Transcriptome Remodeling in Gradual Development of Inverse Resistance between Paclitaxel and Cisplatin in Ovarian Cancer Cells

 , , , ,
, , , ,
Abstract
1. Introduction
- (1)
- Decreased CDDP influx, mediated by the copper transporter: High-affinity Cu transporter CTR1.
- (2)
- Increased CDDP efflux by copper exporters: ATPase copper transporting alpha and beta (ATP7A and ATP7B).
- (3)
- CDDP inactivation by glutathione S-transferase (GST) and cytochrome P450 (CYP) systems.
- (4)
- (1)
- Increased PTX efflux by membrane-bound transporters, particularly by over-expressed proteins from the ABC (ATP-binding cassette) family.
- (2)
- Modulation of the mitochondrial apoptotic pathway, which is induced by a prolonged cell cycle arrest at the G2/M check point after the PTX and/or CDDP treatment. Increased expression of anti-apoptotic proteins, such as BCL2 apoptosis regulator (BCL2) and BCL2-like 1 (BCL-XL); or decreased expression of pro-apoptotic proteins, such as BCL2 associated X, apoptosis regulator (BAX) and caspases, conferring resistance to both PTX and CDDP [8,9].
2. Results
2.1. Development of the A2780 Cell Model with Acquired Resistance to PTX
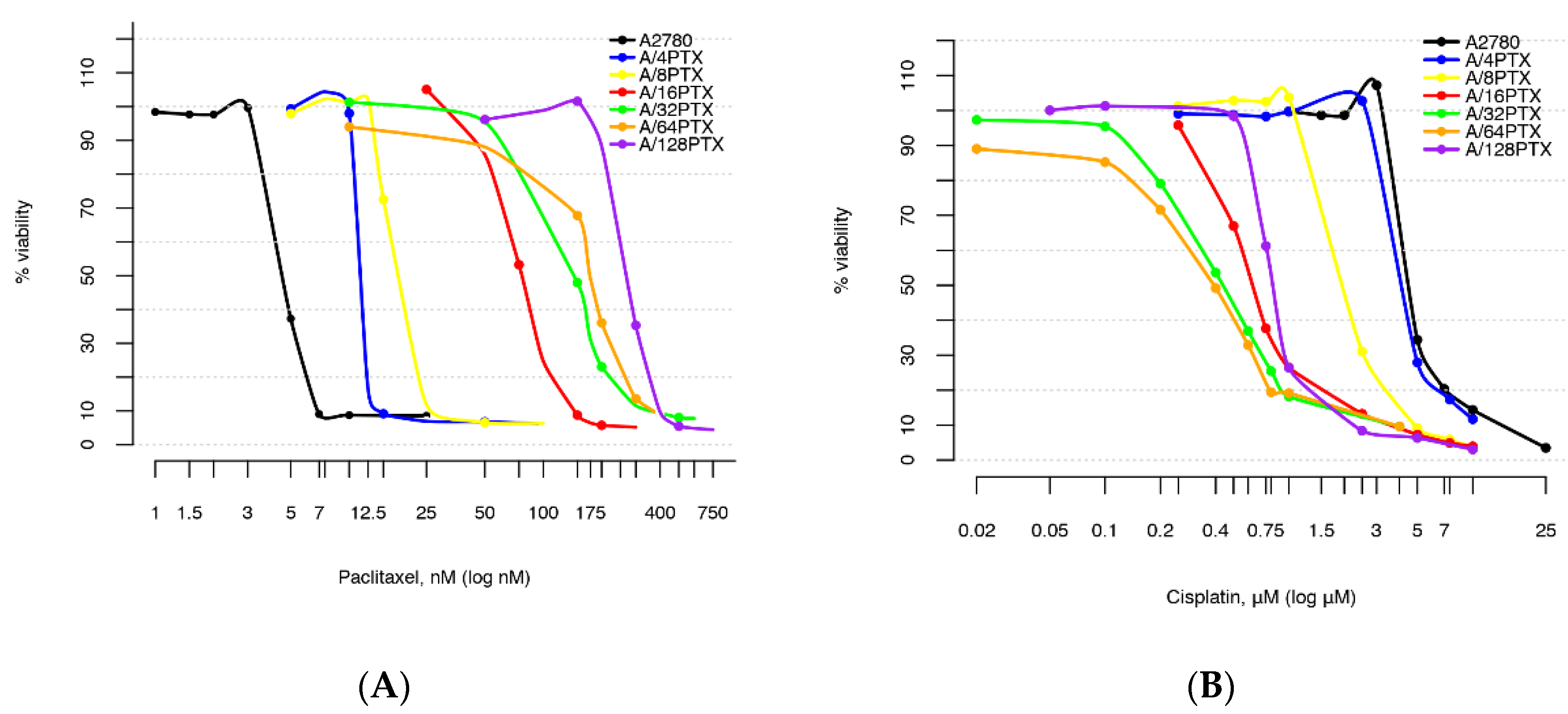
2.2. Comparison of Cell Subline Series with a Very Wide Range of Induced Resistance to PTX and Sensitivity to CDDP
2.2.1. Determination of Drug Concentrations for Selected Values of Viability
- A2780 sublines acquired the inverse resistance to PTX and CDDP—the level of resistance to PTX increased as an effect of the drug treatment and the level of resistance to CDDP decreased as a bystander effect.
- For both PTX and CDDP, the changes in resistance were from a few- to several dozen-fold.
2.2.2. Comparison of Differences in Cell Survival
2.3. Gene Expression Analysis in the A2780 Parental Cell Line and Six Derived Sublines with PTX-Induced Inverse Resistance to PTX and CDDP
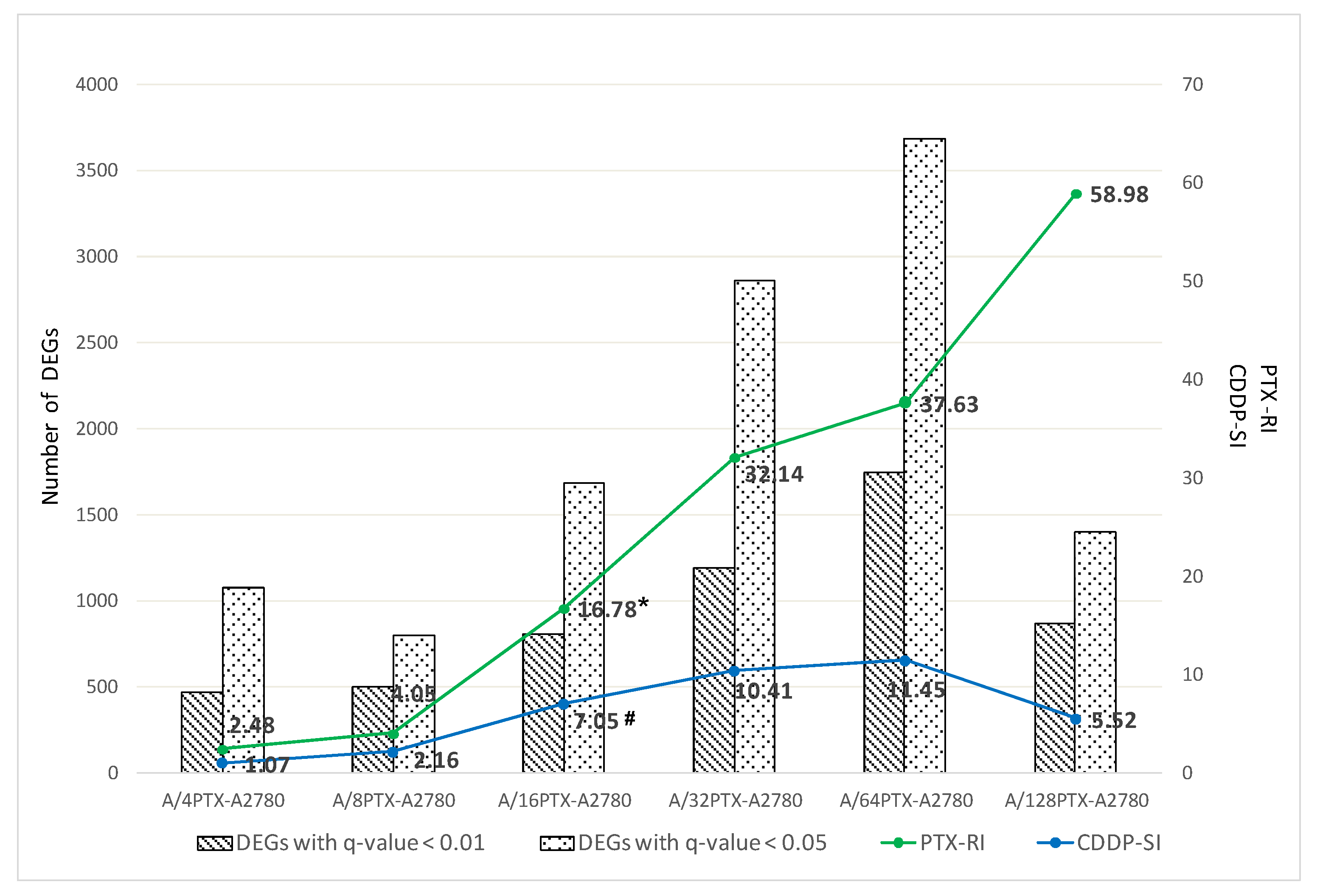
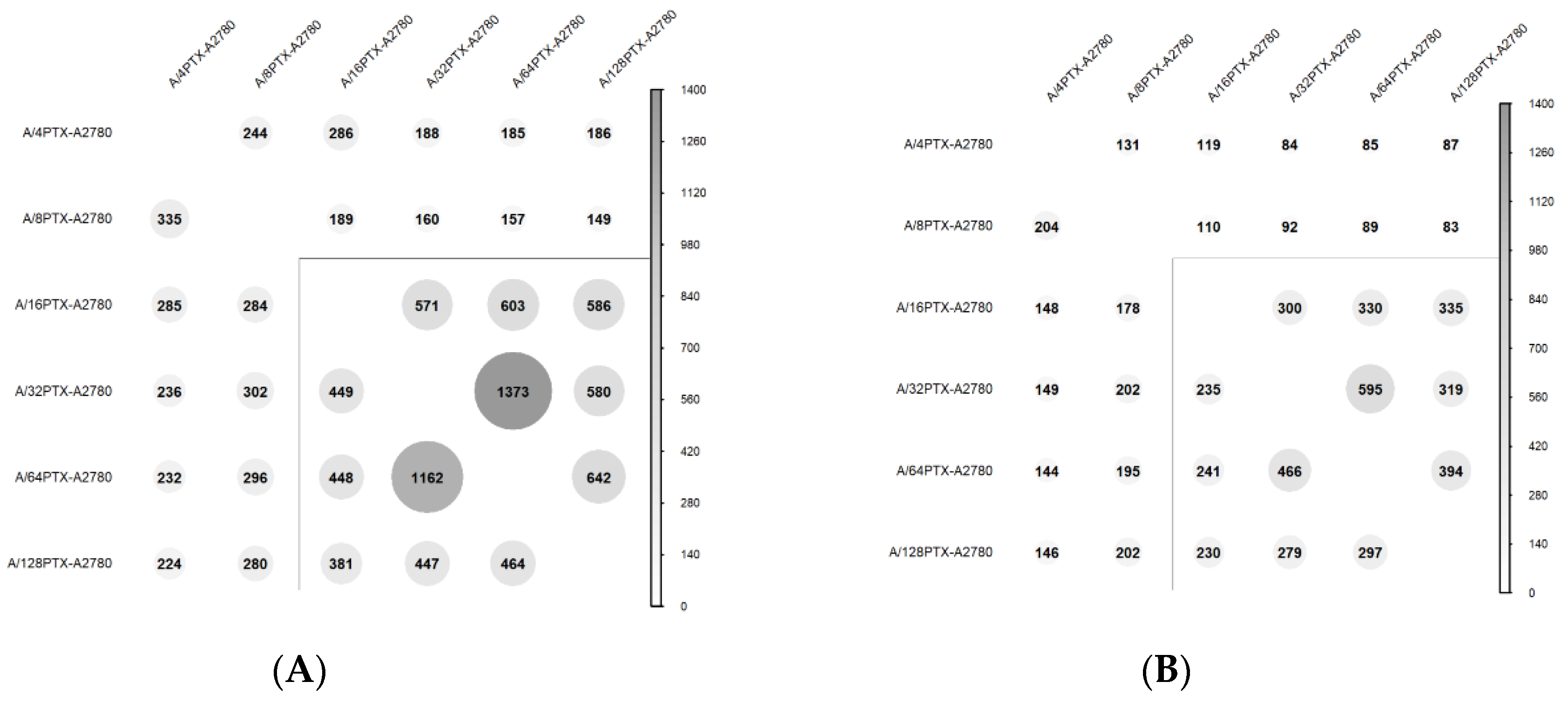
2.3.1. Number of DEGs Is Positively Correlated with the Level of Drug Resistance
2.3.2. Trends in Kinetics of Gene Expression Changes during the Development of Inverse Resistance to PTX and CDDP
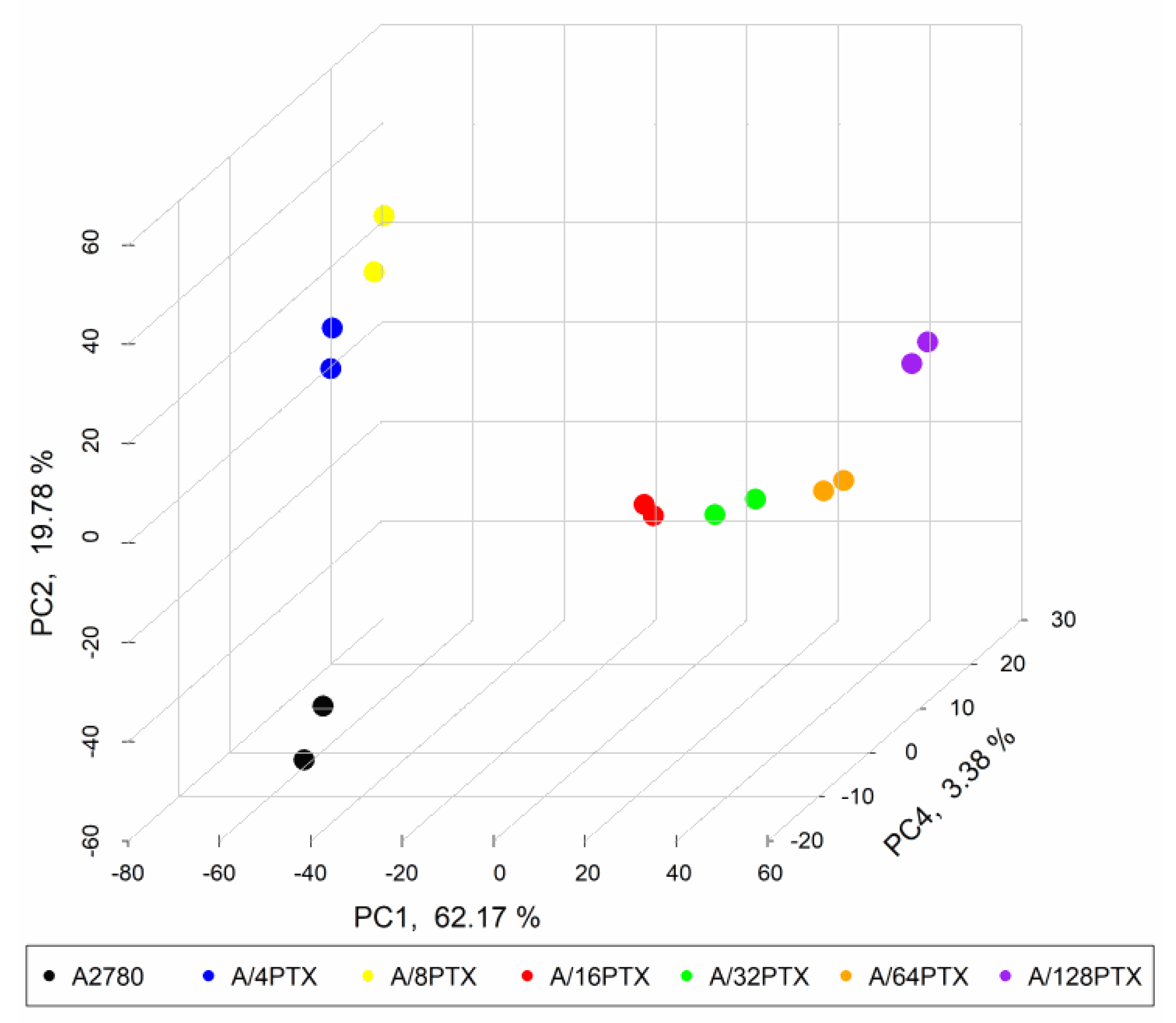
2.3.3. Gene Expression Signature Is Able to Separate the A2780 Parental Cell Line and Six Derived Sublines
2.3.4. Trends in Kinetics of Gene Expression Changes Are Able to Follow the Process of Differentiation of A2780-Derived Sublines during the Development of Inverse Resistance to PTX and CDDP
- 997 DEGs (q-value < 0.001), previously selected for PCA (Figure 7).
- 160 DEGs with q-value < 0.001 and absolute log2 of fold change (FC) > 2 (|log2 FC| > 2) in each pair of cell line comparison (the subset of 997 DEGs) (Figure 8). The term “log2 FC” as used hereafter refers to the ratio of two expression values of a gene, e.g., in a studied cell line vs. control cell line. For example, |log2 FC| values > 2.0 are equivalent with more than the 4-fold expression change.
- DEGs upregulated in six comparisons (consistently upregulated genes).
- DEGs downregulated in six comparisons (consistently downregulated genes).
- DEGs downregulated in the beginning and then upregulated (genes with a rising trend).
- DEGs upregulated in the beginning and then downregulated (genes with a descending trend).
- DEGs with variable expression, which cannot be classified to any of the four classes above (genes with variable expression).
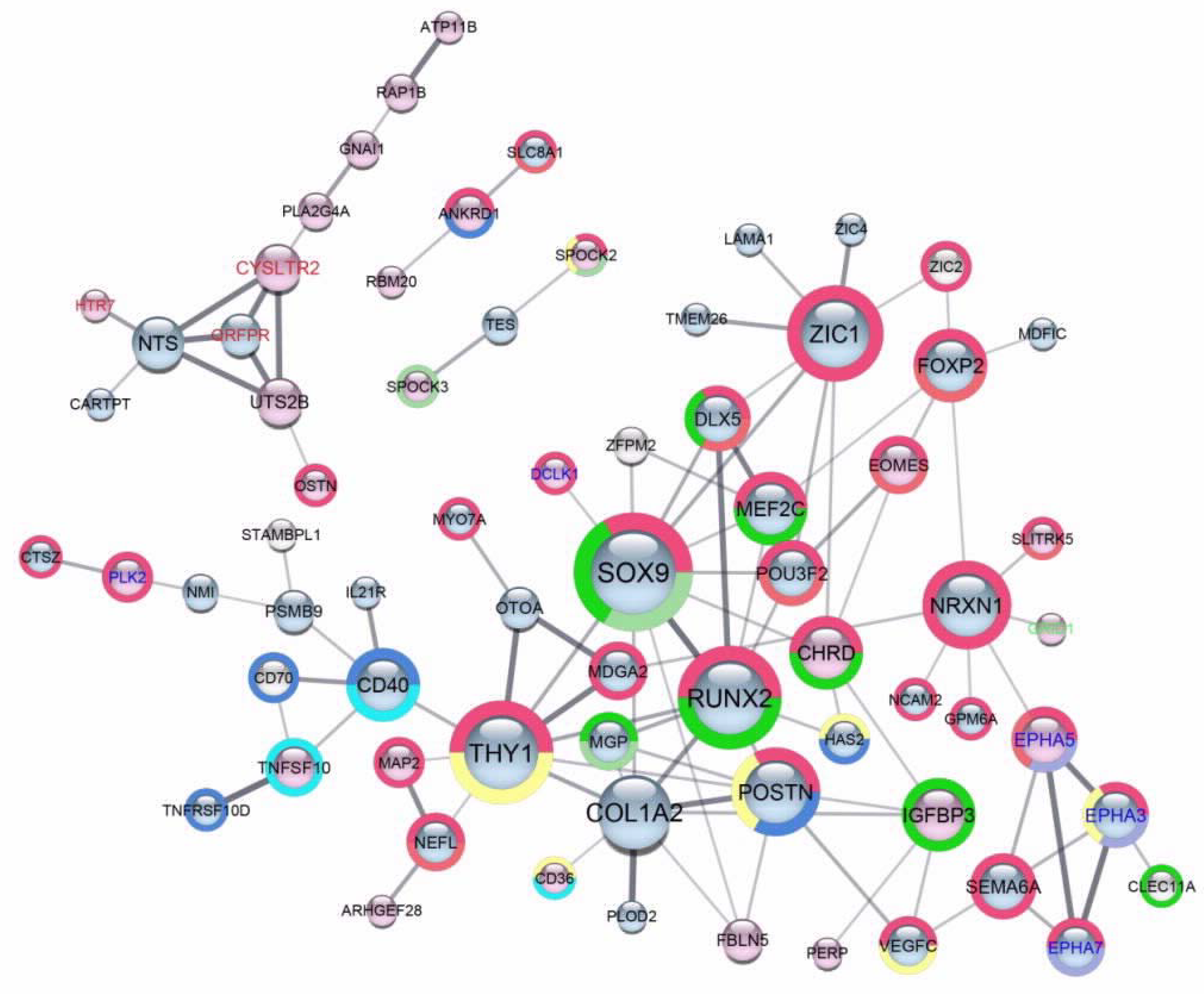
2.4. Scaffold Functional Interaction Networks of Proteins Encoded by DEGs, Identified in Each One of Six A2780-Derived Sublines Compared to the Parental Cell Line
3. Discussion
4. Materials and Methods
4.1. Cell Line and Reagents
4.2. Generation of the Drug Resistant Series of A2780 Sublines
4.3. Cytotoxicity Assay
4.4. RNA-Seq of Transcripts by the NGS Technology
4.4.1. Total RNA Isolation and Assessment of Its Integrity and Concentration
4.4.2. NGS Libraries Preparation and Validation
4.4.3. Transcriptome Sequencing and Data Analysis
4.5. Functional-Interaction Network Analysis
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PTX | Paclitaxel |
| DEG(s) | Differentially expressed gene(s) |
| PCA | Principal Component Analysis |
| CDDP | Cisplatin |
| PPI | Protein-protein interaction network |
| IC50(120h) | Half maximal inhibitory concentration (for 120 h of drug treatment) |
| RI | Resistance index |
| SI | Sensitivity index |
| LMM | Linear mixed models |
| 95% CI | 95% confidential interval |
| NGS | Next generation sequencing |
| FDR | False discovery rate |
| q-value | FDR-adjusted p-value |
| PC | Principal Component |
| RNA-seq | RNA sequencing |
| FC | Fold change |
| GPCR(s) | G protein coupled receptor(s) |
| MSCs | Mesenchymal stem cells |
| CSCs | Cancer stem-like cells |
| ECM | Extracellular matrix |
| MT-MMPs | Membrane-type metalloproteinases |
| alamarBlue | aBlue |
| ECACC | European Collection of Authenticated Cell Cultures |
| PBS | Phosphate-buffered saline |
| RIN | RNA integrity number |
| GO | Gene Ontology |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.; Ueda, Y.; Naka, T.; Enomoto, T. Therapeutic strategies in epithelial ovarian cancer. J. Exp. Clin. Cancer Res. CR 2012, 31, 14. [Google Scholar] [CrossRef] [PubMed]
- Pisano, C.; Bruni, G.S.; Facchini, G.; Marchetti, C.; Pignata, S. Treatment of recurrent epithelial ovarian cancer. Ther. Clin. Risk Manag. 2009, 5, 421–426. [Google Scholar] [PubMed]
- Holmes, D. Ovarian cancer: Beyond resistance. Nature 2015, 527, S217. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.W.; O’Dwyer, P.J. Pharmacology of Cancer Chemotherapy: Cisplatin and Its Analogues. In Principles and Practice of Oncology; DeVita, V.T., Jr., Hellman, S., Rosenberg, S.A., Eds.; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2005; pp. 344–358. [Google Scholar]
- Rowinsky, E.K.; Tolcher, A.W. Pharmacology of Cancer Chemotherapy: Antimicrotubule Agents. In Principles and Practice of Oncology; DeVita, V.T., Jr., Hellman, S., Rosenberg, S.A., Eds.; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2005; pp. 390–416. [Google Scholar]
- Fisher, D.E. Apoptosis in cancer therapy: Crossing the threshold. Cell 1994, 78, 539–542. [Google Scholar] [CrossRef]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug Resistance in Cancer: An Overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef]
- Freimund, A.E.; Beach, J.A.; Christie, E.L.; Bowtell, D.D.L. Mechanisms of Drug Resistance in High-Grade Serous Ovarian Cancer. Hematol. Oncol. Clin. N. Am. 2018, 32, 983–996. [Google Scholar] [CrossRef]
- Stordal, B.; Pavlakis, N.; Davey, R. A systematic review of platinum and taxane resistance from bench to clinic: An inverse relationship. Cancer Treat. Rev. 2007, 33, 688–703. [Google Scholar] [CrossRef]
- Hall, M.D.; Handley, M.D.; Gottesman, M.M. Is resistance useless? Multidrug resistance and collateral sensitivity. Trends Pharmacol. Sci. 2009, 30, 546–556. [Google Scholar] [CrossRef]
- Coley, H.M.; Hatzimichael, E.; Blagden, S.; McNeish, I.; Thompson, A.; Crook, T.; Syed, N. Polo Like Kinase 2 Tumour Suppressor and cancer biomarker: New perspectives on drug sensitivity/resistance in ovarian cancer. Oncotarget 2012, 3, 78–83. [Google Scholar] [CrossRef]
- McDermott, M.; Eustace, A.J.; Busschots, S.; Breen, L.; Crown, J.; Clynes, M.; O’Donovan, N.; Stordal, B. In vitro Development of Chemotherapy and Targeted Therapy Drug-Resistant Cancer Cell Lines: A Practical Guide with Case Studies. Front. Oncol. 2014, 4, 40. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V.; Fojo, T. Molecular effects of paclitaxel: Myths and reality (a critical review). Int. J. Cancer 1999, 83, 151–156. [Google Scholar] [CrossRef]
- Andersen, A.; Warren, D.J.; Brunsvig, P.F.; Aamdal, S.; Kristensen, G.B.; Olsen, H. High sensitivity assays for docetaxel and paclitaxel in plasma using solid-phase extraction and high-performance liquid chromatography with UV detection. BMC Clin. Pharmacol. 2006, 6, 2:1–2:10. [Google Scholar] [CrossRef]
- Gałecki, A.T.; Burzykowski, T. Linear Mixed-Effects Models Using R: A Step-by-Step Approach, 1st ed.; Springer Texts in Statistics; Springer: New York, NY, USA, 2013; ISBN 978-1-4614-3899-1. [Google Scholar]
- Kenakin, T.P. What is Pharmacology? In A Pharmacology Primer. Techniques for More Effective and Strategic Drug Discovery; Elsevier: London, UK, 2014; pp. 16–20. ISBN 978-0-12-407663-1. [Google Scholar]
- Kenakin, T.P. Statistics and Experimental Design. In A Pharmacology Primer. Techniques for More Effective and Strategic Drug Discovery; Elsevier: London, UK, 2014; pp. 322–325. ISBN 978-0-12-407663-1. [Google Scholar]
- Kalayda, G.V.; Wagner, C.H.; Buß, I.; Reedijk, J.; Jaehde, U. Altered localisation of the copper efflux transporters ATP7A and ATP7B associated with cisplatin resistance in human ovarian carcinoma cells. BMC Cancer 2008, 8, 175:1–175:12. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false-discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Parekh, H.; Wiesen, K.; Simpkins, H. Acquisition of taxol resistance via P-glycoprotein- and non-P-glycoprotein-mediated mechanisms in human ovarian carcinoma cells. Biochem. Pharmacol. 1997, 53, 461–470. [Google Scholar] [CrossRef]
- Nakajima, K.; Isonishi, S.; Saito, M.; Tachibana, T.; Ishikawa, H. Characterization of two independent, exposure-time dependent paclitaxel-resistant human ovarian carcinoma cell lines: Exposure-time dependent paclitaxel-resistant cell lines. Hum. Cell 2010, 23, 156–163. [Google Scholar] [CrossRef]
- Yamamoto, K.; Kikuchi, Y.; Kudoh, K.; Nagata, I. Modulation of cisplatin sensitivity by taxol in cisplatin-sensitive and -resistant human ovarian carcinoma cell lines. J. Cancer Res. Clin. Oncol. 2000, 126, 168–172. [Google Scholar] [CrossRef]
- Metzinger, D.S.; Taylor, D.D.; Gercel-Taylor, C. Induction of p53 and drug resistance following treatment with cisplatin or paclitaxel in ovarian cancer cell lines. Cancer Lett. 2006, 236, 302–308. [Google Scholar] [CrossRef]
- Sherman-Baust, C.A.; Becker, K.G.; Wood III, W.H.; Zhang, Y.; Morin, P.J. Gene expression and pathway analysis of ovarian cancer cells selected for resistance to cisplatin, paclitaxel, or doxorubicin. J. Ovarian Res. 2011, 4, 21. [Google Scholar] [CrossRef]
- Januchowski, R.; Wojtowicz, K.; Sujka-Kordowska, P.; Andrzejewska, M.; Zabel, M. MDR gene expression analysis of six drug-resistant ovarian cancer cell lines. BioMed Res. Int. 2013, 2013, 241763. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Luan, Y.; Wang, G.; Tang, B.; Li, D.; Zhang, W.; Li, X.; Zhao, J.; Ding, H.; Reed, E.; et al. Development and characterization of five cell models for chemoresistance studies of human ovarian carcinoma. Int. J. Mol. Med. 2004, 14, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.-N.; Zhao, L.-J.; Wu, L.-N.; He, M.-F.; Qu, J.-W.; Zhao, Y.-B.; Zhao, W.-Z.; Li, J.-S.; Wang, J.-H. Mechanistic analysis of taxol-induced multidrug resistance in an ovarian cancer cell line. Asian Pac. J. Cancer Prev. APJCP 2013, 14, 4983–4988. [Google Scholar] [CrossRef] [PubMed]
- Januchowski, R.; Sterzyńska, K.; Zaorska, K.; Sosińska, P.; Klejewski, A.; Brązert, M.; Nowicki, M.; Zabel, M. Analysis of MDR genes expression and cross-resistance in eight drug resistant ovarian cancer cell lines. J. Ovarian Res. 2016, 9, 65. [Google Scholar] [CrossRef] [PubMed]
- Rutkovskiy, A.; Stensløkken, K.-O.; Vaage, I.J. Osteoblast Differentiation at a Glance. Med. Sci. Monit. Basic Res. 2016, 22, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Komori, T. Runx2, an inducer of osteoblast and chondrocyte differentiation. Histochem. Cell Biol. 2018, 149, 313–323. [Google Scholar] [CrossRef]
- Mevel, R.; Draper, J.E.; Lie-a-Ling, M.; Kouskoff, V.; Lacaud, G. RUNX transcription factors: Orchestrators of development. Development 2019, 146, dev.148296:1–dev.148296:19. [Google Scholar] [CrossRef]
- Lefebvre, V.; Angelozzi, M.; Haseeb, A. SOX9 in cartilage development and disease. Curr. Opin. Cell Biol. 2019, 61, 39–47. [Google Scholar] [CrossRef]
- Aguilar-Medina, M.; Avendaño-Félix, M.; Lizárraga-Verdugo, E.; Bermúdez, M.; Romero-Quintana, J.G.; Ramos-Payan, R.; Ruíz-García, E.; López-Camarillo, C. SOX9 Stem-Cell Factor: Clinical and Functional Relevance in Cancer. J. Oncol. 2019, 2019, 6754040. [Google Scholar] [CrossRef]
- Ito, Y.; Bae, S.-C.; Chuang, L.S.H. The RUNX family: Developmental regulators in cancer. Nat. Rev. Cancer 2015, 15, 81–95. [Google Scholar] [CrossRef]
- Pratap, J.; Lian, J.B.; Javed, A.; Barnes, G.L.; van Wijnen, A.J.; Stein, J.L.; Stein, G.S. Regulatory roles of Runx2 in metastatic tumor and cancer cell interactions with bone. Cancer Metastasis Rev. 2006, 25, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, N.; McDonald, L.; Morris, J.S.; Cameron, E.R.; Blyth, K. RUNX2 in mammary gland development and breast cancer. J. Cell. Physiol. 2013, 228, 1137–1142. [Google Scholar] [CrossRef] [PubMed]
- Browne, G.; Nesbitt, H.; Ming, L.; Stein, G.S.; Lian, J.B.; McKeown, S.R.; Worthington, J. Bicalutamide-induced hypoxia potentiates RUNX2-mediated Bcl-2 expression resulting in apoptosis resistance. Br. J. Cancer 2012, 107, 1714–1721. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Yu, F.; Li, D.; Zhu, X.; Zhang, X.; Lv, Z. MicroRNA-218 regulates cisplatin (DPP) chemosensitivity in non-small cell lung cancer by targeting RUNX2. Tumor Biol. 2016, 37, 1197–1204. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-X.; Ye, F.-G.; Zhang, J.; Li, J.-J.; Chen, Q.-X.; Lin, P.-Y.; Song, C.-G. Serum miR-4530 sensitizes breast cancer to neoadjuvant chemotherapy by suppressing RUNX2. Cancer Manag. Res. 2018, 10, 4393–4400. [Google Scholar] [CrossRef]
- Kanwal, R.; Shukla, S.; Walker, E.; Gupta, S. Acquisition of tumorigenic potential and therapeutic resistance in CD133+ subpopulation of prostate cancer cells exhibiting stem-cell like characteristics. Cancer Lett. 2018, 430, 25–33. [Google Scholar] [CrossRef]
- Song, W.; Kwon, G.Y.; Kim, J.H.; Lim, J.E.; Jeon, H.G.; Il Seo, S.; Jeon, S.S.; Choi, H.Y.; Jeong, B.C.; Lee, H.M. Immunohistochemical staining of ERG and SOX9 as potential biomarkers of docetaxel response in patients with metastatic castration-resistant prostate cancer. Oncotarget 2016, 7, 83735–83743. [Google Scholar] [CrossRef]
- Wang, J.; Xue, X.; Hong, H.; Qin, M.; Zhou, J.; Sun, Q.; Liang, H.; Gao, L. Upregulation of microRNA-524-5p enhances the cisplatin sensitivity of gastric cancer cells by modulating proliferation and metastasis via targeting SOX9. Oncotarget 2017, 8, 574–582. [Google Scholar] [CrossRef]
- Xiao, S.; Li, Y.; Pan, Q.; Ye, M.; He, S.; Tian, Q.; Xue, M. MiR-34c/SOX9 axis regulates the chemoresistance of ovarian cancer cell to cisplatin-based chemotherapy. J. Cell. Biochem. 2019, 120, 2940–2953. [Google Scholar] [CrossRef]
- Xue, M.; Li, G.; Sun, P.; Zhang, D.; Fang, X.; Li, W. MicroRNA-613 induces the sensitivity of gastric cancer cells to cisplatin through targeting SOX9 expression. Am. J. Transl. Res. 2019, 11, 885–894. [Google Scholar]
- Voronkova, M.A.; Rojanasakul, L.W.; Kiratipaiboon, C.; Rojanasakul, Y. The SOX9-Aldehyde Dehydrogenase Axis Determines Resistance to Chemotherapy in Non-Small-Cell Lung Cancer. Mol. Cell. Biol. 2019, 40, e00307-19:1–e00307-19:15. [Google Scholar] [CrossRef] [PubMed]
- Ricard-Blum, S. The collagen family. Cold Spring Harb. Perspect. Biol. 2011, 3, a004978:1–a004978:19. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak-Wielgomas, K.; Dziegiel, P. The role of periostin in neoplastic processes. Folia Histochem. Cytobiol. 2015, 53, 120–132. [Google Scholar] [CrossRef] [PubMed]
- Sterzyńska, K.; Kaźmierczak, D.; Klejewski, A.; Świerczewska, M.; Wojtowicz, K.; Nowacka, M.; Brązert, J.; Nowicki, M.; Januchowski, R. Expression of Osteoblast-Specific Factor 2 (OSF-2, Periostin) Is Associated with Drug Resistance in Ovarian Cancer Cell Lines. Int. J. Mol. Sci. 2019, 20, 3927. [Google Scholar] [CrossRef] [PubMed]
- Hale, J.E.; Fraser, J.D.; Price, P.A. The identification of matrix Gla protein in cartilage. J. Biol. Chem. 1988, 263, 5820–5824. [Google Scholar]
- Luo, G.; Ducy, P.; McKee, M.D.; Pinero, G.J.; Loyer, E.; Behringer, R.R.; Karsenty, G. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature 1997, 386, 78–81. [Google Scholar] [CrossRef]
- Sharma, B.; Albig, A.R. Matrix Gla protein reinforces angiogenic resolution. Microvasc. Res. 2013, 85, 24–33. [Google Scholar] [CrossRef]
- Yue, R.; Shen, B.; Morrison, S.J. Clec11a/osteolectin is an osteogenic growth factor that promotes the maintenance of the adult skeleton. eLife 2016, 5, e18782:1–e18782:27. [Google Scholar] [CrossRef]
- Shen, B.; Vardy, K.; Hughes, P.; Tasdogan, A.; Zhao, Z.; Yue, R.; Crane, G.M.; Morrison, S.J. Integrin alpha11 is an Osteolectin receptor and is required for the maintenance of adult skeletal bone mass. eLife 2019, 8, e42274:1–e42274:31. [Google Scholar] [CrossRef]
- Bornstein, P. Matricellular proteins: An overview. Matrix Biol. J. Int. Soc. Matrix Biol. 2000, 19, 555–556. [Google Scholar] [CrossRef]
- Itoh, Y. Membrane-type matrix metalloproteinases: Their functions and regulations. Matrix Biol. 2015, 44–46, 207–223. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, A.D. Diverse biological functions of the SPARC family of proteins. Int. J. Biochem. Cell Biol. 2012, 44, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Nakada, M.; Miyamori, H.; Yamashita, J.; Sato, H. Testican 2 abrogates inhibition of membrane-type matrix metalloproteinases by other testican family proteins. Cancer Res. 2003, 63, 3364–3369. [Google Scholar] [PubMed]
- Diomede, F.; Marconi, G.D.; Fonticoli, L.; Pizzicanella, J.; Merciaro, I.; Bramanti, P.; Mazzon, E.; Trubiani, O. Functional Relationship between Osteogenesis and Angiogenesis in Tissue Regeneration. Int. J. Mol. Sci. 2020, 21, 3242. [Google Scholar] [CrossRef] [PubMed]
- Kawane, T.; Komori, H.; Liu, W.; Moriishi, T.; Miyazaki, T.; Mori, M.; Matsuo, Y.; Takada, Y.; Izumi, S.; Jiang, Q.; et al. Dlx5 and mef2 regulate a novel runx2 enhancer for osteoblast-specific expression. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2014, 29, 1960–1969. [Google Scholar] [CrossRef]
- Brazil, D.P.; Church, R.H.; Surae, S.; Godson, C.; Martin, F. BMP signalling: Agony and antagony in the family. Trends Cell Biol. 2015, 25, 249–264. [Google Scholar] [CrossRef]
- Moffatt, P.; Thomas, G.P. Osteocrin—Beyond just another bone protein? Cell. Mol. Life Sci. 2009, 66, 1135–1139. [Google Scholar] [CrossRef]
- Spagnoli, A.; Longobardi, L.; O’Rear, L. Cartilage disorders: Potential therapeutic use of mesenchymal stem cells. Endocr. Dev. 2005, 9, 17–30. [Google Scholar]
- Ukena, K.; Osugi, T.; Leprince, J.; Vaudry, H.; Tsutsui, K. Molecular evolution of GPCRs: 26Rfa/GPR103. J. Mol. Endocrinol. 2014, 52, T119–T131. [Google Scholar] [CrossRef]
- Baribault, H.; Danao, J.; Gupte, J.; Yang, L.; Sun, B.; Richards, W.; Tian, H. The G-protein-coupled receptor GPR103 regulates bone formation. Mol. Cell. Biol. 2006, 26, 709–717. [Google Scholar] [CrossRef]
- Rucci, N.; Teti, A. Osteomimicry: How the Seed Grows in the Soil. Calcif. Tissue Int. 2018, 102, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Reissner, C.; Runkel, F.; Missler, M. Neurexins. Genome Biol. 2013, 14, 213. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.J. Thy-1, a Pathfinder Protein for the Post-genomic Era. Front. Cell Dev. Biol. 2018, 6, 173:1–173:12. [Google Scholar] [CrossRef] [PubMed]
- Picke, A.-K.; Campbell, G.M.; Blüher, M.; Krügel, U.; Schmidt, F.N.; Tsourdi, E.; Winzer, M.; Rauner, M.; Vukicevic, V.; Busse, B.; et al. Thy-1 (CD90) promotes bone formation and protects against obesity. Sci. Transl. Med. 2018, 10, eaao6806. [Google Scholar] [CrossRef]
- Saalbach, A.; Anderegg, U. Thy-1: More than a marker for mesenchymal stromal cells. FASEB J. 2019, 33, 6689–6696. [Google Scholar] [CrossRef]
- Kumar, A.; Bhanja, A.; Bhattacharyya, J.; Jaganathan, B.G. Multiple roles of CD90 in cancer. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 2016, 37, 11611–11622. [Google Scholar] [CrossRef]
- Aruga, J. Zic Family Proteins in Emerging Biomedical Studies. In Zic Family; Aruga, J., Ed.; Springer: Singapore, 2018; Volume 1046, pp. 233–248. ISBN 978-981-10-7310-6. [Google Scholar]
- Houtmeyers, R.; Souopgui, J.; Tejpar, S. Deregulation of ZIC Family Members in Oncogenesis. In Zic Family; Aruga, J., Ed.; Springer: Singapore, 2018; Volume 1046, pp. 329–338. ISBN 978-981-10-7310-6. [Google Scholar]
- Kalogeropoulos, M.; Varanasi, S.S.; Olstad, O.K.; Sanderson, P.; Gautvik, V.T.; Reppe, S.; Francis, R.M.; Gautvik, K.M.; Birch, M.A.; Datta, H.K. Zic1 transcription factor in bone: Neural developmental protein regulates mechanotransduction in osteocytes. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2010, 24, 2893–2903. [Google Scholar] [CrossRef]
- Marchini, S.; Poynor, E.; Barakat, R.R.; Clivio, L.; Cinquini, M.; Fruscio, R.; Porcu, L.; Bussani, C.; D’Incalci, M.; Erba, E.; et al. The zinc finger gene ZIC2 has features of an oncogene and its overexpression correlates strongly with the clinical course of epithelial ovarian cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 4313–4324. [Google Scholar] [CrossRef]
- French, L.E.; Tschopp, J. The TRAIL to selective tumor death. Nat. Med. 1999, 5, 146–147. [Google Scholar] [CrossRef]
- Azijli, K.; Weyhenmeyer, B.; Peters, G.J.; de Jong, S.; Kruyt, F.A.E. Non-canonical kinase signaling by the death ligand TRAIL in cancer cells: Discord in the death receptor family. Cell Death Differ. 2013, 20, 858–868. [Google Scholar] [CrossRef]
- Vignati, S.; Codegoni, A.; Polato, F.; Broggini, M. Trail activity in human ovarian cancer cells: Potentiation of the action of cytotoxic drugs. Eur. J. Cancer Oxf. Engl. 1990 2002, 38, 177–183. [Google Scholar]
- Remer, M.; White, A.; Glennie, M.; Al-Shamkhani, A.; Johnson, P. The Use of Anti-CD40 mAb in Cancer. Curr. Top. Microbiol. Immunol. 2017, 405, 165–207. [Google Scholar] [PubMed]
- Jacobs, J.; Deschoolmeester, V.; Zwaenepoel, K.; Rolfo, C.; Silence, K.; Rottey, S.; Lardon, F.; Smits, E.; Pauwels, P. CD70: An emerging target in cancer immunotherapy. Pharmacol. Ther. 2015, 155, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Voorzanger-Rousselot, N.; Alberti, L.; Blay, J.-Y. CD40L induces multidrug resistance to apoptosis in breast carcinoma and lymphoma cells through caspase independent and dependent pathways. BMC Cancer 2006, 6, 75:1–75:12. [Google Scholar] [CrossRef]
- Kania, A.; Klein, R. Mechanisms of ephrin-Eph signalling in development, physiology and disease. Nat. Rev. Mol. Cell Biol. 2016, 17, 240–256. [Google Scholar] [CrossRef]
- Yamada, T.; Yuasa, M.; Masaoka, T.; Taniyama, T.; Maehara, H.; Torigoe, I.; Yoshii, T.; Shinomiya, K.; Okawa, A.; Sotome, S. After repeated division, bone marrow stromal cells express inhibitory factors with osteogenic capabilities, and EphA5 is a primary candidate. Bone 2013, 57, 343–354. [Google Scholar] [CrossRef]
- London, M.; Gallo, E. Critical role of EphA3 in cancer and current state of EphA3 drug therapeutics. Mol. Biol. Rep. 2020, 47, 5523–5533. [Google Scholar] [CrossRef]
- Eckerdt, F.; Yuan, J.; Strebhardt, K. Polo-like kinases and oncogenesis. Oncogene 2005, 24, 267–276. [Google Scholar] [CrossRef]
- Burns, T.F.; Fei, P.; Scata, K.A.; Dicker, D.T.; El-Deiry, W.S. Silencing of the Novel p53 Target Gene Snk/Plk2 Leads to Mitotic Catastrophe in Paclitaxel (Taxol)-Exposed Cells. Mol. Cell. Biol. 2003, 23, 5556–5571. [Google Scholar] [CrossRef]
- Anglesio, M.S.; Wiegand, K.C.; Melnyk, N.; Chow, C.; Salamanca, C.; Prentice, L.M.; Senz, J.; Yang, W.; Spillman, M.A.; Cochrane, D.R.; et al. Type-specific cell line models for type-specific ovarian cancer research. PLoS ONE 2013, 8, e72162. [Google Scholar] [CrossRef]
- Syed, N.; Coley, H.M.; Sehouli, J.; Koensgen, D.; Mustea, A.; Szlosarek, P.; McNeish, I.; Blagden, S.P.; Schmid, P.; Lovell, D.P.; et al. Polo-like Kinase Plk2 Is an Epigenetic Determinant of Chemosensitivity and Clinical Outcomes in Ovarian Cancer. Cancer Res. 2011, 71, 3317–3327. [Google Scholar] [CrossRef] [PubMed]
- Seeburg, D.P.; Pak, D.; Sheng, M. Polo-like kinases in the nervous system. Oncogene 2005, 24, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Gornstein, E.; Schwarz, T.L. The paradox of paclitaxel neurotoxicity: Mechanisms and unanswered questions. Neuropharmacology 2014, 76, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Brewer, J.R.; Morrison, G.; Dolan, M.E.; Fleming, G.F. Chemotherapy-induced peripheral neuropathy: Current status and progress. Gynecol. Oncol. 2016, 140, 176–183. [Google Scholar] [CrossRef]
- Leandro-García, L.J.; Inglada-Pérez, L.; Pita, G.; Hjerpe, E.; Leskelä, S.; Jara, C.; Mielgo, X.; González-Neira, A.; Robledo, M.; Avall-Lundqvist, E.; et al. Genome-wide association study identifies ephrin type A receptors implicated in paclitaxel induced peripheral sensory neuropathy. J. Med. Genet. 2013, 50, 599–605. [Google Scholar] [CrossRef]
- Marcath, L.A.; Kidwell, K.M.; Vangipuram, K.; Gersch, C.L.; Rae, J.M.; Burness, M.L.; Griggs, J.J.; Van Poznak, C.; Hayes, D.F.; Smith, E.M.L.; et al. Genetic variation in EPHA contributes to sensitivity to paclitaxel-induced peripheral neuropathy. Br. J. Clin. Pharmacol. 2020, 86, 880–890. [Google Scholar] [CrossRef]
- Greene, R.F.; Chatterji, D.C.; Hiranaka, P.K.; Gallelli, J.F. Stability of cisplatin in aqueous solution. Am. J. Hosp. Pharm. 1979, 36, 38–43. [Google Scholar] [CrossRef]
- Coley, H.M. Development of drug-resistant models. In Cancer Cell Culture. Methods and Protocols; Langdon, S.P., Ed.; Methods in Molecular Medicine; Humana Press: Totowa, NJ, USA, 2004; Volume 88, pp. 267–273. ISBN 1-58829-079-4. [Google Scholar]
- Lancaster, M.V.; Fields, R.D. alamarBlueAssay. U.S. Patent 5,501,959, 1996. pp. 1–27. [Google Scholar]
- Plumb, J. Cell sensitivity assays: The MTT assay. In Cancer Cell Culture. Methods and Protocols; Langdon, S., Ed.; Methods in Molecular Medicine; Humana Press: Totowa, NJ, USA, 2004; Volume 88, pp. 165–169. [Google Scholar]
- The R Development Core Team R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018; ISBN 3-900051-07-0.
- Bates, D.; Mächler, M.; Bolker, B.; Walker, S. Fitting Linear Mixed-Effects Models Using lme4. J. Stat. Softw. 2015, 67, 1–49. [Google Scholar] [CrossRef]
- Gelman, A.; Su, Y.-S. arm: Data Analysis Using Regression and Multilevel/Hierarchical Models. R Package Version 1.8-6. Available online: https://CRAN.R-project.org/package=arm (accessed on 17 December 2019).
- Wickham, H. Ggplot2—Elegant Graphics for Data Analysis, 2nd ed.; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24275-0. [Google Scholar]
- FASTX-Toolkit. Available online: http://hannonlab.cshl.edu/fastx_toolkit/ (accessed on 29 February 2020).
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, 36:1–36:13. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinforma. Oxf. Engl. 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, 47:1–47:13. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Simko, V. R Package “Corrplot”: Visualization of a Correlation Matrix (Version 0.84). Available online: https://github.com/taiyun/corrplot (accessed on 17 December 2019).
- Ligges, U.; Mächler, M. Scatterplot3d- an R Package for Visualizing Multivariate Data. J. Stat. Softw. 2003, 8, 1–20. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed]
- UniProt Knowledgebase. Available online: https://www.uniprot.org (accessed on 30 June 2020).
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Search Tool for the Retrieval of Interacting Genes. Available online: https://string-db.org (accessed on 30 June 2020).
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Cytoscape. Available online: http://cytoscape.org (accessed on 30 June 2020).
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Gene Ontology Consortium the Gene Ontology: Enhancements for 2011. Nucleic Acids Res. 2012, 40, D559–D564. [CrossRef]
- The Gene Ontology Consortium Gene Ontology Annotations and Resources. Nucleic Acids Res. 2012, 41, D530–D535. [CrossRef]
- The Gene Ontology Resource. Available online: http://geneontology.org (accessed on 30 June 2020).
- PHAROS. Available online: https://pharos.nih.gov/idg/targets (accessed on 30 June 2020).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | IC50(120h) ± SD for PTX (nM) {RI} 1 | IC50(120h) ± SD for CDDP (µM) {SI} 2 |
|---|---|---|
| A2780 | 4.62 ± 0.22 {1} | 4.58 ± 0.14 {1} |
| A/4PTX-63d | 11.47 ± 0.07 {2.48} | 4.27 ± 0.11 {1.07} |
| A/8PTX-33d | 18.71 ± 0.28 {4.05} | 2.12 ± 0.12 {2.16} |
| A/16PTX-56d | 77.54 ± 1.81 {16.78} | 0.65 ± 0.01 {7.05} |
| A/32PTX-48d | 148.47 ± 6.82 {32.14} | 0.44 ± 0.06 {10.41} |
| A/64PTX-30d | 173.86 ± 4.13 {37.63} | 0.40 ± 0.03 {11.45} |
| A/128PTX-37d | 272.47 ± 6.72 {58.98} | 0.83 ± 0.04 {5.52} |
| Pair of Consecutive Cell Lines | PTX-RI 1 | CDDP-SI 2 |
|---|---|---|
| A2780–A/4PTX | 2.48 | 1.07 |
| A/4PTX–A/8PTX | 1.63 | 2.01 |
| A/8PTX–A/16PTX | 4.14 | 3.26 |
| A/16PTX–A/32PTX | 1.91 | 1.48 |
| A/32PTX–A/64PTX | 1.17 | 1.10 |
| A/64PTX–A/128PTX | 1.57 | 0.48 |
| Pair of Cell Lines | p-Value for PTX/CDDP (In LMM) | p-Value for PTX/CDDP (In t-Test) |
|---|---|---|
| A2780–A/4PTX | ***/# | ***/* |
| A2780–A/8PTX | ***/*** | ***/*** |
| A2780–A/16PTX | ***/*** | ***/*** |
| A2780–A/32PTX | ***/*** | ***/*** |
| A2780–A/64PTX | ***/*** | ***/*** |
| A2780–A/128PTX | ***/*** | ***/*** |
| A/4PTX–A/8PTX | ***/*** | ***/*** |
| A/4PTX–A/16PTX | ***/*** | ***/*** |
| A/4PTX–A/32PTX | ***/*** | ***/*** |
| A/4PTX–A/64PTX | ***/*** | ***/*** |
| A/4PTX–A/128PTX | ***/*** | ***/*** |
| A/8PTX–A/16PTX | ***/*** | ***/*** |
| A/8PTX–A/32PTX | ***/*** | ***/*** |
| A/8PTX–A/64PTX | ***/*** | ***/*** |
| A/8PTX–A/128PTX | ***/*** | ***/*** |
| A/16PTX–A/32PTX | ***/*** | ***/* |
| A/16PTX–A/64PTX | ***/*** | ***/** |
| A/16PTX–A/128PTX | ***/# | ***/** |
| A/32PTX–A/64PTX | ***/# | */# |
| A/32PTX–A/128PTX | #/* | ***/** |
| A/64PTX–A/128PTX | **/* | ***/*** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szenajch, J.; Szabelska-Beręsewicz, A.; Świercz, A.; Zyprych-Walczak, J.; Siatkowski, I.; Góralski, M.; Synowiec, A.; Handschuh, L. Transcriptome Remodeling in Gradual Development of Inverse Resistance between Paclitaxel and Cisplatin in Ovarian Cancer Cells. Int. J. Mol. Sci. 2020, 21, 9218. https://doi.org/10.3390/ijms21239218
Szenajch J, Szabelska-Beręsewicz A, Świercz A, Zyprych-Walczak J, Siatkowski I, Góralski M, Synowiec A, Handschuh L. Transcriptome Remodeling in Gradual Development of Inverse Resistance between Paclitaxel and Cisplatin in Ovarian Cancer Cells. International Journal of Molecular Sciences. 2020; 21(23):9218. https://doi.org/10.3390/ijms21239218
Chicago/Turabian StyleSzenajch, Jolanta, Alicja Szabelska-Beręsewicz, Aleksandra Świercz, Joanna Zyprych-Walczak, Idzi Siatkowski, Michał Góralski, Agnieszka Synowiec, and Luiza Handschuh. 2020. "Transcriptome Remodeling in Gradual Development of Inverse Resistance between Paclitaxel and Cisplatin in Ovarian Cancer Cells" International Journal of Molecular Sciences 21, no. 23: 9218. https://doi.org/10.3390/ijms21239218
APA StyleSzenajch, J., Szabelska-Beręsewicz, A., Świercz, A., Zyprych-Walczak, J., Siatkowski, I., Góralski, M., Synowiec, A., & Handschuh, L. (2020). Transcriptome Remodeling in Gradual Development of Inverse Resistance between Paclitaxel and Cisplatin in Ovarian Cancer Cells. International Journal of Molecular Sciences, 21(23), 9218. https://doi.org/10.3390/ijms21239218