Nucleosome Positioning around Transcription Start Site Correlates with Gene Expression Only for Active Chromatin State in Drosophila Interphase Chromosomes


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
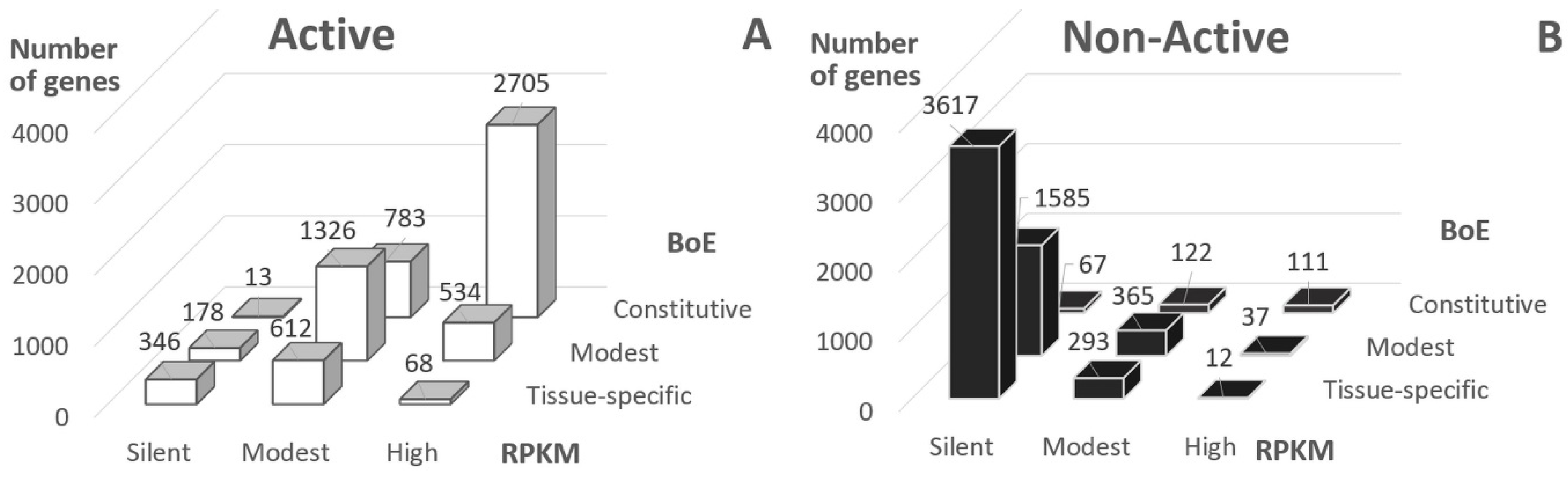
2.1. Gene Classification by Chromatin States and Expression Measures
- Silent (RPKM = 0, 6972 genes);
- Modest (1 ≤ RPKM ≤ 25, 3718);
- High (RPKM > 25, 3584).
- Tissue-specific (BoE ≤ 6, 5534 genes);
- Modest (6 > BoE ≤ 24, 4278);
- Constitutive (BoE >24, 3942).
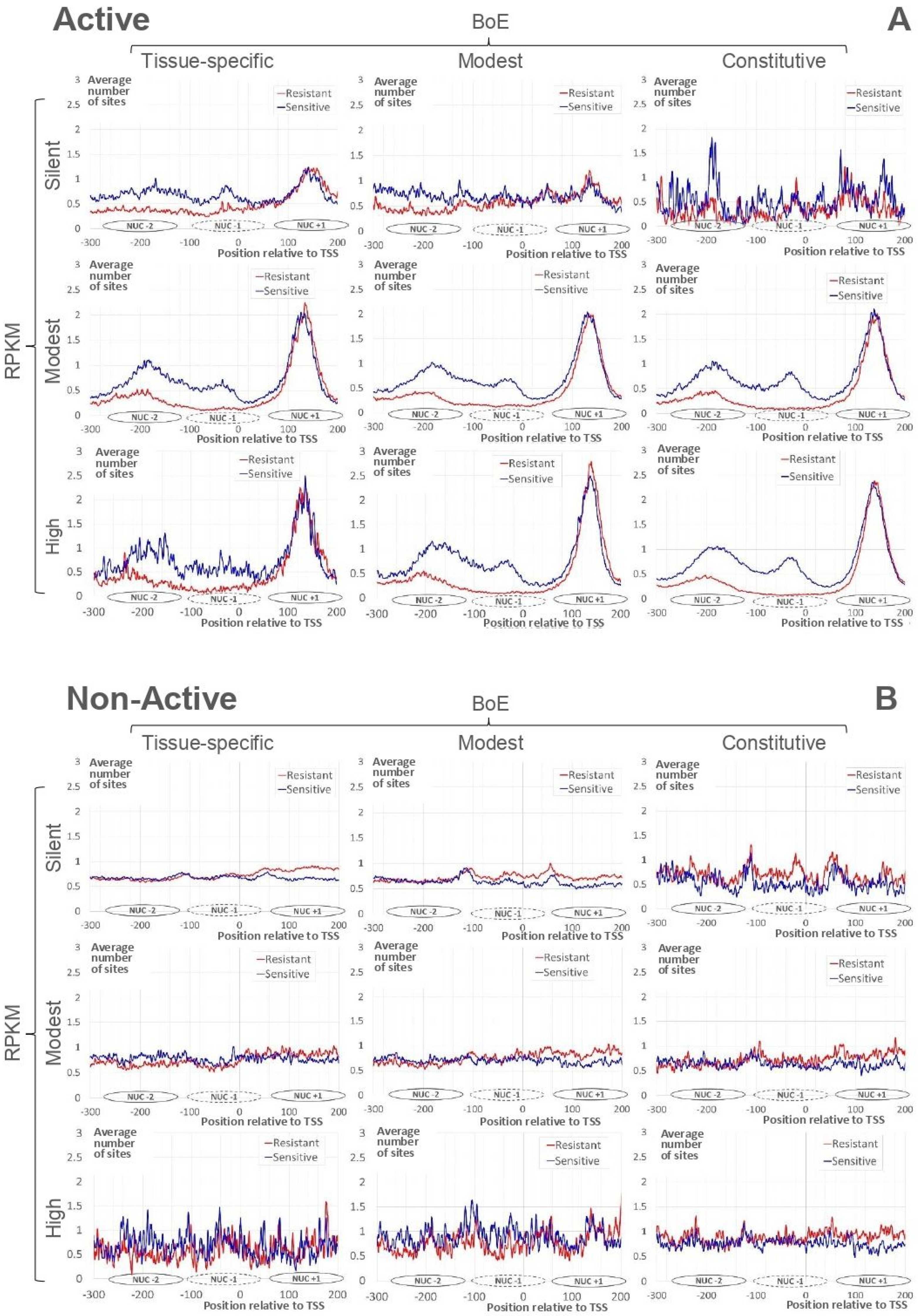
2.2. Irrespective to Expression Measures, Nucleosome Arrangement Is Conservative for Active Genes
- the strong nucleosome formation sites downstream TSS;
- the depletion of resistant nucleosome sites upstream TSS;
- the regular ladder of sensitive nucleosomes upstream TSS.
2.3. Correlations between Expression Measures and Nucleosome Positioning
- strongly significant negative CC for resistant nucleosomal sites around TSS [−120; +80] for tissue-specific and modest; moderate negative CC for sensitive nucleosomal sites for tissue-specific [−30; +50] and modest [+10; +70];
- strongly significant positive CC downstream TSS [+70; +200] for tissue-specific and modest;
- moderately significant positive CC for sensitive nucleosomal sites upstream TSS in tissue-specific [−210; −180]) and modest [−160; −150].
3. Discussion
- about 20% of Active genes were either silent or tissue-specific;
- about 40% of Non-Active genes had at least modest RPKM or BoE.
- It was estimated [37] that almost half of the human genome contained regular arrays of nucleosomes that enriched in active chromatin domains. Since the presence of transcription factor (TF) binding sites (BS) strongly supports gene expression, the analysis examined the positioning of nucleosomes around these BS using ChIP-seq data for 35 different TFs. As a result, TF occupancy was closely related to the strength of nucleosome positioning.
- The genome-wide location of nucleosomes during zebrafish embryogenesis indicated that well-positioned nucleosome arrays appeared on thousands of promoters during genome activation [38]. It is important to remember that both models [23,30,33] of chromatin states were based on whole genome profiles of a number of non-histone proteins, i.e., the pattern of BS of these proteins should be different in Active in Non-Active genes.
- The recent analysis of chromatin folding for various epigenetic states of chromatin [39] supported the importance of chromatin state annotation for analysis of genome-wide nucleosome organization. In particular, the classification of genomic domains in Drosophila cells into transcriptionally active, inactive, or Polycomb-repressed states revealed distinct chromatin organizations for each state.
- for Active state potentiates transcription, i.e., a more regular nucleosome arrangement in promoter points to higher expression level and higher number of expressed tissues;
- for Non-Active state does not influence gene expression, i.e., the expression level/breadth and the nucleosome positioning are independent.
4. Materials and Methods
4.1. Whole Genome Data Analysis
4.2. Statistical Analysis
5. Conclusions
- Active genes demonstrated clear pattern of nucleosome arrangement around TSS; the regular nucleosome positioning correlates with the expression level/breadth;
- Non-Active genes did not show such clear specificity in nucleosome positioning; the correlation between the regular nucleosome positioning and expression level or breadth either is absent, not conservative, or shows even the opposite significant trend compared to that of Active genes.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 4HMM | Four state hidden Markov model |
| BoE | Breadth of expression |
| MNase | Micrococcal nuclease |
| Nuc | Nucleosome |
| NDR | Nucleosome-depleted region |
| RPKM | Reads per kilobase million |
| TSS | Transcription start site |
References
- Hughes, A.L.; Rando, O.J. Mechanisms Underlying Nucleosome Positioning In Vivo. Annu. Rev. Biophys. 2014, 43, 41–63. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Pugh, B.F. Nucleosome positioning and gene regulation: Advances through genomics. Nat. Rev. Genet. 2009, 10, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Morozov, A.V.; Fortney, K.; Gaykalova, D.A.; Studitsky, V.M.; Widom, J.; Siggia, E.D. Using DNA mechanics to predict in vitro nucleosome positions and formation energies. Nucleic Acids Res. 2009, 37, 4707–4722. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.L.; Jin, Y.; Rando, O.J.; Struhl, K. A functional evolutionary approach to identify determinants of nucleosome positioning: A unifying model for establishing the genome-wide pattern. Mol. Cell 2012, 48, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Struhl, K.; Segal, E. Determinants of nucleosome positioning. Nat. Struct. Mol. Biol. 2013, 20, 267–273. [Google Scholar] [CrossRef]
- Moshkin, Y.M.; Chalkley, G.E.; Kan, T.W.; Reddy, B.A.; Ozgur, Z.; Van Ijcken, W.F.J.; Dekkers, D.H.W.; Demmers, J.A.; Travers, A.A.; Verrijzer, C.P. Remodelers Organize Cellular Chromatin by Counteracting Intrinsic Histone-DNA Sequence Preferences in a Class-Specific Manner. Mol. Cell. Biol. 2011, 32, 675–688. [Google Scholar] [CrossRef]
- Zhang, Y.; Moqtaderi, Z.; Rattner, B.P.; Euskirchen, G.; Snyder, M.; Kadonaga, J.T.; Liu, X.S.; Struhl, K. Intrinsic histone-DNA interactions are not the major determinant of nucleosome positions in vivo. Nat. Struct. Mol. Biol. 2009, 16, 847–852. [Google Scholar] [CrossRef]
- Valouev, A.; Johnson, S.M.; Boyd, S.D.; Smith, S.E.; Fire, A.Z.; Sidow, A. Determinants of nucleosome organization in primary human cells. Nat. Cell Biol. 2011, 474, 516–520. [Google Scholar] [CrossRef]
- Radman-Livaja, M.; Rando, O.J. Nucleosome positioning: How is it established, and why does it matter? Dev. Biol. 2010, 339, 258–266. [Google Scholar] [CrossRef]
- Barozzi, I.; Simonatto, M.; Bonifacio, S.; Yang, L.; Rohs, R.; Ghisletti, S.; Natoli, G. Coregulation of Transcription Factor Binding and Nucleosome Occupancy through DNA Features of Mammalian Enhancers. Mol. Cell 2014, 54, 844–857. [Google Scholar] [CrossRef]
- Tsompana, M.; Buck, M.J. Chromatin accessibility: A window into the genome. Epigenet. Chromatin 2014, 7, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Yuan, G.-C.; Liu, Y.-J.; Dion, M.F.; Slack, M.D.; Wu, L.F.; Altschuler, S.J.; Rando, O.J. Genome-Scale Identification of Nucleosome Positions in S. cerevisiae. Science 2005, 309, 626–630. [Google Scholar] [CrossRef] [PubMed]
- Mavrich, T.N.; Ioshikhes, I.P.; Venters, B.J.; Jiang, C.; Tomsho, L.P.; Qi, J.; Schuster, S.C.; Albert, I.; Pugh, B.F. A barrier nucleosome model for statistical positioning of nucleosomes throughout the yeast genome. Genome Res. 2008, 18, 1073–1083. [Google Scholar] [CrossRef]
- Mavrich, T.N.; Jiang, C.; Ioshikhes, I.P.; Li, X.; Venters, B.J.; Zanton, S.J.; Tomsho, L.P.; Qi, J.; Glaser, R.L.; Schuster, S.C.; et al. Nucleosome organization in the Drosophila genome. Nat. Cell Biol. 2008, 453, 358–362. [Google Scholar] [CrossRef]
- Lee, W.; Tillo, D.; Bray, N.; Morse, R.H.; Davis, R.W.; Hughes, T.R.; Nislow, C. A high-resolution atlas of nucleosome occupancy in yeast. Nat. Genet. 2007, 39, 1235–1244. [Google Scholar] [CrossRef]
- Schones, D.E.; Cui, K.; Cuddapah, S.; Roh, T.-Y.; Barski, A.; Wang, Z.; Wei, G.; Zhao, K. Dynamic Regulation of Nucleosome Positioning in the Human Genome. Cell 2008, 132, 887–898. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, W.; Jiang, J. Genome-Wide Nucleosome Occupancy and Positioning and Their Impact on Gene Expression and Evolution in Plants. Plant Physiol. 2015, 168, 1406–1416. [Google Scholar] [CrossRef]
- Li, G.; Liu, S.; Wang, J.; He, J.; Huang, H.; Zhang, Y.; Xu, L. ISWI proteins participate in the genome-wide nucleosome distribution in Arabidopsis. Plant J. 2014, 78, 706–714. [Google Scholar] [CrossRef]
- Liu, M.-J.; Seddon, A.E.; Tsai, Z.T.-Y.; Major, I.T.; Floer, M.; Howe, G.A.; Shiu, S.-H. Determinants of nucleosome positioning and their influence on plant gene expression. Genome Res. 2015, 25, 1182–1195. [Google Scholar] [CrossRef]
- Chereji, R.V.; Kan, T.-W.; Grudniewska, M.; Romashchenko, A.V.; Berezikov, E.; Zhimulev, I.F.; Guryev, V.; Morozov, A.V.; Moshkin, Y.M. Genome-wide profiling of nucleosome sensitivity and chromatin accessibility in Drosophila melanogaster. Nucleic Acids Res. 2015, 44, 1036–1051. [Google Scholar] [CrossRef] [PubMed]
- Van Bakel, H.; Tsui, K.; Gebbia, M.; Mnaimneh, S.; Hughes, T.R.; Nislow, C. A Compendium of Nucleosome and Transcript Profiles Reveals Determinants of Chromatin Architecture and Transcription. PLoS Genet. 2013, 9, e1003479. [Google Scholar] [CrossRef] [PubMed]
- Zhimulev, I.F.; Zykova, T.Y.; Goncharov, F.P.; Khoroshko, V.A.; Demakova, O.V.; Semeshin, V.F.; Pokholkova, G.V.; Boldyreva, L.V.; Demidova, D.S.; Babenko, V.N.; et al. Genetic Organization of Interphase Chromosome Bands and Interbands in Drosophila melanogaster. PLoS ONE 2014, 9, e101631. [Google Scholar] [CrossRef] [PubMed]
- Zykova, T.Y.; Levitsky, V.G.; Belyaeva, E.S.; Zhimulev, I.F. Polytene Chromosomes—A Portrait of Functional Organization of the Drosophila Genome. Curr. Genom. 2018, 19, 179–191. [Google Scholar] [CrossRef]
- Semeshin, V.F.; Demakov, S.A.; Alonso, M.P.; Belyaeva, E.S.; Bonner, J.J.; Zhimulev, I.F. Electron microscopical analysis of Drosophila polytene chromosomes. Chromosoma 1989, 97, 396–412. [Google Scholar] [CrossRef]
- Vatolina, T.Y.; Boldyreva, L.V.; Demakova, O.V.; Demakov, S.A.; Kokoza, E.B.; Semeshin, V.F.; Babenko, V.N.; Goncharov, F.P.; Belyaeva, E.S.; Zhimulev, I.F. Correction: Identical Functional Organization of Nonpolytene and Polytene Chromosomes in Drosophila melanogaster. PLoS ONE 2011, 6, 25960. [Google Scholar] [CrossRef]
- Zhimulev, I.F.; Belyaeva, E.S.; Vatolina, T.Y.; Demakov, S.A. Banding patterns in Drosophila melanogaster polytene chromosomes correlate with DNA-binding protein occupancy. BioEssays 2012, 34, 498–508. [Google Scholar] [CrossRef]
- Celniker, S.E.; modENCODE Consortium; Dillon, L.A.L.; Gerstein, M.B.; Gunsalus, K.C.; Henikoff, S.; Karpen, G.H.; Kellis, M.; Lai, E.C.; Lieb, J.D.; et al. Unlocking the secrets of the genome. Nat. Cell Biol. 2009, 459, 927–930. [Google Scholar] [CrossRef]
- Demakov, S.A.; Zykova, T.Y.; Babenko, V.; Semeshin, V.F.; Belyaeva, E.S.; Zhimulev, I.F. Protein composition of interband regions in polytene and cell line chromosomes of Drosophila melanogaster. BMC Genom. 2011, 12, 566. [Google Scholar] [CrossRef]
- Boldyreva, L.V.; Goncharov, F.P.; Demakova, O.V.; Zykova, T.Y.; Levitsky, V.G.; Kolesnikov, N.N.; Pindyurin, A.V.; Semeshin, V.F.; Zhimulev, I.F. Protein and Genetic Composition of Four Chromatin Types in Drosophila melanogaster Cell Lines. Curr. Genom. 2017, 18, 214–226. [Google Scholar] [CrossRef]
- Brown, J.B.; Boley, N.; Eisman, R.C.; May, G.E.; Stoiber, M.H.; Duff, M.O.; Booth, B.W.; Wen, J.; Park, S.; Suzuki, A.M.; et al. Diversity and dynamics of the Drosophila transcriptome. Nat. Cell Biol. 2014, 512, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Cherbas, L.; Willingham, A.; Zhang, D.; Yang, L.; Zou, Y.; Eads, B.D.; Carlson, J.W.; Landolin, J.M.; Kapranov, P.; Dumais, J.; et al. The transcriptional diversity of 25 Drosophila cell lines. Genome Res. 2011, 21, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Filion, G.J.; Van Bemmel, J.G.; Braunschweig, U.; Talhout, W.; Kind, J.; Ward, L.D.; Brugman, W.; De Castro, I.J.; Kerkhoven, R.M.; Bussemaker, H.J.; et al. Systematic protein location mapping reveals five principal chromatin types in Drosophila cells. Cell 2010, 143, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Kharchenko, P.V.; Alekseyenko, A.A.; Schwartz, Y.B.; Minoda, A.; Riddle, N.C.; Ernst, J.; Sabo, P.J.; Larschan, E.; Gorchakov, A.A.; Gu, T.; et al. Comprehensive analysis of the chromatin landscape in Drosophila melanogaster. Nat. Cell Biol. 2011, 471, 480–485. [Google Scholar] [CrossRef]
- Milon, B.; Sun, Y.; Chang, W.; Creasy, T.; Mahurkar, A.; Shetty, A.; Nurminsky, D.; Nurminskaya, M. Map of open and closed chromatin domains in Drosophila genome. BMC Genom. 2014, 15, 988. [Google Scholar] [CrossRef] [PubMed]
- Graveley, B.R.; Brooks, A.N.; Carlson, J.W.; Duff, M.O.; Landolin, J.M.; Yang, L.; Artieri, C.G.; Van Baren, M.J.; Boley, N.; Booth, B.W.; et al. The developmental transcriptome of Drosophila melanogaster. Nat. Cell Biol. 2011, 471, 473–479. [Google Scholar] [CrossRef]
- Gaffney, D.J.; McVicker, G.; Pai, A.A.; Fondufe-Mittendorf, Y.N.; Lewellen, N.; Michelini, K.; Widom, J.; Gilad, Y.; Pritchard, J.K. Controls of Nucleosome Positioning in the Human Genome. PLoS Genet. 2012, 8, e1003036. [Google Scholar] [CrossRef]
- Zhang, Y.; Vastenhouw, N.L.; Feng, J.; Fu, K.; Wang, C.; Ge, Y.; Pauli, A.; Van Hummelen, P.; Schier, A.F.; Liu, X.S. Canonical nucleosome organization at promoters forms during genome activation. Genome Res. 2013, 24, 260–266. [Google Scholar] [CrossRef]
- Boettiger, A.N.; Bintu, B.; Moffitt, J.R.; Wang, S.; Beliveau, B.J.; Fudenberg, G.; Imakaev, M.V.; Mirny, L.A.; Wu, C.-T.; Zhuang, X. Super-resolution imaging reveals distinct chromatin folding for different epigenetic states. Nat. Cell Biol. 2016, 529, 418–422. [Google Scholar] [CrossRef]
- The Flybase Database RNA-seq Data. Available online: http://fb2014_03.flybase.org/static_pages/downloads/FB2014_03/genes/gene_rpkm_report_fb_2014_03.tsv.gz (accessed on 4 December 2020).
- The Genome of D. Melanogaster, Release 5.50 of FlyBase Database. Available online: ftp://ftp.flybase.net/genomes/Drosophila_melanogaster/dmel_r5.50_FB2013_02 (accessed on 4 December 2020).
- Kendall, M.G. Rank Correlation Methods, 4th ed.; Charles Griffin & Co.: London, UK, 1970; ISBN 978-0852641996. [Google Scholar]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Levitsky, V.G.; Zykova, T.Y.; Moshkin, Y.M.; Zhimulev, I.F. Nucleosome Positioning around Transcription Start Site Correlates with Gene Expression Only for Active Chromatin State in Drosophila Interphase Chromosomes. Int. J. Mol. Sci. 2020, 21, 9282. https://doi.org/10.3390/ijms21239282
Levitsky VG, Zykova TY, Moshkin YM, Zhimulev IF. Nucleosome Positioning around Transcription Start Site Correlates with Gene Expression Only for Active Chromatin State in Drosophila Interphase Chromosomes. International Journal of Molecular Sciences. 2020; 21(23):9282. https://doi.org/10.3390/ijms21239282
Chicago/Turabian StyleLevitsky, Victor G., Tatyana Yu. Zykova, Yuri M. Moshkin, and Igor F. Zhimulev. 2020. "Nucleosome Positioning around Transcription Start Site Correlates with Gene Expression Only for Active Chromatin State in Drosophila Interphase Chromosomes" International Journal of Molecular Sciences 21, no. 23: 9282. https://doi.org/10.3390/ijms21239282
APA StyleLevitsky, V. G., Zykova, T. Y., Moshkin, Y. M., & Zhimulev, I. F. (2020). Nucleosome Positioning around Transcription Start Site Correlates with Gene Expression Only for Active Chromatin State in Drosophila Interphase Chromosomes. International Journal of Molecular Sciences, 21(23), 9282. https://doi.org/10.3390/ijms21239282