.; Birgy, A. KPC beta-lactamases are permissive to insertions and deletions conferring substrate spectrum modifications and resistance to ceftazidime-avibactam. Antimicrob. Agents Chemother. 2020, 64, e01175-20. [Google Scholar] [CrossRef]
- Haidar, G.; Clancy, C.J.; Shields, R.K.; Hao, B.; Cheng, S.; Nguyen, M.H. Mutations in blaKPC-3 that confer ceftazidime-avibactam resistance encode novel KPC-3 variants that function as extended-spectrum β-lactamases. Antimicrob. Agents Chemother. 2017, 61, e02534-16. [Google Scholar] [CrossRef]
- Shields, R.K.; Nguyen, M.H.; Press, E.G.; Chen, L.; Kreiswirth, B.N.; Clancy, C.J. In Vitro Selection of Meropenem Resistance among Ceftazidime-Avibactam-Resistant, Meropenem-Susceptible Klebsiella pneumoniae Isolates with Variant KPC-3 Carbapenemases. Antimicrob. Agents Chemother. 2017, 61, e00079-17. [Google Scholar] [CrossRef]
- Compain, F.; Dorchène, D.; Arthur, M. Combination of Amino Acid Substitutions Leading to CTX-M-15-Mediated Resistance to the Ceftazidime-Avibactam Combination. Antimicrob. Agents Chemother. 2018, 62, e00357-18. [Google Scholar] [CrossRef] [PubMed]
- Nelson, K.; Hemarajata, P.; Sun, D.; Rubio-Aparicio, D.; Tsivkovski, R.; Yang, S.; Sebra, R.; Kasarskis, A.; Nguyen, H.; Hanson, B.M.; et al. Resistance to Ceftazidime-Avibactam Is Due to Transposition of KPC in a Porin-Deficient Strain of Klebsiella pneumoniae with Increased Efflux Activity. Antimicrob. Agents Chemother. 2017, 61, e00989-17. [Google Scholar] [CrossRef] [PubMed]
- Arca-Suárez, J.; Vázquez-Ucha, J.C.; Fraile-Ribot, P.A.; Lence, E.; Cabot, G.; Martínez-Guitián, M.; Lasarte-Monterrubio, C.; Rodríguez-Iglesias, M.; Beceiro, A.; González-Bello, C.; et al. Molecular and biochemical insights into the in vivo evolution of AmpC-mediated resistance to ceftolozane/tazobactam during treatment of an MDR Pseudomonas aeruginosa infection. J. Antimicrob. Chemother. 2020, 75, 3209–3217. [Google Scholar] [CrossRef] [PubMed]
- Fraile-Ribot, P.A.; Mulet, X.; Cabot, G.; Del Barrio-Tofiño, E.; Juan, C.; Pérez, J.L.; Oliver, A. In Vivo Emergence of Resistance to Novel Cephalosporin-β-Lactamase Inhibitor Combinations through the Duplication of Amino Acid D149 from OXA-2 β-Lactamase (OXA-539) in Sequence Type 235 Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2017, 61, e01117-17. [Google Scholar] [CrossRef] [PubMed]
- Slater, C.L.; Winogrodzki, J.; Fraile-Ribot, P.A.; Oliver, A.; Khajehpour, M.; Mark, B.L. Adding Insult to Injury: Mechanistic Basis for How AmpC Mutations Allow Pseudomonas aeruginosa To Accelerate Cephalosporin Hydrolysis and Evade Avibactam. Antimicrob. Agents Chemother. 2020, 64, e00894-20. [Google Scholar] [CrossRef]
- Sun, D.; Rubio-Aparicio, D.; Nelson, K.; Dudley, M.N.; Lomovskaya, O. Meropenem-Vaborbactam Resistance Selection, Resistance Prevention, and Molecular Mechanisms in Mutants of KPC-Producing Klebsiella pneumoniae. Antimicrob. Agents Chemother. 2017, 61, e01694-17. [Google Scholar] [CrossRef] [PubMed]
- Lapuebla, A.; Abdallah, M.; Olafisoye, O.; Cortes, C.; Urban, C.; Landman, D.; Quale, J. Activity of Imipenem with Relebactam against Gram-Negative Pathogens from New York City. Antimicrob. Agents Chemother. 2015, 59, 5029–5031. [Google Scholar] [CrossRef]
- Gomis-Font, M.A.; Cabot, G.; Sánchez-Diener, I.; Fraile-Ribot, P.A.; Juan, C.; Moya, B.; Zamorano, L.; Oliver, A. In Vitro dynamics and mechanisms of resistance development to imipenem and imipenem/relebactam in Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2020, 75, 2508–2515. [Google Scholar] [CrossRef]
- Karlowsky, J.A.; Kazmierczak, K.M.; de Jonge, B.L.M.; Hackel, M.A.; Sahm, D.F.; Bradford, P.A. In Vitro Activity of Aztreonam-Avibactam against Enterobacteriaceae and Pseudomonas aeruginosa Isolated by Clinical Laboratories in 40 Countries from 2012 to 2015. Antimicrob. Agents Chemother. 2017, 61, e00472-17. [Google Scholar] [CrossRef]
- Shields, R.K.; Doi, Y. Aztreonam Combination Therapy: An Answer to Metallo-β-Lactamase-Producing Gram-Negative Bacteria? Clin. Infect. Dis. 2020, 71, 1099–1101. [Google Scholar] [CrossRef]
- Lee, M.; Abbey, T.; Biagi, M.; Wenzler, E. Activity of aztreonam in combination with ceftazidime-avibactam against serine- and metallo-β-lactamase-producing Pseudomonas aeruginosa. Diagn. Microbiol. Infect. Dis. 2020, 99, 115227. [Google Scholar] [CrossRef] [PubMed]
- Wenzler, E.; Deraedt, M.F.; Harrington, A.T.; Danizger, L.H. Synergistic activity of ceftazidime-avibactam and aztreonam against serine and metallo-β-lactamase-producing gram-negative pathogens. Diagn. Microbiol. Infect. Dis. 2017, 88, 352–354. [Google Scholar] [CrossRef] [PubMed]
- Shaw, E.; Rombauts, A.; Tubau, F.; Padullés, A.; Càmara, J.; Lozano, T.; Cobo-Sacristán, S.; Sabe, N.; Grau, I.; Rigo-Bonnin, R.; et al. Clinical outcomes after combination treatment with ceftazidime/avibactam and aztreonam for NDM-1/OXA-48/CTX-M-15-producing Klebsiella pneumoniae infection. J. Antimicrob. Chemother. 2018, 73, 1104–1106. [Google Scholar] [CrossRef] [PubMed]
- Benchetrit, L.; Mathy, V.; Armand-Lefevre, L.; Bouadma, L.; Timsit, J.-F. Successful treatment of septic shock due to NDM-1-producing Klebsiella pneumoniae using ceftazidime/avibactam combined with aztreonam in solid organ transplant recipients: Report of two cases. Int. J. Antimicrob. Agents 2020, 55, 105842. [Google Scholar] [CrossRef]
- Sieswerda, E.; van den Brand, M.; van den Berg, R.B.; Sträter, J.; Schouls, L.; van Dijk, K.; Budding, A.E. Successful rescue treatment of sepsis due to a pandrug-resistant, NDM-producing Klebsiella pneumoniae using aztreonam powder for nebulizer solution as intravenous therapy in combination with ceftazidime/avibactam. J. Antimicrob. Chemother. 2020, 75, 773–775. [Google Scholar] [CrossRef]
- Moya, B.; Barcelo, I.M.; Bhagwat, S.; Patel, M.; Bou, G.; Papp-Wallace, K.M.; Bonomo, R.A.; Oliver, A. Potent β-Lactam Enhancer Activity of Zidebactam and WCK 5153 against Acinetobacter baumannii, Including Carbapenemase-Producing Clinical Isolates. Antimicrob. Agents Chemother. 2017, 61, e01238-17. [Google Scholar] [CrossRef]
- Karlowsky, J.A.; Hackel, M.A.; Bouchillon, S.K.; Sahm, D.F. In Vitro Activity of WCK 5222 (Cefepime-Zidebactam) against Worldwide Collected Gram-Negative Bacilli Not Susceptible to Carbapenems. Antimicrob. Agents Chemother. 2020. [Google Scholar] [CrossRef]
- Asli, A.; Brouillette, E.; Krause, K.M.; Nichols, W.W.; Malouin, F. Distinctive Binding of Avibactam to Penicillin-Binding Proteins of Gram-Negative and Gram-Positive Bacteria. Antimicrob. Agents Chemother. 2016, 60, 752–756. [Google Scholar] [CrossRef]
- Shapiro, A.B.; Gao, N.; Jahić, H.; Carter, N.M.; Chen, A.; Miller, A.A. Reversibility of Covalent, Broad-Spectrum Serine β-Lactamase Inhibition by the Diazabicyclooctenone ETX2514. ACS Infect. Dis. 2017, 3, 833–844. [Google Scholar] [CrossRef]
- McLeod, S.M.; Shapiro, A.B.; Moussa, S.H.; Johnstone, M.; McLaughlin, R.E.; de Jonge, B.L.M.; Miller, A.A. Frequency and Mechanism of Spontaneous Resistance to Sulbactam Combined with the Novel β-Lactamase Inhibitor ETX2514 in Clinical Isolates of Acinetobacter baumannii. Antimicrob. Agents Chemother. 2018, 62, e01576-17. [Google Scholar] [CrossRef]
- Higgins, P.G.; Wisplinghoff, H.; Stefanik, D.; Seifert, H. In Vitro activities of the beta-lactamase inhibitors clavulanic acid, sulbactam, and tazobactam alone or in combination with beta-lactams against epidemiologically characterized multidrug-resistant Acinetobacter baumannii strains. Antimicrob. Agents Chemother. 2004, 48, 1586–1592. [Google Scholar] [CrossRef] [PubMed]
- Kuo, S.-C.; Lee, Y.-T.; Yang Lauderdale, T.-L.; Huang, W.-C.; Chuang, M.-F.; Chen, C.-P.; Su, S.-C.; Lee, K.-R.; Chen, T.-L. Contribution of Acinetobacter-derived cephalosporinase-30 to sulbactam resistance in Acinetobacter baumannii. Front. Microbiol. 2015, 6, 231. [Google Scholar] [CrossRef] [PubMed]
- Seifert, H.; Müller, C.; Stefanik, D.; Higgins, P.G.; Miller, A.; Kresken, M. In Vitro activity of sulbactam/durlobactam against global isolates of carbapenem-resistant Acinetobacter baumannii. J. Antimicrob. Chemother. 2020, 75, 2616–2621. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Xu, Y.; Jia, P.; Zhu, Y.; Zhang, J.; Zhang, G.; Deng, J.; Hackel, M.; Bradford, P.A.; Reinhart, H. In Vitro activity of sulbactam/durlobactam against clinical isolates of Acinetobacter baumannii collected in China. J. Antimicrob. Chemother. 2020, 75, 1833–1839. [Google Scholar] [CrossRef]
- McLeod, S.M.; Moussa, S.H.; Hackel, M.A.; Miller, A.A. In Vitro Activity of Sulbactam-Durlobactam against Acinetobacter baumannii-calcoaceticus Complex Isolates Collected Globally in 2016 and 2017. Antimicrob. Agents Chemother. 2020, 64, e02534-19. [Google Scholar] [CrossRef]
- Mushtaq, S.; Vickers, A.; Woodford, N.; Haldimann, A.; Livermore, D.M. Activity of nacubactam (RG6080/OP0595) combinations against MBL-producing Enterobacteriaceae. J. Antimicrob. Chemother. 2019, 74, 953–960. [Google Scholar] [CrossRef]
- Okujava, R.; Garcia-Alcalde, F.; Haldimann, A.; Zampaloni, C.; Morrissey, I.; Magnet, S.; Kothari, N.; Harding, I.; Bradley, K. 1359. Activity of meropenem/nacubactam combination against Gram-negative clinical isolates: ROSCO global surveillance 2017. Open Forum Infect. Dis. 2018, 5, S416. [Google Scholar] [CrossRef]
- Asempa, T.E.; Motos, A.; Abdelraouf, K.; Bissantz, C.; Zampaloni, C.; Nicolau, D.P. Meropenem-nacubactam activity against AmpC-overproducing and KPC-expressing Pseudomonas aeruginosa in a neutropenic murine lung infection model. Int. J. Antimicrob. Agents 2020, 55, 105838. [Google Scholar] [CrossRef]
- Livermore, D.M.; Mushtaq, S.; Warner, M.; Woodford, N. Activity of OP0595/β-lactam combinations against Gram-negative bacteria with extended-spectrum, AmpC and carbapenem-hydrolysing β-lactamases. J. Antimicrob. Chemother. 2015, 70, 3032–3041. [Google Scholar] [CrossRef]
- Durand-Réville, T.F.; Comita-Prevoir, J.; Zhang, J.; Wu, X.; May-Dracka, T.L.; Romero, J.A.C.; Wu, F.; Chen, A.; Shapiro, A.B.; Carter, N.M.; et al. Discovery of an Orally Available Diazabicyclooctane Inhibitor (ETX0282) of Class A, C, and D Serine β-Lactamases. J. Med. Chem. 2020, 63, 12511–12525. [Google Scholar] [CrossRef]
- Mushtaq, S.; Vickers, A.; Woodford, N.; Livermore, D.M. WCK 4234, a novel diazabicyclooctane potentiating carbapenems against Enterobacteriaceae, Pseudomonas and Acinetobacter with class A, C and D β-lactamases. J. Antimicrob. Chemother. 2017, 72, 1688–1695. [Google Scholar] [CrossRef] [PubMed]
- Iregui, A.; Khan, Z.; Landman, D.; Quale, J. Activity of Meropenem with a Novel Broader-Spectrum β-Lactamase Inhibitor, WCK 4234, against Gram-Negative Pathogens Endemic to New York City. Antimicrob. Agents Chemother. 2019, 64, e01666-19. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.P.; Pinto, N.A.; Vu, T.N.; Lee, H.; Cho, Y.L.; Byun, J.-H.; D’Souza, R.; Yong, D. In Vitro Activity of a Novel Siderophore-Cephalosporin, GT-1 and Serine-Type β-Lactamase Inhibitor, GT-055, against Escherichia coli, Klebsiella pneumoniae and Acinetobacter spp. Panel Strains. Antibiotics 2020, 9, 267. [Google Scholar] [CrossRef]
- Brascoy, B.D.; Trang, M.; Conde, H.; Bhavnani, S.M.; Biek, D.; Hannah, B.; Thye, D.; Ambrose, P.G. Pharmacokinetics-pharmacodynamics of the novel beta-lactamase inhibitor GT-055 in combination with the siderophore cephalosporin GT-1. In Proceedings of the ECCMID 2019, Amsterdam, The Netherlands, 13–16 April 2019. [Google Scholar]
- Wang, X.; Zhao, C.; Wang, Q.; Wang, Z.; Liang, X.; Zhang, F.; Zhang, Y.; Meng, H.; Chen, H.; Li, S.; et al. In Vitro activity of the novel β-lactamase inhibitor taniborbactam (VNRX-5133), in combination with cefepime or meropenem, against MDR Gram-negative bacterial isolates from China. J. Antimicrob. Chemother. 2020, 75, 1850–1858. [Google Scholar] [CrossRef] [PubMed]
- John, K.J.; Chatwin, C.L.; Hamrick, J.C.; Moeck, G.; Pevear, D.C. Rescue of Ceftibuten Activity by the Oral β-Lactamase Inhibitor VNRX-7145 against Enterobacteriaceae Expressing Class A, C and/or D β-Lactamases. In Proceedings of the ASM 2019, San Francisco, CA, USA, 20–24 June 2019. [Google Scholar]
- Hamrick, J.; Cassandra, C.; John, K.; Burns, C.; Xerri, L.; Moeck, G.; Pevear, D.C. Selection of ceftibuten as the partner antibiotic for the oral beta-lactamase inhibitor VNRX-7145. In Proceedings of the ECCMID 2019, Amsterdam, The Netherlands, 13–16 April 2019. [Google Scholar]
- Lomovskaya, O.; Tsivkovski, R.; Nelson, K.; Rubio-Aparicio, D.; Sun, D.; Totrov, M.; Dudley, M.N. Spectrum of Beta-Lactamase Inhibition by the Cyclic Boronate QPX7728, an Ultrabroad-Spectrum Beta-Lactamase Inhibitor of Serine and Metallo-Beta-Lactamases: Enhancement of Activity of Multiple Antibiotics against Isogenic Strains Expressing Single Beta-La. Antimicrob. Agents Chemother. 2020, 64, e00212-20. [Google Scholar] [CrossRef]
- Nelson, K.; Rubio-Aparicio, D.; Sun, D.; Dudley, M.; Lomovskaya, O. In Vitro Activity of the Ultrabroad-Spectrum-Beta-Lactamase Inhibitor QPX7728 against Carbapenem-Resistant Enterobacterales with Varying Intrinsic and Acquired Resistance Mechanisms. Antimicrob. Agents Chemother. 2020, 64, e00757-20. [Google Scholar] [CrossRef]
- Lomovskaya, O.; Nelson, K.; Rubio-Aparicio, D.; Tsivkovski, R.; Sun, D.; Dudley, M.N. Impact of Intrinsic Resistance Mechanisms on Potency of QPX7728, a New Ultrabroad-Spectrum Beta-Lactamase Inhibitor of Serine and Metallo-Beta-Lactamases in Enterobacteriaceae, Pseudomonas aeruginosa, and Acinetobacter baumannii. Antimicrob. Agents Chemother. 2020, 64, e00552-20. [Google Scholar] [CrossRef]
- Sabet, M.; Tarazi, Z.; Griffith, D.C. In Vivo Activity of QPX7728, an Ultrabroad-Spectrum Beta-Lactamase Inhibitor, in Combination with Beta-Lactams against Carbapenem-Resistant Klebsiella pneumoniae. Antimicrob. Agents Chemother. 2020, 64, e01267-20. [Google Scholar] [CrossRef]
- Rubio-Aparicio, D.; Nelson, K.; Griffith, D.C.; Dudley, M.N.; Lomovskaya, O. QPX7728: In Vitro Activity in Combination with Oral Beta-Lactam Antibiotics against Enterobacteriaceae. In Proceedings of the ASM 2019, San Francisco, CA, USA, 20–24 June 2019. [Google Scholar]
- Tselepis, L.; Langley, G.W.; Aboklaish, A.F.; Widlake, E.; Jackson, D.E.; Walsh, T.R.; Schofield, C.J.; Brem, J.; Tyrrell, J.M. In Vitro efficacy of imipenem-relebactam and cefepime-AAI101 against a global collection of ESBL-positive and carbapenemase-producing Enterobacteriaceae. Int. J. Antimicrob. Agents 2020, 56, 105925. [Google Scholar] [CrossRef]
- Crandon, J.L.; Nicolau, D.P. In Vitro Activity of Cefepime/AAI101 and Comparators against Cefepime Non-susceptible Enterobacteriaceae. Pathogens 2015, 4, 620–625. [Google Scholar] [CrossRef]
- Mushtaq, S.; Chaudhry, A.; Adkin, R.; Woodford, N.; Benedict, N.; Pypstra, R.; Shapiro, S. In-vitro activity of diverse β-lactam/aai101 combinations vs. Multidrug-resistant gram-negative clinical strains. In Proceedings of the ECCMID 2014, Barcelona, Spain, 24 May 2014. [Google Scholar]
- Nordmann, P.; Girlich, D.; Benedict, N.; Pypstra, R.; Shapiro, S. Characterization of b-lactamase inhibition by aai101. In Proceedings of the ECCMID 2014, Barcelona, Spain, 24 May 2014. [Google Scholar]
- Morrissey, I.; Magnet, S.; Hawser, S.; Shapiro, S.; Knechtle, P. In Vitro Activity of Cefepime-Enmetazobactam against Gram-Negative Isolates Collected from U.S. and European Hospitals during 2014-2015. Antimicrob. Agents Chemother. 2019, 63, e00514-19. [Google Scholar] [CrossRef] [PubMed]
- Crandon, J.L.; Nicolau, D.P. In vivo activities of simulated human doses of cefepime and cefepime-AAI101 against multidrug-resistant Gram-negative Enterobacteriaceae. Antimicrob. Agents Chemother. 2015, 59, 2688–2694. [Google Scholar] [CrossRef] [PubMed]
- Buynak, J.D.; Rao, A.S.; Doppalapudi, V.R.; Adam, G.; Petersen, P.J.; Nidamarthy, S.D. The synthesis and evaluation of 6-alkylidene-2′-beta-substituted penam sulfones as beta-lactamase inhibitors. Bioorg. Med. Chem. Lett. 1999, 9, 1997–2002. [Google Scholar] [CrossRef]
- Kalp, M.; Sheri, A.; Buynak, J.D.; Bethel, C.R.; Bonomo, R.A.; Carey, P.R. Efficient inhibition of class A and class D beta-lactamases by Michaelis complexes. J. Biol. Chem. 2007, 282, 21588–21591. [Google Scholar] [CrossRef]
- Buynak, J.D. The discovery and development of modified penicillin- and cephalosporin-derived beta-lactamase inhibitors. Curr. Med. Chem. 2004, 11, 1951–1964. [Google Scholar] [CrossRef] [PubMed]
- Drawz, S.M.; Bethel, C.R.; Doppalapudi, V.R.; Sheri, A.; Pagadala, S.R.R.; Hujer, A.M.; Skalweit, M.J.; Anderson, V.E.; Chen, S.G.; Buynak, J.D.; et al. Penicillin sulfone inhibitors of class D beta-lactamases. Antimicrob. Agents Chemother. 2010, 54, 1414–1424. [Google Scholar] [CrossRef]
- Bou, G.; Santillana, E.; Sheri, A.; Beceiro, A.; Sampson, J.M.; Kalp, M.; Bethel, C.R.; Distler, A.M.; Drawz, S.M.; Pagadala, S.R.R.; et al. Design, synthesis, and crystal structures of 6-alkylidene-2′-substituted penicillanic acid sulfones as potent inhibitors of Acinetobacter baumannii OXA-24 carbapenemase. J. Am. Chem. Soc. 2010, 132, 13320–13331. [Google Scholar] [CrossRef]
- Vázquez-Ucha, J.C.; Martínez-Guitián, M.; Maneiro, M.; Conde-Pérez, K.; Álvarez-Fraga, L.; Torrens, G.; Oliver, A.; Buynak, J.D.; Bonomo, R.A.; Bou, G.; et al. Therapeutic Efficacy of LN-1-255 in Combination with Imipenem in Severe Infection Caused by Carbapenem-Resistant Acinetobacter baumannii. Antimicrob. Agents Chemother. 2019, 63, e01092-19. [Google Scholar] [CrossRef]
- Rodríguez, D.; Maneiro, M.; Vázquez-Ucha, J.C.; Beceiro, A.; González-Bello, C. 6-Arylmethylidene Penicillin-Based Sulfone Inhibitors for Repurposing Antibiotic Efficiency in Priority Pathogens. J. Med. Chem. 2020, 63, 3737–3755. [Google Scholar] [CrossRef]
- Everett, M.; Sprynski, N.; Coelho, A.; Castandet, J.; Bayet, M.; Bougnon, J.; Lozano, C.; Davies, D.T.; Leiris, S.; Zalacain, M.; et al. Discovery of a Novel Metallo-β-Lactamase Inhibitor That Potentiates Meropenem Activity against Carbapenem-Resistant Enterobacteriaceae. Antimicrob. Agents Chemother. 2018, 62, e00074-18. [Google Scholar] [CrossRef]
- Buynak, J.D.; Chen, H.; Vogeti, L.; Gadhachanda, V.R.; Buchanan, C.A.; Palzkill, T.; Shaw, R.W.; Spencer, J.; Walsh, T.R. Penicillin-derived inhibitors that simultaneously target both metallo- and serine-beta-lactamases. Bioorg. Med. Chem. Lett. 2004, 14, 1299–1304. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, A.M.; Gu, Y.; Dos Santos, O.; Abe, T.; Agarwal, A.; Yang, Y.; Petersen, P.J.; Weiss, W.J.; Mansour, T.S.; Nukaga, M.; et al. Structure-activity relationship of 6-methylidene penems bearing tricyclic heterocycles as broad-spectrum beta-lactamase inhibitors: Crystallographic structures show unexpected binding of 1,4-thiazepine intermediates. J. Med. Chem. 2004, 47, 6556–6568. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, A.M.; Agarwal, A.; Abe, T.; Ushirogochi, H.; Yamamura, I.; Ado, M.; Tsuyoshi, T.; Dos Santos, O.; Gu, Y.; Sum, F.-W.; et al. Structure-activity relationship of 6-methylidene penems bearing 6,5 bicyclic heterocycles as broad-spectrum beta-lactamase inhibitors: Evidence for 1,4-thiazepine intermediates with C7 R stereochemistry by computational methods. J. Med. Chem. 2006, 49, 4623–4637. [Google Scholar] [CrossRef] [PubMed]
- Weiss, W.J.; Petersen, P.J.; Murphy, T.M.; Tardio, L.; Yang, Y.; Bradford, P.A.; Venkatesan, A.M.; Abe, T.; Isoda, T.; Mihira, A.; et al. In Vitro and in vivo activities of novel 6-methylidene penems as beta-lactamase inhibitors. Antimicrob. Agents Chemother. 2004, 48, 4589–4596. [Google Scholar] [CrossRef] [PubMed]
- Nagano, R.; Adachi, Y.; Imamura, H.; Yamada, K.; Hashizume, T.; Morishima, H. Carbapenem derivatives as potential inhibitors of various beta-lactamases, including class B metallo-beta-lactamases. Antimicrob. Agents Chemother. 1999, 43, 2497–2503. [Google Scholar] [CrossRef] [PubMed]
- Nagano, R.; Adachi, Y.; Hashizume, T.; Morishima, H. In Vitro antibacterial activity and mechanism of action of J-111,225, a novel 1beta-methylcarbapenem, against transferable IMP-1 metallo-beta-lactamase producers. J. Antimicrob. Chemother. 2000, 45, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Nagano, R.; Shibata, K.; Adachi, Y.; Imamura, H.; Hashizume, T.; Morishima, H. In Vitro activities of novel trans-3,5-disubstituted pyrrolidinylthio-1beta-methylcarbapenems with potent activities against methicillin-resistant Staphylococcus aureus and Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2000, 44, 489–495. [Google Scholar] [CrossRef]
- Ganta, S.R.; Perumal, S.; Pagadala, S.R.R.; Samuelsen, O.; Spencer, J.; Pratt, R.F.; Buynak, J.D. Approaches to the simultaneous inactivation of metallo- and serine-beta-lactamases. Bioorg. Med. Chem. Lett. 2009, 19, 1618–1622. [Google Scholar] [CrossRef]
- González, M.M.; Kosmopoulou, M.; Mojica, M.F.; Castillo, V.; Hinchliffe, P.; Pettinati, I.; Brem, J.; Schofield, C.J.; Mahler, G.; Bonomo, R.A.; et al. Bisthiazolidines: A Substrate-Mimicking Scaffold as an Inhibitor of the NDM-1 Carbapenemase. ACS Infect. Dis. 2015, 1, 544–554. [Google Scholar] [CrossRef]
- Chen, A.Y.; Thomas, P.W.; Stewart, A.C.; Bergstrom, A.; Cheng, Z.; Miller, C.; Bethel, C.R.; Marshall, S.H.; Credille, C.V.; Riley, C.L.; et al. Dipicolinic Acid Derivatives as Inhibitors of New Delhi Metallo-β-lactamase-1. J. Med. Chem. 2017, 60, 7267–7283. [Google Scholar] [CrossRef]
- Muhammad, Z.; Skagseth, S.; Boomgaren, M.; Akhter, S.; Fröhlich, C.; Ismael, A.; Christopeit, T.; Bayer, A.; Leiros, H.-K.S. Structural studies of triazole inhibitors with promising inhibitor effects against antibiotic resistance metallo-β-lactamases. Bioorg. Med. Chem. 2020, 28, 115598. [Google Scholar] [CrossRef] [PubMed]
- Spicer, T.; Minond, D.; Enogieru, I.; Saldanha, S.A.; Allais, C.; Liu, Q.; Mercer, B.A.; Roush, W.R.; Hodder, P. ML302, a Novel Beta-lactamase (BLA) Inhibitor. In Probe Reports from the NIH Molecular Libraries Program; NCBI: Bethesda, MD, USA, 2010. [Google Scholar]
- Palacios, A.R.; Rossi, M.-A.; Mahler, G.S.; Vila, A.J. Metallo-β-Lactamase Inhibitors Inspired on Snapshots from the Catalytic Mechanism. Biomolecules 2020, 10, 854. [Google Scholar] [CrossRef] [PubMed]
- Reck, F.; Bermingham, A.; Blais, J.; Casarez, A.; Colvin, R.; Dean, C.R.; Furegati, M.; Gamboa, L.; Growcott, E.; Li, C.; et al. IID572: A New Potentially Best-In-Class β-Lactamase Inhibitor. ACS Infect. Dis. 2019, 5, 1045–1051. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.-H.; Li, G.; Li, G.-B. Principles and current strategies targeting metallo-β-lactamase mediated antibacterial resistance. Med. Res. Rev. 2020, 40, 1558–1592. [Google Scholar] [CrossRef]
- Sharma, S.; Sharma, S.; Singh, P.P.; Khan, I.A. Potential Inhibitors against NDM-1 Type Metallo-β-Lactamases: An Overview. Microb. Drug Resist. 2020. [Google Scholar] [CrossRef]
- Ju, L.-C.; Cheng, Z.; Fast, W.; Bonomo, R.A.; Crowder, M.W. The Continuing Challenge of Metallo-β-Lactamase Inhibition: Mechanism Matters. Trends Pharmacol. Sci. 2018, 39, 635–647. [Google Scholar] [CrossRef]
- Docquier, J.-D.; Mangani, S. An update on β-lactamase inhibitor discovery and development. Drug Resist. Updat. Rev. Comment. Antimicrob. Anticancer Chemother. 2018, 36, 13–29. [Google Scholar] [CrossRef]
- Somboro, A.M.; Osei Sekyere, J.; Amoako, D.G.; Kumalo, H.M.; Khan, R.; Bester, L.A.; Essack, S.Y. In Vitro potentiation of carbapenems with tannic acid against carbapenemase-producing Enterobacteriaceae: Exploring natural products as potential carbapenemase inhibitors. J. Appl. Microbiol. 2019, 126, 452–467. [Google Scholar] [CrossRef]
- Asempa, T.E.; Abdelraouf, K.; Nicolau, D.P. Metallo-β-lactamase resistance in Enterobacteriaceae is an artefact of currently utilized antimicrobial susceptibility testing methods. J. Antimicrob. Chemother. 2020, 75, 997–1005. [Google Scholar] [CrossRef]
- Solgi, H.; Shahcheraghi, F.; Bolourchi, N.; Ahmadi, A. Molecular characterization of carbapenem-resistant serotype K1 hypervirulent Klebsiella pneumoniae ST11 harbouring bla(NDM-1) and bla(OXA-48) carbapenemases in Iran. Microb. Pathog. 2020, 149, 104507. [Google Scholar] [CrossRef]
- Spyrakis, F.; Santucci, M.; Maso, L.; Cross, S.; Gianquinto, E.; Sannio, F.; Verdirosa, F.; De Luca, F.; Docquier, J.-D.; Cendron, L.; et al. Virtual screening identifies broad-spectrum β-lactamase inhibitors with activity on clinically relevant serine- and metallo-carbapenemases. Sci. Rep. 2020, 10, 12763. [Google Scholar] [CrossRef] [PubMed]
- Meletis, G. Carbapenem resistance: Overview of the problem and future perspectives. Ther. Adv. Infect. Dis. 2016, 3, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Sader, H.S.; Farrell, D.J.; Flamm, R.K.; Jones, R.N. Variation in potency and spectrum of tigecycline activity against bacterial strains from U.S. medical centers since its approval for clinical use (2006 to 2012). Antimicrob. Agents Chemother. 2014, 58, 2274–2280. [Google Scholar] [CrossRef] [PubMed]
- Corcione, S.; Lupia, T.; Maraolo, A.E.; Mornese Pinna, S.; Gentile, I.; De Rosa, F.G. Carbapenem-sparing strategy: Carbapenemase, treatment, and stewardship. Curr. Opin. Infect. Dis. 2019, 32, 663–673. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Mechanism of Action | Class | Carbapenemase | More Common Enzymes | |||
|---|---|---|---|---|---|---|
| Serine-β-lactamases | A | KPC | KPC-2 | KPC-3 | ||
| GES | GES-2 | GES-5 | GES-6 | |||
| D | OXA | OXA-23 | OXA-24/40 | OXA-58 | OXA-48 | |
| Metallo-β-lactamases | B | IMP | IMP-1 | IMP-6 | IMP-7 | |
| VIM | VIM-1 | VIM-2 | ||||
| NDM | NDM-1 | NDM-4 | NDM-5 | |||
| Antibiotic/Inhibitor Combination | ClinicalTrials.gov Identifier | Phase | Title | Status | Start Date |
|---|---|---|---|---|---|
| Diazabicyclooctane-derived | |||||
| Ceftazidime/Avibactam | NCT04040621 | I | Single-dose PK Study of Ceftazidime-Avibactam in Hospitalized Children Receiving Systemic Antibiotics for Nosocomial Pneumonia | Recruiting | June, 2020 |
| Ceftazidime/Avibactam | NCT02504827 | IV | Steady-State Pharmacokinetics of Ceftazidime/Avibactam in Cystic Fibrosis | Completed | September, 2015 |
| Imipenem /Relebactam | NCT02452047 | III | Efficacy and Safety of Imipenem + Cilastatin/Relebactam (MK-7655A) Versus Colistimethate Sodium+Imipenem+Cilastatin in Imipenem-Resistant Bacterial Infection (MK-7655A-013) | Completed | September, 2017 |
| Imipenem /Relebactam | NCT02493764 | III | Imipenem/Relebactam/Cilastatin Versus Piperacillin/Tazobactam for Treatment of Participants with Bacterial Pneumonia (MK-7655A-014) | Completed | April, 2019 |
| Meropenem/Vaborbactam | NCT02166476 | III | Efficacy/Safety of Meropenem-Vaborbactam Compared to Piperacillin-Tazobactam in Adults with cUTI and AP | Completed | November, 2014 |
| Meropenem/Vaborbactam | NCT02168946 | III | Efficacy, Safety, Tolerability of Vabomere Compared to Best Available Therapy in Treating Serious Infections in Adults | Completed | July, 2014 |
| Aztreonam/Avibactam | NCT01689207 | I | To Investigate the Safety and Tolerability of Aztreonam-Avibactam (ATM-AVI) | Completed | September, 2012 |
| Aztreonam/Ceftazidime/Avibactam | NCT03978091 | I | A Trial to Evaluate the Pharmacokinetics and Safety of AVYCAZ(R) in Combination with Aztreonam | Recruiting | June, 2019 |
| Aztreonam/Avibactam | NCT04486625 | I | Pharmacokinetic Study of Aztreonam-Avibactam in Severe Renal Impairment | Recruiting | August, 2020 |
| Aztreonam/Avibactam | NCT02655419 | II | Determine the PK and Safety and Tolerability of ATM-AVI for the Treatment of cIAIs in Hospitalized Adults (REJUVENATE) | Completed | May, 2016 |
| Aztreonam/Avibactam/Metronidazole | NCT03329092 | III | A Study to Determine the Efficacy, Safety and Tolerability of Aztreonam-Avibactam (ATM-AVI) ± Metronidazole (MTZ) Versus Meropenem (MER) ± Colistin (COL) for the Treatment of Serious Infections due to Gram-Negative Bacteria (REVISIT) | Recruiting | April, 2018 |
| Aztreonam/Avibactam | NCT03580044 | III | Efficacy, Safety, and Tolerability of ATM-AVI in the Treatment of Serious Infection Due to MBL-producing Gram-negative Bacteria | Not yet recruiting | December, 2020 |
| Zidebactam | NCT02674347 | I | MAD Study to Evaluate the Safety, Tolerability and Pharmacokinetics of Intravenous Zidebactam in Healthy Adults | Completed | February, 2016 |
| Cefepime/Zidebactam | NCT02707107 | I | MED Study to Evaluate the Safety, Tolerability and Pharmacokinetics of Intravenous WCK 5222 (Zidebactam and Cefepime) in Healthy Volunteers | Completed | March, 2016 |
| Cefepime/Zidebactam | NCT02942810 | I | To Investigate the Pharmacokinetics of Intravenous WCK 5222 (FEP-ZID) in Patients with Renal Impairment | Completed | October, 2016 |
| Cefepime/Zidebactam | NCT03554304 | I | Evaluate the Effect of WCK 5222 on the QT/QTc Interval in Healthy Volunteers | Completed | February, 2017 |
| Cefepime/Zidebactam | NCT03630094 | I | Plasma and Intrapulmonary Concentrations Study of WCK 5222 | Completed | March, 2017 |
| Cefepime/Zidebactam | NCT02532140 | I | Study to Evaluate the Safety, Tolerability, and Pharmacokinetics of WCK 5107 Alone and in Combination with Cefepime | Completed | August, 2017 |
| Durlobactam | NCT02971423 | I | Evaluation of the Safety, Tolerability and Pharmacokinetics of Intravenous ETX2514 Administered in Healthy Subjects | Completed | October, 2016 |
| Durlobactam | NCT03985410 | I | Study Evaluating the Effect of ETX2514 on Cardiac Repolarization in Healthy Male or Female Volunteers | Completed | May, 2019 |
| Durlobactam | NCT04018950 | I | Study to Determine the Excretion and Metabolism of 14C-ETX2514 Administered Intravenously in Healthy Male Subjects | Completed | June, 2019 |
| Sulbactam/Durlobactam | NCT03303924 | I | Study to Determine and Compare Plasma and Intrapulmonary Concentrations of ETX2514 and Sulbactam in Healthy Subjects | Completed | August, 2017 |
| Sulbactam/Durlobactam | NCT03310463 | I | Evaluation of the Pharmacokinetics, Safety, and Tolerability of Intravenous ETX2514 and Sulbactam Administered Concurrently to Subjects with Various Degrees of Renal Impairment and Healthy Matched Control Subjects | Completed | October, 2017 |
| Sulbactam/Durlobactam | NCT03445195 | II | Evaluation of Safety and Efficacy of Intravenous Sulbactam-ETX2514 in the Treatment of Hospitalized Adults with Complicated Urinary Tract Infections | Completed | January, 2018 |
| Sulbactam/Durlobactam/Imipinem/Cilastatin | NCT03894046 | III | Study to Evaluate the Efficacy and Safety of Intravenous Sulbactam-ETX2514 in the Treatment of Patients with Infections Caused by Acinetobacter bumannii-calcoaceticus Complex (ATTACK) | Recruiting | April, 2019 |
| Nacubactam | NCT02134834 | I | A Phase I Study to Assess Safety, Tolerability and Pharmacokinetics of OP0595 | Completed | May, 2014 |
| Meropenem/Nacubactam | NCT03182504 | I | A Study to Investigate the Intrapulmonary Lung Penetration of Nacubactam in Healthy Participants | Completed | June, 2017 |
| ETX0282 | NCT03491748 | I | A Study to Evaluate the Safety, Tolerability, and Pharmacokinetics (PK, the Measure of How the Human Body Processes a Substance) of ETX0282 when Administered Orally to Healthy Participants | Completed | March, 2018 |
| Boronic acid-derived | |||||
| Taniborbactam | NCT02955459 | I | VNRX-5133 SAD/MAD Safety and PK in Healthy Adult Volunteers | Completed | November, 2016 |
| Cefepime/Taniborbactam | NCT03332732 | I | VNRX-5133 Drug–Drug Interaction in Healthy Adult Volunteers | Completed | October, 2017 |
| Cefepime/Taniborbactam | NCT03690362 | I | VNRX-5133 with VNRX-5022 in Subjects with Varying Degrees of Renal Impairment | Completed | April, 2018 |
| Cefepime/Taniborbactam | NCT03870490 | I | Safety and Pharmacokinetics of VNRX-5133 in the Epithelial Lining Fluid of Healthy Adult Subjects | Completed | March, 2019 |
| Cefepime/Taniborbactam | NCT03840148 | III | Safety and Efficacy Study of Cefepime/VNRX-5133 in Patients with Complicated Urinary Tract Infections | Recruiting | August, 2019 |
| VNRX-5236 | NCT04243863 | I | VNRX-7145 SAD/MAD Safety and PK in Healthy Adult Volunteers | Recruiting | January, 2020 |
| QPX7728 | NCT04380207 | I | P1 Single and Multiple Ascending Dose (SAD/MAD) Study of IV QPX7728 Alone and Combined with QPX2014 in NHV | Not yet recruiting | November, 2020 |
| β-lactam-derived | |||||
| Enmetazobactam | NCT03775668 | I | Single Dose Mass Balance Study with C14—Labeled AAI101 in Healthy Male Volunteers | Completed | November, 2018 |
| Cefepime or Piperacillin/Enmetazobactam | NCT03685084 | I | Investigation of AAI101 Safety, Tolerability and PK in Healthy Volunteers | Completed | October, 2013 |
| Cefepime/Enmetazobactam | NCT03680378 | I | Lung Pharmacokinetics (PK) in Epithelial Lining Fluid (ELF) | Completed | July, 2017 |
| Cefepime/Enmetazobactam | NCT03680352 | I | Pharmacokinetics of Cefepime and AAI101 in Subjects with Renal Insufficiency and Healthy Subjects | Unknown | September, 2017 |
| Cefepime/Enmetazobactam | NCT03680612 | II | Cefepime/AAI101 Phase 2 Study in Hospitalized Adults with cUTI | Completed | September, 2017 |
| Cefepime/Enmetazobactam | NCT03687255 | III | Safety and Efficacy Study of Cefepime-AAI101 in the Treatment of Complicated Urinary Tract Infections | Completed | September, 2018 |
| Inhibitor | Clinically Most Relevant Carbapenemases | |||||||
|---|---|---|---|---|---|---|---|---|
| KPC-2 | IMP-1 | VIM-1 | NDM-1 | OXA-23 | OXA-24/40 | OXA-48 | ||
| Diazabicyclooctane-derived | ||||||||
| Avibactam | IC50 (nM) | 170 [30] */22 [31]/60 [32] | >1.6 × 105 [31]/>1.0 × 105 [32] | >1.6 × 105 [31] | >1.6 × 105 [31]/>1.0 × 105 [32] | 3.1 × 103 [31]/8.9 × 103 [33] | 1.60 × 104 [30]/2.23 × 104 [33] | 180 [31]/550 [32] |
| Kiapp (nM) | 900 [34]/11 [35] | >4.0 × 104 [35] | >4.0 × 104 [35] | >4.0 × 104 [35] | >1.0 × 105 [34]/1.7 × 103 [35] | >1.0 × 105 [34]/1.54 × 105 [33] | 3.0 × 104 [34]/27 [35] | |
| Relebactam | IC50 (nM) | 82 [31]/230 [36] | >1.6 × 105 [31] | >1.6 × 105 [31] | >1.6 × 105 [31] | 9 × 104 [31] | ||
| Kiapp (nM) | 2.2 × 103 [34]/1.2 × 103 [36] | >1.0 × 105 [34] | >1.0 × 105 [34] | >1.0 × 105 [34] | ||||
| Zidebactam | IC50 (nM) | |||||||
| Kiapp (nM) | 4.5 × 103 [34] | >1.00 × 105 (VIM-2) [37] | >1.0 × 105 [34] | >1.0 × 105 [34] | >1.0 × 105 [34] | |||
| Durlobactam | IC50 (nM) | 4 [30] | 190 [30] | |||||
| Kiapp (nM) | ||||||||
| Nacubactam | IC50 (nM) | 869 [38] | >3.0 × 105 [38] | 4.64 × 104 [38] | ||||
| Kiapp (nM) | 3.1 × 104 [39] | |||||||
| ETX1317 | IC50 (nM) | 43 [40] | 540 [40] | 77 [40] | ||||
| Kiapp (nM) | ||||||||
| WCK 4234 | IC50 (nM) | |||||||
| Kiapp (nM) | 320 [34] | 8.0 × 103 [34] | 5.0 × 103 [34] | 290 [34] | ||||
| Boronic acid-derived | ||||||||
| Vaborbactam | IC50 (nM) | 110 [31]/100 [32] | >1.6 × 105 [31]/>1.0 × 105 [32] | >1.6 × 105 [31] | >1.6 × 105 [31]/>1.0 × 105 [32] | 1.2 × 105 [31] | 6.9 × 103 [31]/3.88 × 104 [32] | |
| Kiapp (nM) | 22 [41]/56 [35] | >3.0 × 104 [41]/>4.0 × 104 [35] | >4.0 × 104 [35] | >3.0 × 104 [41]/>4.0 × 104 [35] | >4.0 × 104 [35] | 350 [41]/1.4 × 104 [35] | ||
| Taniborbactam | IC50 (nM) | 30 [32] | 2.51 × 103 [42]/3.98 × 104 [32] | 7.9 [42] | 10 [42]/190 [32] | 537 [42]/420 [32] | ||
| Kiapp (nM) | 4 [41] | >3.0 × 104 [41] | 19 (VIM-2) [41] | 81 [41] | 350 [41] | |||
| VNRX-5236 | IC50 (nM) | 80 [43] | >1.0 × 105 [43] | 9.04 × 103 (VIM-2) [43] | 3.81 × 104 [43] | 317 [43] | ||
| Kiapp (nM) | 110 [43] | |||||||
| QPX7728 | IC50 (nM) | 2.9 [31] | 610 [31] | 14 [31] | 55 [31] | 1.2 [31] | 1.1 [31] | |
| Kiapp (nM) | 1.9 [35] | 220 [35] | 8 [35] | 32 [35] | 0.74 [35] | 0.28 [35] | ||
| β-lactam-derived | ||||||||
| Enmetazobactam | IC50 (nM) | 360 a [44] | 1.1 × 104 a [44] | |||||
| Kiapp (nM) | ||||||||
| LN-1-255 | IC50 (nM) | 12 [33] | 15 [33] | 3 [45] | ||||
| Kiapp (nM) | 88 [33] | 289 [33] | 170 [45] | |||||
| Pyridine-2-carboxylic acid | ||||||||
| ANT2681 | IC50 (nM) | |||||||
| Kiapp (nM) | 3.81 × 103 [46] | 630 [46] | 40 [46] | |||||
| Inhibitor | Carbapenemase | ||||||
|---|---|---|---|---|---|---|---|
| Class A | Class B | Class D | |||||
| KPC | NDM | VIM | IMP | OXA-23 | OXA-24/40 | OXA-48 | |
| Diazabicyclooctane-derived | |||||||
| Relebactam | ✓ | ✗ | ✗ | ✗ | ✗ | ✗ | |
| Avibactam | ✓ | ✗ | ✗ | ✗ | ✗ | ✗ | ✓ |
| Zidebactam | ✓ a | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ |
| Durlobactam | ✓ | ✗ | ✗ | ✓ | ✓ | ✓ | |
| Nacubactam | ✓ b | ✗ | ✗ | ✗ | ✗ | ✗ | |
| ETX1317 | ✓ | ✓ | |||||
| WCK 4234 | ✓ | ✗ | ✗ | ✗ | ✓ c | ✓ d | ✓ |
| Boronic acid derived | |||||||
| Vaborbactam | ✓ | ✗ | ✗ | ✗ | ✗ | ✗ | |
| Taniborbactam | ✓ | ✓ | ✓ | ✗ | ✓ | ||
| VNRX-5236 | ✓ e | ✗ | ✗ | ✗ | ✓ | ||
| QPX7728 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | |
| β-lactam-derived | |||||||
| Enmetazobactam | ✓ | ✓ f | |||||
| LN-1-255 | ✓ | ✓ | ✓ | ||||
| New B-lactam/B-lactamase Inhibitor | Main Bacterial Targets | |||||
|---|---|---|---|---|---|---|
| Carbapenem Resistant A. Baumannii | Carbapenem Resistant P. Aeruginosa | Carbapenem Resistant Enterobacterales | ||||
| SBLs | MBLs | SBLs | MBLs | SBLs | MBLs | |
| Diazabicyclooctane derived inhibitors | ||||||
| Ceftazidime/avibactam | ✓ | ✓ | ||||
| Imipenem/relebactam | ||||||
| Aztreonam/avibactam | ✓ | ✓ | ||||
| Cefepime/zidebactam | ✓ | ✓ | ✓ | ✓ | ||
| Sulbactam/durlobactam | ✓ | |||||
| Meropenem(or cefepime, or aztreonem)/nacubactam | ✓ | ✓ | ✓ | |||
| Cefpodoxime/ETX1317 | ✓ | ✓ | ||||
| Meropenem/WCK 4234 | ✓ | ✗ | ✓ | ✗ | ||
| GT-1/GT-055 a | ✓ | ✓ b | ✓ | ✓ | ||
| Boronic acid derivative inhibitors | ||||||
| Meropenem/vaborbactam | ||||||
| Cefepime (or meropenem)/ taniborbactam | ✓ | ✓ | ✓ | ✓ | ||
| VNRX-7145/ceftibuten | ✓ | |||||
| Meropenem/QPX7728 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| β-lactam-derived inhibitors | ||||||
| Cefepime/Enmetazobactam | ✓ | |||||
| Imipenem/LN-1-255 | ✓ | ✓ | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vázquez-Ucha, J.C.; Arca-Suárez, J.; Bou, G.; Beceiro, A. New Carbapenemase Inhibitors: Clearing the Way for the β-Lactams. Int. J. Mol. Sci. 2020, 21, 9308. https://doi.org/10.3390/ijms21239308
Vázquez-Ucha JC, Arca-Suárez J, Bou G, Beceiro A. New Carbapenemase Inhibitors: Clearing the Way for the β-Lactams. International Journal of Molecular Sciences. 2020; 21(23):9308. https://doi.org/10.3390/ijms21239308
Chicago/Turabian StyleVázquez-Ucha, Juan C., Jorge Arca-Suárez, Germán Bou, and Alejandro Beceiro. 2020. "New Carbapenemase Inhibitors: Clearing the Way for the β-Lactams" International Journal of Molecular Sciences 21, no. 23: 9308. https://doi.org/10.3390/ijms21239308
APA StyleVázquez-Ucha, J. C., Arca-Suárez, J., Bou, G., & Beceiro, A. (2020). New Carbapenemase Inhibitors: Clearing the Way for the β-Lactams. International Journal of Molecular Sciences, 21(23), 9308. https://doi.org/10.3390/ijms21239308