Epigenetic Targets for Oligonucleotide Therapies of Pulmonary Arterial Hypertension


Abstract
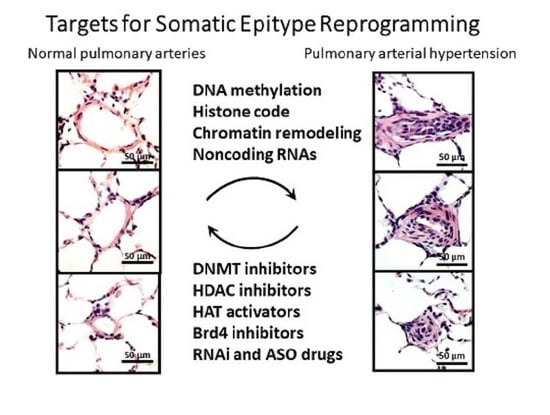
1. Introduction
2. Pulmonary Vascular Remodeling in PAH
3. Epigenetic Targets for Novel Therapy of PAH
3.1. DNA Methylation and Inhibitors
3.2. Histone Modifications and Inhibitors
3.2.1. Histone Deacetylases
3.2.2. Histone Acetyltransferases
3.2.3. Histone Methylation
3.3. Bromodomain Proteins
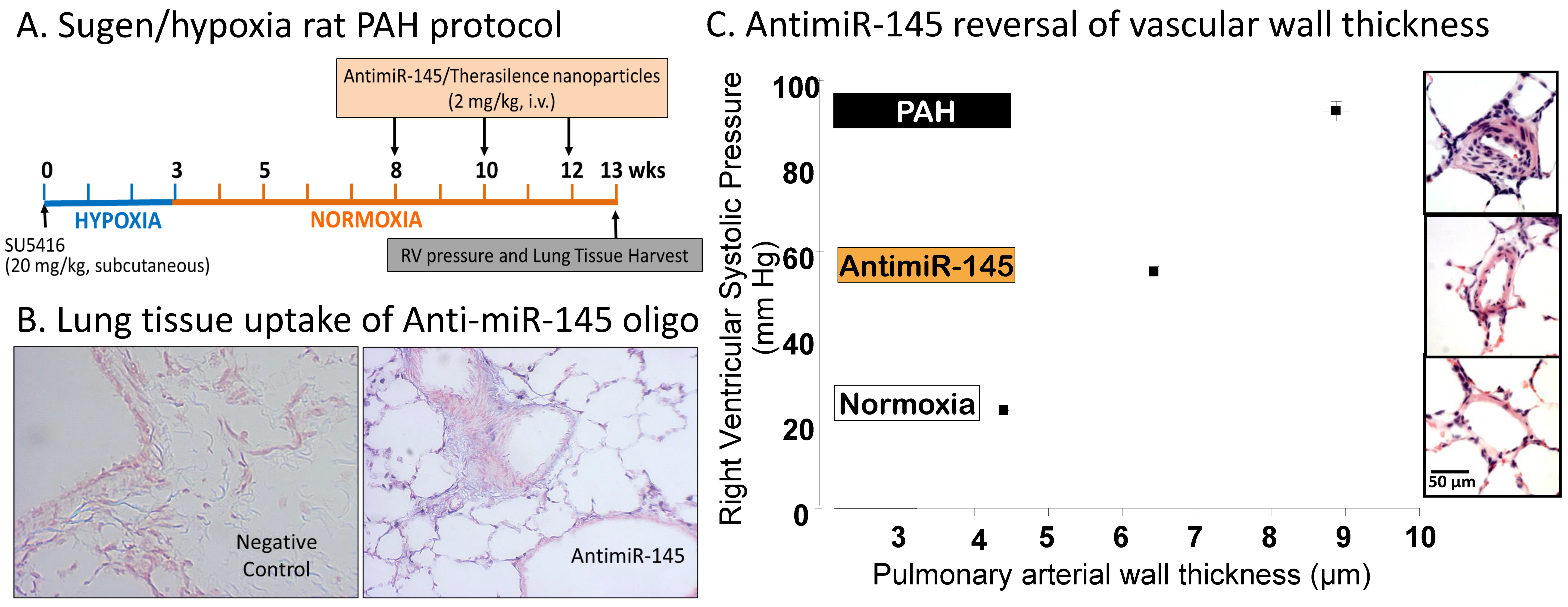
3.4. MicroRNAs
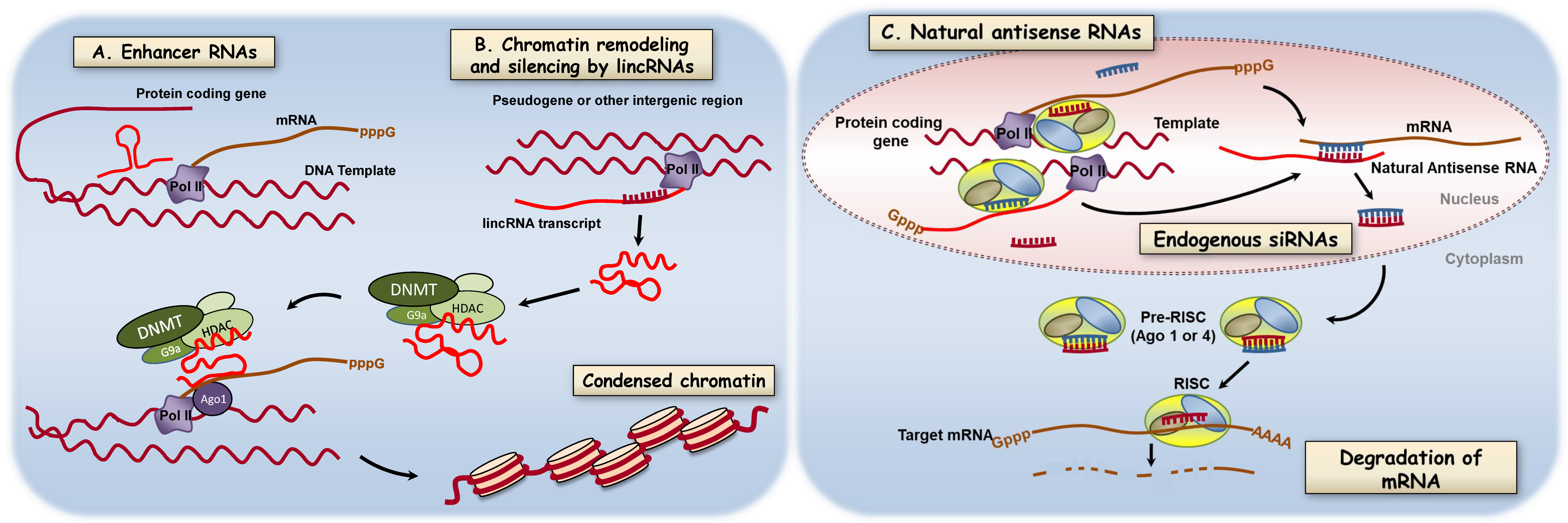
3.5. Long Noncoding RNAs
4. Summary and Future Directions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| DNMT | DNA methyltransferase |
| HAT | histone acetyltransferase |
| HDAC | histone deacetylase |
| lncRNA | long noncoding RNA |
| MALAT1 | metastasis associated lung adenocarcinoma transcript 1 |
| miRNA | microRNA |
| NSD2 | nuclear receptor binding SET domain 2 |
| PAH | pulmonary arterial hypertension |
| RVSP | right ventricular systolic pressure |
References
- Lahiri, D.K.; Maloney, B. Genes are not our destiny: The somatic epitype bridges between the genotype and the phenotype. Nat. Rev. Neurosci. 2006, 7, 976. [Google Scholar] [CrossRef]
- Chelladurai, P.; Seeger, W.; Pullamsetti, S.S. Epigenetic mechanisms in pulmonary arterial hypertension: The need for global perspectives. Eur. Respir. Rev. 2016, 25, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Luna, R.C.P.; de Oliveira, Y.; Lisboa, J.V.C.; Chaves, T.R.; de Araujo, T.A.M.; de Sousa, E.E.; Miranda Neto, M.; Pirola, L.; Braga, V.A.; de Brito Alves, J.L. Insights on the epigenetic mechanisms underlying pulmonary arterial hypertension. Braz. J. Med. Biol. Res. 2018, 51, e7437. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yan, L.; Zhang, Z.; Prado, E.; Fu, L.; Xu, X.; Du, L. Epigenetic Regulation and Its Therapeutic Potential in Pulmonary Hypertension. Front. Pharmacol. 2018, 9, 241. [Google Scholar] [CrossRef] [PubMed]
- Chelladurai, P.; Boucherat, O.; Stenmark, K.; Kracht, M.; Seeger, W.; Bauer, U.M.; Bonnet, S.; Pullamsetti, S.S. Targeting histone acetylation in pulmonary hypertension and right ventricular hypertrophy. Br. J. Pharmacol. 2019. [Google Scholar] [CrossRef]
- Humbert, M.; Guignabert, C.; Bonnet, S.; Dorfmuller, P.; Klinger, J.R.; Nicolls, M.R.; Olschewski, A.J.; Pullamsetti, S.S.; Schermuly, R.T.; Stenmark, K.R.; et al. Pathology and pathobiology of pulmonary hypertension: State of the art and research perspectives. Eur. Respir. J. 2019, 53, 1801887. [Google Scholar] [CrossRef]
- Dhanak, D.; Jackson, P. Development and Classes of Epigenetic Drugs for Cancer. Biochem. Biophys. Res. Commun. 2014, 455, 58–69. [Google Scholar] [CrossRef]
- Haldar, S.M.; McKinsey, T.A. BET-ting on chromatin-based therapeutics for heart failure. J. Mol. Cell Cardiol. 2014, 74C, 98–102. [Google Scholar] [CrossRef]
- Natarajan, R. Drugs targeting epigenetic histone acetylation in vascular smooth muscle cells for restenosis and atherosclerosis. Arter. Thromb. Vasc. Biol. 2011, 31, 725–727. [Google Scholar] [CrossRef]
- Tao, H.; Shi, K.H.; Yang, J.J.; Huang, C.; Zhan, H.Y.; Li, J. Histone deacetylases in cardiac fibrosis: Current perspectives for therapy. Cell Signal. 2014, 26, 521–527. [Google Scholar] [CrossRef]
- Doudna, J.A. The promise and challenge of therapeutic genome editing. Nature 2020, 578, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Prins, K.W.; Thenappan, T. World Health Organization Group I Pulmonary Hypertension: Epidemiology and Pathophysiology. Cardiol. Clin. 2016, 34, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Morrell, N.W.; Archer, S.L.; Defelice, A.; Evans, S.; Fiszman, M.; Martin, T.; Saulnier, M.; Rabinovitch, M.; Schermuly, R.; Stewart, D.; et al. Anticipated classes of new medications and molecular targets for pulmonary arterial hypertension. Pulm. Circ. 2013, 3, 226–244. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M.; Archer, S.L.; Dorfmuller, P.; Erzurum, S.C.; Guignabert, C.; Michelakis, E.; Rabinovitch, M.; Schermuly, R.; Stenmark, K.R.; Morrell, N.W. Relevant issues in the pathology and pathobiology of pulmonary hypertension. J. Am. Coll. Cardiol. 2013, 62 (Suppl. 25), D4–D12. [Google Scholar] [CrossRef]
- Stratton, M.S.; Farina, F.M.; Elia, L. Epigenetics and vascular diseases. J. Mol. Cell. Cardiol. 2019, 133, 148–163. [Google Scholar] [CrossRef]
- White, K.; Loscalzo, J.; Chan, S.Y. Holding our breath: The emerging and anticipated roles of microRNA in pulmonary hypertension. Pulm. Circ. 2012, 2, 278–290. [Google Scholar] [CrossRef]
- Archer, S.L.; Marsboom, G.; Kim, G.H.; Zhang, H.J.; Toth, P.T.; Svensson, E.C.; Dyck, J.R.; Gomberg-Maitland, M.; Thebaud, B.; Husain, A.N.; et al. Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: A basis for excessive cell proliferation and a new therapeutic target. Circulation 2010, 121, 2661–2671. [Google Scholar] [CrossRef]
- Xu, X.F.; Ma, X.L.; Shen, Z.; Wu, X.L.; Cheng, F.; Du, L.Z. Epigenetic regulation of the endothelial nitric oxide synthase gene in persistent pulmonary hypertension of the newborn rat. J. Hypertens. 2010, 28, 2227–2235. [Google Scholar] [CrossRef]
- Pullamsetti, S.S.; Perros, F.; Chelladurai, P.; Yuan, J.; Stenmark, K. Transcription factors, transcriptional coregulators, and epigenetic modulation in the control of pulmonary vascular cell phenotype: Therapeutic implications for pulmonary hypertension (2015 Grover Conference series). Pulm. Circ. 2016, 6, 448–464. [Google Scholar] [CrossRef]
- Kim, J.D.; Lee, A.; Choi, J.; Park, Y.; Kang, H.; Chang, W.; Lee, M.S.; Kim, J. Epigenetic modulation as a therapeutic approach for pulmonary arterial hypertension. Exp. Mol. Med. 2015, 47, e175. [Google Scholar] [CrossRef]
- Gros, C.; Fahy, J.; Halby, L.; Dufau, I.; Erdmann, A.; Gregoire, J.M.; Ausseil, F.; Vispe, S.; Arimondo, P.B. DNA methylation inhibitors in cancer: Recent and future approaches. Biochimie 2012, 94, 2280–2296. [Google Scholar] [CrossRef] [PubMed]
- Perros, F.; Cohen-Kaminsky, S.; Gambaryan, N.; Girerd, B.; Raymond, N.; Klingelschmitt, I.; Huertas, A.; Mercier, O.; Fadel, E.; Simonneau, G.; et al. Cytotoxic cells and granulysin in pulmonary arterial hypertension and pulmonary veno-occlusive disease. Am. J. Respir. Crit. Care Med. 2013, 187, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kahaleh, B. Epigenetic repression of bone morphogenetic protein receptor II expression in scleroderma. J. Cell. Mol. Med. 2013, 17, 1291–1299. [Google Scholar] [CrossRef] [PubMed]
- Pousada, G.; Baloira, A.; Valverde, D. Methylation Analysis of the BMPR2 Gene Promoter Region in Patients With Pulmonary Arterial Hypertension. Arch. Bronconeumol. 2016, 52, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Yan, Y.; Chen, J.W.; Yuan, P.; Wang, X.J.; Jiang, R.; Wang, L.; Zhao, Q.H.; Wu, W.H.; Simonneau, G.; et al. Hypermethylation of BMPR2 Promoter Occurs in Patients with Heritable Pulmonary Arterial Hypertension and Inhibits BMPR2 Expression. Am. J. Respir. Crit. Care Med. 2017, 196, 925–928. [Google Scholar] [CrossRef] [PubMed]
- Potus, F.; Pauciulo, M.W.; Cook, E.K.; Zhu, N.; Hsieh, A.; Welch, C.L.; Shen, Y.; Tian, L.; Lima, P.; Mewburn, J.; et al. Novel Mutations and Decreased Expression of the Epigenetic Regulator TET2 in Pulmonary Arterial Hypertension. Circulation 2020, 141, 1986–2000. [Google Scholar] [CrossRef]
- Kato, N.; Loh, M.; Takeuchi, F.; Verweij, N.; Wang, X.; Zhang, W.; Kelly, T.N.; Saleheen, D.; Lehne, B.; Mateo Leach, I.; et al. Trans-ancestry genome-wide association study identifies 12 genetic loci influencing blood pressure and implicates a role for DNA methylation. Nat. Genet. 2015, 47, 1282–1293. [Google Scholar] [CrossRef]
- Pechalrieu, D.; Etievant, C.; Arimondo, P.B. DNA methyltransferase inhibitors in cancer: From pharmacology to translational studies. Biochem. Pharmacol. 2017, 129, 1–13. [Google Scholar] [CrossRef]
- Rudenko, A.; Dawlaty, M.M.; Seo, J.; Cheng, A.W.; Meng, J.; Le, T.; Faull, K.F.; Jaenisch, R.; Tsai, L.H. Tet1 is critical for neuronal activity-regulated gene expression and memory extinction. Neuron 2013, 79, 1109–1122. [Google Scholar] [CrossRef]
- Feng, J.; Zhou, Y.; Campbell, S.L.; Le, T.; Li, E.; Sweatt, J.D.; Silva, A.J.; Fan, G. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat. Neurosci. 2010, 13, 423–430. [Google Scholar] [CrossRef]
- Southwell, A.L.; Skotte, N.H.; Bennett, C.F.; Hayden, M.R. Antisense oligonucleotide therapeutics for inherited neurodegenerative diseases. Trends Mol. Med. 2012, 18, 634–643. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Paredes, M.; Esteller, M. Cancer epigenetics reaches mainstream oncology. Nat. Med. 2011, 17, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Zhang, L. Serotonylation: A novel histone H3 marker. Signal Transduct. Target. Ther. 2019, 4, 15. [Google Scholar] [CrossRef] [PubMed]
- Lepack, A.E.; Werner, C.T.; Stewart, A.F.; Fulton, S.L.; Zhong, P.; Farrelly, L.A.; Smith, A.C.W.; Ramakrishnan, A.; Lyu, Y.; Bastle, R.M.; et al. Dopaminylation of histone H3 in ventral tegmental area regulates cocaine seeking. Science 2020, 368, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Riddle, S.R.; Frid, M.G.; El Kasmi, K.C.; McKinsey, T.A.; Sokol, R.J.; Strassheim, D.; Meyrick, B.; Yeager, M.E.; Flockton, A.R.; et al. Emergence of fibroblasts with a proinflammatory epigenetically altered phenotype in severe hypoxic pulmonary hypertension. J. Immunol. 2011, 187, 2711–2722. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Chen, C.N.; Hajji, N.; Oliver, E.; Cotroneo, E.; Wharton, J.; Wang, D.; Li, M.; McKinsey, T.A.; Stenmark, K.R.; et al. Histone deacetylation inhibition in pulmonary hypertension: Therapeutic potential of valproic acid and suberoylanilide hydroxamic acid. Circulation 2012, 126, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Li, X.; Aquadro, E.; Haigh, S.; Zhou, J.; Stepp, D.W.; Weintraub, N.L.; Barman, S.A.; Fulton, D.J. Inhibition of histone deacetylase reduces transcription of NADPH oxidases and ROS production and ameliorates pulmonary arterial hypertension. Free Radic. Biol. Med. 2016, 99, 167–178. [Google Scholar] [CrossRef]
- Boucherat, O.; Chabot, S.; Paulin, R.; Trinh, I.; Bourgeois, A.; Potus, F.; Lampron, M.C.; Lambert, C.; Breuils-Bonnet, S.; Nadeau, V.; et al. HDAC6: A Novel Histone Deacetylase Implicated in Pulmonary Arterial Hypertension. Sci. Rep. 2017, 7, 4546. [Google Scholar] [CrossRef]
- Mumby, S.; Gambaryan, N.; Meng, C.; Perros, F.; Humbert, M.; Wort, S.J.; Adcock, I.M. Bromodomain and extra-terminal protein mimic JQ1 decreases inflammation in human vascular endothelial cells: Implications for pulmonary arterial hypertension. Respirology 2017, 22, 157–164. [Google Scholar] [CrossRef]
- Strassheim, D.; Riddle, S.R.; Burke, D.L.; Geraci, M.W.; Stenmark, K.R. Prostacyclin inhibits IFN-gamma-stimulated cytokine expression by reduced recruitment of CBP/p300 to STAT1 in a SOCS-1-independent manner. J. Immunol. 2009, 183, 6981–6988. [Google Scholar] [CrossRef]
- Cedar, H.; Bergman, Y. Linking DNA methylation and histone modification: Patterns and paradigms. Nat. Rev. Genet. 2009, 10, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Bennett, R.L.; Swaroop, A.; Troche, C.; Licht, J.D. The Role of Nuclear Receptor-Binding SET Domain Family Histone Lysine Methyltransferases in Cancer. Cold Spring Harb. Perspect. Med. 2017, 7, a026708. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.L.; Liu, Z.B.; Zhu, R.R.; Huang, H.; Xu, Q.R.; Xu, H.; Zeng, L.; Li, Y.Y.; Huang, C.H.; Wu, Q.C.; et al. NSD2 silencing alleviates pulmonary arterial hypertension by inhibiting trehalose metabolism and autophagy. Clin. Sci. 2019, 133, 1085–1096. [Google Scholar] [CrossRef] [PubMed]
- Arrowsmith, C.H.; Bountra, C.; Fish, P.V.; Lee, K.; Schapira, M. Epigenetic protein families: A new frontier for drug discovery. Nat. Rev. Drug Discov. 2012, 11, 384–400. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Prakash, C.; Sum, C.; Gong, Y.; Li, Y.; Kwok, J.J.T.; Thiessen, N.; Pettersson, S.; Jones, S.J.M.; Knapp, S.; et al. Bromodomain-containing protein 4 (BRD4) regulates RNA polymerase II serine 2 phosphorylation in human CD4+ T cells. J. Biol. Chem. 2012, 287, 43137–43155. [Google Scholar] [CrossRef] [PubMed]
- Meloche, J.; Renard, S.; Provencher, S.; Bonnet, S. Anti-inflammatory and immunosuppressive agents in PAH. Handb. Exp. Pharmacol. 2013, 218, 437–476. [Google Scholar] [PubMed]
- Van der Feen, D.E.; Kurakula, K.; Tremblay, E.; Boucherat, O.; Bossers, G.P.; Szulcek, R.; Bourgeois, A.; Lampron, M.C.; Habbout, K.; Martineau, S.; et al. Multicenter Preclinical Validation of BET Inhibition for the Treatment of Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2019, 200, 910–920. [Google Scholar] [CrossRef]
- Piquereau, J.; Boet, A.; Pechoux, C.; Antigny, F.; Lambert, M.; Gressette, M.; Ranchoux, B.; Gambaryan, N.; Domergue, V.; Mumby, S.; et al. The BET Bromodomain Inhibitor I-BET-151 Induces Structural and Functional Alterations of the Heart Mitochondria in Healthy Male Mice and Rats. Int. J. Mol. Sci. 2019, 20, 1527. [Google Scholar] [CrossRef]
- Brock, M.; Trenkmann, M.; Gay, R.E.; Michel, B.A.; Gay, S.; Fischler, M.; Ulrich, S.; Speich, R.; Huber, L.C. Interleukin-6 modulates the expression of the bone morphogenic protein receptor type II through a novel STAT3-microRNA cluster 17/92 pathway. Circ. Res. 2009, 104, 1184–1191. [Google Scholar] [CrossRef]
- Caruso, P.; MacLean, M.R.; Khanin, R.; McClure, J.; Soon, E.; Southgate, M.; MacDonald, R.A.; Greig, J.A.; Robertson, K.E.; Masson, R.; et al. Dynamic changes in lung microRNA profiles during the development of pulmonary hypertension due to chronic hypoxia and monocrotaline. Arter. Thromb. Vasc. Biol. 2010, 30, 716–723. [Google Scholar] [CrossRef] [PubMed]
- Courboulin, A.; Paulin, R.; Giguere, N.J.; Saksouk, N.; Perreault, T.; Meloche, J.; Paquet, E.R.; Biardel, S.; Provencher, S.; Cote, J.; et al. Role for miR-204 in human pulmonary arterial hypertension. J. Exp. Med. 2011, 208, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Caruso, P.; Dempsie, Y.; Stevens, H.C.; McDonald, R.A.; Long, L.; Lu, R.; White, K.; Mair, K.M.; McClure, J.D.; Southwood, M.; et al. A role for miR-145 in pulmonary arterial hypertension: Evidence from mouse models and patient samples. Circ. Res. 2012, 111, 290–300. [Google Scholar] [CrossRef]
- Bockmeyer, C.L.; Maegel, L.; Janciauskiene, S.; Rische, J.; Lehmann, U.; Maus, U.A.; Nickel, N.; Haverich, A.; Hoeper, M.M.; Golpon, H.A.; et al. Plexiform vasculopathy of severe pulmonary arterial hypertension and microRNA expression. J. Heart Lung Transplant. 2012, 31, 764–772. [Google Scholar] [CrossRef]
- Comer, B.S.; Ba, M.; Singer, C.A.; Gerthoffer, W.T. Epigenetic targets for novel therapies of lung diseases. Pharmacol. Ther. 2015, 147, 91–110. [Google Scholar] [CrossRef] [PubMed]
- Boucherat, O.; Potus, F.; Bonnet, S. microRNA and Pulmonary Hypertension. Adv. Exp. Med. Biol. 2015, 888, 237–252. [Google Scholar] [PubMed]
- Pullamsetti, S.S.; Doebele, C.; Fischer, A.; Savai, R.; Kojonazarov, B.; Dahal, B.K.; Ghofrani, H.A.; Weissmann, N.; Grimminger, F.; Bonauer, A.; et al. Inhibition of microRNA-17 improves lung and heart function in experimental pulmonary hypertension. Am. J. Respir. Crit Care Med. 2012, 185, 409–419. [Google Scholar] [CrossRef]
- Parikh, V.N.; Jin, R.C.; Rabello, S.; Gulbahce, N.; White, K.; Hale, A.; Cottrill, K.A.; Shaik, R.S.; Waxman, A.B.; Zhang, Y.Y.; et al. MicroRNA-21 integrates pathogenic signaling to control pulmonary hypertension: Results of a network bioinformatics approach. Circulation 2012, 125, 1520–1532. [Google Scholar] [CrossRef]
- Brock, M.; Samillan, V.J.; Trenkmann, M.; Schwarzwald, C.; Ulrich, S.; Gay, R.E.; Gassmann, M.; Ostergaard, L.; Gay, S.; Speich, R.; et al. AntagomiR directed against miR-20a restores functional BMPR2 signalling and prevents vascular remodelling in hypoxia-induced pulmonary hypertension. Eur. Heart J. 2014, 35, 3203–3211. [Google Scholar] [CrossRef]
- McLendon, J.M.; Joshi, S.R.; Sparks, J.; Matar, M.; Fewell, J.G.; Abe, K.; Oka, M.; McMurtry, I.F.; Gerthoffer, W.T. Lipid nanoparticle delivery of a microRNA-145 inhibitor improves experimental pulmonary hypertension. J. Control. Release 2015, 210, 67–75. [Google Scholar] [CrossRef]
- Gubrij, I.B.; Pangle, A.K.; Pang, L.; Johnson, L.G. Reversal of microRNA dysregulation in an animal model of pulmonary hypertension. PLoS ONE 2016, 11, e0147827. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kang, Y.; Kojima, Y.; Lighthouse, J.K.; Hu, X.; Aldred, M.A.; McLean, D.L.; Park, H.; Comhair, S.A.; Greif, D.M.; et al. An endothelial apelin-FGF link mediated by miR-424 and miR-503 is disrupted in pulmonary arterial hypertension. Nat. Med. 2013, 19, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Chen, Z.; Chen, Y.; Lv, H.; Lu, H.; Yan, F.; Li, L.; Zhang, W.; Shi, J. Long non-coding RNA CASC2 suppresses pulmonary artery smooth muscle cell proliferation and phenotypic switch in hypoxia-induced pulmonary hypertension. Respir. Res. 2019, 20, 53. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Xu, X.; Yan, C.; Shi, Y.; Hu, Y.; Dong, L.; Ying, S.; Ying, K.; Zhang, R. LncRNA H19 promotes the proliferation of pulmonary artery smooth muscle cells through AT1R via sponging let-7b in monocrotaline-induced pulmonary arterial hypertension. Respir. Res. 2018, 19, 254. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, Y.; Yan, L.; Wang, S.; Zhang, M.; Ma, C.; Zheng, X.; Chen, H.; Zhu, D. Long noncoding RNA Hoxaas3 contributes to hypoxia-induced pulmonary artery smooth muscle cell proliferation. Cardiovasc. Res. 2019, 115, 647–657. [Google Scholar] [CrossRef]
- Wei, C.; Henderson, H.; Spradley, C.; Li, L.; Kim, I.K.; Kumar, S.; Hong, N.; Arroliga, A.C.; Gupta, S. Circulating miRNAs as potential marker for pulmonary hypertension. PLoS ONE 2013, 8, e64396. [Google Scholar] [CrossRef]
- Chinnappan, M.; Gunewardena, S.; Chalise, P.; Dhillon, N.K. Analysis of lncRNA-miRNA-mRNA Interactions in Hyper-proliferative Human Pulmonary Arterial Smooth Muscle Cells. Sci. Rep. 2019, 9, 10533. [Google Scholar] [CrossRef]
- Kang, K.; Peng, X.; Zhang, X.; Wang, Y.; Zhang, L.; Gao, L.; Weng, T.; Zhang, H.; Ramchandran, R.; Raj, J.U.; et al. MicroRNA-124 suppresses the transactivation of nuclear factor of activated T cells by targeting multiple genes and inhibits the proliferation of pulmonary artery smooth muscle cells. J. Biol. Chem. 2013, 288, 25414–25427. [Google Scholar] [CrossRef]
- Chen, J.; Guo, J.; Cui, X.; Dai, Y.; Tang, Z.; Qu, J.; Raj, J.U.; Hu, Q.; Gou, D. The Long Noncoding RNA LnRPT Is Regulated by PDGF-BB and Modulates the Proliferation of Pulmonary Artery Smooth Muscle Cells. Am. J. Respir. Cell Mol. Biol. 2018, 58, 181–193. [Google Scholar] [CrossRef]
- Oldham, W.M. The Long Noncoding RNA LnRPT Puts the Brakes on Pulmonary Artery Smooth Muscle Cell Proliferation. Am. J. Respir. Cell Mol. Biol. 2018, 58, 138–139. [Google Scholar] [CrossRef]
- Wang, D.; Xu, H.; Wu, B.; Jiang, S.; Pan, H.; Wang, R.; Chen, J. Long noncoding RNA MALAT1 sponges miR1243p.1/KLF5 to promote pulmonary vascular remodeling and cell cycle progression of pulmonary artery hypertension. Int. J. Mol. Med. 2019, 44, 871–884. [Google Scholar] [PubMed]
- Mou, X.; Wang, J.; Wang, L.; Wang, S. Correlation Between Single Nucleotide Polymorphisms of the rs664589 Locus in the Long-Chain Noncoding RNA Lung Adenocarcinoma Metastasis-Associated Gene 1, Hypertension, and Its Mechanism. Genet. Test. Mol. Biomark. 2020, 24, 120–130. [Google Scholar] [CrossRef]
- Yang, L.; Liang, H.; Shen, L.; Guan, Z.; Meng, X. LncRNA Tug1 involves in the pulmonary vascular remodeling in mice with hypoxic pulmonary hypertension via the microRNA-374c-mediated Foxc1. Life Sci. 2019, 237, 116769. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.T.; Sun, R.L.; Yin, Y.L.; Quan, J.P.; Song, P.; Xu, J.; Zhang, M.X.; Li, P. Long noncoding RNA UCA1 promotes the proliferation of hypoxic human pulmonary artery smooth muscle cells. Pflug. Arch. 2019, 471, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Jalali, S.; Ramanathan, G.K.; Parthasarathy, P.T.; Aljubran, S.; Galam, L.; Yunus, A.; Garcia, S.; Cox, R.R., Jr.; Lockey, R.F.; Kolliputi, N. Mir-206 regulates pulmonary artery smooth muscle cell proliferation and differentiation. PLoS ONE 2012, 7, e46808. [Google Scholar] [CrossRef]
- Freilich, R.W.; Woodbury, M.E.; Ikezu, T. Integrated expression profiles of mRNA and miRNA in polarized primary murine microglia. PLoS ONE 2013, 8, e79416. [Google Scholar] [CrossRef]
- Collison, A.; Mattes, J.; Plank, M.; Foster, P.S. Inhibition of house dust mite-induced allergic airways disease by antagonism of microRNA-145 is comparable to glucocorticoid treatment. J. Allergy Clin. Immunol. 2011, 128, 160–167. [Google Scholar] [CrossRef]
- Kim, R.Y.; Horvat, J.C.; Pinkerton, J.W.; Starkey, M.R.; Essilfie, A.T.; Mayall, J.R.; Nair, P.M.; Hansbro, N.G.; Jones, B.; Haw, T.J.; et al. MicroRNA-21 drives severe, steroid-insensitive experimental asthma by amplifying phosphoinositide 3-kinase-mediated suppression of histone deacetylase 2. J. Allergy Clin. Immunol. 2017, 139, 519–532. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, H.; Li, M.; Frid, M.G.; Flockton, A.R.; McKeon, B.A.; Yeager, M.E.; Fini, M.A.; Morrell, N.W.; Pullamsetti, S.S.; et al. MicroRNA-124 controls the proliferative, migratory, and inflammatory phenotype of pulmonary vascular fibroblasts. Circ. Res. 2014, 114, 67–78. [Google Scholar] [CrossRef]
- Natoli, G.; Andrau, J.C. Noncoding transcription at enhancers: General principles and functional models. Annu. Rev. Genet. 2012, 46, 1–19. [Google Scholar] [CrossRef]
- Ulitsky, I.; Bartel, D.P. lincRNAs: Genomics, evolution, and mechanisms. Cell 2013, 154, 26–46. [Google Scholar] [CrossRef] [PubMed]
- Cabili, M.N.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011, 25, 1915–1927. [Google Scholar] [CrossRef] [PubMed]
- Rinn, J.L.; Chang, H.Y. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef] [PubMed]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef]
- Zong, X.; Tripathi, V.; Prasanth, K.V. RNA splicing control: Yet another gene regulatory role for long nuclear noncoding RNAs. RNA Biol. 2011, 8, 968–977. [Google Scholar] [CrossRef]
- Kretz, M.; Siprashvili, Z.; Chu, C.; Webster, D.E.; Zehnder, A.; Qu, K.; Lee, C.S.; Flockhart, R.J.; Groff, A.F.; Chow, J.; et al. Control of somatic tissue differentiation by the long non-coding RNA TINCR. Nature 2013, 493, 231–235. [Google Scholar] [CrossRef]
- Hacisuleyman, E.; Goff, L.A.; Trapnell, C.; Williams, A.; Henao-Mejia, J.; Sun, L.; McClanahan, P.; Hendrickson, D.G.; Sauvageau, M.; Kelley, D.R.; et al. Topological organization of multichromosomal regions by the long intergenic noncoding RNA Firre. Nat. Struct. Mol. Biol. 2014, 21, 198–206. [Google Scholar] [CrossRef]
- Wang, X.; Yan, C.; Xu, X.; Dong, L.; Su, H.; Hu, Y.; Zhang, R.; Ying, K. Long noncoding RNA expression profiles of hypoxic pulmonary hypertension rat model. Gene 2016, 579, 23–28. [Google Scholar] [CrossRef]
- Wahlestedt, C. Targeting long non-coding RNA to therapeutically upregulate gene expression. Nat. Rev. Drug Discov. 2013, 12, 433–446. [Google Scholar] [CrossRef]
- Michalik, K.M.; You, X.; Manavski, Y.; Doddaballapur, A.; Zornig, M.; Braun, T.; John, D.; Ponomareva, Y.; Chen, W.; Uchida, S.; et al. Long noncoding RNA MALAT1 regulates endothelial cell function and vessel growth. Circ. Res. 2014, 114, 1389–1397. [Google Scholar] [CrossRef]
- Hassoun, P.M. Inflammation in chronic thromboembolic pulmonary hypertension: Accomplice or bystander in altered angiogenesis? Eur. Respir. J. 2015, 46, 303–306. [Google Scholar] [CrossRef] [PubMed]
- Li, C.H.; Chen, Y. Targeting long non-coding RNAs in cancers: Progress and prospects. Int. J. Biochem. Cell Biol. 2013, 45, 1895–1910. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M.; Marecki, J.C.; Richter, A.; Fijalkowska, I.; Flores, S. Pathology of pulmonary hypertension. Clin. Chest Med. 2007, 28, 23–42. [Google Scholar] [CrossRef] [PubMed]
- Boucherat, O.; Vitry, G.; Trinh, I.; Paulin, R.; Provencher, S.; Bonnet, S. The cancer theory of pulmonary arterial hypertension. Pulm. Circ. 2017, 7, 285–299. [Google Scholar] [CrossRef]
- Sitbon, O.; Gomberg-Maitland, M.; Granton, J.; Lewis, M.I.; Mathai, S.C.; Rainisio, M.; Stockbridge, N.L.; Wilkins, M.R.; Zamanian, R.T.; Rubin, L.J. Clinical trial design and new therapies for pulmonary arterial hypertension. Eur. Respir. J. 2019, 53, 1801908. [Google Scholar] [CrossRef]
- Sommer, N.; Ghofrani, H.A.; Pak, O.; Bonnet, S.; Provencher, S.; Sitbon, O.; Rosenkranz, S.; Hoeper, M.M.; Kiely, D.G. Current and future treatments of pulmonary arterial hypertension. Br. J. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Barros, S.A.; Gollob, J.A. Safety profile of RNAi nanomedicines. Adv. Drug Deliv. Rev. 2012, 64, 1730–1737. [Google Scholar] [CrossRef]
- Subhan, M.A.; Torchilin, V.P. siRNA based drug design, quality, delivery and clinical translation. Nanomedicine 2020, 29, 102239. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNAs | Long Noncoding RNAs |
|---|---|
| let-7f/miR-22/30 [51] | CASC2 [63] |
| miR-17/92 cluster [50,57] | H19 [64] |
| miR-21 [51] | Hoxaas3 [65] |
| miR-23b/130a/191/451/1246 3 [66] | HOXB1, CBL, GDF7, RND1 [67] |
| miR-124 [68] | LnRPT [69,70] |
| miR-143 [54] | MALAT1 [71], [72] |
| miR-145 [53] | Tug1 [73] |
| miR-204 [52] | UCA1 [74] |
| miR-206 [75] | |
| miR-322/451 [51] | |
| miR-424/503 [62] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gerthoffer, W. Epigenetic Targets for Oligonucleotide Therapies of Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2020, 21, 9222. https://doi.org/10.3390/ijms21239222
Gerthoffer W. Epigenetic Targets for Oligonucleotide Therapies of Pulmonary Arterial Hypertension. International Journal of Molecular Sciences. 2020; 21(23):9222. https://doi.org/10.3390/ijms21239222
Chicago/Turabian StyleGerthoffer, William. 2020. "Epigenetic Targets for Oligonucleotide Therapies of Pulmonary Arterial Hypertension" International Journal of Molecular Sciences 21, no. 23: 9222. https://doi.org/10.3390/ijms21239222
APA StyleGerthoffer, W. (2020). Epigenetic Targets for Oligonucleotide Therapies of Pulmonary Arterial Hypertension. International Journal of Molecular Sciences, 21(23), 9222. https://doi.org/10.3390/ijms21239222