Abstract
Caloric restriction (CR), the reduction of caloric intake without inducing malnutrition, is the most reproducible method of extending health and lifespan across numerous organisms, including humans. However, with nearly one-third of the world’s population overweight, it is obvious that caloric restriction approaches are difficult for individuals to achieve. Therefore, identifying compounds that mimic CR is desirable to promote longer, healthier lifespans without the rigors of restricting diet. Many compounds, such as rapamycin (and its derivatives), metformin, or other naturally occurring products in our diets (nutraceuticals), induce CR-like states in laboratory models. An alternative to CR is the removal of specific elements (such as individual amino acids) from the diet. Despite our increasing knowledge of the multitude of CR approaches and CR mimetics, the extent to which these strategies overlap mechanistically remains unclear. Here we provide an update of CR and CR mimetic research, summarizing mechanisms by which these strategies influence genome function required to treat age-related pathologies and identify the molecular fountain of youth.
1. Introduction
Aging is an inevitable process experienced by all life on this planet. In humans (and numerous other species), aging comes with a battery of age-linked pathologies, including type 2 diabetes (T2D), cardiovascular disease (CVD), atherosclerosis, osteoarthritis, neurodegenerative diseases (ND), and cancer [1,2,3,4]. These pathologies are often the combination of nature and nurture; genetics play an important role, but environmental conditions (including exercise and diet) also have a significant impact on the rate at which individuals acquire disease and age. Excessive calorie intake increases the risk of many of the aforementioned age-linked pathologies, deleteriously promoting chronic inflammation and signaling insensitivities. Does aging have to be fraught with deteriorating health and increasingly poor quality of life until death? Or could healthy aging be feasible? Those that exercise and have a moderate diet are already shown to be at lower risk of numerous diseases, but could this effect be enhanced to promote longer, healthier lives? Addressing this question could have a tremendous societal impact, decreasing the socioeconomic burden of an aging population and, as a result, is an intensively researched area of biology.
A key area of research for maintaining longer, healthier lives is caloric restriction (CR). CR is the reduction of nutritional intake to 60–80% ad libitum without inducing malnutrition or starvation. This approach has been successful and reproducible across laboratory-model organisms in extending lifespan (from yeast to nonhuman primates) [5,6,7,8,9]. Short-term trials of CR in humans have also proven effective in preventing and improving the treatment outcomes of age-linked pathologies such as cancer [10,11]. Despite the promise of CR, this regime is unlikely to be achieved, particularly as large percentages of individuals already struggle with obesity and thus moderating their diets (World Health Organization, 2020). Therefore, the question is: could alternative dietary regimes that may not require decreased food consumption achieve the same beneficial effects as CR? Or is there a pharmacological mimetic of CR, a “magic pill,” that will alter how the body senses energy inputs and reproduces a CR-like state?
Amino acid restriction (AAR) is a potential CR alternative, with current work focusing on the efficacy of total AAR or the restriction of specific amino acids in promoting health and lifespan [12,13]. Similarly, the immunosuppressant rapamycin [14] (and the structurally related family of compounds called rapalogs [15]), the polyphenol resveratrol (found in the skin of red fruits, such as grapes) [16,17], and the T2D treatment metformin [18,19] have been examined for their ability to reproduce CR-like effects without dietary alterations. Although it is generally accepted that these compounds can promote health and lifespan, exactly how this is achieved is still being investigated. Therefore, it is necessary to understand how these compounds modulate cell function to better comprehend the aging process but also to identify other potential pro-longevity strategies. Here, we will discuss the theories of aging in relation to CR and how CR is sensed at the cellular level before discussing AAR and potential CR mimetics (CRMs) in the same contexts.
2. Caloric Restriction Attenuates Features and Processes of Cellular Aging
Aging is caused by the accumulation of numerous cellular alterations that induce cells to senescence, which consequently secrete inflammatory proteins that spread this state in neighboring cells. The accumulation of senescent cells contributes to age-associated diseases, and the clearance of these cells has been shown to delay or improve age-related phenotypes [20,21,22]. Key alterations associated with promoting aging (including the accumulation of senescent cells) include increased genomic instability, telomere shortening, changes in the epigenome, loss of protein homeostasis (proteostasis), deregulated nutrient sensing, mitochondrial dysfunction, stem cell exhaustion, and altered intracellular signaling [23]. CR has shown promise in attenuating age-promoting factors, reducing the number of senescent cells in a culture and model organisms [24]. CR was first reported in 1935 [5] and since then has become the most reproducible and reliable method of extending health and lifespan across model organisms, ranging from unicellular to nonhuman primates (yeast, [9], fruit flies [25], nematodes [26], crustaceans [27,28], spiders [29], lab mouse/rats [30], and monkeys [6,8,31]. At the molecular level, CR mitigates and reverses features of cellular aging. The free-radical theory of aging proposes that increased energy intake can result in more ROS as a result of oxidative phosphorylation in the mitochondria, contributing to lipid, protein, and DNA damage and advancing age (reviewed in [32,33]). CR counteracts this by lowering metabolic rates [34,35], improving DNA damage repair [36,37,38,39,40], and decreasing oxidative stress [41,42,43]. Although this theory is generally accepted, free-radical production and oxidative stress are not always decreased in response to CR [44,45]; however, increased expression of antioxidant enzymes may act as a compensatory mechanism for this.
In connection with ROS and mitochondrial dysfunction, the insulin sensitivity of cells decreases with age, increasing levels of serum glucose and insulin required for cellular responses. As a result, glucose metabolism is upregulated, increasing cell replication, decreasing the time between cell divisions, and permitting less time for the maintenance and repair of DNA [46,47]. Together, these processes push cells toward senescence. CR decreases levels of circulating glucose and insulin, increases insulin sensitivity, and extends health and lifespan [48,49,50,51]. The upregulation of insulin-like growth factor-1 (IGF-1) also occurs with age, again promoting cell growth, division, and metabolism. CR decreases the activity of the IGF-1 signaling pathway, promoting cellular maintenance, and repair in mice [52]. However, human clinical trial data have been less consistent, with two-year CR having no impact on circulating IGF-1 levels [53,54]. Despite these findings, alternative analyses of the same trial concluded that CR decreased the rate of biological aging [55].
The hormesis model of aging proposes that low levels of stress primes cells, enabling them to prepare cellular processes required to appropriately counteract larger stress [56]. CR increases the expression of proteins involved with DNA repair, free-radical scavenging, and stress, as well as mediating ROS-producing pathways [24,40,42,43]. This model is often favored for explaining the health and lifespan benefits of CR, whereby CR activates transcription factors and mechanisms involved in regulating gene expression and protein translation to better mediate cellular stress responses [57,58] (reviewed in [59]).
2.1. How are Nutrients Sensed at the Cellular Level?
Key in promoting health and lifespan at the cellular and organismal level is the manipulation of how nutrients are detected. The target of the rapamycin (TOR) pathway is a highly conserved nutrient-sensing pathway comprising multiple proteins [60]. In mammals, the mammalian (m)TOR signaling cascade indirectly detects inputs from multiple sources, including growth signals, amino acids, and energy levels. There are two mTOR complexes: mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). mTORC1 is directly involved in mediating cellular responses to nutrients, although recently, mTORC2 has also been shown as nutrient-sensitive [61]. Multiple growth factor pathways (such as the insulin/IGF-1 pathway) converge upstream of mTORC1 at the tuberous sclerosis complex (TSC; a heterotrimeric complex of TSC1, TSC2, and TBC1D7 [62]). In the presence of growth factors, the phosphoinositide 3-kinase/3-phosphoinositide-dependent protein kinase 1/protein kinase B (PI3K/PDK1/AKT) pathway inhibits TSC by the AKT-dependent multisite phosphorylation of TSC2. This induces the dissociation of TSC from the lysosomal membrane [63], inactivating the RAS homolog enriched in brain (Rheb), which is also partially localized to the lysosome. Subsequently, mTORC1 is phosphorylated and activated by Rheb, promoting downstream cellular growth and proliferation (reviewed by [64]). Other growth factors functioning via TSC inhibition to activate mTORC1 include receptor tyrosine kinase-dependent Ras signaling, wingless-related integration site (WNT), and tumor necrosis factor α (TNFα) pathways (reviewed in [64,65]). Exactly how the TSC integrates the input of these various signals is unknown. When active, mTORC1 promotes various processes, including nucleotide, lipid, and protein synthesis, which stimulate cell growth and proliferation. In the absence of growth factors, TSC sequesters Rheb at the lysosomal membrane [66,67], inhibiting mTORC1, and shifting cellular processes toward maintenance and repair, upregulating autophagy (recycling of cellular components) and inhibiting the synthesis of new molecules.
2.2. What Downstream Effects Does Caloric Restriction Have on Genome Function?
CR does not directly inhibit mTORC1 but modulates upstream energy-sensing complexes. Adenosine monophosphate kinase (AMPK) is activated (phosphorylated by liver kinase B1; LKB1) in response to increased AMP/ATP ratios, whereas Sirtuin 1 (SIRT1) is activated by increased NAD+ levels, indicating low cellular energy availability [68,69,70]. AMPK and SIRT1 also activate one another, although AMPK is active much sooner (seconds/minutes) following CR when compared to SIRT1 activation (4–12 h) [71]. Perhaps SIRT1 functions later in response to CR in order to maintain AMPK activity. AMPK is also regulated by increases in intracellular Ca2+ (likely the result of hormonal changes), specifically via phosphorylation by calcium/calmodulin-dependent protein kinase kinase B (CamKKB) [72,73]. Once activated, SIRT1 deacetylates downstream targets such as p53, inhibiting p53-mediated gene expression [74,75]. However, there is potential for additional feedback as SIRT1 expression depends on interaction with Forkhead Box O3 (FOXO3a) and p53 under CR, whereas FOXO family members are also involved in regulating IGF-1 signaling upstream of mTOR [70,76]. Moreover, SIRT1 deacetylates components of nuclear factor (NF)-κB to decrease the transcription of proinflammatory genes [77,78,79,80]. SIRT1 also deacetylates other mTOR-linked proteins, such as hypoxia-inducible factor 1α (HIF1α) [81,82], which is usually increased with age [83,84,85]. The deacetylation of HIF1α decreases the expression of genes involved in cell cycle progression, angiogenesis, and glucose metabolism [86].
The impact of complex signaling in response to CR and how it could potentially reverse age-linked transcriptomic changes have been examined through numerous transcriptome-wide studies across model organisms, including humans [87,88,89,90,91,92,93,94,95]. In general, whole-genome studies (either by microarray or RNA sequencing) have concluded that CR was able to upregulate “antiaging” genes, including genes that inhibit oxidative stress, decrease age-linked inflammation, decrease metabolism, and decrease genes involved in pathways such as IGF-1 signaling and cell cycle progression (Table 1). These findings are striking given the consensus of several publications linking pathways associated with aging to modulation by CR. Furthermore, these data highlight the importance of age at CR initiation and CR duration. For example, short-term CR induces different transcriptome profiles to long-term CR [96,97], whereas the age of CR onset also impacts its efficacy [98,99]. Analyses have been conducted over a period spanning two decades; therefore, in addition to the promising results and concordant conclusions, additional meta-analyses of datasets may provide new insights into processes governing the progression of aging as knowledge of genes and pathways evolves.

Table 1.
Summary of the impact of calorie restriction on aging-related pathways of calorie restriction.
Aging is the result of numerous genetic and environmental factors that may not all be controllable in laboratory experiments. Furthermore, tissues and organs exhibit unique aging processes [100]. For example, some tissues age faster and secrete substantially more inflammatory factors. Mitochondria-rich tissues may exhibit greater susceptibility to DNA-damage-linked aging, whereas other tissues respond more to the loss of insulin and IGF-1 sensitivity. Recently, Ma and colleagues examined seven different tissues from rats for age-linked changes and their potential for reversal by CR from midlife [101]. Ma et al. also examined the composition of cell types, determining that, in accordance with changes in gene expression linked to inflammation/the immune system, cell-type composition within the rat tissues changed. Although CR rescued many age-associated changes at the genomic level, there were also CR-induced gene expression changes unique to CR in each tissue [101], corroborating other studies in mouse liver and flies [97,102]. In addition to tissue-specific changes, treatment length and dosage effects need to be considered, given that chronic or more stringent CR potentially has a different impact than shorter-term or less stringent regimes (Table 1).
Although understanding the aging transcriptome has progressed to the development of a transcriptomic clock (software that can predict tissue age based on transcript profiles), mechanisms behind these changes are still lacking, and pathway analyses are still vague [103]. It is important to go beyond the aging transcriptome and the impact of CR to understand how other regulatory elements of the genome are influenced. This information will further aid in the understanding of potential CR mimetics, their similarity to CR, and their ability to increase health and lifespan.
3. Amino Acid Restriction: An Alternative to Caloric Restriction?
Although CR has been studied for over a century, removing specific elements of diets is a more recently evolved area of longevity research. Amino acid restriction (AAR) centers around the theory that it is not CR per se that is responsible for increasing health and lifespan but the reduction of amino acids specifically from the diet. This consideration, combined with biochemical evidence that the presence/absence of amino acids alone is sufficient to modulate mTORC1 function [104,105,106,107], supports the proposal that AAR could be an alternative to CR.
3.1. Does Amino Acid Restriction Extend Health and Lifespan?
The restriction of all amino acids has been shown to extend lifespan in yeast [9,108], ants [109], and flies [108,110]. Additionally, the restriction of specific amino acids, such as branched-chain amino acids (BCAAs) in mice, can be as beneficial as total amino acid restriction [111], whereas in rats, the restriction of individual essential amino acids (leucine, lysine, methionine, and threonine) and total AAR reduced IGF-1 levels [112]. Tryptophan restriction increased the lifespan of rodents [113]; however, not all AAR is successful, with glutamic acid deprivation decreasing yeast lifespan [114]. Leucine restriction improves metabolic health, glucose tolerance, and insulin sensing in mice [115,116]. Protein restriction prior to surgery exhibited physiological stress protection in mice [113], whereas low-protein diets in humans under 65 years of age resulted in an overall reduction in IGF-1 levels, cancer, and mortality [117]. Furthermore, high-protein diets are linked with higher mortality rates when compared to diets based on low-carbohydrate and high vegetable content [118]. There is also an increased association with red meat and diabetes, cancer, and ischemic heart disease [119,120], whereas reduction in protein intake reduces circulating IGF-1 levels [54]. AAR-linked reductions in IGF-1 levels and improved glucose sensitivity are parallel to those observed in CR and important in extending health and lifespan. The AAR of specific amino acids further influences the post-translational modification of proteins. For example, BCAAs are important in the formation of acetic acid, a molecule used in the acetylation of histones, as well as in the regulation of autophagy, supporting findings of BCAAs in promoting lifespan-extending biological states [111,112,121]. This effect on epigenetic modification is not unique to BCAAs, with a decrease in methionine leading to decreased S-adenosyl-methionine (SAM) and reduced histone methylation [122]. Therefore, the combination of amino acids in AAR is likely important in directing which pathways are influenced within an organism and subsequent genomic and epigenomic responses. It is likely many of the beneficial impacts of total AAR, or the restriction of specific amino acids, are the result of mTORC1 inhibition and upregulated autophagy. AAR has been shown to upregulate autophagosomal–lysosomal pathways, promoting the degradation of cytoplasmic proteins and organelles, thereby providing free amino acids for protein synthesis [123,124].
Unlike CR, AAR is more complex and can be the result of restricting single or multiple amino acids. AAR strategies focusing on methionine restriction are the best defined to-date; however, this is only effective if cystine is also restricted and is therefore referred to as sulfur amino acid restriction (SAAR). In addition to increasing lifespan [114,125,126,127], SAAR lowers levels of serum IGF-1, insulin, glucose, and thyroid hormone, increasing insulin sensitivity, decreasing body weight, improving resistance to oxidative stress in various mice and rat strains [128,129,130,131,132,133], and decreasing mitochondrial ROS production and protein oxidation [134,135]. Additionally, in mice, SAAR prevents the onset of T2D [136] and extends the lifespan of progeroid-models [137,138]. Therefore, as with CR, SAAR counteracts many of the molecular processes associated with aging.
Although AAR is capable of extending health and lifespan, the deprivation of different amino acids differentially impacts the transcriptome. When compared to 15 other amino acids in breast and prostate cancer cells, the restriction of methionine was able to elicit the largest transcriptional changes, likely due in part to additional impacts on the epigenome [122]. As cancer cells are often hyperproliferative and have dysregulated cell cycles, to better understand the impact of AAR in aging, the replication of these studies in noncancerous human cell lines would be beneficial. Evidence for amino-acid-specific effects is provided by methionine restriction, which extends the lifespan of Drosophila melanogaster, albeit not to the same degree as total AAR [108]. Similarly, leucine restriction exerts similar benefits as methionine restriction (although not as consistently) [116]. In flies, the restriction of tryptophan or arginine required the general control nonderepressible 2 (GCN2) protein (a protein kinase that specifically responds to amino acid levels) to achieve health-promoting effects, whereas others (histidine and arginine) did not [139]. Finally, protein-restricted mice supplemented with different BCAAs exhibited varied metabolic profiles. [111,140]. This may be due to the different sensitivity of mTORC1 to each amino acid, in particular leucine and arginine [104]. Although there is much evidence regarding the benefits of either total AAR or SAAR restriction, it will be important to determine how healthy tissues and organisms respond to the removal of individual amino acids. Furthermore, understanding genomic changes and mechanisms mediating these processes will aid in the development of diets in which amino acid levels can be manipulated to treat many age-linked diseases without reducing overall caloric intake.
3.2. Is Amino Acid Sensing also Regulated through mTOR?
Although some organisms can synthesize all amino acids, mammals must rely on extracellular sources for a subset of amino acids (termed essential amino acids). Amino acid levels can also be used to modulate mTOR activity to direct subsequent cellular processes [104,105,106,107]. Amino acid sensing by mTOR is complex, with potentially distinct upstream receptors for each amino acid. Few of these have been identified; however, in general, amino acid depletion downregulates mTORC1, resulting in decreased protein synthesis and cell proliferation. This inactivation also promotes autophagy, catabolizing pre-existing molecules for the amino acids required to prevent starvation [141]. If enough autophagy occurs, mTORC1 is upregulated.
mTORC1 is localized to the lysosomal surface in the presence of amino acids. In general, amino acid sensors indirectly promote stable dimerization between RagA/B and RagC/D, which are targeted to the lysosome via Ragulator (as the RagA/B/C/D GTPases do not have a lipid-targeting signal, this acts as an intermediate). To interact with mTORC1 (via the raptor subunit), RagA/B must be GTP loaded and RagC/D must be GDP loaded. Rheb then activates mTOR (mTORC1) in a Rag-dependent manner [142,143,144]. If artificially anchored to the lysosome, mTORC1 becomes insensitive to amino acids while still responding to insulin levels [142]. Therefore, it is likely that Rheb and Rag form part of a regulatory network that enables appropriate mTORC1 activation in response to a balance of amino acids and growth factors.
Upstream of the Rag GTPases are cellular amino acid sensors. Of these, the sensors for arginine and leucine (sestrin2 and cytosolic arginine sensor for mTORC1 (CASTOR)) [145,146,147,148] bind their respective amino acids, disrupting interactions with and activating GTPase-activating protein toward Rags (GATOR)2, inhibiting GATOR1, and activating RagA/B and, ultimately, mTORC1 [145,149,150] (Figure 1C). In the absence of arginine/leucine, their respective sensors are bound to GATOR2, inhibiting it as well as mTORC1 (Figure 1D). Unlike arginine/leucine, methionine is sensed by the S-adenysyl-methionine (SAM) sensor (SAMTOR) via the presence of its metabolite SAM. SAM binds SAMTOR, dissociating it from GATOR1. Amino acids are also sensed from the lysosome; for example, SLC39A9 is a lysosomal arginine sensor that interacts with the Rag-GTPase-Ragulator-v-ATPase complex to activate mTORC1 (reviewed by [60]). This process may couple the release of essential amino acids from the lysosome to activation of mTORC1 and cell growth.
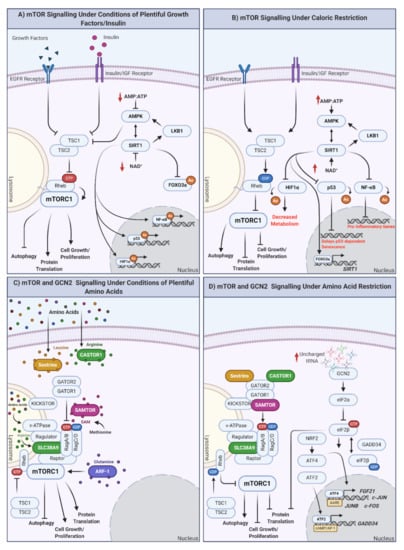
Figure 1.
How are nutrients sensed at the cellular level? The mTOR and GCN2 pathways. (A) mTOR signaling in response to plentiful nutrient (growth hormone, insulin) supply. Under conditions of plentiful nutrients, growth factors and insulin inhibit TSC1/2, leading to the inhibition of Rheb and the induction of mTORC1 at the lysosomal surface. mTORC1 upregulation results in autophagy inhibition and the promotion of protein translation, cell growth, and proliferation. Under these conditions, cellular ATP levels increase, reflective of plentiful nutrient status. ATP increase inhibits AMPK, modulating the AMPK-SIRT1-LKB1 feedback loop and inducing mTORC1. Independently of mTORC1, SIRT1 also downregulates the expression of FOXO3a and promotes the synthesis of genes/proteins involved in the cell cycle, as well as response inflammation and cellular stress (when required). (B) mTOR signaling under conditions of caloric restriction. In the absence of nutrients (or under conditions of caloric restriction; CR) TSC1/2 are activated, Rheb is GDP loaded, and mTORC1 is inhibited by undocking from the lysosomal surface. This promotes autophagy and inhibits protein translation, cell growth, and proliferation. Simultaneously, ATP levels decrease, increasing the AMP/ATP ratio indicative of low cellular energy status, activating AMPK, and modulating the AMPK-SIRT1-LKB1 feedback loop. In addition, proinflammatory transcription/translation is inhibited, metabolism is decreased, and longevity promoting FOXO3a is translocated to the nucleus to regulate its target genes. (C) mTOR signaling under plentiful amino acid availability. In the presence of amino acids, mTORC1 integrates signals from multiple amino acid receptors: (inhibited sestrin (leucine), castor (arginine), or samtor (methionine) complex formation), glutamine sensor ARF-1, lysosomal arginine sensor SLC38A9, and lysosomal amino acid sensor v-ATPase. In the presence of all amino acids, mTORC1 is active and promotes cellular growth and proliferation. If individual amino acids are absent, this could result in mTORC1 inhibition and shift cells to a status of amino acid restriction (AAR). (D) mTOR and GCN2 under conditions of amino acid restriction (AAR). In the absence of amino acids, mTORC1 is inhibited, promoting the previously mentioned prolongevity factors, including autophagy and preventing cell proliferation. GCN2 is activated under conditions of AAR by the binding of uncharged tRNAs. eIF2α is then converted to eIF2β, which cycles between GTP- and GDP-loaded states, regulated by GADD34. eIF2B promotes translocation of ATF4 to the nucleus, where it regulates subsequent expression of genes with amino acid response elements (AARE). NRF2 is also upregulated in response to AAR and can promote upregulation of ATF4. ATF2 similarly binds cAMP/AP-1 motifs to promote the expression of GADD34. Created with BioRender.com.
Beyond utilizing specific amino acid receptors, mTORC1 also responds to amino acid signaling via Rag-GTPase-independent methods. Glutamine requires lysosomal H+ATPase (v-ATPase) and vesicle trafficking ADP ribosylation factor (Arf-1 GTPase) [151,152]. Glutamine has also been reported to activate mTORC1 through a Rag-dependent mechanism [153]. The ability of AAR to modulate mTOR signaling to inhibit mTORC1 and, subsequently, upregulate similar downstream processes as seen with CR, makes it a strong candidate as an alternative to CR.
4. Are Amino Acid Restriction and Caloric Restriction the Same?
In addition to modulating mTORC1 function, AAR also mediates mTORC1-independent functions via the integrated stress response (ISR). The ISR rapidly assimilates extracellular stress signals, such as AAR, through protein kinases that function to prevent the exhaustion of cellular resources. This process also likely occurs in response to CR, thereby reducing numerous energy status indicators, including levels of amino acids and growth factors. As intracellular amino acid levels decrease, so do levels of charged tRNA. Uncharged tRNA then bind and activate GCN2, which then phosphorylates eukaryotic initiation factor 2 (eIF2)α, which is converted into an inhibitor of eIF2β. The inhibition of eIF2β will slow the rate at which eIF2α is reloaded with GTP, ultimately reducing GTP-GDP exchange rates on eIF2α, necessary for mRNA translation re-initiation and decreasing global translation. Although global translation is decreased, the translation of specific transcription factors such as activating transcription factor (ATF) 2, ATF4, ATF5, and growth arrest and DNA -damage-inducible protein (GADD34) are increased [154] (reviewed by [155]). ATF4 translocates to the nucleus where it binds amino acid response elements (AAREs), upregulating the expression of amino acid transporters, metabolic regulators, antioxidant defenses, and other homeostatic processes to promote the re-establishment of homeostatic cellular conditions. GADD34 enables feedback signaling to permit the translation of upregulated stress-response genes [156]. Some ATF4-regulated genes require the phosphorylation of ATF2 to upregulate transcription. ATF2 specifically targets genes that do not respond to other amino acid response linked transcription factors [157]. Therefore, AAR can be regulated independently of the mTOR pathway. In addition, GCN2 is required to extend lifespan in yeast and flies in response to AAR [114,158], with GCN2-deficient organisms displaying aging phenotypes. As downstream processes regulated by GCN2 and mTORC1 significantly reduce protein translation, this is one potential mechanism by which longevity is promoted. GCN2 signaling is responsive to CR likely through a decrease in amino acids and therefore plays an important role in mediating the response to CR, albeit more specific to amino acid sensing. The role of mTORC1 in response to AAR and CR has been extensively examined; however, the role of GCN2 in promoting health and lifespan remains relatively unexplored.
The IRS and mTOR pathways feedback on one another, linking cellular responses to CR and AAR in promoting health and lifespan. One important molecule involved in this feedback is fibroblast growth factor 21 (FGF21), which is upregulated by ATF2/4. The FGF21 promoter contains an AARE [159,160], and FGF21 is implicated in promoting the beneficial effects of CR and AAR/SAAR [161,162]. Additionally, the overexpression of FGF21 increases lifespan [163] and prevents cellular senescence via SIRT1 [164], demonstrating its significant role in longevity and well-established longevity-promoting pathways. Counterintuitively to its upregulation in response to CR and AAR, FGF21 levels are also increased with age [165,166,167]. It has been proposed that this could be due to insensitivity to FGF21 levels/loss of FGF21 receptors [166,168,169]; however, findings are mixed [167,170]. Although the reason for an increase in FGF21 levels in aging, as well as in response to CR, is unclear, there is a consensus that this molecule upregulates metabolism and reduces the impacts of chronic disease (reviewed by [171]) by preventing insulin resistance ([172,173]). FGF21 also activates the mTORC1-S6K axis via mitogen activator protein kinase (MAPK), perhaps as the result of feedback signaling from increased amino acid availability due to autophagy, with increased mTORC1 levels demonstrated to mediate the beneficial impacts of FGF21 in vitro [174]. As a molecule primarily secreted from the liver, FGF21 could act as a signal of global energy status across many tissues, promoting energy-saving states in other cells.
5. Caloric Restriction Mimetics
CR clearly has positive impacts on health and lifespan. However, with 39% of the world’s population already overweight (World Health Organization, 2020), this is a difficult strategy to implement. Alternatively, a variety of compounds exist that mimic CR without having to restrict food intake. Caloric restriction mimetics (CRMs) often function via similar pathways to CR, such as the mTOR signaling cascade, and have the potential to be used as treatments to prevent the development of age-related pathologies and promote healthy aging.
5.1. Rapamycin
Rapamycin, also known as sirolimus, was initially isolated in 1964 from soil containing Streptomyces hygroscopicus on Rapa Nui (Easter Island) during a Canadian expedition to the South Pacific. First described as an antifungal, the immunosuppressive and antitumor properties of rapamycin led to its frequent use in clinical settings [175]. In 2006, it was proposed that rapamycin could be used to slow aging and improve many physiological functions that deteriorate with age in humans, making this compound one of the first CRMs [176]. Rapamycin forms a complex with the FK506-binding protein 12 (FKBP12), which induces inhibition of mTORC1 and subsequently the mTOR pathway (Figure 2A) [177,178]. Similar to CR, the rapamycin-mediated inhibition of mTORC1 decreases cellular growth, proliferation, and protein translation [179,180,181] and upregulates autophagy [182,183], thus promoting health and lifespan.
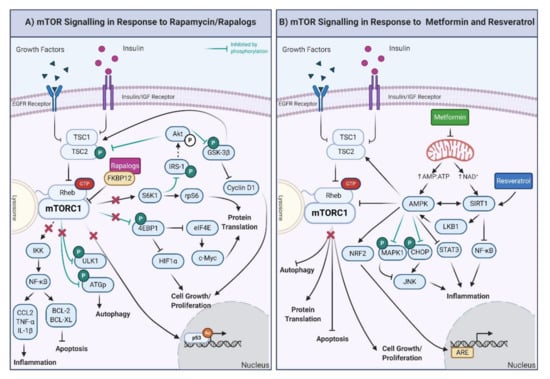
Figure 2.
(A) Downstream effects of mTOR signaling in response to rapamycin and rapalogs. The inhibition of mTORC1 results in the decrease of the inflammatory response and promotion of apoptosis through regulation of IκB kinase. ULK1 is also inhibited, promoting autophagy. Furthermore, the activity of the eukaryotic translation initiation factor 4E-binding protein 1 (4EBP1) and S6K1 proteins is decreased, which results in diminished cell growth, proliferation, and protein translation. The inhibition of mTORC1 also decreases p53-mediated gene expression. Moreover, through the inhibition of S6K1, Akt activity is upregulated, generating a feedback loop to the products of decreased mTORC1 activity, specifically via the inhibition of TSC1/2 and GSK-3β. IRS-1: insulin receptor substrate 1; CCL-2: C-C motif chemokine ligand 2, BCL-2: B-cell lymphoma 2; BCL-XL: B-cell lymphoma-extra-large. (B) Downstream effects of mTOR, AMPK, and SIRT1 signaling in response to metformin and resveratrol. Metformin exposure decreases mitochondrial function, increasing the AMP/ATP ratio and NAD+ levels. The increased AMP/ATP ratio activates AMPK, which, in turn, decreases protein translation, cell growth/proliferation, and stimulates autophagy and apoptosis. AMP also activates the nuclear factor erythroid 2-related factor 2 (NRF2), which triggers the synthesis of genes involved in the antioxidant response (ARE). Through inhibition by phosphorylation of the mitogen-activated protein kinase (MAPK1) and CCAAT-enhancer-binding homologous protein, AMPK inhibits and activates the c-Jun N-terminal kinase (which is involved in inflammation, JNK). Furthermore, AMPK also reduces the inflammatory response through the inhibition of the signal transducer and activator of transcription (STAT3). In the presence of resveratrol, the most potent inducer of SIRT1, inflammatory proteins are decreased by the inhibition of NF-κB. Finally, AMPK is also activated by phosphorylation of the liver kinase B1 (LKB1) and deacetylation of SIRT1, creating a feedback loop. Created with BioRender.com.
In addition to its lifespan-extending properties, additional studies have shown that rapamycin can ameliorate age-related disease phenotypes in cell-based models of cancer [184,185,186], CVD [187,188], premature aging diseases (e.g., HGPS [189,190,191]), and ND [192]. These findings can be recapitulated at the organismal level, including in ND (e.g., amyotrophic lateral sclerosis [193], tauopathies [194], Alzheimer’s disease [195,196,197], Parkinson’s disease [198]), cancer [199,200], obesity [201], disrupted circadian clock [202,203], and laminopathies [204,205]. Furthermore, rapamycin pretreatment improves the success of pancreatic islet cell engraftment through the inhibition of inflammatory chemokines in mice models and human patients with type I diabetes [206]. The results of these studies have encouraged the exploration of rapamycin in humans beyond its use as an immunosuppressant following organ transplantation [207,208,209]. For example, when applied topically, rapamycin can improve the clinical appearance of the skin [210], but, more importantly, it can prevent insulin resistance [211]. Therefore, like CR, rapamycin has numerous benefits in various disease states to promote healthier aging across species.
There is evidence indicating that rapamycin in a bona fide CRM due to its induction of autophagy. This is supported by combinatorial studies of CR and rapamycin, in which the same pathway is targeted [212]. However, there is also evidence that, unlike CR, which improves insulin sensitivity and glucose tolerance, rapamycin promotes insulin resistance [213]. This could be the result of chronic rapamycin treatment, which induces starvation pseudodiabetes (SPD), also triggered in response to prolonged fasting [214,215]. It is possible that in models where rapamycin induces SPD, the dose is too high and mimics starvation rather than CR. To further support that rapamycin functions alternatively to CR, Unnikrishnan et al. [14] demonstrated that CR, in combination with rapamycin, improved glucose and insulin intolerance when compared to control or rapamycin alone, indicating that CR could alleviate the secondary effect of rapamycin on glucose metabolism Although this may be a cause for concern in considering rapamycin as a lifespan-promoting compound, or even a CRM, there are numerous experimental considerations. For example, the increase in insulin insensitivity is not observed in all mouse strains and treatment conditions are often not consistent across studies (e.g., dose and length). Nevertheless, rapamycin-fed model organisms frequently live longer, healthier lives.
To establish whether rapamycin is a CRM, it is important to consider changes in specific genes that are markers of CRM function. Cyclin D1, a cell cycle marker, increased in response to both CR and rapamycin in mouse liver tissue. However, transcript levels of other proteins involved in the same cellular process, such as p16, p21, and p53, were decreased [212,216] (Table 2). Unnikrishnan and colleagues also identified transcriptomic changes in the genome of mouse livers under regimes of ad libitum, CR, and rapamycin treatment [14]. Rapamycin was found to have overlapping, but distinct, gene expression profiles to those of ad libitum or CR [217], supporting that rapamycin is not a CRM. This could also provide evidence that to achieve health and lifespan extension, a subset of transcriptomic changes in response to CR/AAR/CRM is required, and other changes may be condition- or compound-specific and not essential for the desired effects. Furthermore, when combined, rapamycin and CR have greater impacts on both the transcriptome and metabolome of mice than when applied separately [217], with <20% of mRNA transcripts shared by CR and rapamycin. Moreover, of genes changing by CR, 74% matched genes previously identified in response to CR (using the GenDR database), but only 7.5% of rapamycin-induced gene expression changes were found in the same database. In comparing rapamycin and CR in white adipose tissue (WAT) of mice, fewer transcripts changed in response to rapamycin [216]. Similarly, CR induced greater changes in the yeast transcriptome than rapamycin, with CR upregulating genes for autophagy and β-oxidation, whereas rapamycin increased the expression of enzymes involved in the degradation of energy-storage molecules [218]. Both rapamycin and CR had beneficial impacts on reproduction in mice (increased preservation of ovarian primordial follicular reserves); however, the two treatments again exhibited divergent metabolic effects. Regardless, both were associated with higher transcript levels encoding the pro-longevity factor FOXO3a [219]. Therefore, although CR and rapamycin both function to extend health and lifespan consistently across organisms, the impacts of these compounds are transcriptomically varied.
Table 2.
Summary of the effects of rapamycin and rapalogs on aging-related pathways.
5.2. Rapalogs: Rapamycin Analogs
Rapamycin has many benefits; however, its poor solubility, bioavailability, and pharmacokinetics, as well as side- and off-target effects [228], have led to the development of rapalogs. Rapalogs are synthetic analogs of rapamycin that aim to improve clinical outcomes of rapamycin, with fewer off-target effects. These rapalogs, including everolimus, temsirolimus, and ridaforolimus, have been examined to determine if they can also extend health and lifespan.
Everolimus (RAD001) is an orally administered rapalog that has been reported to have the same therapeutic properties as rapamycin with higher thermal stability, greater solubility, and improved pharmacokinetics [229]. Everolimus binds to FKBP12, inhibiting mTORC1, and results in the downregulation of the PI3K/AKT/mTOR pathway (Figure 2A). In some human cancers, this exerts an antiproliferative effect, inhibiting migration and angiogenesis [230], parallel to CR, AAR, and rapamycin [230]. Everolimus alone, or combined with other anticancer agents, has been shown to attenuate cancer cell growth, promote increased cell death, and increase patient survival [221,224,225,231,232]. Sabine et al. [233] observed that patients with early-stage breast cancer had decreased expression of genes involved in cell cycle and proliferation, pyrimidine metabolism, and p53 signaling pathways in response to everolimus. However, the genes involved in the mTOR pathway were not differentially expressed after everolimus treatment, indicating that this compound can modulate other pathways in addition to mTOR [233]. Genes involved in the PI3K/AKT/mTOR pathway are highly responsive to everolimus [234], and as such, everolimus is routinely used in cancer treatments in Europe and the United States. The widespread use of everolimus in clinics highlights the benefits of this compound over rapamycin.
As a derivative of rapamycin, everolimus has also been proposed as a pro-longevity compound and CRM. Everolimus prolongs the survival of HGPS cell models and rescues age-linked cellular defects [227]. In mice, everolimus attenuates neurological decline and vascular dementia [235] as well as decreasing neuronal loss and oxidative stress [236]. Additionally, everolimus reduced amyloid precursor protein (APP) levels, the amyloidogenic Aβ peptide, as well as levels of phosphorylated tau, which are hallmarks of AD pathology [222]. In aged rats, everolimus reversed 37% of age-linked transcriptional changes in kidney tissue, including the reversal of the upregulation of inflammatory response, interferon alpha/gamma response, and apoptosis [226] and, in mice, downregulated glucose metabolism [237] (Table 2). Everolimus also reduces cytokine production [231,238] in liver transplant patients, improves immune function, and prevents immune-induced senescence in elderly humans [223]. In addition to these longevity-promoting effects, everolimus downregulated the protein c-Myc [226]. Increased c-Myc levels are associated with aging [239] and have been reported to respond to rapamycin treatments [240,241] but rarely to CR [242]. c-Myc and SIRT1 function in a negative feedback loop; c-Myc binds the SIRT1 promoter to increase expression, with SIRT1 deacetylating c-Myc to decrease its DNA binding activity [243]. This feedback loop functions independently of mTOR to regulate downstream targets associated with health and lifespan [241]. Furthermore, c-Myc can upregulate ATF4 via GCN2, linking this protein to AAR responses [244]. These complex mechanistic relationships are still under investigation; however, they support the divergence between rapamycin/rapalogs and CR while also highlighting the importance of pathways other than mTOR in regulating the response to longevity-promoting interventions (LPIs).
Both temsirolimus and ridaforolimus have improved solubility and pharmacokinetics over rapamycin [245,246]. Both compounds have been shown to inhibit the growth of cancer cells, with ridaforolimus currently being tested in phase I and II clinical trials. In addition to its potential application as a cancer therapy, temsirolimus ameliorates age-linked phenotypes in a HGPS cell model [247], attenuates tauopathies [248], and clears AD-related Aβ by inducing autophagy in preclinical models [249]. Although promising, information on molecular mechanisms or transcriptomics of these rapalogs is limited.
Rapalogs are associated with numerous beneficial effects linked with healthier aging, such as decreasing inflammation and circulating IGF-1 levels; however, like rapamycin, these derivatives also induce glucose insensitivity. Regardless, these compounds have been documented to reverse age-associated phenotypes in HGPS cell models and ND; benefits that may outweigh potential negative impacts. Notably, most of the research done for these derivatives has been conducted in cancer-based models; therefore, the impact they could have on normal cells/organisms is unclear. Furthermore, the impact of these compounds on genome function is poorly defined, making it difficult to establish whether rapalogs are CRMs; however, although these compounds overlap in their function via inhibition of mTOR, they also likely influence pathways distinct to those altered by CR.
5.3. Resveratrol
Resveratrol (3,5,4′-trihydroxystilbene) is a polyphenol produced by plants in response to physiological damage and is found in foods such as grapes, berries, pine nuts, and legumes [250]. Mediterranean diets are rich in resveratrol (as well as many other polyphenols) and have been associated with promoting positive markers of health. For example, the consumption of resveratrol via red wine has been linked to the lower incidence of CVD in France [251] despite high-fat diets. This proposed effect on CVD is due to resveratrol’s antioxidant and anti-inflammatory effects, which have been recapitulated in nonhuman primates [252,253] and have further been associated with the alleviation of age-related diseases [254]. As with other CRMs, resveratrol inhibits cancer cell growth (leukemia [255], breast [256], liver [257], and gastric [258]) and has anticancer effects in multiple animal models ([259,260,261,262,263,264,265]) as well as the capacity to suppress metastasis (reviewed in [266]). Additionally, resveratrol has been documented to decrease age-dependent cognitive decline and AD-like pathologies [267]. In human trials, resveratrol has been shown to regulate neuroinflammation in AD patients [267], improve cognitive function in adults with T2D [268], improve insulin sensitivity [269], and suppress postprandial glucagon [270,271] in overweight individuals. Moreover, in healthy humans, resveratrol improves cerebral blood flow and cognitive performance [272] and decreases IGF-1 levels [273] (Table 3). Since the mentioned pathologies (including cancer and T2D) are often associated with age, resveratrol has been proposed as a CRM and antiaging drug, with evidence demonstrating lifespan increases [274,275,276,277,278,279].
Table 3.
Summary of the effects of metformin and resveratrol on aging-related pathways.
Resveratrol generally functions via interaction with, and the activation of, SIRT1, although at higher doses, it also interacts with, and activates, AMPK [280] (Figure 2B). As noted previously, SIRT1 and AMPK feedback to one another in response to CR and proposed CRMs. SIRT1 deacetylates proteins, such as p53, FOXO3a, NF-κB, and peroxisome proliferator-activated receptor gamma coactivator 1α (PGC-1α) through a NAD+ substrate that is converted to NAM (reviewed by [281]). Given the role of NAD+ as a potent activator of SIRT1, this molecule is key in regulating the energy metabolism and cellular stress mediation effects of resveratrol [279]. To compare the impact of CR to resveratrol, Li and colleagues found that both treatments can upregulate SIRT1, increasing the expression of the pro-longevity protein FOXO3a and the activity of telomerase leading to inhibition of senescence and apoptosis; notably, the impact of resveratrol was greater than that of CR [16]. Additionally, resveratrol reduced the expression of genes encoding inflammatory markers, including IL-6, chemokine (C-C motif) ligand 3 (CCL3), IL-1β, and TNF-α [282] in humans. Analysis of microarray data from breast cancer cells found that 48 h of resveratrol treatment downregulated key genes related to the cell cycle, DNA metabolic process, cellular response to stress, and regulation of cell death [283]. The downregulation of these pathways is similar to that observed in response to CR, maintaining insulin sensitivity [17] and decreasing circulating IGF-1 levels [284]. However, resveratrol was not able to recapitulate the effects of CR in mice [285] and induced differing metabolomes [286]. Limited whole-transcriptome impacts of resveratrol have been generated. In aged mice, resveratrol had minimal effects (note: the mice in question were fasting during treatment, which may impact the interpretation of the data). However, changes in neuron synaptosome function [287] show the potential of resveratrol in treating ND diseases. Resveratrol promotes antioxidant enzyme function, maintains mitochondrial function, and reduces inflammation more than other potential CRMs. Therefore, it is also unlikely an exact mimetic of CR. More information on the impact of resveratrol on the genome, and potential transcription factors regulating these genome-wide changes could provide interesting mechanistic insights into resveratrol’s antiaging properties.
As with other CRMs, resveratrol can exhibit harmful side effects linked to dosage concentration and duration. For example, in indomethacin-induced gastric ulcerated mice, low-dose resveratrol was protective, whereas higher doses delayed healing [288]. Additionally, resveratrol concentrations that induce cancer cell death are also cytotoxic to healthy cells and tissues (reviewed in [289]). Resveratrol metabolites, produced under certain conditions (e.g., o-Quinone), also exhibit detrimental effects, inducing oxidative stress and alkylation [290,291]. It has been suggested that these harmful effects are the result of resveratrol modulating oxidative stress, functioning as an antioxidant or pro-oxidant compound dependent on dosage concentration, duration, and environment. Specifically, high concentrations of resveratrol inhibit DNA repair pathways, triggering ROS production, and inducing cell death (the opposite of which is true for low-dose treatments in promoting health and lifespan) (reviewed in [289]). Further research into the conditions that regulate resveratrol function as a pro- or antioxidant is required to better understand its potential in extending health and lifespan.
5.4. Metformin
Metformin (N,N-dimethylimidodicarbonimidic diamide), derived from the French Lilac (G. officinalis), is a commonly used treatment for T2D [296]. Metformin increases AMP/ATP ratios, indicating low cellular energy status and activating AMPK and sirtuin proteins [297]. Surprisingly, despite its widespread use, the exact mechanism by which metformin alters AMP/ATP ratios remains unclear. Metformin likely influences AMPK levels by modulating mitochondrial function and ATP production, triggering AMPK activation and indirectly inhibiting mTORC1 [298], which may parallel CR (Figure 2B). Consequently, the potential of metformin as a CRM relies on its mediation of mitochondrial metabolism and insulin signaling [299,300]. As with CR/AAR and other CRMs, metformin has shown antitumoral effects in various cancers [294,301,302,303,304], neuroprotective effects in ND diseases [305,306], and benefit in treating epilepsy [307]. Metformin also shows potential as an adjuvant cancer therapy [308,309] and is associated with decreased cancer incidence in the general population [310]. Furthermore, metformin has been documented to upregulate longevity-associated phenotypes [311] and extend lifespan in multiple organisms [307,312,313,314,315].
Metformin mediates levels of cytokines linked with inflammation [316,317,318], increases insulin sensitivity [319], and decreases IGF-1 levels [320] (Table 3). This cytokine mediation has been linked to the pro-longevity FOXO3a and the activator protein (AP)-1 transcription factor pathway, both of which can regulate cytokine levels. Interestingly, the ATF2 network was also enriched in response to metformin in human fibroblasts [316], indicating metformin may also function to mimic AAR; therefore, working via multiple pathways to promote longevity. Although cytokine–cytokine receptor signaling was also previously identified in response to rapamycin treatment in normal human fibroblasts, meta-analyses comparing metformin and rapamycin in the same cell line identified divergent transcriptomes [220,316], suggesting that this alteration in cytokine signaling is a downstream impact of these compounds to promote health and lifespan, as opposed to key mechanisms driving response to these proposed CRMs.
Similar to CR, metformin decreases levels of p53, likely via AMPK-induced activation of SIRT1 function [298]. CR and metformin have common physiological impacts; for example, obese mice under CR and treated with metformin exhibit mitophagy and reduced oxidative stress [298]. Notably, expression of ER stress markers (such as ATF6 and X-box binding protein 1 (XBP-1)) were also decreased. XBP1 has been proposed as a transcription factor for regulating changes in gene expression to CR, whereas ATF6 is linked to the ISR in response to AAR and attenuating global protein translation to counteract cellular stressors. Song et al. [298] also demonstrated that Parkin, a tumor suppressor protein involved in mitophagy, was translocated to the mitochondria in response to CR and metformin in obese mice, inducing autophagy, indicating that both antiaging treatments can lead to the same results albeit by different paths. More research is needed to determine the similarities between metformin and CR mechanisms, at the transcriptomic and epigenomic levels; however, it is unlikely that this compound directly mimics CR.
6. Conclusions
Since discovering the ability of CR to extend the lifespan of rodents, research into CR and CRMs has increased exponentially. In parallel, investigations into the basis of aging have also increased, demonstrating the link between getting older and numerous pathologies (cancer, CVD, T2D, ND). As a result, a plethora of research now demonstrates the ability of CR and CRMs to promote longevity and decrease incidence/delay age-linked diseases. Moreover, it is now clear that CR and various CRMs are able to delay/reverse molecular mechanisms associated with aging processes; however, some CR/CRMs are better at delaying/reversing specific age-linked effects than others. For example, CR and metformin consistently decrease IGF-1 levels and increase insulin sensitivity, whereas resveratrol acts more frequently to promote antioxidant pathways. The complexity of feedback signaling at the cellular and organismal levels indicates that to further pursue mechanisms in promoting longevity, it will be important to consider these effects in future experiments, with current data likely exhibiting controversy due to the lack of the elucidation of feedback loops and time required for these to take effect.
As whole-transcriptome profiling becomes more widely used, the availability of data regarding the impact of CR, AAR, and CRMs on the genome also increases. These data are beginning to reveal that it is unlikely any of these interventions directly mimic one another, but instead implement divergent mechanisms to arrive at the same endpoint, even when considering the usual factors underlying data variation (species, tissue type, dosage, time). As such, it is important that in the future, researchers are cautious of terminology, and perhaps it is timely to propose that this long-established class of supposed CRMs be referred to as longevity-promoting interventions (LPIs): diets, regimes, and compounds that function to achieve the same goal, health, and lifespan extension, but may do so in divergent manners. Finally, it is well established that CR, AAR, and CRMs (those discussed here, as well as others) extend health and lifespan. Although decreased mTOR function is often used as the benchmark for this, new findings, in part derived from whole-transcriptome analyses, enable the identification of other pathways that may exert influence. Moving forward, examining the crosstalk among these pathways will advance understanding of how to promote healthy aging.
Funding
We would like to acknowledge the funding agencies that helped support the individuals involved in the production of this manuscript. C.A.-P. is supported by a scholarship provided by the College of Graduate and Postdoctoral Studies (U of S). D.D.M. is the Saskatchewan Research Chair in Alzheimer Disease and Related Dementias funded by the Alzheimer Society of Saskatchewan and the Saskatchewan Health Research Foundation. C.H.E. is supported through Natural Sciences and Engineering Council of Canada (NSERC) and the Canadian Institutes of Health Research (CIHR) funding. Z.E.G. is supported by a Vanier Canada Graduate Scholarship.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AAR | Amino acid restriction |
| AARE | Amino acid response element |
| AD | Alzheimer’s disease |
| ADAM10 | Disintegrin and metalloproteinase domain-containing protein 10 |
| AP-1 | Activator protein 1 |
| APP | Amyloid precursor protein |
| AKT | Protein Kinase B |
| AMP | Adenosine monophosphate |
| AMPK | Adenosine monophosphate kinase |
| APP | Amyloid precursor protein |
| ARE | Antioxidant response elements |
| Arf-1 GTPase | ADP ribosylation factor |
| α-SMA | Alpha smooth muscle actin |
| ATF | Activating transcription factor |
| ATG | Autophagy-related genes |
| ATP | Adenosine triphosphate |
| AZF1P | Nuclear-localized zinc-finger |
| BACE | Beta-secretase 1 |
| BAT | Brown adipose tissue |
| BCAA | Branched-chain amino acids |
| BCL-2 | B-cell lymphoma 2 |
| BCL-XL | B-cell lymphoma-extra-large |
| BDNF | Brain-derived neurotrophic factor |
| CA-9 | Carbonic anhydrase 9 |
| CamKKB | Calcium/calmodulin-dependent protein kinase kinase B |
| CASTOR | Cytosolic arginine sensor for mTORC1 |
| CAT | Catalase |
| CCL3 | Chemokine (C-C motif) ligand 3 |
| Cpsf6 | Cleavage and polyadenylation specific factor 6 |
| CK19 | Cytokeratin-19 |
| CR | Caloric restriction |
| CRMs | Caloric restriction mimetic |
| CVD | Cardiovascular disease |
| CYP1 | Cytochrome P450 |
| DMTF | Drosophila metal-responsive transcription factor |
| eIF2 | Eukaryotic initiation factor 2 |
| ER | Endoplasmic reticulum |
| FGF21 | Fibroblast growth factor 21 |
| FOXO3a | Forkhead box O3a |
| GADD34 | Growth arrest and DNA damage-inducible protein 34 |
| GATOR | GTPase-activating protein toward rags |
| GCN2 | General control nonderepressible 2 |
| GDP | Guanosine diphosphate |
| GHS | Glycinyl-histidinyl-serine |
| GLUT1 | Glucose transporter 1 |
| GREB1 | Growth-regulating estrogen receptor binding 1 |
| GSK-3β | Glycogen synthase kinase-3β |
| GTP | Guanosine triphosphate |
| HGPS | Hutchinson–Gilford progeria syndrome |
| HIF1α | Hypoxia-inducible factor-1α |
| HSF1P | Heat shock factor |
| IGF-1 | Insulin-like growth factor-1 |
| IL | Interleukin |
| IRS-1 | Insulin receptor substrate 1 |
| ISR | Integrated stress response |
| JNK | C-jun N-terminal kinase |
| LKB1 | Liver kinase B1 |
| LMX1b | LIM homeobox transcription factor 1-β |
| LPIs | Longevity-promoting interventions |
| MAPK | Mitogen activator protein kinase |
| MEF3 | Mitochondrial editing factor 3 |
| MMP9 | Matrix metallopeptidase 9 |
| Mt | Metallothionein |
| mTOR | Mammalian target of rapamycin |
| mTORC1 | Mammalian target of rapamycin complex 1 |
| mTORC2 | Mammalian target of rapamycin complex 2 |
| NAD+ | Nicotinamide adenine dinucleotide |
| NAM | Nicotinamide |
| ND | Neurodegenerative diseases |
| NF-κB | Nuclear factor-κB |
| Nfκbia | NFκB inhibitor α |
| NRF2 | Nuclear receptor factor 2 |
| Pax6 | Paired box 6 |
| PDK1 | 3-phosphoinositide-dependent protein kinase 1 |
| PGC-1α | Peroxisome proliferator-activated receptor gamma coactivator-1α |
| PI3K | Phosphoinositide 3-kinase |
| Pitx2 | pituitary homeobox 2 |
| PRDM14 | PR domain zinc finger protein 14 |
| RagA/B | Ras-related GTP-binding protein A and B |
| RagC/D | ras-related GTP-binding protein C and D |
| Rheb | RAS homolog enriched in brain |
| ROS | Reactive oxygen species |
| S6K1 | Ribosomal protein S6 kinase |
| SAAR | Sulfur amino acid restriction |
| SAM | S-adenosyl-methionine |
| SAMTOR | SAM sensor upstream of mTORC1 |
| Sfpq | Splicing factor proline and glutamine rich |
| Sfrs | Serine- and arginine-rich splicing factor |
| SIRT1 | Sirtuin 1 |
| SOD | Superoxide dismutase |
| SPD | Starvation pseudodiabetes |
| STAT3 | Signal transducer and activator of transcription 3 |
| STAT5A/B | Signal transducer and activator of transcription 5A/B |
| T2D | Type 2 diabetes |
| Timp3 | Metalloproteinase inhibitor 3 |
| TNFα | Tumor necrosis factor-α |
| TSC | Tuberous sclerosis complex |
| Txnip | Thioredoxin interacting protein |
| ULK1 | Unc-51 Like Autophagy Activating Kinase 1 |
| VEGFR | Vascular endothelial growth factor receptor |
| WAT | White adipose tissue |
| WNT | Wingless-related integration site |
| XBP-1 | X-box-binding protein 1 |
| YBX1 | Y-Box 1 |
| Zbt16 | Zinc finger and BTB domain-containing protein 16 |
| Zscan1 | Zinc finger and SCAN domain-containing 1 |
References
- Seals, D.R.; Jablonski, K.L.; Donato, A.J. Aging and vascular endothelial function in humans. Clin. Sci. 2011, 120, 357–375. [Google Scholar] [CrossRef]
- Del Zoppo, G.J. Aging and the neurovascular unit. Ann. N. Y. Acad. Sci. 2012, 1268, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.Y.; Cesari, M.; Anton, S.; Marzetti, E.; Giovannini, S.; Seo, A.Y.; Carter, C.; Yu, B.P.; Leeuwenburgh, C. Molecular inflammation: Underpinnings of aging and age-related diseases. Ageing Res. Rev. 2009, 8, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Ahima, R.S. Connecting obesity, aging and diabetes. Nat. Med. 2009, 15, 996–997. [Google Scholar] [CrossRef]
- McCay, C.M.; Crowell, M.F.; Maynard, L.A. The effect of retarded growth upon the length of life span and upon the ultimate body size. J. Nutr. 1935, 10, 63–79. [Google Scholar] [CrossRef]
- Mattison, J.A.; Colman, R.J.; Beasley, T.M.; Allison, D.B.; Kemnitz, J.W.; Roth, G.S.; Ingram, D.K.; Weindruch, R.; De Cabo, R.; Anderson, R.M. Caloric restriction improves health and survival of rhesus monkeys. Nat. Commun. 2017, 8, 14063. [Google Scholar] [CrossRef]
- Mattison, J.A.; Roth, G.S.; Beasley, T.M.; Tilmont, E.M.; Handy, A.M.; Herbert, R.L.; Longo, D.L.; Allison, D.B.; Young, J.E.; Bryant, M.; et al. Impact of caloric restriction on health and survival in rhesus monkeys from the nia study. Nature 2012, 489, 318–321. [Google Scholar] [CrossRef] [PubMed]
- Colman, R.J.; Beasley, T.M.; Kemnitz, J.W.; Johnson, S.C.; Weindruch, R.; Anderson, R.M. Caloric restriction reduces age-related and all-cause mortality in rhesus monkeys. Nat. Commun. 2014, 5, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.C.; Jaruga, E.; Repnevskaya, M.V.; Jazwinski, S.M. An intervention resembling caloric restriction prolongs life span and retards aging in yeast. FASEB J. 2000, 14, 2135–2137. [Google Scholar] [CrossRef]
- Brandhorst, S.; Choi, I.Y.; Wei, M.; Cheng, C.W.; Sedrakyan, S.; Navarrete, G.; Dubeau, L.; Yap, L.P.; Park, R.; Vinciguerra, M.; et al. A periodic diet that mimics fasting promotes multi-system regeneration, enhanced cognitive performance, and healthspan. Cell Metab. 2015, 22, 86–99. [Google Scholar] [CrossRef]
- Mercken, E.M.; Crosby, S.D.; Lamming, D.W.; JeBailey, L.; Krzysikwalker, S.M.; Villareal, D.T.; Capri, M.; Franceschi, C.; Zhang, Y.; Becker, K.G.; et al. Calorie restriction in humans inhibits the pi3k/akt pathway and induces a younger transcription profile. Aging Cell 2013, 12, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, H.; Suarez, J.A.; Longo, V.D. Protein and amino acid restriction, aging and disease: From yeast to humans. Trends Endocrinol. Metab. TEM 2014, 25, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Jewell, J.L.; Russell, R.C.; Guan, K.L. Amino acid signalling upstream of mtor. Nat. Rev. Mol. Cell Biol. 2013, 14, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Unnikrishnan, A.; Kurup, K.; Salmon, A.B.; Richardson, A. Is rapamycin a dietary restriction mimetic? J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2020, 75, 4–13. [Google Scholar] [CrossRef]
- Lamming, D.W.; Ye, L.; Sabatini, D.M.; Baur, J.A. Rapalogs and mtor inhibitors as anti-aging therapeutics. J. Clin. Investig. 2013, 123, 980–989. [Google Scholar] [CrossRef]
- Li, J.; Zhang, C.; Liu, Y.-M.; Chen, K.-L.; Chen, G. A comparative study of anti-aging properties and mechanism: Resveratrol and caloric restriction. Oncotarget 2017, 8, 65717–65729. [Google Scholar] [CrossRef]
- Chung, J.H.; Manganiello, V.; Dyck, J.R. Resveratrol as a calorie restriction mimetic: Therapeutic implications. Trends Cell Biol. 2012, 22, 546–554. [Google Scholar] [CrossRef]
- Mouchiroud, L.; Molin, L.; Dalliere, N.; Solari, F. Life span extension by resveratrol, rapamycin, and metformin: The promise of dietary restriction mimetics for an healthy aging. BioFactors 2010, 36, 377–382. [Google Scholar] [CrossRef]
- Onken, B.; Driscoll, M. Metformin induces a dietary restriction-like state and the oxidative stress response to extend c. Elegans healthspan via ampk, lkb1, and skn-1. PLoS ONE 2010, 5, 1–13. [Google Scholar] [CrossRef]
- Walaszczyk, A.; Dookun, E.; Redgrave, R.; Tual-Chalot, S.; Victorelli, S.; Spyridopoulos, I.; Owens, A.; Arthur, H.M.; Passos, J.F.; Richardson, G.D. Pharmacological clearance of senescent cells improves survival and recovery in aged mice following acute myocardial infarction. Aging Cell 2019, 18, e12945. [Google Scholar] [CrossRef]
- Baar, M.P.; Brandt, R.M.C.; Putavet, D.A.; Klein, J.D.; Derks, K.W.; Bourgeois, B.R.; Stryeck, S.; Rijksen, Y.; van Willigenburg, H.; Feijtel, D.A.; et al. Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell 2017, 169, 132–147.e116. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; Lebrasseur, N.K.; Childs, B.G.; Van De Sluis, B.; Kirkland, J.L.; Van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nat. Cell Biol. 2011, 479, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Aunan, J.R.; Watson, M.M.; Hagland, H.R.; Soreide, K. Molecular and biological hallmarks of ageing. Br. J. Sci. 2016, 103, e29–e46. [Google Scholar] [CrossRef] [PubMed]
- Csiszar, A.; Labinskyy, N.; Jiménez, R.; Pinto, J.T.; Ballabh, P.; Losonczy, G.; Pearson, K.J.; De Cabo, R.; Ungvari, Z. Anti-oxidative and anti-inflammatory vasoprotective effects of caloric restriction in aging: Role of circulating factors and sirt1. Mech. Ageing Dev. 2009, 130, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Mair, W.; Morantte, I.; Rodrigues, A.P.C.; Manning, G.; Montminy, M.; Shaw, R.J.; Dillin, A. Lifespan extension induced by ampk and calcineurin is mediated by crtc-1 and creb. Nature 2011, 470, 404–408. [Google Scholar] [CrossRef] [PubMed]
- Lakowski, B.; Hekimi, S. The genetics of caloric restriction in caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 1998, 95, 13091–13096. [Google Scholar] [CrossRef] [PubMed]
- Ingle, L.; Wood, T.R.; Banta, A.M. A study of longevity, growth, reproduction and heart rate in daphnia longispina as influenced by limitations in quantity of food. J. Exp. Zool. 1937, 76, 325–353. [Google Scholar] [CrossRef]
- Weeks, S.C. Effects of dietary restriction on lifespan, growth, and reproduction of the clam shrimp eulimnadia texana. Hydrobiologia 2020, 847, 3067–3076. [Google Scholar] [CrossRef]
- Austad, S.N. Life extension by dietary restriction in the bowl and doily spider, frontinella pyramitela. Exp. Gerontol. 1989, 24, 83–92. [Google Scholar] [CrossRef]
- Weindruch, R.; Walford, R.L.; Fligiel, S.; Guthrie, D. The retardation of aging in mice by dietary restriction: Longevity, cancer, immunity and lifetime energy intake. J. Nutr. 1986, 116, 641–654. [Google Scholar] [CrossRef]
- Colman, R.J.; Anderson, R.M.; Johnson, S.C.; Kastman, E.K.; Kosmatka, K.J.; Beasley, T.M.; Allison, D.B.; Cruzen, C.; Simmons, H.A.; Kemnitz, J.W.; et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science 2009, 325, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Yegorov, Y.E. Healthy aging: Antioxidants, uncouplers and/or telomerase? Mol. Biol. 2020, 54, 311–316. [Google Scholar] [CrossRef]
- Barja, G. Towards a unified mechanistic theory of aging. Exp. Gerontol. 2019, 124, 110627. [Google Scholar] [CrossRef]
- Hambly, C.; Speakman, J.R. Contribution of different mechanisms to compensation for energy restriction in the mouse. Obes. Res. 2005, 13, 1548–1557. [Google Scholar] [CrossRef] [PubMed]
- Blanc, S.; Schoeller, D.; Kemnitz, J.; Weindruch, R.; Colman, R.; Newton, W.; Wink, K.; Baum, S.; Ramsey, J. Energy expenditure of rhesus monkeys subjected to 11 years of dietary restriction. J. Clin. Endocrinol. Metab. 2003, 88, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Cabelof, D.C.; Yanamadala, S.; Raffoul, J.J.; Guo, Z.; Soofi, A.; Heydari, A.R. Caloric restriction promotes genomic stability by induction of base excision repair and reversal of its age-related decline. DNA Repair 2003, 2, 295–307. [Google Scholar] [CrossRef]
- Um, J.H.; Kim, S.J.; Kim, D.W.; Ha, M.Y.; Jang, J.H.; Chung, B.S.; Kang, C.D.; Kim, S.H. Tissue-specific changes of DNA repair protein ku and mthsp70 in aging rats and their retardation by caloric restriction. Mech. Ageing Dev. 2003, 124, 967–975. [Google Scholar] [CrossRef]
- Vermeij, W.P.; Dollé, M.E.T.; Reiling, E.; Jaarsma, D.; Payan-Gomez, C.; Bombardieri, C.R.; Wu, H.; Roks, H.W.A.J.M.; Botter, S.M.; Van Der Eerden, B.C.; et al. Restricted diet delays accelerated ageing and genomic stress in DNA-repair-deficient mice. Nature 2016, 537, 427–431. [Google Scholar] [CrossRef]
- Ke, Z.; Firsanov, D.; Spencer, B.; Seluanov, A.; Gorbunova, V. Short-term calorie restriction enhances DNA repair by non-homologous end joining in mice. NPJ Aging Mech. Dis. 2020, 6, 9. [Google Scholar] [CrossRef]
- Wang, W.; Cai, G.; Chen, X. Dietary restriction delays the secretion of senescence associated secretory phenotype by reducing DNA damage response in the process of renal aging. Exp. Gerontol. 2018, 107, 4–10. [Google Scholar] [CrossRef]
- Il’yasova, D.; Fontana, L.; Bhapkar, M.; Pieper, C.F.; Spasojevic, I.; Redman, L.M.; Das, S.K.; Huffman, K.M.; Kraus, W.E.; CALERIE Study Investigators. Effects of 2 years of caloric restriction on oxidative status assessed by urinary f2-isoprostanes: The calerie 2 randomized clinical trial. Aging Cell 2018, 17. [Google Scholar] [CrossRef] [PubMed]
- Redman, L.M.; Smith, S.R.; Burton, J.H.; Martin, C.K.; Il’yasova, D.; Ravussin, E. Metabolic slowing and reduced oxidative damage with sustained caloric restriction support the rate of living and oxidative damage theories of aging. Cell Metab. 2018, 27, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Heilbronn, L.K.; De Jonge, L.; Frisard, M.I.; DeLany, J.P.; Larson-Meyer, D.E.; Rood, J.; Nguyen, T.; Martin, C.K.; Volaufova, J.; Most, M.M.; et al. Effect of 6-month calorie restriction on biomarkers of longevity, metabolic adaptation, and oxidative stress in overweight individuals: A randomized controlled trial. JAMA 2006, 295, 1539–1548. [Google Scholar] [CrossRef] [PubMed]
- Miwa, S.; Riyahi, K.; Partridge, L.; Brand, M.D. Lack of Correlation between Mitochondrial Reactive Oxygen Species Production and Life Span in Drosophila. Ann. N. Y. Acad. Sci. 2004, 1019, 388–391. [Google Scholar] [CrossRef] [PubMed]
- Kabil, H.; Partridge, L.; Harshman, L.G. Superoxide dismutase activities in long-lived Drosophila melanogaster females: Chico 1 genotypes and dietary dilution. Biogerontology 2007, 8, 201–208. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wei, M.L.; Duan, P.; Wang, Z.-M.; Ding, M.; Tu, P. High glucose and high insulin conditions promote MCF-7 cell proliferation and invasion by upregulating IRS1 and activating the Ras/Raf/ERK pathway. Mol. Med. Rep. 2017, 16, 6690–6696. [Google Scholar] [CrossRef]
- Ito, M.; Makino, N.; Matsuda, A.; Ikeda, Y.; Kakizaki, Y.; Saito, Y.; Ueno, Y.; Kawata, S. High Glucose Accelerates Cell Proliferation and Increases the Secretion and mRNA Expression of Osteopontin in Human Pancreatic Duct Epithelial Cells. Int. J. Mol. Sci. 2017, 18, 807. [Google Scholar] [CrossRef]
- Kannan, K.; Fridell, Y.-W.C. Functional implications of Drosophila insulin-like peptides in metabolism, aging, and dietary restriction. Front. Physiol. 2013, 4, 288. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, F. Tissue-specific insulin signaling in the regulation of metabolism and aging. IUBMB Life 2014, 66, 485–495. [Google Scholar] [CrossRef]
- Dao, M.C.; Sokolovska, N.; Brazeilles, R.; Affeldt, S.; Pelloux, V.; Prifti, E.; Chilloux, J.; Verger, E.O.; Kayser, B.D.; Aron-Wisnewsky, J.; et al. A Data Integration Multi-Omics Approach to Study Calorie Restriction-Induced Changes in Insulin Sensitivity. Front. Physiol. 2019, 9. [Google Scholar] [CrossRef]
- Granado, M.; Amor, S.; Martín-Carro, B.; Guerra-Menéndez, L.; Tejera-Muñoz, A.; González-Hedström, D.; Rubio, C.; Carrascosa, J.M.; García-Villalón, Á.L. Caloric restriction attenuates aging-induced cardiac insulin resistance in male Wistar rats through activation of PI3K/Akt pathway. Nutr. Metab. Cardiovasc. Dis. 2019, 29, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.J.; Madrigal-Matute, J.; Scheibye-Knudsen, M.; Fang, E.; Aon, M.; González-Reyes, J.A.; Cortassa, S.; Kaushik, S.; Gonzalez-Freire, M.; Patel, B.; et al. Effects of Sex, Strain, and Energy Intake on Hallmarks of Aging in Mice. Cell Metab. 2016, 23, 1093–1112. [Google Scholar] [CrossRef] [PubMed]
- Fontana, L.; Villareal, D.T.; Das, S.K.; Smith, S.R.; Meydani, S.N.; Pittas, A.G.; Klein, S.; Bhapkar, M.V.; Rochon, J.; Ravussin, E.; et al. Effects of 2-year calorie restriction on circulating levels of IGF-1, IGF-binding proteins and cortisol in nonobese men and women: A randomized clinical trial. Aging Cell 2016, 15, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Fontana, L.; Weiss, E.P.; Villareal, D.T.; Klein, S.; Holloszy, J.O. Long-term effects of calorie or protein restriction on serum IGF-1 and IGFBP-3 concentration in humans. Aging Cell 2008, 7, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Belsky, D.W.; Huffman, K.M.; Pieper, C.F.; Shalev, I.; Kraus, W.E. Change in the Rate of Biological Aging in Response to Caloric Restriction: CALERIE Biobank Analysis. J. Gerontol. Ser. A Biol. Sci. Med Sci. 2018, 73, 4–10. [Google Scholar] [CrossRef]
- Rattan, S.I. Hormesis in aging. Ageing Res. Rev. 2008, 7, 63–78. [Google Scholar] [CrossRef]
- Calabrese, V.; Cornelius, C.; Cuzzocrea, S.; Iavicoli, I.; Rizzarelli, E.; Calabrese, E.J. Hormesis, cellular stress response and vitagenes as critical determinants in aging and longevity. Mol. Asp. Med. 2011, 32, 279–304. [Google Scholar] [CrossRef]
- Matai, L.; Sarkar, G.C.; Chamoli, M.; Malik, Y.; Kumar, S.S.; Rautela, U.; Jana, N.R.; Chakraborty, K.; Mukhopadhyay, A. Dietary restriction improves proteostasis and increases life span through endoplasmic reticulum hormesis. Proc. Natl. Acad. Sci. USA 2019, 116, 17383–17392. [Google Scholar] [CrossRef]
- Santoro, A.; Martucci, M.; Conte, M.; Capri, M.; Franceschi, C.; Salvioli, S. Inflammaging, hormesis and the rationale for anti-aging strategies. Ageing Res. Rev. 2020, 64, 101142. [Google Scholar] [CrossRef]
- Kim, J.; Guan, K.-L. mTOR as a central hub of nutrient signalling and cell growth. Nat. Cell Biol. 2019, 21, 63–71. [Google Scholar] [CrossRef]
- Kazyken, D.; Magnuson, B.; Bodur, C.; Acosta-Jaquez, H.A.; Zhang, D.; Tong, X.; Barnes, T.M.; Steinl, G.K.; Patterson, N.E.; Altheim, C.H.; et al. Ampk directly activates mtorc2 to promote cell survival during acute energetic stress. Sci. Signal. 2019, 12, eaav3249. [Google Scholar] [CrossRef]
- Dibble, C.C.; Elis, W.; Menon, S.; Menon, S.; Qin, W.; Klekota, J.; Asara, J.M.; Finan, P.M.; Kwiatkowski, D.J.; Murphy, L.O.; et al. Tbc1d7 is a third subunit of the tsc1-tsc2 complex upstream of mtorc1. Mol. Cell. 2012, 47, 535–546. [Google Scholar] [CrossRef]
- Menon, S.; Dibble, C.C.; Talbott, G.; Hoxhaj, G.; Valvezan, A.J.; Takahashi, H.; Cantley, L.C.; Manning, B.D. Spatial control of the tsc complex integrates insulin and nutrient regulation of mtorc1 at the lysosome. Cell 2014, 156, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.Y.; Sabatini, D.M. Mtor at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. Mtor signaling in growth, metabolism, and disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Brugarolas, J.; Lei, K.; Hurley, R.L.; Manning, B.D.; Reiling, J.H.; Hafen, E.; Witters, L.A.; Ellisen, L.W.; Kaelin, W.G., Jr. Regulation of mtor function in response to hypoxia by redd1 and the tsc1/tsc2 tumor suppressor complex. Genes Dev. 2004, 18, 2893–2904. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Li, Y.; Xu, T.; Guan, K.-L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003, 17, 1829–1834. [Google Scholar] [CrossRef] [PubMed]
- B’Chir, W.; Maurin, A.C.; Carraro, V.; Averous, J.; Jousse, C.; Muranishi, Y.; Parry, L.; Stepien, G.; Fafournoux, P.; Bruhat, A. The eif2alpha/atf4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013, 41, 7683–7699. [Google Scholar] [CrossRef]
- Cohen, H.Y.; Miller, C.; Bitterman, K.J.; Wall, N.R.; Hekking, B.; Kessler, B.; Howitz, K.T.; Gorospe, M.; De Cabo, R.; Sinclair, D.A. Calorie Restriction Promotes Mammalian Cell Survival by Inducing the SIRT1 Deacetylase. Science 2004, 305, 390–392. [Google Scholar] [CrossRef]
- Nemoto, S.; Fergusson, M.M.; Finkel, T. Nutrient availability regulates sirt1 through a forkhead-dependent pathway. Science 2004, 306, 2105–2108. [Google Scholar] [CrossRef]
- Canto, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Woods, A.; Johnstone, S.R.; Dickerson, K.; Leiper, F.C.; Fryer, L.G.; Neumann, D.; Schlattner, U.; Wallimann, T.; Carlson, M.; Carling, D. LKB1 Is the Upstream Kinase in the AMP-Activated Protein Kinase Cascade. Curr. Biol. 2003, 13, 2004–2008. [Google Scholar] [CrossRef] [PubMed]
- Hawley, S.A.; Pan, D.A.; Mustard, K.J.; Ross, L.; Bain, J.; Edelman, A.M.; Frenguelli, B.G.; Hardie, D.G. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for amp-activated protein kinase. Cell Metab. 2005, 2, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Giannakou, M.E.; Partridge, L. The interaction between FOXO and SIRT1: Tipping the balance towards survival. Trends Cell Biol. 2004, 14, 408–412. [Google Scholar] [CrossRef] [PubMed]
- Demidenko, Z.N.; Zubova, S.V.; Bukreeva, E.I.; Pospelov, V.A.; Pospelova, T.V.; Blagosklonny, M.V. Rapamycin decelerates cellular senescence. Cell Cycle 2009, 8, 1888–1895. [Google Scholar] [CrossRef] [PubMed]
- Furuyama, T.; Yamashita, H.; Kitayama, K.; Higami, Y.; Shimokawa, I.; Mori, N. Effects of aging and caloric restriction on the gene expression of Foxo1, 3, and 4 (FKHR, FKHRL1, and AFX) in the rat skeletal muscles. Microsc. Res. Tech. 2002, 59, 331–334. [Google Scholar] [CrossRef]
- Opalach, K.; Rangaraju, S.; Madorsky, I.; Leeuwenburgh, C.; Notterpek, L. Lifelong Calorie Restriction Alleviates Age-Related Oxidative Damage in Peripheral Nerves. Rejuvenation Res. 2010, 13, 65–74. [Google Scholar] [CrossRef]
- Spaulding, C.C.; Walford, R.L.; Effros, R.B. Calorie restriction inhibits the age-related dysregulation of the cytokines tnf-a and il-6 in c3b10rf1 mice. Mech. Ageing Dev. 1997, 93, 87–94. [Google Scholar] [CrossRef]
- Kalani, R.; Judge, S.; Carter, C.; Pahor, M.; Leeuwenburgh, C. Effects of Caloric Restriction and Exercise on Age-Related Chronic Inflammation Assessed by C-Reactive Protein and Interleukin-6. J. Gerontol. Ser. A Biol. Sci. Med Sci. 2006, 61, 211–217. [Google Scholar] [CrossRef][Green Version]
- Meydani, S.; Das, S.; Piper, C.; Lewis, M.; Dixit, V.; Gupta, A.; Villareal, D.; Klein, S.; Bhapkar, M.; Huang, M.; et al. Effects of prolonged calorie restriction on inflammation and immune function: A randomized controlled trial in non-obese humans. FASEB J. 2014, 28, 40–44. [Google Scholar]
- Chen, D.; Thomas, E.L.; Kapahi, P. HIF-1 Modulates Dietary Restriction-Mediated Lifespan Extension via IRE-1 in Caenorhabditis elegans. PLoS Genet. 2009, 5, e1000486. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.H.; Lee, Y.-M.; Chun, Y.-S.; Chen, J.; Kim, J.-E.; Park, J.-W. Sirtuin 1 Modulates Cellular Responses to Hypoxia by Deacetylating Hypoxia-Inducible Factor 1alpha. Mol. Cell 2010, 38, 864–878. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.J.; Kim, H.J.; Kim, H.K.; Kim, H.J.; Kim, H.K.; Lee, J.Y.; Kim, D.H.; Jung, K.J.; Kim, K.W.; Baik, H.S.; et al. The effect of age and calorie restriction on HIF-1-responsive genes in aged liver. Biogerontology 2005, 6, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, R.; Guernah, I.; Jin, D.; Grisendi, S.; Alimonti, A.; Teruya-Feldstein, J.; Cordon-Cardo, C.; Simon, M.C.; Rafii, S.; Pandolfi, P.P. PML inhibits HIF-1alpha translation and neoangiogenesis through repression of mTOR. Nature 2006, 442, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Hui, A.S.; Bauer, A.L.; Striet, J.B.; Schnell, P.O.; Czyzyk-Krzeska, M.F. Calcium signaling stimulates translation of HIF-alpha during hypoxia. FASEB J. 2006, 20, 466–475. [Google Scholar] [CrossRef]
- Hong, S.S.; Lee, H.; Kim, K.-W. HIF-1a: A Valid Therapeutic Target for Tumor Therapy. Cancer Res. Treat. 2004, 36, 343–353. [Google Scholar] [CrossRef]
- Weindruch, R.; Kayo, T.; Lee, C.H.; Prolla, T.A. Microarray profiling of gene expression in aging and its alteration by caloric restriction in mice. J. Nutr. 2001, 131, 918S–923S. [Google Scholar] [CrossRef]
- Kim, S.S.; Choi, K.-M.; Kim, S.; Park, T.; Cho, I.-C.; Lee, J.-W.; Lee, C.-K. Whole-transcriptome analysis of mouse adipose tissue in response to short-term caloric restriction. Mol. Genet. Genom. 2016, 291, 831–847. [Google Scholar] [CrossRef]
- Lee, C.K.; Allison, D.B.; Brand, J.; Weindruch, R.; Prolla, T.A. Transcriptional profiles associated with aging and middle age-onset caloric restriction in mouse hearts. Proc. Natl. Acad. Sci. USA 2002, 99, 14988–14993. [Google Scholar] [CrossRef]
- Kayo, T.; Allison, D.B.; Weindruch, R.; Prolla, T.A. Influences of aging and caloric restriction on the transcriptional profile of skeletal muscle from rhesus monkeys. Proc. Natl. Acad. Sci. USA 2001, 98, 5093–5098. [Google Scholar] [CrossRef]
- Plank, M.; Wuttke, D.; van Dam, S.; Clarke, S.A.; De Magalhães, J.P. A meta-analysis of caloric restriction gene expression profiles to infer common signatures and regulatory mechanisms. Mol. BioSyst. 2012, 8, 1339–1349. [Google Scholar] [CrossRef] [PubMed]
- Swindell, W.R. Dietary restriction in rats and mice: A meta-analysis and review of the evidence for genotype-dependent effects on lifespan. Ageing Res. Rev. 2012, 11, 254–270. [Google Scholar] [CrossRef]
- Choi, K.M.; Kwon, Y.Y.; Lee, C.K. Characterization of global gene expression during assurance of lifespan extension by caloric restriction in budding yeast. Exp. Gerontol. 2013, 48, 1455–1468. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Licastro, D.; Cava, E.; Veronese, N.; Spelta, F.; Rizza, W.; Bertozzi, B.; Villareal, D.T.; Hotamisligil, G.S.; Holloszy, J.O.; et al. Long-term calorie restriction enhances cellular quality-control processes in human skeletal muscle. Cell Rep. 2016, 14, 422–428. [Google Scholar] [CrossRef]
- Rhoads, T.W.; Burhans, M.S.; Chen, V.B.; Hutchins, P.D.; Rush, M.J.; Clark, J.P.; Stark, J.L.; McIlwain, S.J.; Eghbalnia, H.R.; Pavelec, D.M.; et al. Caloric restriction engages hepatic RNA processing mechanisms in rhesus monkeys. Cell Metab. 2018, 27, 677–688.e675. [Google Scholar] [CrossRef] [PubMed]
- Ulu, Z.O.; Ulu, S.; Dogan, S.; Tuna, B.G.; Ozgenturk, N.O. Transcriptome analysis of the thymus in short-term calorie-restricted mice using RNA-seq. Int. J. Genom. 2018, 2018. [Google Scholar]
- Cao, S.X.; Dhahbi, J.M.; Mote, P.L.; Spindler, S.R. Genomic profiling of short- and long-term caloric restriction effects in the liver of aging mice. Proc. Natl. Acad. Sci. USA 2001, 98, 10630–10635. [Google Scholar] [CrossRef] [PubMed]
- Van Bussel, I.P.; Jolink-Stoppelenburg, A.; De Groot, L.C.P.G.M.; Müller, M.; Afman, L.A. Differences in genome-wide gene expression response in peripheral blood mononuclear cells between young and old men upon caloric restriction. Genes Nutr. 2016, 11, 13. [Google Scholar] [CrossRef]
- Whitaker, R.; Gil, M.P.; Ding, F.; Tatar, M.; Helfand, S.L.; Neretti, N. Dietary switch reveals fast coordinated gene expression changes in Drosophila melanogaster. Aging 2014, 6, 355–368. [Google Scholar] [CrossRef]
- Khan, S.S.; Singer, B.D.; Vaughan, D.E. Molecular and physiological manifestations and measurement of aging in humans. Aging Cell 2017, 16, 624–633. [Google Scholar] [CrossRef]
- Ma, S.; Sun, S.; Geng, L.; Song, M.; Wang, W.; Ye, Y.; Ji, Q.; Zou, Z.; Wang, S.; He, X.; et al. Caloric Restriction Reprograms the Single-Cell Transcriptional Landscape of Rattus Norvegicus Aging. Cell 2020, 180, 984–1001.e22. [Google Scholar] [CrossRef] [PubMed]
- Pletcher, S.D.; Macdonald, S.J.; Marguerie, R.; Certa, U.; Stearns, S.C.; Goldstein, D.B.; Partridge, L. Genome-Wide Transcript Profiles in Aging and Calorically Restricted Drosophila melanogaster. Curr. Biol. 2002, 12, 712–723. [Google Scholar] [CrossRef]
- Fleischer, J.G.; Schulte, R.; Tsai, H.H.; Tyagi, S.; Ibarra, A.; Shokhirev, M.N.; Huang, L.; Hetzer, M.W.; Navlakha, S. Predicting age from the transcriptome of human dermal fibroblasts. Genome Biol. 2018, 19, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hara, K.; Yonezawa, K.; Weng, Q.-P.; Kozlowski, M.T.; Belham, C.; Avruch, J. Amino Acid Sufficiency and mTOR Regulate p70 S6 Kinase and eIF-4E BP1 through a Common Effector Mechanism. J. Biol. Chem. 1998, 273, 14484–14494. [Google Scholar] [CrossRef]
- Long, X.; Ortiz-Vega, S.; Lin, Y.; Avruch, J. Rheb Binding to Mammalian Target of Rapamycin (mTOR) Is Regulated by Amino Acid Sufficiency. J. Biol. Chem. 2005, 280, 23433–23436. [Google Scholar] [CrossRef]
- Beugnet, A.; Tee, A.R.; Taylor, P.M.; Proud, C.G. Regulation of targets of mTOR (mammalian target of rapamycin) signalling by intracellular amino acid availability. Biochem. J. 2003, 372, 555–566. [Google Scholar] [CrossRef]
- Carroll, B.; Maetzel, D.; Maddocks, O.D.K.; Otten, G.; Ratcliff, M.; Smith, G.R.; Dunlop, E.A.; Passos, J.F.; Davies, O.R.; Jaenisch, R.; et al. Control of TSC2-Rheb signaling axis by arginine regulates mTORC1 activity. eLife 2016, 5, e11058. [Google Scholar] [CrossRef]
- Cheon Lee, B.; Kaya, A.; Ma, S.; Kim, G.; Gerashchenko, M.V.; Yim, S.H.; Hu, Z.; Harshman, L.G.; Gladyshev, V.N. Methionine restriction extends lifespan of Drosophila melanogaster under conditions of low amino-acid status. Nat. Commun. 2014, 5, 1–12. [Google Scholar] [CrossRef]
- Arganda, S.; Bouchebti, S.; Bazazi, S.; Le Hesran, S.; Puga, C.; Latil, G.; Simpson, S.J.; Dussutour, A. Parsing the life-shortening effects of dietary protein: Effects of individual amino acids. Proc. R. Soc. B Biol. Sci. 2017, 284, 20162052. [Google Scholar] [CrossRef]
- Min, K.-J.; Tatar, M. Restriction of amino acids extends lifespan in Drosophila melanogaster. Mech. Ageing Dev. 2006, 127, 643–646. [Google Scholar] [CrossRef]
- Xiao, F.; Yu, J.; Guo, Y.; Deng, J.; Li, K.; Du, Y.; Chen, S.; Zhu, J.; Sheng, H.; Guo, F. Effects of individual branched-chain amino acids deprivation on insulin sensitivity and glucose metabolism in mice. Metabolism 2014, 63, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, A.; Oki, N.; Takahashi, S.-I.; Noguchi, T. Dietary Restriction of Single Essential Amino Acids Reduces Plasma Insulin-Like Growth Factor-I (IGF-I) but Does Not Affect Plasma IGF-Binding Protein-1 in Rats. Am. Soc. Nutr. Sci. 2000, 130, 2910–2914. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Robertson, L.; Gallinetti, J.; Mejia, P.; Vose, S.; Charlip, A.; Chu, T.; Mitchell, J.R. Surgical Stress Resistance Induced by Single Amino Acid Deprivation Requires Gcn2 in Mice. Sci. Transl. Med. 2012, 4, 118ra11. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Song, L.; Liu, S.Q.; Huang, D. Independent and Additive Effects of Glutamic Acid and Methionine on Yeast Longevity. PLoS ONE 2013, 8, e79319. [Google Scholar] [CrossRef]
- Wanders, D.; Stone, K.P.; Dille, K.; Simon, J.; Pierse, A.; Gettys, T.W. Metabolic responses to dietary leucine restriction involve remodeling of adipose tissue and enhanced hepatic insulin signaling. BioFactors 2015, 41, 391–402. [Google Scholar] [CrossRef]
- Lees, E.K.; Banks, R.; Cook, C.; Hill, S.; Morrice, N.; Grant, L.; Mody, N.; Delibegović, M. Direct comparison of methionine restriction with leucine restriction on the metabolic health of C57BL/6J mice. Sci. Rep. 2017, 7, 9977. [Google Scholar] [CrossRef]
- Levine, M.E.; Suarez, J.A.; Brandhorst, S.; Balasubramanian, P.; Cheng, C.-W.; Madia, F.; Fontana, L.; Mirisola, M.G.; Guevara-Aguirre, J.; Wan, J.; et al. Low Protein Intake Is Associated with a Major Reduction in IGF-1, Cancer, and Overall Mortality in the 65 and Younger but Not Older Population. Cell Metab. 2014, 19, 407–417. [Google Scholar] [CrossRef]
- Fung, T.T.; van Dam, R.M.; Hankinson, S.E.; Stampfer, M.; Willett, W.C.; Hu, F.B. Low-carbohydrate diets and all-cause and cause-specific mortality: Two cohort studies. Ann. Intern. Med. 2010, 153, 289–298. [Google Scholar] [CrossRef]
- Bernstein, A.M.; Sun, Q.; Hu, F.B.; Stampfer, M.J.; Manson, J.E.; Willett, W.C. Major Dietary Protein Sources and Risk of Coronary Heart Disease in Women. Circulation 2010, 122, 876–883. [Google Scholar] [CrossRef]
- Steinbrecher, A.; Erber, E.; Grandinetti, A.; Kolonel, L.N.; Maskarinec, G. Meat consumption and risk of type 2 diabetes: The Multiethnic Cohort. Public Health Nutr. 2011, 14, 568–574. [Google Scholar] [CrossRef]
- Grandison, R.C.; Piper, M.D.W.; Partridge, L. Amino-acid imbalance explains extension of lifespan by dietary restriction in Drosophila. Nat. Cell Biol. 2009, 462, 1061–1064. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Keenan, M.M.; Wu, J.; Lin, C.-A.; Dubois, L.; Thompson, J.W.; Freedland, S.J.; Murphy, S.K.; Chi, J.-T. Comprehensive Profiling of Amino Acid Response Uncovers Unique Methionine-Deprived Response Dependent on Intact Creatine Biosynthesis. PLoS Genet. 2015, 11, e1005158. [Google Scholar] [CrossRef] [PubMed]
- Mortimore, G.E. Intracellular protein catabolism and its control during nutrient deprivation and supply. Ann. Rev. Nutr. 1987, 7, 539–564. [Google Scholar] [CrossRef] [PubMed]
- Kuma, A.; Hatano, M.; Matsui, M.; Yamamoto, A.; Nakaya, H.; Yoshimori, T.; Ohsumi, Y.; Tokuhisa, T.; Mizushima, N. The role of autophagy during the early neonatal starvation period. Nat. Cell Biol. 2004, 432, 1032–1036. [Google Scholar] [CrossRef] [PubMed]
- Orentreich, N.; Matias, J.R.; DeFelice, A.; Zimmerman, J.A. Low methionine ingestion by rats extends life span. J. Nutr. 1993, 123, 269–274. [Google Scholar] [PubMed]
- Richie, J.P., Jr.; Leutzinger, Y.; Parthasarathy, S.; Maixoy, V.; Orentreich, N.; Zimmerman, J.A. Methionine restriction increases blood glutathione and longevity in F344 rats. FASEB J. 1994, 8, 1302–1307. [Google Scholar] [CrossRef]
- Zimmerman, J.A. Nutritional control of aging. Exp. Gerontol. 2003, 38, 47–52. [Google Scholar] [CrossRef]
- Miller, R.A.; Buehner, G.; Chang, Y.; Harper, J.M.; Sigler, R.; Smith-Wheelock, M. Methionine-deficient diet extends mouse lifespan, slows immune and lens aging, alters glucose, T4, IGF-I and insulin levels, and increases hepatocyte MIF levels and stress resistance. Aging Cell 2005, 4, 119–125. [Google Scholar] [CrossRef]
- Malloy, V.L.; Krajcik, R.A.; Bailey, S.J.; Hristopoulos, G.; Plummer, J.D.; Orentreich, N. Methionine restriction decreases visceral fat mass and preserves insulin action in aging male Fischer 344 rats independent of energy restriction. Aging Cell 2006, 5, 305–314. [Google Scholar] [CrossRef]
- Hasek, B.E.; Stewart, L.K.; Henagan, T.M.; Boudreau, A.; Lenard, N.R.; Black, C.; Shin, J.; Huypens, P.; Malloy, V.L.; Plaisance, E.P.; et al. Dietary methionine restriction enhances metabolic flexibility and increases uncoupled respiration in both fed and fasted states. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 299, R728–R739. [Google Scholar] [CrossRef]
- Plaisance, E.P.; Henagan, T.M.; Echlin, H.; Boudreau, A.; Hill, K.L.; Lenard, N.R.; Hasek, B.E.; Orentreich, N.; Gettys, T.W. Role of beta-adrenergic receptors in the hyperphagic and hypermetabolic responses to dietary methionine restriction. Am. J. Physiol. Integr. Comp. Physiol. 2010, 299, R740–R750. [Google Scholar] [CrossRef] [PubMed]
- Ables, G.P.; Perrone, C.E.; Orentreich, D.; Orentreich, N. Methionine-Restricted C57BL/6J Mice Are Resistant to Diet-Induced Obesity and Insulin Resistance but Have Low Bone Density. PLoS ONE 2012, 7, e51357. [Google Scholar] [CrossRef] [PubMed]
- Maddineni, S.; Nichenametla, S.; Sinha, R.; Wilson, R.P.; Richie, J.J.P. Methionine restriction affects oxidative stress and glutathione-related redox pathways in the rat. Exp. Biol. Med. (Maywood) 2013, 238, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Sanz, A.; Caro, P.; Ayala, V.; Portero-Otin, M.; Pamplona, R.; Barja, G. Methionine restriction decreases mitochondrial oxygen radical generation and leak as well as oxidative damage to mitochondrial DNA and proteins. FASEB J. 2006, 20, 1064–1073. [Google Scholar] [CrossRef]
- Caro, P.; Gómez, J.; López-Torres, M.; Sánchez, I.; Naudí, A.; Jove, M.; Pamplona, R.; Barja, G. Forty percent and eighty percent methionine restriction decrease mitochondrial ROS generation and oxidative stress in rat liver. Biogerontology 2008, 9, 183–196. [Google Scholar] [CrossRef]
- Castaño-Martinez, T.; Schumacher, F.; Schumacher, S.; Kochlik, B.; Weber, D.; Grune, T.; Biemann, R.; McCann, A.; Abraham, K.; Weikert, C.; et al. Methionine restriction prevents onset of type 2 diabetes in NZO mice. FASEB J. 2019, 33, 7092–7102. [Google Scholar] [CrossRef]
- Bárcena, C.; López-Otín, C.; Kroemer, G. Methionine restriction for improving progeria: Another autophagy-inducing anti-aging strategy? Autophagy 2019, 15, 558–559. [Google Scholar] [CrossRef]
- Bárcena, C.; Quirós, P.M.; Durand, S.; Mayoral, P.; Rodríguez, F.; Caravia, X.M.; Mariño, G.; Garabaya, C.; Fernández-García, M.T.; Kroemer, G.; et al. Methionine Restriction Extends Lifespan in Progeroid Mice and Alters Lipid and Bile Acid Metabolism. Cell Rep. 2018, 24, 2392–2403. [Google Scholar] [CrossRef]
- Zaborske, J.M.; Wu, X.; WEK, R.C.; Pan, T. Selective control of amino acid metabolism by the GCN2 eIF2 kinase pathway in Saccharomyces cerevisiae. BMC Biochem. 2010, 11, 29. [Google Scholar] [CrossRef]
- Xiao, F.; Huang, Z.; Li, H.; Yu, J.; Wang, C.; Chen, S.; Meng, Q.; Cheng, Y.; Gao, X.; Li, J.; et al. Leucine Deprivation Increases Hepatic Insulin Sensitivity via GCN2/mTOR/S6K1 and AMPK Pathways. Diabetes 2011, 60, 746–756. [Google Scholar] [CrossRef]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010, 465, 942–946. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D. Ragulator-Rag Complex Targets mTORC1 to the Lysosomal Surface and Is Necessary for Its Activation by Amino Acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The Rag GTPases Bind Raptor and Mediate Amino Acid Signaling to mtorc1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Goraksha-Hicks, P.; Li, L.; Neufeld, T.P.; Guan, K.-L. Regulation of TORC1 by Rag GTPases in nutrient response. Nat. Cell Biol. 2008, 10, 935–945. [Google Scholar] [CrossRef]
- Wolfson, R.L.; Chantranupong, L.; Wyant, G.A.; Gu, X.; Orozco, J.M.; Shen, K.; Condon, K.J.; Petri, S.; Kedir, J.; Scaria, S.M.; et al. KICSTOR recruits GATOR1 to the lysosome and is necessary for nutrients to regulate mTORC1. Nature 2017, 543, 438–442. [Google Scholar] [CrossRef]
- Saxton, R.A.; Chantranupong, L.; Knockenhauer, K.E.; Schwartz, T.U.; Sabbatin, T.M. Mechanism of arginine sensing by castor1 upstream of mtorc1. Nature 2016, 536, 229–233. [Google Scholar] [CrossRef]
- Saxton, R.A.; Knockenhauer, K.E.; Wolfson, R.L.; Chantranupong, L.; Pacold, M.E.; Wang, T.; Schwartz, T.U.; Sabatini, D. Structural basis for leucine sensing by the Sestrin2-mTORC1 pathway. Science 2016, 351, 53–58. [Google Scholar] [CrossRef]
- Chantranupong, L.; Scaria, S.M.; Saxton, R.A.; Gygi, M.P.; Shen, K.; Wyant, G.A.; Wang, T.; Harper, J.W.; Gygi, S.P.; Sabatini, D.M. The CASTOR Proteins Are Arginine Sensors for the mTORC1 Pathway. Cell 2016, 165, 153–164. [Google Scholar] [CrossRef]
- Chantranupong, L.; Wolfson, R.L.; Orozco, J.M.; Saxton, R.A.; Scaria, S.M.; Bar-Peled, L.; Spooner, E.; Isasa, M.; Gygi, S.P.; Sabatini, D.M. The Sestrins Interact with GATOR2 to Negatively Regulate the Amino-Acid-Sensing Pathway Upstream of mTORC1. Cell Rep. 2014, 9, 1–8. [Google Scholar] [CrossRef]
- Parmigiani, A.; Nourbakhsh, A.; Ding, B.; Wang, W.; Kim, Y.C.; Akopiants, K.; Guan, K.-L.; Karin, M.; Budanov, A.V. Sestrins Inhibit mTORC1 Kinase Activation through the GATOR Complex. Cell Rep. 2014, 9, 1281–1291. [Google Scholar] [CrossRef]
- Stracka, D.; Jozefczuk, S.; Rudroff, F.; Sauer, U.; Hall, M.N. Nitrogen Source Activates TOR (Target of Rapamycin) Complex 1 via Glutamine and Independently of Gtr/Rag Proteins. J. Biol. Chem. 2014, 289, 25010–25020. [Google Scholar] [CrossRef] [PubMed]
- Jewell, J.L.; Kim, Y.C.; Russell, R.C.; Yu, F.-X.; Park, H.W.; Plouffe, S.W.; Tagliabracci, V.S.; Guan, K.-L. Differential regulation of mTORC1 by leucine and glutamine. Science 2015, 347, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Durán, R.V.; Oppliger, W.; Robitaille, A.M.; Heiserich, L.; Skendaj, R.; Gottlieb, E.; Hall, M.N. Glutaminolysis activates rag-mtorc1 signaling. Mol. Cell 2012, 47, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-Y.; Cevallos, R.C.; Jan, E. An Upstream Open Reading Frame Regulates Translation of GADD34 during Cellular Stresses That Induce eIF2alpha Phosphorylation. J. Biol. Chem. 2009, 284, 6661–6673. [Google Scholar] [CrossRef]
- Kilberg, M.S.; Balasubramanian, M.; Fu, L.; Shan, J. The Transcription Factor Network Associated With the Amino Acid Response in Mammalian Cells. Adv. Nutr. 2012, 3, 295–306. [Google Scholar] [CrossRef]
- Novoa, I.; Zhang, Y.; Zeng, H.; Jungreis, R.; Harding, H.P.; Ron, D. Stress-induced gene expression requires programmed recovery from translational repression. EMBO J. 2003, 22, 1180–1187. [Google Scholar] [CrossRef]
- Chaveroux, C.; Jousse, C.; Cherasse, Y.; Maurin, A.-C.; Parry, L.; Carraro, V.; Derijard, B.; Bruhat, A.; Fafournoux, P. Identification of a Novel Amino Acid Response Pathway Triggering ATF2 Phosphorylation in Mammals. Mol. Cell. Biol. 2009, 29, 6515–6526. [Google Scholar] [CrossRef]
- Rousakis, A.; Vlassis, A.; Vlanti, A.; Patera, S.; Thireos, G.; Syntichaki, P. The general control nonderepressible-2 kinase mediates stress response and longevity induced by target of rapamycin inactivation inCaenorhabditis elegans. Aging Cell 2013, 12, 742–751. [Google Scholar] [CrossRef]
- De Sousa-Coelho, A.L.; Marrero, P.F.; Haro, D. Activating transcription factor 4-dependent induction of FGF21 during amino acid deprivation. Biochem. J. 2012, 443, 165–171. [Google Scholar] [CrossRef]
- Maruyama, R.; Shimizu, M.; Li, J.; Inoue, J.; Sato, R. Fibroblast growth factor 21 induction by activating transcription factor 4 is regulated through three amino acid response elements in its promoter region. Biosci. Biotechnol. Biochem. 2016, 80, 929–934. [Google Scholar] [CrossRef]
- Lees, E.K.; Król, E.; Grant, L.; Shearer, K.; Wyse, C.; Moncur, E.; Bykowska, A.S.; Mody, N.; Gettys, T.W.; Delibegovic, M. Methionine restriction restores a younger metabolic phenotype in adult mice with alterations in fibroblast growth factor 21. Aging Cell 2014, 13, 817–827. [Google Scholar] [CrossRef] [PubMed]
- Laeger, T.; Albarado, D.C.; Burke, S.J.; Trosclair, L.; Hedgepeth, J.W.; Berthoud, H.-R.; Gettys, T.W.; Collier, J.J.; Münzberg, H.; Morrison, C.D. Metabolic Responses to Dietary Protein Restriction Require an Increase in FGF21 that Is Delayed by the Absence of GCN2. Cell Rep. 2016, 16, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xie, Y.; Berglund, E.D.; Coate, K.C.; He, T.T.; Katafuchi, T.; Xiao, G.; Potthoff, M.J.; Wei, W.; Wan, Y.; et al. The starvation hormone, fibroblast growth factor-21, extends lifespan in mice. eLife 2012, 1, e00065. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Wang, J.; Huang, H.; Huang, Y.; Mi, T.; Zhang, C.; Zhang, L. Fibroblast growth factor 21 delayed endothelial replicative senescence and protected cells from h2o2-induced premautre senescence through sirt1. Am. J. Transl. Res. 2017, 9, 4492–4501. [Google Scholar] [PubMed]
- Hanks, L.J.; Gutiérrez, O.M.; Bamman, M.M.; Ashraf, A.; McCormick, K.L.; Casazza, K. Circulating levels of fibroblast growth factor-21 increase with age independently of body composition indices among healthy individuals. J. Clin. Transl. Endocrinol. 2015, 2, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Fujii, N.; Uta, S.; Kobayashi, M.; Sato, T.; Okita, N.; Higami, Y. Impact of aging and caloric restriction on fibroblast growth factor 21 signaling in rat white adipose tissue. Exp. Gerontol. 2019, 118, 55–64. [Google Scholar] [CrossRef]
- Villarroya, J.; Gallego-Escuredo, J.M.; Delgado-Anglés, A.; Cairó, M.; Moure, R.; Mateo, M.G.; Domingo, J.C.; Domingo, P.; Giralt, M.; Villarroya, F. Aging is associated with increased FGF21 levels but unaltered FGF21 responsiveness in adipose tissue. Aging Cell 2018, 17, e12822. [Google Scholar] [CrossRef]
- So, W.Y.; Cheng, Q.; Xu, A.; Lam, K.S.L.; Leung, P.S. Loss of fibroblast growth factor 21 action induces insulin resistance, pancreatic islet hyperplasia and dysfunction in mice. Cell Death Dis. 2015, 6, e1707. [Google Scholar] [CrossRef]
- Fisher, F.M.; Chui, P.C.; Antonellis, P.J.; Bina, H.A.; Kharitonenkov, A.; Flier, J.S.; Maratos-Flier, E. Obesity Is a Fibroblast Growth Factor 21 (FGF21)-Resistant State. Diabetes 2010, 59, 2781–2789. [Google Scholar] [CrossRef]
- Hale, C.; Chen, M.M.; Stanislaus, S.; Chinookoswong, N.; Hager, T.; Wang, M.; Veéniant, M.M.; Xu, J. Lack of Overt FGF21 Resistance in Two Mouse Models of Obesity and Insulin Resistance. Endocrinology 2012, 153, 69–80. [Google Scholar] [CrossRef]
- Opoku, Y.K.; Liu, Z.; Afrifa, J.; Khoso, M.H.; Ren, G.; Li, D.-S. Therapeutic Role of Fibroblast Growth Factor 21 (FGF21) in the Amelioration of Chronic Diseases. Int. J. Pept. Res. Ther. 2019, 26, 107–119. [Google Scholar] [CrossRef]
- Lee, D.V.; Li, D.; Yan, Q.; Zhu, Y.; Goodwin, B.; Calle, R.A.; Brenner, M.B.; Talukdar, S. Fibroblast Growth Factor 21 Improves Insulin Sensitivity and Synergizes with Insulin in Human Adipose Stem Cell-Derived (hASC) Adipocytes. PLoS ONE 2014, 9, e111767. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wu, G.; Fang, Q.; Zhang, M.; Hui, X.; Sheng, B.; Wu, L.; Bao, Y.; Li, P.; Xu, A.; et al. Fibroblast growth factor 21 increases insulin sensitivity through specific expansion of subcutaneous fat. Nat. Commun. 2018, 9, 272. [Google Scholar] [CrossRef] [PubMed]
- Minard, A.Y.; Tan, S.-X.; Yang, P.; Fazakerley, D.J.; Domanova, W.; Parker, B.L.; Humphrey, S.J.; Jothi, R.; Stöckli, J.; James, D.E. mTORC1 Is a Major Regulatory Node in the FGF21 Signaling Network in Adipocytes. Cell Rep. 2016, 17, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Martel, R.R.; Klicius, J.; Galet, S. Inhibition of the immune response by rapamycin, a new antifungal antibiotic. Can. J. Physiol. Pharmacol. 1976, 55, 48–51. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. Aging and Immortality: Quasi-Programmed Senescence and Its Pharmacologic Inhibition. Cell Cycle 2006, 5, 2087–2102. [Google Scholar] [CrossRef]
- Heitman, J.; Movva, N.R.; Hall, M.N. Targets for cell cycle arrest by the immunosuppresant rapamycin in yeast. Science 1991, 253, 905–909. [Google Scholar] [CrossRef]
- Van Duyne, G.D.; Standaert, R.F.; Schreiber, S.L.; Clardy, J. Atomic structure of the rapamycin human immunophilin FKBP-12 complex. J. Am. Chem. Soc. 1991, 113, 7433–7434. [Google Scholar] [CrossRef]
- Hay, N.; Sonenberg, N. Upstream and downstream of mtor. Genes Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef]
- Weichhart, T. mTOR as Regulator of Lifespan, Aging, and Cellular Senescence: A Mini-Review. Gerontology 2018, 64, 127–134. [Google Scholar] [CrossRef]
- Jansova, D.; Koncicka, M.; Tetkova, A.; Cerna, R.; Malik, R.; Del Llano, E.; Kubelka, M.; Susor, A. Regulation of 4E-BP1 activity in the mammalian oocyte. Cell Cycle 2017, 16, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Alvers, A.L.; Fishwick, L.K.; Wood, M.S.; Hu, D.; Chung, H.S.; Jr, W.A.D.; Aris, J.P. Autophagy and amino acid homeostasis are required for chronological longevity in Saccharomyces cerevisiae. Aging Cell 2009, 8, 353–369. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Lu, J.; Gong, P.; Chen, X.; Liang, C.; Zhang, J. Rapamycin induces autophagy to alleviate acute kidney injury following cerebral ischemia and reperfusion via the mtorc1/atg13/ulk1 signaling pathway. Mol. Med. Rep. 2018, 18, 5445–5454. [Google Scholar] [CrossRef] [PubMed]
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting mTOR for cancer therapy. J. Hematol. Oncol. 2019, 12, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Imrali, A.; Mao, X.; Yeste-Velasco, M.; Shamash, J.; Lu, Y. Rapamycin inhibits prostate cancer cell growth through cyclin D1 and enhances the cytotoxic efficacy of cisplatin. Am. J. Cancer Res. 2016, 6, 1772–1784. [Google Scholar] [PubMed]
- Karthik, G.-M.; Ma, R.; Lövrot, J.; Kis, L.L.; Lindh, C.; Blomquist, L.; Fredriksson, I.; Bergh, J.; Hartman, J. mTOR inhibitors counteract tamoxifen-induced activation of breast cancer stem cells. Cancer Lett. 2015, 367, 76–87. [Google Scholar] [CrossRef]
- Gao, G.; Chen, W.; Yan, M.; Liu, J.; Luo, H.; Wang, C.; Yang, P. Rapamycin regulates the balance between cardiomyocyte apoptosis and autophagy in chronic heart failure by inhibiting mtor signaling. Int. J. Mol. Med. 2019, 45, 195–209. [Google Scholar] [CrossRef]
- Quarles, E.; Basisty, N.; Chiao, Y.A.; Merrihew, G.; Gu, H.; Sweetwyne, M.T.; Fredrickson, J.; Nguyen, N.; Razumova, M.; Kooiker, K.; et al. Rapamycin persistently improves cardiac function in aged, male and female mice, even following cessation of treatment. Aging Cell 2020, 19, e13086. [Google Scholar] [CrossRef]
- Bikkul, M.U.; Clements, C.S.; Godwin, L.S.; Goldberg, M.W.; Kill, I.R.; Bridger, J.M. Farnesyltransferase inhibitor and rapamycin correct aberrant genome organisation and decrease DNA damage respectively, in Hutchinson–Gilford progeria syndrome fibroblasts. Biogerontology 2018, 19, 579–602. [Google Scholar] [CrossRef]
- Graziotto, J.J.; Cao, K.; Collins, F.S.; Krainc, D. Rapamycin activates autophagy in hutchinson-gilford progeria syndrome: Implications for normal aging and age-dependent neurodegenerative disorders. Autophagy 2012, 8, 147–151. [Google Scholar] [CrossRef]
- Kawakami, Y.; Hambright, W.S.; Takayama, K.; Mu, X.; Lu, A.; Cummins, J.H.; Matsumoto, T.; Yurube, T.; Kuroda, R.; Kurosaka, M.; et al. Rapamycin Rescues Age-Related Changes in Muscle-Derived Stem/Progenitor Cells from Progeroid Mice. Mol. Ther. Methods Clin. Dev. 2019, 14, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Boyer, L.; Jin, M.; Kim, Y.; Fan, W.; Bardy, C.; Berggren, T.; Evans, R.M.; Gage, F.H.; Hunter, T. Alleviation of neuronal energy deficiency by mTOR inhibition as a treatment for mitochondria-related neurodegeneration. eLife 2016, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Staats, K.A.; Hernandez, S.; Schönefeldt, S.; Bento-Abreu, A.; Dooley, J.; Van Damme, P.; Liston, A.; Robberecht, W.; Bosch, L.V.D. Rapamycin increases survival in ALS mice lacking mature lymphocytes. Mol. Neurodegener. 2013, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ma, Q.; Ma, X.; Zhang, Z.; Liu, N.; Wang, M. Role of mammalian target of rapamycin signaling in autophagy and the neurodegenerative process using a senescence accelerated mouse-prone 8 model. Exp. Ther. Med. 2017, 14, 1051–1057. [Google Scholar] [CrossRef]
- Kaeberlein, M.; Galvan, V. Rapamycin and Alzheimer’s disease: Time for a clinical trial? Sci. Transl. Med. 2019, 11, eaar4289. [Google Scholar] [CrossRef]
- Tramutola, A.; Lanzillotta, C.; Barone, E.; Arena, A.; Zuliani, I.; Mosca, L.; Blarzino, C.; Butterfield, D.A.; Perluigi, M.; Di Domenico, F. Intranasal rapamycin ameliorates Alzheimer-like cognitive decline in a mouse model of Down syndrome. Transl. Neurodegener. 2018, 7, 28. [Google Scholar] [CrossRef]
- Caccamo, A.; Majumder, S.; Richardson, A.; Strong, R.; Oddo, S. Molecular interplay between mammalian target of rapamycin (mtor), amyloid-beta, and tau: Effects on cognitive impairments. J. Biol. Chem. 2010, 285, 13107–13120. [Google Scholar] [CrossRef]
- Pupyshev, A.B.; Korolenko, T.A.; Tikhonova, M.A. Effects and Mechanisms of Rapamycin Action on Experimental Neurodegeneration. Neurochem. J. 2018, 12, 347–358. [Google Scholar] [CrossRef]
- Anisimov, V.N.; Zabezhinski, M.A.; Popovich, I.G.; Piskunova, T.S.; Semenchenko, A.V.; Tyndyk, M.L.; Yurova, M.N.; Antoch, M.P.; Blagosklonny, M.V. Rapamycin Extends Maximal Lifespan in Cancer-Prone Mice. Am. J. Pathol. 2010, 176, 2092–2097. [Google Scholar] [CrossRef]
- Komarova, E.A.; Antoch, M.P.; Novototskaya, L.R.; Chernova, O.B.; Paszkiewicz, G.; Leontieva, O.V.; Blagosklonny, M.V.; Gudkov, A.V. Rapamycin extends lifespan and delays tumorigenesis in heterozygous p53+/- mice. Aging 2012, 4, 709–714. [Google Scholar] [CrossRef]
- Hebert, M.; Licursi, M.; Jensen, B.; Baker, A.; Milway, S.; Malsbury, C.; Grant, V.L.; Adamec, R.; Hirasawa, M.; Blundell, J. Single Rapamycin Administration Induces Prolonged Downward Shift in Defended Body Weight in Rats. PLoS ONE 2014, 9, e93691. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Kennedy, B.K.; Liao, C.-Y. Mechanistic target of rapamycin signaling in mouse models of accelerated aging. J. Gerontol. Ser. A Biol. Sci. Med Sci. 2020, 75, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Khapre, R.V.; Kondratova, A.A.; Patel, S.; Dubrovsky, Y.; Wrobel, M.; Antoch, M.P.; Kondratov, R.V. BMAL1-dependent regulation of the mTOR signaling pathway delays aging. Aging 2014, 6, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.-Y.; Anderson, S.S.; Chicoine, N.H.; Mayfield, J.R.; Academia, E.C.; Wilson, J.A.; Pongkietisak, C.; Thompson, M.A.; Lagmay, E.P.; Miller, D.M.; et al. Rapamycin Reverses Metabolic Deficits in Lamin A/C-Deficient Mice. Cell Rep. 2016, 17, 2542–2552. [Google Scholar] [CrossRef]
- Cao, K.; Graziotto, J.J.; Blair, C.D.; Mazzulli, J.R.; Erdos, M.R.; Krainc, D.; Collins, F.S. Rapamycin Reverses Cellular Phenotypes and Enhances Mutant Protein Clearance in Hutchinson-Gilford Progeria Syndrome Cells. Sci. Transl. Med. 2011, 3, 89ra58. [Google Scholar] [CrossRef] [PubMed]
- Melzi, R.; Maffi, P.; Nano, R.; Sordi, V.; Mercalli, A.; Scavini, M.; Secchi, A.; Bonifacio, E.; Piemonti, L. Rapamycin does not adversely affect intrahepatic islet engraftment in mice and improves early islet engraftment in humans. Islets 2009, 1, 42–49. [Google Scholar] [CrossRef]
- Piemonti, L.; Maffi, P.; Monti, L.; Lampasona, V.; Perseghin, G.; Magistretti, P.; Secchi, A.; Bonifacio, E. Beta cell function during rapamycin monotherapy in long-term type 1 diabetes. Diabetologia 2010, 54, 433–439. [Google Scholar] [CrossRef]
- Groth, C.G.; Bäckman, L.; Morales, J.M.; Calne, R.; Kreis, H.; Lang, P.; Touraine, J.L.; Claesson, K.; Campistol, J.M.; Durand, D.; et al. Sirolimus (rapamycin)-based therapy in human renal transplantation: Similar efficacy and different toxicity compared with cyclosporine. Transplantation 1999, 67, 1036–1042. [Google Scholar] [CrossRef]
- Lamming, D.W. Inhibition of the mechanistic target of rapamycin (mtor)-rapamycin and beyond. Cold Spring Harb. Perspect. Med. 2016, 6, a025924. [Google Scholar] [CrossRef]
- Chung, C.L.; Lawrence, I.; Hoffman, M.; Elgindi, D.; Nadhan, K.; Potnis, M.; Jin, A.; Sershon, C.; Binnebose, R.; Lorenzini, A.; et al. Topical rapamycin reduces markers of senescence and aging in human skin: An exploratory, prospective, randomized trial. GeroScience 2019, 41, 861–869. [Google Scholar] [CrossRef]
- Krebs, M.; Brunmair, B.; Brehm, A.; Artwohl, M.; Szendroedi, J.; Nowotny, P.; Roth, E.; Fürnsinn, C.; Promintzer, M.; Anderwald, C.; et al. The Mammalian Target of Rapamycin Pathway Regulates Nutrient-Sensitive Glucose Uptake in Man. Diabetes 2007, 56, 1600–1607. [Google Scholar] [CrossRef] [PubMed]
- Fok, W.C.Y.; Zhang, Y.; Salmon, A.B.; Bhattacharya, A.; Gunda, R.; Jones, D.; Ward, W.; Fisher, K.; Richardson, A.; Pérez, V.I. Short-Term Treatment With Rapamycin and Dietary Restriction Have Overlapping and Distinctive Effects in Young Mice. J. Gerontol. Ser. A Biol. Sci. Med Sci. 2013, 68, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Lamming, D.W.; Ye, L.; Katajisto, P.; Goncalves, M.D.; Saitoh, M.; Stevens, D.M.; Davis, J.G.; Salmon, A.B.; Richardson, A.; Ahima, R.S.; et al. Rapamycin-Induced Insulin Resistance Is Mediated by mTORC2 Loss and Uncoupled from Longevity. Science 2012, 335, 1638–1643. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. Fasting and rapamycin: Diabetes versus benevolent glucose intolerance. Cell Death Dis. 2019, 10, 1–10. [Google Scholar] [CrossRef]
- Shivaswamy, V.; McClure, M.; Passer, J.; Frahm, C.; Ochsner, L.; Erickson, J.; Bennett, R.G.; Hamel, F.G.; Larsen, J.L. Hyperglycemia induced by tacrolimus and sirolimus is reversible in normal sprague–dawley rats. Endocrine 2010, 37, 489–496. [Google Scholar] [CrossRef]
- Fok, W.C.; Livi, C.B.; Bokov, A.; Yu, Z.; Chen, Y.; Richardson, A.; Pérez, V.I. Short-term rapamycin treatment in mice has few effects on the transcriptome of white adipose tissue compared to dietary restriction. Mech. Ageing Dev. 2014, 140, 23–29. [Google Scholar] [CrossRef]
- Fok, W.C.; Bokov, A.; Gelfond, J.; Yu, Z.; Zhang, Y.; Doderer, M.; Chen, Y.; Javors, M.; Wood, W.H., III; Zhang, Y.; et al. Combined treatment of rapamycin and dietary restriction has a larger effect on the transcriptome and metabolome off liver. Aging Cell 2014, 13, 311–319. [Google Scholar] [CrossRef]
- Choi, K.-M.; Hong, S.-J.; Van Deursen, J.M.; Kim, S.; Kim, K.H.; Lee, C.-K. Caloric Restriction and Rapamycin Differentially Alter Energy Metabolism in Yeast. J. Gerontol. Ser. A Biol. Sci. Med Sci. 2017, 73, 29–38. [Google Scholar] [CrossRef]
- Garcia, D.N.; Saccon, T.D.; Pradiee, J.; Rincón, J.A.A.; Andrade, K.R.S.; Rovani, M.T.; Mondadori, R.G.; Cruz, L.A.X.; Barros, C.C.; Masternak, M.M.; et al. Effect of caloric restriction and rapamycin on ovarian aging in mice. GeroScience 2019, 41, 395–408. [Google Scholar] [CrossRef]
- Gillespie, Z.E.; Mackay, K.; Sander, M.; Trost, B.; Dawicki, W.; Wickramarathna, A.; Gordon, J.; Eramian, M.; Kill, I.R.; Bridger, J.M.; et al. Rapamycin reduces fibroblast proliferation without causing quiescence and induces STAT5A/B-mediated cytokine production. Nucleus 2015, 6, 490–506. [Google Scholar] [CrossRef]
- Ciołczyk-Wierzbicka, D.; Gil, D.; Zarzycka, M.; Laidler, P. mTOR inhibitor everolimus reduces invasiveness of melanoma cells. Hum. Cell 2020, 33, 88–97. [Google Scholar] [CrossRef]
- Cassano, T.; Magini, A.; Giovagnoli, S.; Polchi, A.; Calcagnini, S.; Pace, L.; Lavecchia, M.A.; Scuderi, C.; Bronzuoli, M.R.; Ruggeri, L.; et al. Early intrathecal infusion of everolimus restores cognitive function and mood in a murine model of Alzheimer’s disease. Exp. Neurol. 2019, 311, 88–105. [Google Scholar] [CrossRef] [PubMed]
- Mannick, J.B.; Morris, M.; Hockey, H.-U.; Roma, G.; Beibel, M.; Kulmatycki, K.; Watkins, M.; Shavlakadze, T.; Zhou, W.; Quinn, D.; et al. TORC1 inhibition enhances immune function and reduces infections in the elderly. Sci. Transl. Med. 2018, 10, eaaq1564. [Google Scholar] [CrossRef]
- Nawroth, R.; Stellwagen, F.; Stoehr, R.; Hartmann, A.; Krause, B.J.; Gschwend, J.E.; Retz, M. S6k1 and 4e-bp1 are independent regulated and control cellular growth in bladder cancer. PLoS ONE 2011, 6, e27509. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.R. Are we missing the mtor target in breast cancer? Breast Cancer Res. Treat. 2011, 128, 607–611. [Google Scholar] [CrossRef][Green Version]
- Shavlakadze, T.; Zhu, J.; Wang, S.; Zhou, W.; Morin, B.; Egerman, M.A.; Fan, L.; Wang, Y.; Iartchouk, O.; Meyer, A.; et al. Short-term Low-Dose mTORC1 Inhibition in Aged Rats Counter-Regulates Age-Related Gene Changes and Blocks Age-Related Kidney Pathology. J. Gerontol. Ser. A Biol. Sci. Med Sci. 2018, 73, 845–852. [Google Scholar] [CrossRef] [PubMed]
- DuBose, A.J.; Lichtenstein, S.T.; Petrash, N.M.; Erdos, M.R.; Gordon, L.B.; Collins, F.S. Everolimus rescues multiple cellular defects in laminopathy-patient fibroblasts. Proc. Natl. Acad. Sci. USA 2018, 115, 4206–4211. [Google Scholar] [CrossRef]
- Salmon, A.B. About-face on the metabolic side effects of rapamycin. Oncotarget 2015, 6, 2585–2586. [Google Scholar] [CrossRef]
- Kirchner, G.I.; Meier-Widenbach, I.; Manns, M.P. Clinical pharmacokinetics of everolimus. Clin. Pharmacokinet. 2004, 43, 83–95. [Google Scholar] [CrossRef]
- Kuo, C.-T.; Chen, C.-L.; Li, C.-C.; Huang, G.-S.; Ma, W.-Y.; Hsu, W.-F.; Lin, C.-H.; Lu, Y.-S.; Wo, A.M. Immunofluorescence can assess the efficacy of mTOR pathway therapeutic agent Everolimus in breast cancer models. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Saran, U.; Foti, M.; Dufour, J.-F.F. Cellular and molecular effects of the mTOR inhibitor everolimus. Clin. Sci. (London) 2015, 129, 895–914. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Campone, M.; Piccart-Gebhart, M.; Burris, H.A.; Rugo, H.S.; Sahmoud, T.; Noguchi, S.; Gnant, M.; Pritchard, K.I.; Lebrun, F.; et al. Everolimus in Postmenopausal Hormone-Receptor–Positive Advanced Breast Cancer. N. Engl. J. Med. 2012, 366, 520–529. [Google Scholar] [CrossRef] [PubMed]
- Sabine, V.S.; Sims, A.H.; Macaskill, E.J.; Renshaw, L.; Thomas, J.S.; Dixon, J.M.; Bartlett, J.M.S. Gene expression profiling of response to mTOR inhibitor everolimus in pre-operatively treated post-menopausal women with oestrogen receptor-positive breast cancer. Breast Cancer Res. Treat. 2010, 122, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Granata, S.; Gassa, A.D.; Carraro, A.; Brunelli, M.; Stallone, G.; Lupo, A.; Zaza, G. Sirolimus and Everolimus Pathway: Reviewing Candidate Genes Influencing Their Intracellular Effects. Int. J. Mol. Sci. 2016, 17, 735. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, Y.; Li, D.; Zhang, N.; Liu, R.; Han, B.; Wei, C.; Liu, H.; Xu, X.; Hao, J. Everolimus (RAD001) ameliorates vascular cognitive impairment by regulating microglial function via the mTORC1 signaling pathway. J. Neuroimmunol. 2016, 299, 164–171. [Google Scholar] [CrossRef]
- Fanoudi, S.; Hosseini, M.; Alavi, M.S.; Boroushaki, M.T.; Hosseini, A.; Sadeghnia, H.R. Everolimus, a mammalian target of rapamycin inhibitor, ameliorated streptozotocin-induced learning and memory deficits via neurochemical alterations in male rats. EXCLI J. 2018, 17, 999–1017. [Google Scholar]
- Yoshida, K.; Imamura, C.K.; Hara, K.; Mochizuki, M.; Kenmotsu, H. Effect of everolimus on the glucose metabolic pathway in mouse skeletal muscle cells (C2C12). Metabolomics 2017, 13, 98. [Google Scholar] [CrossRef]
- Martínez, J.; Pulido, L.; Bellido, C.; Usero, D.; Tallón-Aguilar, L.; Moreno, J.; Artacho, G.; Diez-Canedo, J.; Gómez, L.; Bravo, M. Rescue Immunosuppression With Mammalian Target of Rapamycin Inhibitor Drugs in Liver Transplantation. Transplant. Proc. 2010, 42, 641–643. [Google Scholar] [CrossRef]
- Hofmann, J.W.; Zhao, X.; De Cecco, M.; Peterson, A.L.; Pagliaroli, L.; Manivannan, J.; Hubbard, G.B.; Ikeno, Y.; Zhang, Y.; Bin Feng, B.; et al. Reduced Expression of MYC Increases Longevity and Enhances Healthspan. Cell 2015, 160, 477–488. [Google Scholar] [CrossRef]
- Wall, M.; Poortinga, G.; Hannan, K.M.; Pearson, R.B.; Hannan, R.D.; McArthur, G.A. Translational control of c-MYC by rapamycin promotes terminal myeloid differentiation. Blood 2008, 112, 2305–2317. [Google Scholar] [CrossRef]
- Jimenez, R.H.; Lee, J.-S.; Francesconi, M.; Castellani, G.; Neretti, N.; Sanders, J.A.; Sedivy, J.; Gruppuso, P.A. Regulation of Gene Expression in Hepatic Cells by the Mammalian Target of Rapamycin (mTOR). PLoS ONE 2010, 5, e9084. [Google Scholar] [CrossRef] [PubMed]
- Volk, M.J.; Pugh, T.D.; Kim, M.; Frith, C.H.; Daynes, R.A.; Ershler, W.B.; Weindruch, R. Dietary restriction from middle age attenuates age-associated lymphoma development and interleukin 6 dysregulation in C57BL/6 mice. Cancer Res. 1994, 54, 3054–3061. [Google Scholar] [PubMed]
- Yuan, J.; Minter-Dykhouse, K.; Lou, Z. A c-Myc–SIRT1 feedback loop regulates cell growth and transformation. J. Cell Biol. 2009, 185, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Tameire, F.; Verginadis, I.I.; Leli, N.M.; Polte, C.; Conn, C.S.; Ojha, R.; Salinas, C.S.; Chinga, F.; Monroy, A.M.; Fu, W.; et al. ATF4 couples MYC-dependent translational activity to bioenergetic demands during tumour progression. Nat. Cell Biol. 2019, 21, 889–899. [Google Scholar] [CrossRef]
- Klümpen, H.-J.; Beijnen, J.H.; Gurney, H.; Schellens, J.H. Inhibitors of mTOR. Oncologist 2010, 15, 1262–1269. [Google Scholar] [CrossRef]
- Schreiber, K.H.; O’Leary, M.N.; Kennedy, B.K. The mTOR Pathway and Aging. In The Handbook of the Biology of Aging; Academic Press: Cambridge, MA, USA, 2016; pp. 55–81. [Google Scholar] [CrossRef]
- Gabriel, D.; Gordon, L.B.; Djabali, K. Temsirolimus Partially Rescues the Hutchinson-Gilford Progeria Cellular Phenotype. PLoS ONE 2016, 11, e0168988. [Google Scholar] [CrossRef]
- Jiang, T.; Yu, J.-T.; Zhu, X.-C.; Zhang, Q.-Q.; Cao, L.; Wang, H.-F.; Tan, M.-S.; Gao, Q.; Qin, H.; Zhang, Y.-D.; et al. Temsirolimus attenuates tauopathy in vitro and in vivo by targeting tau hyperphosphorylation and autophagic clearance. Neuropharmacology 2014, 85, 121–130. [Google Scholar] [CrossRef]
- Jiang, T.; Yu, J.T.; Zhu, X.C.; Tan, M.S.; Wang, H.F.; Cao, L.; Zhang, Q.Q.; Shi, J.Q.; Gao, L.; Qin, H.; et al. Temsirolimus promotes autophagic clearance of amyloid-beta and provides protective effects in cellular and animal models of Alzheimer’s disease. Pharmacol. Res. 2014, 81, 54–63. [Google Scholar] [CrossRef]
- Burns, J.; Yokota, T.; Ashihara, H.; Lean, M.E.J.; Crozier, A. Plant Foods and Herbal Sources of Resveratrol. J. Agric. Food Chem. 2002, 50, 3337–3340. [Google Scholar] [CrossRef]
- Renaud, S.; De Lorgeril, M. Wine, alcohol, platelets, and the French paradox for coronary heart disease. Lancet 1992, 339, 1523–1526. [Google Scholar] [CrossRef]
- Fiori, J.L.; Shin, Y.K.; Kim, W.; Krzysik-Walker, S.M.; González-Mariscal, I.; Carlson, O.D.; Sanghvi, M.; Moaddel, R.; Farhang, K.; Gadkaree, S.K.; et al. Resveratrol prevents b-cell dedifferentiation in nonhuman primates given a high-fat/high-sugar diet. Diabetes 2013, 62, 3500–3513. [Google Scholar] [CrossRef] [PubMed]
- Mattison, J.A.; Wang, M.; Bernier, M.; Zhang, J.; Park, S.-S.; Maudsley, S.; An, S.S.; Santhanam, L.; Martin, B.; Faulkner, S.; et al. Resveratrol Prevents High Fat/Sucrose Diet-Induced Central Arterial Wall Inflammation and Stiffening in Nonhuman Primates. Cell Metab. 2014, 20, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Gülçin, I. Antioxidant properties of resveratrol: A structure–activity insight. Innov. Food Sci. Emerg. Technol. 2010, 11, 210–218. [Google Scholar] [CrossRef]
- Li, Y.; Yuan, Y.Y.; Meeran, S.M.; Tollefsbol, T.O. Synergistic epigenetic reactivation of estrogen receptor-alpha (eralpha) by combined green tea polyphenol and histone deacetylase inhibitor in eralpha-negative breast cancer cells. Mol. Cancer 2010, 9, 274. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.-Y.; Su, Y.-C.; Chen, N.-C.; Hsieh, H.-S.; Chen, K.-S. Resveratrol inhibits migration and invasion of human breast-cancer cells. Mol. Nutr. Food Res. 2008, 52, 683–691. [Google Scholar] [CrossRef]
- Ciolino, H.P.; Yeh, G.C. Inhibition of aryl hydrocarbon-induced cytochrome P-450 1A1 enzyme activity and CYP1A1 expression by resveratrol. Mol. Pharmacol. 1999, 56, 760–767. [Google Scholar]
- Peng, T.-L.; Chen, J.; Mao, W.; Song, X.; Chen, M.-H. Aryl hydrocarbon receptor pathway activation enhances gastric cancer cell invasiveness likely through a c-Jun-dependent induction of matrix metalloproteinase-9. BMC Cell Biol. 2009, 10, 27. [Google Scholar] [CrossRef]
- Kumar, S.; Eroglu, E.; Stokes, J.A., III; Scissum-Gunn, K.; Saldanha, S.N.; Singh, U.P.; Manne, U.; Ponnazhagan, S.; Mishra, M.K. Resveratrol induces mitochondria-mediated, caspase-independent apoptosis in murine prostate cancer cells. Oncotarget 2017, 8, 20895–20908. [Google Scholar] [CrossRef]
- Hao, Y.; Huang, W.; Liao, M.; Zhu, Y.; Liu, H.; Hao, C.; Liu, G.; Zhang, G.; Feng, H.; Ning, X.; et al. The inhibition of resveratrol to human skin squamous cell carcinoma A431 xenografts in nude mice. Fitoterapia 2013, 86, 84–91. [Google Scholar] [CrossRef]
- Mohapatra, P.; Satapathy, S.R.; Das, D.; Siddharth, S.; Choudhuri, T.; Kundu, C.N. Resveratrol mediated cell death in cigarette smoke transformed breast epithelial cells is through induction of p21Waf1/Cip1 and inhibition of long patch base excision repair pathway. Toxicol. Appl. Pharmacol. 2014, 275, 221–231. [Google Scholar] [CrossRef]
- Huderson, A.C.; Devi, P.V.R.; Niaz, M.S.; Adunyah, S.E.; Ramesh, A. Alteration of benzo(a)pyrene biotransformation by resveratrol in apc(min/+) mouse model of colon carcinogenesis. Investig. N. Drugs 2019, 37, 238–251. [Google Scholar] [CrossRef] [PubMed]
- Monteillier, A.; Voisin, A.; Furrer, P.; Allémann, E.; Cuendet, M. Intranasal administration of resveratrol successfully prevents lung cancer in A/J mice. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tong, L.; Luo, Y.; Li, X.; Chen, G.; Wang, Y. Resveratrol inhibits the proliferation and induces the apoptosis in ovarian cancer cells via inhibiting glycolysis and targeting AMPK/mTOR signaling pathway. J. Cell. Biochem. 2018, 119, 6162–6172. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, H.; Teimouri, M.; Shabani, M.; Koushki, M.; Khorzoughi, R.B.; Namvarjah, F.; Izadi, P.; Meshkani, R. Resveratrol alleviates non-alcoholic fatty liver disease through epigenetic modification of the Nrf2 signaling pathway. Int. J. Biochem. Cell Biol. 2020, 119, 105667. [Google Scholar] [CrossRef]
- Elshaer, M.; Chen, Y.; Wang, X.J.; Tang, X. Resveratrol: An overview of its anti-cancer mechanisms. Life Sci. 2018, 207, 340–349. [Google Scholar] [CrossRef]
- Moussa, C.; Hebron, M.; Huang, X.; Ahn, J.; Rissman, R.A.; Aisen, P.S.; Turner, R.S. Resveratrol regulates neuro-inflammation and induces adaptive immunity in Alzheimer’s disease. J. Neuroinflamm. 2017, 14, 1–10. [Google Scholar] [CrossRef]
- Wong, R.H.X.; Raederstorff, D.; Howe, P.R.C. Acute Resveratrol Consumption Improves Neurovascular Coupling Capacity in Adults with Type 2 Diabetes Mellitus. Nutrients 2016, 8, 425. [Google Scholar] [CrossRef]
- Crandall, J.P.; Barzilai, N. Exploring the Promise of Resveratrol: Where Do We Go From Here? Diabetes 2013, 62, 1022–1023. [Google Scholar] [CrossRef][Green Version]
- Szkudelski, T.; Szkudelska, K. Resveratrol and diabetes: From animal to human studies. Biochim. Biophys. Acta 2015, 1852, 1145–1154. [Google Scholar] [CrossRef]
- Knop, F.K.; Konings, E.; Timmers, S.; Schrauwen, P.; Holst, J.J.; Blaak, E.E. Thirty days of resveratrol supplementation does not affect postprandial incretin hormone responses, but suppresses postprandial glucagon in obese subjects. Diabet. Med. 2013, 30, 1214–1218. [Google Scholar] [CrossRef]
- Kennedy, D.O.; Wightman, E.L.; Reay, J.L.; Lietz, G.; Okello, E.J.; Wilde, A.; Haskell, C.F. Effects of resveratrol on cerebral blood flow variables and cognitive performance in humans: A double-blind, placebo-controlled, crossover investigation. Am. J. Clin. Nutr. 2010, 91, 1590–1597. [Google Scholar] [CrossRef] [PubMed]
- Brown, V.A.; Patel, K.R.; Viskaduraki, M.; Crowell, J.A.; Perloff, M.; Booth, T.D.; Vasilinin, G.; Sen, A.; Schinas, A.M.; Piccirilli, G.; et al. Repeat Dose Study of the Cancer Chemopreventive Agent Resveratrol in Healthy Volunteers: Safety, Pharmacokinetics, and Effect on the Insulin-like Growth Factor Axis. Cancer Res. 2010, 70, 9003–9011. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Kesimer, M.; Tolun, G.; Zheng, X.; Xu, Q.; Lu, J.; Sheehan, J.K.; Griffith, J.D.; Li, X. The nad(+)-dependent protein deacetylase activity of SIRT1 is regulated by its oligomeric status. Sci. Rep. 2012, 2, 640. [Google Scholar] [CrossRef] [PubMed]
- Abolaji, A.O.; Adedara, A.O.; Adie, M.A.; Vicente-Crespo, M.; Farombi, E.O. Resveratrol prolongs lifespan and improves 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced oxidative damage and behavioural deficits in Drosophila melanogaster. Biochem. Biophys. Res. Commun. 2018, 503, 1042–1048. [Google Scholar] [CrossRef] [PubMed]
- Bass, T.M.; Weinkove, D.; Houthoofd, K.; Gems, D.; Partridge, L. Effects of resveratrol on lifespan in Drosophila melanogaster and Caenorhabditis elegans. Mech. Ageing Dev. 2007, 128, 546–552. [Google Scholar] [CrossRef] [PubMed]
- Rascón, B.; Hubbard, B.P.; Sinclair, D.A.; Amdam, G.V. The lifespan extension effects of resveratrol are conserved in the honey bee and may be driven by a mechanism related to caloric restriction. Aging 2012, 4, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Baur, J.A.; Pearson, K.J.; Price, N.L.; Jamieson, H.A.; Lerin, C.; Kalra, A.; Prabhu, V.V.; Allard, J.S.; Lopez-Lluch, G.; Lewis, K.; et al. Resveratrol improves health and surival of mice on high-calorie diet. Nature 2006, 444, 337–342. [Google Scholar] [CrossRef]
- Howitz, K.T.; Bitterman, K.J.; Cohen, H.Y.; Lamming, D.W.; Lavu, S.; Wood, J.G.; Zipkin, R.E.; Chung, P.; Kisielewski, A.; Zhang, L.-L.; et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 2003, 425, 191–196. [Google Scholar] [CrossRef]
- Price, N.L.; Gomes, A.P.; Ling, A.J.; Duarte, F.V.; Martin-Montalvo, A.; North, B.J.; Agarwal, B.; Ye, L.; Ramadori, G.; Teodoro, J.S.; et al. SIRT1 Is Required for AMPK Activation and the Beneficial Effects of Resveratrol on Mitochondrial Function. Cell Metab. 2012, 15, 675–690. [Google Scholar] [CrossRef]
- Hubbard, B.P.; Sinclair, D.A. Small molecule SIRT1 activators for the treatment of aging and age-related diseases. Trends Pharmacol. Sci. 2014, 35, 146–154. [Google Scholar] [CrossRef]
- Cione, E.; La Torre, C.; Cannataro, R.; Caroleo, M.C.; Plastina, P.; Gallelli, L. Quercetin, Epigallocatechin Gallate, Curcumin, and Resveratrol: From Dietary Sources to Human MicroRNA Modulation. Molecules 2019, 25, 63. [Google Scholar] [CrossRef] [PubMed]
- Medina-Aguilar, R.; Marchat, L.A.; Aréchaga-Ocampo, E.; Gariglio, P.; Mena, J.G.; Sepúlveda, N.V.; Castillo, M.M.; López-Camarillo, C. Resveratrol inhibits cell cycle progression by targeting Aurora kinase A and Polo-like kinase 1 in breast cancer cells. Oncol. Rep. 2016, 35, 3696–3704. [Google Scholar] [CrossRef] [PubMed]
- Arablou, T.; Delbandi, A.; Khodaverdi, S.; Arefi, S.; Kolahdouz-Mohammadi, R.; Heidari, S.; Mohammadi, T.; Aryaeian, N. Resveratrol reduces the expression of insulin-like growth factor-1 and hepatocyte growth factor in stromal cells of women with endometriosis compared with nonendometriotic women. Phytotherapy Res. 2019, 33, 1044–1054. [Google Scholar] [CrossRef] [PubMed]
- Pallauf, K.; Chin, D.; Günther, I.; Birringer, M.; Lüersen, K.; Schultheiß, G.; Vieten, S.; Krauß, J.; Bracher, F.; Danylec, N.; et al. Resveratrol, lunularin and dihydroresveratrol do not act as caloric restriction mimetics when administered intraperitoneally in mice. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Günther, I.; Rimbach, G.; Mack, C.I.; Weinert, C.H.; Danylec, N.; Lüersen, K.; Birringer, M.; Bracher, F.; Soukup, S.T.; Kulling, S.E.; et al. The Putative Caloric Restriction Mimetic Resveratrol has Moderate Impact on Insulin Sensitivity, Body Composition, and the Metabolome in Mice. Mol. Nutr. Food Res. 2020, 64, 1901116. [Google Scholar] [CrossRef]
- Zhou, J.; Liao, Z.; Jia, J.; Chen, J.-L.; Xiao, Q. The effects of resveratrol feeding and exercise training on the skeletal muscle function and transcriptome of aged rats. PeerJ 2019, 7, e7199. [Google Scholar] [CrossRef]
- Dey, A.; Guha, P.; Chattopadhyay, S.; Bandyopadhyay, S.K. Biphasic activity of resveratrol on indomethacin-induced gastric ulcers. Biochem. Biophys. Res. Commun. 2009, 381, 90–95. [Google Scholar] [CrossRef]
- Shaito, A.; Posadino, A.M.; Younes, N.; Hasan, H.; Halabi, S.; Alhababi, D.; Al-Mohannadi, A.; Abdel-Rahman, W.M.; Eid, A.H.; Nasrallah, G.K.; et al. Potential Adverse Effects of Resveratrol: A Literature Review. Int. J. Mol. Sci. 2020, 21, 2084. [Google Scholar] [CrossRef]
- Ito, S.; Fujiki, Y.; Matsui, N.; Ojika, M.; Wakamatsu, K. Tyrosinase-catalyzed oxidation of resveratrol produces a highly reactive ortho -quinone: Implications for melanocyte toxicity. Pigment Cell Melanoma Res. 2019, 32, 766–776. [Google Scholar] [CrossRef]
- Bolton, J.L.; Dunlap, T. Formation and Biological Targets of Quinones: Cytotoxic versus Cytoprotective Effects. Chem. Res. Toxicol. 2017, 30, 13–37. [Google Scholar] [CrossRef]
- Porquet, D.; Griñán-Ferré, C.; Ferrer, I.; Camins, A.; Sanfeliu, C.; Del Valle, J.; Pallàs, M. Neuroprotective role of trans-resveratrol in a murine model of familial Alzheimer’s disease. J. Alzheimers Dis. 2014, 42, 1209–1220. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.H.; Hurh, Y.J.; Na, H.K.; Kim, J.H.; Chun, Y.J.; Kim, D.H.; Kang, K.S.; Cho, M.H.; Surh, Y.J. Resveratrol inhibits tcdd-induced expression of cyp1a1 and cyp1b1 and catechol estrogen-mediated oxidative DNA damage in cultured human mammary epitheilial cells. Carcinogenesis 2004, 25, 2005–2013. [Google Scholar] [CrossRef] [PubMed]
- Cauchy, F.; Mebarki, M.; Leporq, B.; Laouirem, S.; Albuquerque, M.; Lambert, S.; Bourgoin, P.; Soubrane, O.; Van Beers, B.E.; Faivre, S.; et al. Strong antineoplastic effects of metformin in preclinical models of liver carcinogenesis. Clin. Sci. (London) 2017, 131, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.; Wu, F.; Li, D.; Yang, L.; Li, Q.; Li, R. Metformin improves obesity-associated inflammation by altering macrophages polarization. Mol. Cell. Endocrinol. 2018, 461, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef]
- Ren, H.; Shao, Y.; Wu, C.; Ma, X.; Lv, C.; Wang, Q. Metformin alleviates oxidative stress and enhances autophagy in diabetic kidney disease via AMPK/SIRT1-FoxO1 pathway. Mol. Cell. Endocrinol. 2020, 500, 110628. [Google Scholar] [CrossRef]
- Song, Y.M.; Lee, W.K.; Lee, Y.H.; Kang, E.S.; Cha, B.S.; Lee, B.W. Metformin restores parkin-mediated mitophagy, suppressed by cytosolic p53. Int. J. Mol. Sci. 2016, 17, 122. [Google Scholar] [CrossRef]
- Rotermund, C.; Machetanz, G.; Fitzgerald, J.C. The Therapeutic Potential of Metformin in Neurodegenerative Diseases. Front. Endocrinol. 2018, 9, 400. [Google Scholar] [CrossRef]
- Andrzejewski, S.; Gravel, S.-P.; Pollak, M.; St-Pierre, J. Metformin directly acts on mitochondria to alter cellular bioenergetics. Cancer Metab. 2014, 2, 12. [Google Scholar] [CrossRef]
- Al-Zaidan, L.; Abu El Ruz, R.; Malki, A. Screening Novel Molecular Targets of Metformin in Breast Cancer by Proteomic Approach. Front. Public Health 2017, 5, 277. [Google Scholar] [CrossRef]
- De Santi, M.; Baldelli, G.; Diotallevi, A.; Galluzzi, L.; Schiavano, G.F.; Brandi, G. Metformin prevents cell tumorigenesis through autophagy-related cell death. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Qian, W.; Jiang, Z.; Cheng, L.; Liang, C.; Sun, L.; Zhou, C.; Gao, L.; Lei, M.; Yan, B.; et al. Metformin suppresses cancer initiation and progression in genetic mouse models of pancreatic cancer. Mol. Cancer 2017, 16, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Quinn, B.J.; Dallos, M.; Kitagawa, H.; Kunnumakkara, A.B.; Memmott, R.M.; Hollander, M.C.; Gills, J.J.; Dennis, P.A. Inhibition of Lung Tumorigenesis by Metformin Is Associated with Decreased Plasma IGF-I and Diminished Receptor Tyrosine Kinase Signaling. Cancer Prev. Res. (Philadelphia) 2013, 6, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, J.C.; Zimprich, A.; Berrio, D.A.C.; Schindler, K.M.; Maurer, B.; Schulte, C.; Bus, C.; Hauser, A.-K.; Kübler, M.; Lewin, R.; et al. Metformin reverses TRAP1 mutation-associated alterations in mitochondrial function in Parkinson’s disease. Brain J. Neurol. 2017, 140, 2444–2459. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.; Jain, P.; Ghumatkar, P.; Tambe, R.; Sathaye, S. Neuroprotective effect of metformin in MPTP-induced Parkinson’s disease in mice. Neuroscience 2014, 277, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ou, Y.; Li, Y.; Hu, S.; Shao, L.W.; Liu, Y. Metformin extends c. Elegans lifespan through lysosomal pathway. eLife 2017, 6, 1–17. [Google Scholar] [CrossRef]
- Jiralerspong, S.; Palla, S.L.; Giordano, S.H.; Meric-Bernstam, F.; Liedtke, C.; Barnett, C.M.; Hsu, L.; Hung, M.-C.; Hortobagyi, G.N.; Gonzalez-Angulo, A.M. Metformin and Pathologic Complete Responses to Neoadjuvant Chemotherapy in Diabetic Patients With Breast Cancer. J. Clin. Oncol. 2009, 27, 3297–3302. [Google Scholar] [CrossRef]
- Wan, G.; Yu, X.; Chen, P.; Wang, X.; Pan, D.; Wang, X.; Li, L.; Cai, X.; Cao, F. Metformin therapy associated with survival benefit in lung cancer patients with diabetes. Oncotarget 2016, 7, 35437–35445. [Google Scholar] [CrossRef]
- Sonnenblick, A.A.; Agbor-Tarh, D.D.; Bradbury, I.I.; Di Cosimo, S.; Azim, H.H.A.; Fumagalli, D.; Sarp, S.S.; Wolff, A.A.C.A.; Andersson, M.M.; Kroep, J.J.; et al. Impact of Diabetes, Insulin, and Metformin Use on the Outcome of Patients With Human Epidermal Growth Factor Receptor 2–Positive Primary Breast Cancer: Analysis From the ALTTO Phase III Randomized Trial. J. Clin. Oncol. 2017, 35, 1421–1429. [Google Scholar] [CrossRef]
- Kulkarni, A.S.; Brutsaert, E.F.; Anghel, V.; Zhang, K.; Bloomgarden, N.; Pollak, M.; Mar, J.C.; Hawkins, M.; Crandall, J.P.; Barzilai, N. Metformin regulates metabolic and nonmetabolic pathways in skeletal muscle and subcutaneous adipose tissues of older adults. Aging Cell 2018, 17, e12723. [Google Scholar] [CrossRef]
- Song, J.; Jiang, G.; Zhang, J.; Guo, J.; Li, Z.; Hao, K.; Liu, L.; Cheng, Z.; Tong, X.; Dai, F. Metformin prolongs lifespan through remodeling the energy distribution strategy in silkworm, Bombyx mori. Aging 2019, 11, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Du, D.; Chen, Q.; Wu, M.; Wu, T.; Wen, J.; Jin, Y.; Zhang, J.; Wang, S. Metformin prevents murine ovarian aging. Aging 2019, 11, 3785–3794. [Google Scholar] [CrossRef] [PubMed]
- Martin-Montalvo, A.; Mercken, E.M.; Mitchell, S.J.; Palacios, H.H.; Mote, P.L.; Scheibye-Knudsen, M.; Gomes, A.P.; Ward, T.M.; Minor, R.K.; Blouin, M.-J.; et al. Metformin improves healthspan and lifespan in mice. Nat. Commun. 2013, 4, 2192. [Google Scholar] [CrossRef] [PubMed]
- Cabreiro, F.; Au, C.; Leung, K.-Y.; Vergara-Irigaray, N.; Cochemé, H.M.; Noori, T.; Weinkove, D.; Schuster, E.; Greene, N.D.; Gems, D. Metformin Retards Aging in C. elegans by Altering Microbial Folate and Methionine Metabolism. Cell 2013, 153, 228–239. [Google Scholar] [CrossRef]
- Gillespie, Z.E.; Wang, C.; Vadan, F.; Yu, T.Y.; Ausió, J.; Kusalik, A.; Eskiw, C.H. Metformin induces the AP-1 transcription factor network in normal dermal fibroblasts. Sci. Rep. 2019, 9, 5369. [Google Scholar] [CrossRef]
- Saisho, Y. Metformin and Inflammation: Its Potential Beyond Glucose-lowering Effect. Endocrine Metab. Immune Disord. Drug Targets 2015, 15, 196–205. [Google Scholar] [CrossRef]
- Ustinova, M.; Silamikelis, I.; Kalnina, I.; Ansone, L.; Rovite, V.; Elbere, I.; Radovica-Spalvina, I.; Fridmanis, D.; Aladyeva, J.; Konrade, I.; et al. Metformin strongly affects transcriptome of peripheral blood cells in healthy individuals. PLoS ONE 2019, 14, e0224835. [Google Scholar] [CrossRef]
- Cree-Green, M.; Bergman, B.C.; Cengiz, E.; Fox, L.A.; Hannon, T.S.; Miller, K.M.; Nathan, B.; Pyle, L.; Kahn, D.; Tansey, M.; et al. Metformin Improves Peripheral Insulin Sensitivity in Youth With Type 1 Diabetes. J. Clin. Endocrinol. Metab. 2019, 104, 3265–3278. [Google Scholar] [CrossRef]
- Yang, X.; Kord-Varkaneh, H.; Talaei, S.; Clark, C.C.; Zanghelini, F.; Tan, S.C.; Zarezadeh, M.; Mousavi, S.M.; Rahmani, J.; Zhang, Y. The influence of metformin on IGF-1 levels in humans: A systematic review and meta-analysis. Pharmacol. Res. 2020, 151, 104588. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).