Innate Immunity and Biological Therapies for the Treatment of Sjögren’s Syndrome


Abstract
1. Introduction
2. Lacrimal and Salivary Glands Structure and Function
3. Pathogenesis of Sjögren’s Syndrome
4. Innate Immune Cells in SS Disease
4.1. Epithelial Cells
4.2. B Cells (B Lyphocytes)
4.2.1. Development and Function of B Cells
4.2.2. B Cells in Sjögren’s Syndrome
4.3. T Cells (T Lymphocytes)
4.3.1. T Cell Development and Function
4.3.2. T Cells in Sjögren’s Syndrome
4.3.3. Th1 and Th2 Cells
4.3.4. Th17 Cells
4.3.5. T Regulatory Cells (Tregs)
4.3.6. Follicular Helper T Cells (Tfh) and Follicular Regulatory T Cells (Tfr)
4.4. Dendritic Cells (DCs)
4.5. Natural Killers (NK) Cells
4.6. Macrophages
5. Biological Therapies for the Treatment of Sjögren’s Syndrome
5.1. B Cell Targeting
5.1.1. CD20 Targeting
5.1.2. CD22 Targeting
5.1.3. BAFF and APRIL Targeting
5.1.4. Lymphotoxin β Receptor Targeting
5.2. T Cells Targeting
5.3. Mesenchyme Stem Cells Transplantation
5.4. Cytokines as a Therapeutic Target
5.4.1. TNF Family
5.4.2. IFN Family
5.4.3. IL-1 Family
5.4.4. IL-2 Family
5.4.5. IL-6 and IL-12 Family
5.4.6. IL-10 Family
5.4.7. Gene Therapy
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Moutsopoulos, H.M. Sjogren’s syndrome: Autoimmune epithelitis. Clin. Immunol. Immunopathol. 1994, 72, 162–165. [Google Scholar] [CrossRef]
- Brandt, J.E.; Priori, R.; Valesini, G.; Fairweather, D. Sex differences in Sjogren’s syndrome: A comprehensive review of immune mechanisms. Biol. Sex Differ. 2015, 6, 19. [Google Scholar] [CrossRef] [PubMed]
- Helmick, C.G.; Felson, D.T.; Lawrence, R.C.; Gabriel, S.; Hirsch, R.; Kwoh, C.K.; Liang, M.H.; Kremers, H.M.; Mayes, M.D.; Merkel, P.A.; et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part I. Arthritis Rheum. 2008, 58, 15–25. [Google Scholar] [CrossRef]
- Malladi, A.S.; Sack, K.E.; Shiboski, S.C.; Shiboski, C.H.; Baer, A.N.; Banushree, R.; Dong, Y.; Helin, P.; Kirkham, B.W.; Li, M.; et al. Primary Sjogren’s syndrome as a systemic disease: A study of participants enrolled in an international Sjogren’s syndrome registry. Arthritis Care. Res. (Hoboken) 2012, 64, 911–918. [Google Scholar] [CrossRef]
- Ienopoli, S.; Carsons, S.E. Extraglandular manifestations of primary Sjögren’s syndrome. Oral. Maxillofac. Surg. Clin. N. Am. 2014, 26, 91–99. [Google Scholar] [CrossRef]
- Sebastian, A.; Szachowicz, A.; Wiland, P. Classification criteria for secondary Sjogren’s syndrome. Current state of knowledge. Reumatologia 2019, 57, 277–280. [Google Scholar] [CrossRef]
- Goules, A.V.; Tzioufas, A.G. Primary Sjogren’s syndrome: Clinical phenotypes, outcome and the development of biomarkers. Autoimmun. Rev. 2016, 15, 695–703. [Google Scholar] [CrossRef]
- Lin, D.F.; Yan, S.M.; Zhao, Y.; Zhang, W.; Li, M.T.; Zeng, X.F.; Zhang, F.C.; Dong, Y. Clinical and prognostic characteristics of 573 cases of primary Sjögren’s syndrome. Chin. Med. J. 2010, 123, 3252–3257. [Google Scholar]
- Fox, R.I. Sjogren’s syndrome. Lancet 2005, 366, 321–331. [Google Scholar] [CrossRef]
- Kramer, J.M. Early events in Sjogren’s Syndrome pathogenesis: The importance of innate immunity in disease initiation. Cytokine 2014, 67, 92–101. [Google Scholar] [CrossRef]
- Conrady, C.D.; Joos, Z.P.; Patel, B.C. Review: The Lacrimal Gland and Its Role in Dry Eye. J. Ophthalmol. 2016, 2016, 7542929. [Google Scholar] [CrossRef] [PubMed]
- Chilla, R.; Niemann, H.; Arglebe, C.; Domagk, G.F. Age-dependent changes in the alpha-isoamylase pattern of human and rat parotid glands. Orl. J. Otorhinolaryngol. Relat. Spec. 1974, 36, 372–382. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, I.; Seguchi, H.; Okada, T.; Kobayashi, T.; Jin, Q.S.; Jiang, X.D. Fine structure of the acinar and duct cell components in the parotid and submandibular salivary glands of the rat: A TEM, SEM, and HRSEM study. Histol. Histopathol. 1996, 11, 103–110. [Google Scholar]
- Amano, O.; Mizobe, K.; Bando, Y.; Sakiyama, K. Anatomy and histology of rodent and human major salivary glands: Overview of the Japan salivary gland society-sponsored workshop. Acta. Histochem. Cytochem. 2012, 45, 241–250. [Google Scholar] [CrossRef]
- Cha, S.; Peck, A.B.; Humphreys-Beher, M.G. Progress in understanding autoimmune exocrinopathy using the non-obese diabetic mouse: An update. Crit. Rev. Oral. Biol. Med. 2002, 13, 5–16. [Google Scholar] [CrossRef]
- Peck, A.B.; Saylor, B.T.; Nguyen, L.; Sharma, A.; She, J.X.; Nguyen, C.Q.; McIndoe, R.A. Gene expression profiling of early-phase Sjogren’s syndrome in C57BL/6.NOD-Aec1Aec2 mice identifies focal adhesion maturation associated with infiltrating leukocytes. Investig. Ophthalmol. Vis. Sci. 2011, 52, 5647–5655. [Google Scholar] [CrossRef]
- Cha, S.; van Blockland, S.C.; Versnel, M.A.; Homo-Delarche, F.; Nagashima, H.; Brayer, J.; Peck, A.B.; Humphreys-Beher, M.G. Abnormal organogenesis in salivary gland development may initiate adult onset of autoimmune exocrinopathy. Exp. Clin. Immunogenet. 2001, 18, 143–160. [Google Scholar] [CrossRef]
- Wanchu, A.; Khullar, M.; Sud, A.; Bambery, P. Elevated nitric oxide production in patients with primary Sjögren’s syndrome. Clin. Rheumatol. 2000, 19, 360–364. [Google Scholar] [CrossRef]
- Caulfield, V.L.; Balmer, C.; Dawson, L.J.; Smith, P.M. A role for nitric oxide-mediated glandular hypofunction in a non-apoptotic model for Sjogren’s syndrome. Rheumatology (Oxford) 2009, 48, 727–733. [Google Scholar] [CrossRef][Green Version]
- Shaalan, A.; Carpenter, G.; Proctor, G. Inducible nitric oxide synthase-mediated injury in a mouse model of acute salivary gland dysfunction. Nitric. Oxide. 2018, 78, 95–102. [Google Scholar] [CrossRef]
- Ambe, K.; Watanabe, H.; Takahashi, S.; Nakagawa, T.; Sasaki, J. Production and physiological role of NO in the oral cavity. Jpn. Dent. Sci. Rev. 2016, 52, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Makarenkova, H.P.; Dartt, D.A. Myoepithelial Cells: Their Origin and Function in Lacrimal Gland Morphogenesis, Homeostasis, and Repair. Curr. Mol. Biol. Rep. 2015, 1, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Hawley, D.; Tang, X.; Zyrianova, T.; Shah, M.; Janga, S.; Letourneau, A.; Schicht, M.; Paulsen, F.; Hamm-Alvarez, S.; Makarenkova, H.P.; et al. Myoepithelial cell-driven acini contraction in response to oxytocin receptor stimulation is impaired in lacrimal glands of Sjogren’s syndrome animal models. Sci. Rep. 2018, 8, 9919. [Google Scholar] [CrossRef] [PubMed]
- Soinila, J.; Nuorva, K.; Soinila, S. Nitric oxide synthase in human salivary glands. Histochem. Cell Biol. 2006, 125, 717–723. [Google Scholar] [CrossRef]
- Rosignoli, F.; Roca, V.; Meiss, R.; Leceta, J.; Gomariz, R.P.; Perez Leiros, C. Defective signalling in salivary glands precedes the autoimmune response in the non-obese diabetic mouse model of sialadenitis. Clin. Exp. Immunol. 2005, 142, 411–418. [Google Scholar] [CrossRef]
- Kong, L.; Robinson, C.P.; Peck, A.B.; Vela-Roch, N.; Sakata, K.M.; Dang, H.; Talal, N.; Humphreys-Beher, M.G. Inappropriate apoptosis of salivary and lacrimal gland epithelium of immunodeficient NOD-scid mice. Clin. Exp. Rheumatol. 1998, 16, 675–681. [Google Scholar]
- Economopoulou, P.; Hanby, A.; Odell, E.W. Expression of E-cadherin, cellular differentiation and polarity in epithelial salivary neoplasms. Oral. Oncol. 2000, 36, 515–518. [Google Scholar] [CrossRef]
- Segawa, A.; Sahara, N.; Suzuki, K.; Yamashina, S. Destruction of cell surface polarity by colchicine in rat salivary gland acinar cells: Reevaluation of the microtubular function. J. Electron. Microsc. (Tokyo) 1988, 37, 81–85. [Google Scholar]
- Bahamondes, V.; Albornoz, A.; Aguilera, S.; Alliende, C.; Molina, C.; Castro, I.; Urzua, U.; Quest, A.F.; Barrera, M.J.; Gonzalez, S.; et al. Changes in Rab3D expression and distribution in the acini of Sjogren’s syndrome patients are associated with loss of cell polarity and secretory dysfunction. Arthritis Rheum. 2011, 63, 3126–3135. [Google Scholar] [CrossRef]
- Wakamatsu, T.H.; Dogru, M.; Matsumoto, Y.; Kojima, T.; Kaido, M.; Ibrahim, O.M.A.; Sato, E.A.; Igarashi, A.; Ichihashi, Y.; Satake, Y.; et al. Evaluation of Lipid Oxidative Stress Status in Sjögren Syndrome Patients. Investig. Ophthalmol. Vis. Sci. 2013, 54, 201–210. [Google Scholar] [CrossRef]
- Uchino, Y.; Kawakita, T.; Miyazawa, M.; Ishii, T.; Onouchi, H.; Yasuda, K.; Ogawa, Y.; Shimmura, S.; Ishii, N.; Tsubota, K. Oxidative stress induced inflammation initiates functional decline of tear production. PLoS ONE 2012, 7, e45805. [Google Scholar] [CrossRef] [PubMed]
- Kurimoto, C.; Kawano, S.; Tsuji, G.; Hatachi, S.; Jikimoto, T.; Sugiyama, D.; Kasagi, S.; Komori, T.; Nakamura, H.; Yodoi, J.; et al. Thioredoxin may exert a protective effect against tissue damage caused by oxidative stress in salivary glands of patients with Sjogren’s syndrome. J. Rheumatol. 2007, 34, 2035–2043. [Google Scholar] [PubMed]
- Dogru, M.; Kojima, T.; Simsek, C.; Tsubota, K. Potential Role of Oxidative Stress in Ocular Surface Inflammation and Dry Eye Disease. Investig. Ophthalmol. Vis. Sci. 2018, 59, Des163–Des168. [Google Scholar] [CrossRef]
- Xuan, J.; Shen, L.; Malyavantham, K.; Pankewycz, O.; Ambrus, J.L., Jr.; Suresh, L. Temporal histological changes in lacrimal and major salivary glands in mouse models of Sjogren’s syndrome. BMC Oral. Health. 2013, 13, 51. [Google Scholar] [CrossRef] [PubMed]
- Williamson, J.; Gibson, A.A.; Wilson, T.; Forrester, J.V.; Whaley, K.; Dick, W.C. Histology of the lacrimal gland in keratoconjunctivitis sicca. Br. J. Ophthalmol. 1973, 57, 852–858. [Google Scholar] [CrossRef]
- Greenspan, J.S.; Daniels, T.E.; Talal, N.; Sylvester, R.A. The histopathology of Sjogren’s syndrome in labial salivary gland biopsies. Oral. Surg. Oral. Med. Oral. Pathol. 1974, 37, 217–229. [Google Scholar] [CrossRef]
- Barone, F.; Campos, J.; Bowman, S.; Fisher, B.A. The value of histopathological examination of salivary gland biopsies in diagnosis, prognosis and treatment of Sjogren’s Syndrome. Swiss Med. Wkly. 2015, 145, w14168. [Google Scholar] [CrossRef]
- Umazume, T.; Thomas, W.M.; Campbell, S.; Aluri, H.; Thotakura, S.; Zoukhri, D.; Makarenkova, H.P. Lacrimal Gland Inflammation Deregulates Extracellular Matrix Remodeling and Alters Molecular Signature of Epithelial Stem/Progenitor Cells. Investig. Ophthalmol. Vis. Sci. 2015, 56, 8392–8402. [Google Scholar] [CrossRef]
- Shatos, M.A.; Hodges, R.R.; Morinaga, M.; McNay, D.E.; Islam, R.; Bhattacharya, S.; Li, D.; Turpie, B.; Makarenkova, H.P.; Masli, S.; et al. Alteration in cellular turnover and progenitor cell population in lacrimal glands from thrombospondin 1(-/-) mice, a model of dry eye. Exp. Eye Res. 2016, 153, 27–41. [Google Scholar] [CrossRef]
- Zoukhri, D. Effect of inflammation on lacrimal gland function. Exp. Eye Res. 2006, 82, 885–898. [Google Scholar] [CrossRef]
- Schenke-Layland, K.; Xie, J.; Magnusson, M.; Angelis, E.; Li, X.; Wu, K.; Reinhardt, D.P.; Maclellan, W.R.; Hamm-Alvarez, S.F. Lymphocytic infiltration leads to degradation of lacrimal gland extracellular matrix structures in NOD mice exhibiting a Sjogren’s syndrome-like exocrinopathy. Exp. Eye Res. 2010, 90, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Tzioufas, A.G.; Kapsogeorgou, E.K.; Moutsopoulos, H.M. Pathogenesis of Sjogren’s syndrome: What we know and what we should learn. J. Autoimmun. 2012, 39, 4–8. [Google Scholar] [CrossRef]
- Manoussakis, M.N.; Kapsogeorgou, E.K. The role of intrinsic epithelial activation in the pathogenesis of Sjogren’s syndrome. J. Autoimmun. 2010, 35, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.T.; Lazarev, S.; Bahrami, A.F.; Noble, L.B.; Chen, F.Y.; Zhou, D.; Gallup, M.; Yadav, M.; McNamara, N.A. Interleukin-1 receptor mediates the interplay between CD4+ T cells and ocular resident cells to promote keratinizing squamous metaplasia in Sjogren’s syndrome. Lab. Investig. 2012, 92, 556–570. [Google Scholar] [CrossRef]
- Manoussakis, M.N.; Kapsogeorgou, E.K. The role of epithelial cells in the pathogenesis of Sjogren’s syndrome. Clin. Rev. Allergy. Immunol. 2007, 32, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Shaalan, A.; Liefers, S.; Coudenys, J.; Elewaut, D.; Proctor, G.B.; Bootsma, H.; Kroese, F.G.M.; Pringle, S. Dysregulation of NF-kB in glandular epithelial cells results in Sjogren’s-like features. PLoS ONE 2018, 13, e0200212. [Google Scholar] [CrossRef]
- Asam, S.; Neag, G.; Berardicurti, O.; Gardner, D.; Barone, F. The role of stroma and epithelial cells in primary Sjogren’s syndrome. Rheumatology (Oxford) 2019. (ahead of print). [Google Scholar] [CrossRef]
- Ohlsson, M.; Jonsson, R.; Brokstad, K.A. Subcellular redistribution and surface exposure of the Ro52, Ro60 and La48 autoantigens during apoptosis in human ductal epithelial cells: A possible mechanism in the pathogenesis of Sjogren’s syndrome. Scand. J. Immunol. 2002, 56, 456–469. [Google Scholar] [CrossRef]
- Zhang, X.; M, V.J.; Qu, Y.; He, X.; Ou, S.; Bu, J.; Jia, C.; Wang, J.; Wu, H.; Liu, Z.; et al. Dry Eye Management: Targeting the Ocular Surface Microenvironment. Int. J. Mol. Sci. 2017, 18, 1398. [Google Scholar] [CrossRef]
- Xanthou, G.; Polihronis, M.; Tzioufas, A.G.; Paikos, S.; Sideras, P.; Moutsopoulos, H.M. “Lymphoid” chemokine messenger RNA expression by epithelial cells in the chronic inflammatory lesion of the salivary glands of Sjogren’s syndrome patients: Possible participation in lymphoid structure formation. Arthritis Rheum. 2001, 44, 408–418. [Google Scholar] [CrossRef]
- Ogawa, N.; Ping, L.; Zhenjun, L.; Takada, Y.; Sugai, S. Involvement of the interferon-gamma-induced T cell-attracting chemokines, interferon-gamma-inducible 10-kd protein (CXCL10) and monokine induced by interferon-gamma (CXCL9), in the salivary gland lesions of patients with Sjogren’s syndrome. Arthritis Rheum. 2002, 46, 2730–2741. [Google Scholar] [CrossRef]
- Kramer, J.M.; Klimatcheva, E.; Rothstein, T.L. CXCL13 is elevated in Sjogren’s syndrome in mice and humans and is implicated in disease pathogenesis. J. Leukoc. Biol. 2013, 94, 1079–1089. [Google Scholar] [CrossRef]
- Jin, J.O.; Shinohara, Y.; Yu, Q. Innate immune signaling induces interleukin-7 production from salivary gland cells and accelerates the development of primary Sjogren’s syndrome in a mouse model. PLoS ONE 2013, 8, e77605. [Google Scholar] [CrossRef]
- Bombardieri, M.; Barone, F.; Pittoni, V.; Alessandri, C.; Conigliaro, P.; Blades, M.C.; Priori, R.; McInnes, I.B.; Valesini, G.; Pitzalis, C. Increased circulating levels and salivary gland expression of interleukin-18 in patients with Sjogren’s syndrome: Relationship with autoantibody production and lymphoid organization of the periductal inflammatory infiltrate. Arthritis Res. Ther. 2004, 6, R447–R456. [Google Scholar] [CrossRef]
- Ciccia, F.; Guggino, G.; Rizzo, A.; Ferrante, A.; Raimondo, S.; Giardina, A.; Dieli, F.; Campisi, G.; Alessandro, R.; Triolo, G. Potential involvement of IL-22 and IL-22-producing cells in the inflamed salivary glands of patients with Sjogren’s syndrome. Ann. Rheum. Dis. 2012, 71, 295–301. [Google Scholar] [CrossRef]
- Ittah, M.; Miceli-Richard, C.; Eric Gottenberg, J.; Lavie, F.; Lazure, T.; Ba, N.; Sellam, J.; Lepajolec, C.; Mariette, X. B cell-activating factor of the tumor necrosis factor family (BAFF) is expressed under stimulation by interferon in salivary gland epithelial cells in primary Sjogren’s syndrome. Arthritis Res. Ther. 2006, 8, R51. [Google Scholar] [CrossRef]
- Erdinest, N.; Aviel, G.; Moallem, E.; Anteby, I.; Yahalom, C.; Mechoulam, H.; Ovadia, H.; Solomon, A. Expression and activation of toll-like receptor 3 and toll-like receptor 4 on human corneal epithelial and conjunctival fibroblasts. J. Inflamm. (Lond) 2014, 11, 3. [Google Scholar] [CrossRef]
- Redfern, R.L.; Barabino, S.; Baxter, J.; Lema, C.; McDermott, A.M. Dry eye modulates the expression of toll-like receptors on the ocular surface. Exp. Eye Res. 2015, 134, 80–89. [Google Scholar] [CrossRef]
- Spachidou, M.P.; Bourazopoulou, E.; Maratheftis, C.I.; Kapsogeorgou, E.K.; Moutsopoulos, H.M.; Tzioufas, A.G.; Manoussakis, M.N. Expression of functional Toll-like receptors by salivary gland epithelial cells: Increased mRNA expression in cells derived from patients with primary Sjogren’s syndrome. Clin. Exp. Immunol. 2007, 147, 497–503. [Google Scholar] [CrossRef]
- Manoussakis, M.N.; Spachidou, M.P.; Maratheftis, C.I. Salivary epithelial cells from Sjogren’s syndrome patients are highly sensitive to anoikis induced by TLR-3 ligation. J. Autoimmun. 2010, 35, 212–218. [Google Scholar] [CrossRef]
- Nandula, S.R.; Dey, P.; Corbin, K.L.; Nunemaker, C.S.; Bagavant, H.; Deshmukh, U.S. Salivary gland hypofunction induced by activation of innate immunity is dependent on type I interferon signaling. J. Oral. Pathol. Med. 2013, 42, 66–72. [Google Scholar] [CrossRef]
- Nandula, S.R.; Scindia, Y.M.; Dey, P.; Bagavant, H.; Deshmukh, U.S. Activation of innate immunity accelerates sialoadenitis in a mouse model for Sjogren’s syndrome-like disease. Oral. Dis. 2011, 17, 801–807. [Google Scholar] [CrossRef]
- Stern, M.E.; Gao, J.; Schwalb, T.A.; Ngo, M.; Tieu, D.D.; Chan, C.C.; Reis, B.L.; Whitcup, S.M.; Thompson, D.; Smith, J.A. Conjunctival T-cell subpopulations in Sjogren’s and non-Sjogren’s patients with dry eye. Investig. Ophthalmol. Vis. Sci. 2002, 43, 2609–2614. [Google Scholar]
- Mak, T.W.; Saunders, M.E.; Jett, B.D. (Eds.) The B Cell Receptor. In Primer to the Immune Response; Academic Cell: Cambridge, MA, USA, 2014; pp. 85–110. [Google Scholar]
- LeBien, T.W.; Tedder, T.F. B lymphocytes: How they develop and function. Blood 2008, 112, 1570–1580. [Google Scholar] [CrossRef]
- Brack, C.; Hirama, M.; Lenhard-Schuller, R.; Tonegawa, S. A complete immunoglobulin gene is created by somatic recombination. Cell 1978, 15, 1–14. [Google Scholar] [CrossRef]
- Perez-Andres, M.; Paiva, B.; Nieto, W.G.; Caraux, A.; Schmitz, A.; Almeida, J.; Vogt, R.F., Jr.; Marti, G.E.; Rawstron, A.C.; Van Zelm, M.C.; et al. Human peripheral blood B-cell compartments: A crossroad in B-cell traffic. Cytom. B. Clin. Cytom. 2010, 78 (Suppl. 1), S47–S60. [Google Scholar] [CrossRef]
- Cesta, M.F. Normal structure, function, and histology of the spleen. Toxicol. Pathol. 2006, 34, 455–465. [Google Scholar] [CrossRef]
- Lanzavecchia, A. Antigen-specific interaction between T and B cells. Nature 1985, 314, 537–539. [Google Scholar] [CrossRef]
- Elgueta, R.; Benson, M.J.; de Vries, V.C.; Wasiuk, A.; Guo, Y.; Noelle, R.J. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol. Rev. 2009, 229, 152–172. [Google Scholar] [CrossRef]
- Nutt, S.L.; Hodgkin, P.D.; Tarlinton, D.M.; Corcoran, L.M. The generation of antibody-secreting plasma cells. Nat. Rev. Immunol. 2015, 15, 160–171. [Google Scholar] [CrossRef]
- Berkowska, M.A.; Driessen, G.J.; Bikos, V.; Grosserichter-Wagener, C.; Stamatopoulos, K.; Cerutti, A.; He, B.; Biermann, K.; Lange, J.F.; van der Burg, M.; et al. Human memory B cells originate from three distinct germinal center-dependent and -independent maturation pathways. Blood 2011, 118, 2150–2158. [Google Scholar] [CrossRef]
- Rosser, E.C.; Mauri, C. Regulatory B cells: Origin, phenotype, and function. Immunity 2015, 42, 607–612. [Google Scholar] [CrossRef]
- Mauri, C.; Bosma, A. Immune regulatory function of B cells. Annu. Rev. Immunol. 2012, 30, 221–241. [Google Scholar] [CrossRef]
- Vazquez, M.I.; Catalan-Dibene, J.; Zlotnik, A. B cells responses and cytokine production are regulated by their immune microenvironment. Cytokine 2015, 74, 318–326. [Google Scholar] [CrossRef]
- Varin, M.M.; Guerrier, T.; Devauchelle-Pensec, V.; Jamin, C.; Youinou, P.; Pers, J.O. In Sjogren’s syndrome, B lymphocytes induce epithelial cells of salivary glands into apoptosis through protein kinase C delta activation. Autoimmun. Rev. 2012, 11, 252–258. [Google Scholar] [CrossRef]
- Gottenberg, J.E.; Seror, R.; Miceli-Richard, C.; Benessiano, J.; Devauchelle-Pensec, V.; Dieude, P.; Dubost, J.J.; Fauchais, A.L.; Goeb, V.; Hachulla, E.; et al. Serum levels of beta2-microglobulin and free light chains of immunoglobulins are associated with systemic disease activity in primary Sjogren’s syndrome. Data at enrollment in the prospective ASSESS cohort. PLoS ONE 2013, 8, e59868. [Google Scholar] [CrossRef]
- Singh, N.; Chin, I.; Gabriel, P.; Blaum, E.; Masli, S. Dysregulated Marginal Zone B Cell Compartment in a Mouse Model of Sjogren’s Syndrome with Ocular Inflammation. Int. J. Mol. Sci. 2018, 19, 3117. [Google Scholar] [CrossRef]
- Bohnhorst, J.O.; Bjorgan, M.B.; Thoen, J.E.; Natvig, J.B.; Thompson, K.M. Bm1-Bm5 classification of peripheral blood B cells reveals circulating germinal center founder cells in healthy individuals and disturbance in the B cell subpopulations in patients with primary Sjogren’s syndrome. J. Immunol. 2001, 167, 3610–3618. [Google Scholar] [CrossRef]
- Hansen, A.; Gosemann, M.; Pruss, A.; Reiter, K.; Ruzickova, S.; Lipsky, P.E.; Dorner, T. Abnormalities in peripheral B cell memory of patients with primary Sjogren’s syndrome. Arthritis Rheum. 2004, 50, 1897–1908. [Google Scholar] [CrossRef]
- Hamza, N.; Bos, N.A.; Kallenberg, C.G. B-cell populations and sub-populations in Sjogren’s syndrome. Presse. Med. 2012, 41, e475–e483. [Google Scholar] [CrossRef]
- Roberts, M.E.; Kaminski, D.; Jenks, S.A.; Maguire, C.; Ching, K.; Burbelo, P.D.; Iadarola, M.J.; Rosenberg, A.; Coca, A.; Anolik, J.; et al. Primary Sjogren’s syndrome is characterized by distinct phenotypic and transcriptional profiles of IgD+ unswitched memory B cells. Arthritis Rheumatol. 2014, 66, 2558–2569. [Google Scholar] [CrossRef]
- Szabo, K.; Papp, G.; Szanto, A.; Tarr, T.; Zeher, M. A comprehensive investigation on the distribution of circulating follicular T helper cells and B cell subsets in primary Sjogren’s syndrome and systemic lupus erythematosus. Clin. Exp. Immunol. 2016, 183, 76–89. [Google Scholar] [CrossRef]
- Ibrahem, H.M. B cell dysregulation in primary Sjogren’s syndrome: A review. Jpn. Dent. Sci. Rev. 2019, 55, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Nocturne, G.; Mariette, X. B cells in the pathogenesis of primary Sjögren syndrome. Nat. Rev. Rheumatol. 2018, 14, 133–145. [Google Scholar] [CrossRef]
- Van der Gaag, R. Immunological responses in the eyelid and orbit. Eye 1988, 2, 158–163. [Google Scholar] [CrossRef][Green Version]
- Belfort, R., Jr.; Mendes, N.F. Identification of T and B lymphocytes in the human conjunctiva and lacrimal gland in ocular diseases. Br. J. Ophthalmol. 1980, 64, 217–219. [Google Scholar] [CrossRef]
- Parkin, B.; Chew, J.B.; White, V.A.; Garcia-Briones, G.; Chhanabhai, M.; Rootman, J. Lymphocytic Infiltration and Enlargement of the Lacrimal Glands. Ophthalmology 2005, 112, 2040–2047. [Google Scholar] [CrossRef]
- Fletcher, C.A.; Sutherland, A.P.; Groom, J.R.; Batten, M.L.; Ng, L.G.; Gommerman, J.; Mackay, F. Development of nephritis but not sialadenitis in autoimmune-prone BAFF transgenic mice lacking marginal zone B cells. Eur. J. Immunol. 2006, 36, 2504–2514. [Google Scholar] [CrossRef]
- Nguyen, C.Q.; Kim, H.; Cornelius, J.G.; Peck, A.B. Development of Sjogren’s syndrome in nonobese diabetic-derived autoimmune-prone C57BL/6.NOD-Aec1Aec2 mice is dependent on complement component-3. J. Immunol. 2007, 179, 2318–2329. [Google Scholar] [CrossRef]
- Shen, L.; Zhang, C.; Wang, T.; Brooks, S.; Ford, R.J.; Lin-Lee, Y.C.; Kasianowicz, A.; Kumar, V.; Martin, L.; Liang, P.; et al. Development of autoimmunity in IL-14alpha-transgenic mice. J. Immunol. 2006, 177, 5676–5686. [Google Scholar] [CrossRef]
- Shen, L.; Gao, C.; Suresh, L.; Xian, Z.; Song, N.; Chaves, L.D.; Yu, M.; Ambrus, J.L., Jr. Central role for marginal zone B cells in an animal model of Sjogren’s syndrome. Clin. Immunol. 2016, 168, 30–36. [Google Scholar] [CrossRef]
- Blair, P.A.; Norena, L.Y.; Flores-Borja, F.; Rawlings, D.J.; Isenberg, D.A.; Ehrenstein, M.R.; Mauri, C. CD19(+)CD24(hi)CD38(hi) B cells exhibit regulatory capacity in healthy individuals but are functionally impaired in systemic Lupus Erythematosus patients. Immunity 2010, 32, 129–140. [Google Scholar] [CrossRef]
- Lemoine, S.; Morva, A.; Youinou, P.; Jamin, C. Human T cells induce their own regulation through activation of B cells. J. Autoimmun. 2011, 36, 228–238. [Google Scholar] [CrossRef]
- Wang, R.X.; Yu, C.R.; Dambuza, I.M.; Mahdi, R.M.; Dolinska, M.B.; Sergeev, Y.V.; Wingfield, P.T.; Kim, S.H.; Egwuagu, C.E. Interleukin-35 induces regulatory B cells that suppress autoimmune disease. Nat. Med. 2014, 20, 633–641. [Google Scholar] [CrossRef]
- Shen, P.; Roch, T.; Lampropoulou, V.; O’Connor, R.A.; Stervbo, U.; Hilgenberg, E.; Ries, S.; Dang, V.D.; Jaimes, Y.; Daridon, C.; et al. IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature 2014, 507, 366–370. [Google Scholar] [CrossRef]
- Lin, Y.; Huang, Y.; Lu, Z.; Luo, C.; Shi, Y.; Zeng, Q.; Cao, Y.; Liu, L.; Wang, X.; Ji, Q. Decreased plasma IL-35 levels are related to the left ventricular ejection fraction in coronary artery diseases. PLoS ONE 2012, 7, e52490. [Google Scholar] [CrossRef]
- Fogel, O.; Riviere, E.; Seror, R.; Nocturne, G.; Boudaoud, S.; Ly, B.; Gottenberg, J.E.; Le Guern, V.; Dubost, J.J.; Nititham, J.; et al. Role of the IL-12/IL-35 balance in patients with Sjogren syndrome. J. Allergy. Clin. Immunol. 2018, 142, 258–268.e5. [Google Scholar] [CrossRef]
- Sene, D.; Ismael, S.; Forien, M.; Charlotte, F.; Kaci, R.; Cacoub, P.; Diallo, A.; Dieude, P.; Liote, F. Ectopic Germinal Center-Like Structures in Minor Salivary Gland Biopsy Tissue Predict Lymphoma Occurrence in Patients with Primary Sjogren’s Syndrome. Arthritis Rheumatol. 2018, 70, 1481–1488. [Google Scholar] [CrossRef]
- He, J.; Jin, Y.; Zhang, X.; Zhou, Y.; Li, R.; Dai, Y.; Sun, X.; Zhao, J.; Guo, J.; Li, Z. Characteristics of germinal center-like structures in patients with Sjogren’s syndrome. Int. J. Rheum. Dis. 2017, 20, 245–251. [Google Scholar] [CrossRef]
- Karabiyik, A.; Peck, A.B.; Nguyen, C.Q. The important role of T cells and receptor expression in Sjogren’s syndrome. Scand. J. Immunol. 2013, 78, 157–166. [Google Scholar] [CrossRef]
- Bombardieri, M.; Lewis, M.; Pitzalis, C. Ectopic lymphoid neogenesis in rheumatic autoimmune diseases. Nat. Rev. Rheumatol. 2017, 13, 141–154. [Google Scholar] [CrossRef]
- Salomonsson, S.; Jonsson, M.V.; Skarstein, K.; Brokstad, K.A.; Hjelmstrom, P.; Wahren-Herlenius, M.; Jonsson, R. Cellular basis of ectopic germinal center formation and autoantibody production in the target organ of patients with Sjogren’s syndrome. Arthritis Rheum. 2003, 48, 3187–3201. [Google Scholar] [CrossRef]
- Chtanova, T.; Tangye, S.G.; Newton, R.; Frank, N.; Hodge, M.R.; Rolph, M.S.; Mackay, C.R. T follicular helper cells express a distinctive transcriptional profile, reflecting their role as non-Th1/Th2 effector cells that provide help for B cells. J. Immunol. 2004, 173, 68–78. [Google Scholar] [CrossRef]
- Mariette, X.; Roux, S.; Zhang, J.; Bengoufa, D.; Lavie, F.; Zhou, T.; Kimberly, R. The level of BLyS (BAFF) correlates with the titre of autoantibodies in human Sjogren’s syndrome. Ann. Rheum. Dis. 2003, 62, 168–171. [Google Scholar] [CrossRef]
- Lavie, F.; Miceli-Richard, C.; Quillard, J.; Roux, S.; Leclerc, P.; Mariette, X. Expression of BAFF (BLyS) in T cells infiltrating labial salivary glands from patients with Sjogren’s syndrome. J. Pathol. 2004, 202, 496–502. [Google Scholar] [CrossRef]
- Daridon, C.; Devauchelle, V.; Hutin, P.; Le Berre, R.; Martins-Carvalho, C.; Bendaoud, B.; Dueymes, M.; Saraux, A.; Youinou, P.; Pers, J.O. Aberrant expression of BAFF by B lymphocytes infiltrating the salivary glands of patients with primary Sjogren’s syndrome. Arthritis Rheum. 2007, 56, 1134–1144. [Google Scholar] [CrossRef]
- Petrie, H.T.; Zuniga-Pflucker, J.C. Zoned out: Functional mapping of stromal signaling microenvironments in the thymus. Annu. Rev. Immunol. 2007, 25, 649–679. [Google Scholar] [CrossRef]
- Laurent, J.; Bosco, N.; Marche, P.N.; Ceredig, R. New insights into the proliferation and differentiation of early mouse thymocytes. Int. Immunol. 2004, 16, 1069–1080. [Google Scholar] [CrossRef]
- Li, X.; von Boehmer, H. Notch Signaling in T-Cell Development and T-ALL. ISRN Hematol. 2011, 2011, 921706. [Google Scholar] [CrossRef]
- Dik, W.A.; Pike-Overzet, K.; Weerkamp, F.; de Ridder, D.; de Haas, E.F.; Baert, M.R.; van der Spek, P.; Koster, E.E.; Reinders, M.J.; van Dongen, J.J.; et al. New insights on human T cell development by quantitative T cell receptor gene rearrangement studies and gene expression profiling. J. Exp. Med. 2005, 201, 1715–1723. [Google Scholar] [CrossRef]
- Klein, L.; Hinterberger, M.; Wirnsberger, G.; Kyewski, B. Antigen presentation in the thymus for positive selection and central tolerance induction. Nat. Rev. Immunol. 2009, 9, 833–844. [Google Scholar] [CrossRef]
- Koch, U.; Radtke, F. Mechanisms of T cell development and transformation. Annu. Rev. Cell. Dev. Biol. 2011, 27, 539–562. [Google Scholar] [CrossRef]
- Kumar, B.V.; Connors, T.J.; Farber, D.L. Human T Cell Development, Localization, and Function throughout Life. Immunity 2018, 48, 202–213. [Google Scholar] [CrossRef]
- Crotty, S. A brief history of T cell help to B cells. Nat. Rev. Immunol. 2015, 15, 185–189. [Google Scholar] [CrossRef]
- Luckheeram, R.V.; Zhou, R.; Verma, A.D.; Xia, B. CD4+T Cells: Differentiation and Functions. Clin. Dev. Immunol. 2012, 2012, 925135. [Google Scholar] [CrossRef]
- Volpe, E.; Sambucci, M.; Battistini, L.; Borsellino, G. Fas-Fas Ligand: Checkpoint of T Cell Functions in Multiple Sclerosis. Front. Immunol. 2016, 7, 382. [Google Scholar] [CrossRef]
- Elemans, M.; Seich Al Basatena, N.K.; Asquith, B. The efficiency of the human CD8+ T cell response: How should we quantify it, what determines it, and does it matter? Plos. Comput. Biol. 2012, 8, e1002381. [Google Scholar] [CrossRef]
- Omilusik, K.D.; Goldrath, A.W. The origins of memory T cells. Nature. 2017, 552, 337–339. [Google Scholar] [CrossRef]
- Vignali, D.A.A.; Collison, L.W.; Workman, C.J. How regulatory T cells work. Nat. Rev. Immunol. 2008, 8, 523–532. [Google Scholar] [CrossRef]
- Tupin, E.; Kinjo, Y.; Kronenberg, M. The unique role of natural killer T cells in the response to microorganisms. Nat. Rev. Microbiol. 2007, 5, 405–417. [Google Scholar] [CrossRef]
- Adamson, T.C., 3rd; Fox, R.I.; Frisman, D.M.; Howell, F.V. Immunohistologic analysis of lymphoid infiltrates in primary Sjogren’s syndrome using monoclonal antibodies. J. Immunol. 1983, 130, 203–208. [Google Scholar]
- Roescher, N.; Tak, P.P.; Illei, G.G. Cytokines in Sjogren’s syndrome. Oral. Dis. 2009, 15, 519–526. [Google Scholar] [CrossRef]
- Kwok, S.K.; Lee, J.; Yu, D.; Kang, K.Y.; Cho, M.L.; Kim, H.R.; Ju, J.H.; Lee, S.H.; Park, S.H.; Kim, H.Y. A pathogenetic role for IL-21 in primary Sjogren syndrome. Nat. Rev. Rheumatol. 2015, 11, 368–374. [Google Scholar] [CrossRef]
- Niederkorn, J.Y.; Stern, M.E.; Pflugfelder, S.C.; De Paiva, C.S.; Corrales, R.M.; Gao, J.; Siemasko, K. Desiccating stress induces T cell-mediated Sjogren’s Syndrome-like lacrimal keratoconjunctivitis. J. Immunol. 2006, 176, 3950–3957. [Google Scholar] [CrossRef]
- Verstappen, G.M.; Kroese, F.G.M.; Bootsma, H. T cells in primary Sjogren’s syndrome: Targets for early intervention. Rheumatology (Oxford) 2019. (ahead of print). [Google Scholar] [CrossRef]
- Barr, J.Y.; Wang, X.; Meyerholz, D.K.; Lieberman, S.M. CD8 T cells contribute to lacrimal gland pathology in the nonobese diabetic mouse model of Sjogren syndrome. Immunol. Cell. Biol. 2017, 95, 684–694. [Google Scholar] [CrossRef] [PubMed]
- Pflugfelder, S.C.; Corrales, R.M.; de Paiva, C.S. T helper cytokines in dry eye disease. Exp. Eye Res. 2013, 117, 118–125. [Google Scholar] [CrossRef]
- Mosmann, T.R.; Coffman, R.L. TH1 and TH2 cells: Different patterns of lymphokine secretion lead to different functional properties. Annu. Rev. Immunol. 1989, 7, 145–173. [Google Scholar] [CrossRef]
- Pflugfelder, S.C.; De Paiva, C.S.; Moore, Q.L.; Volpe, E.A.; Li, D.Q.; Gumus, K.; Zaheer, M.L.; Corrales, R.M. Aqueous Tear Deficiency Increases Conjunctival Interferon-gamma (IFN-gamma) Expression and Goblet Cell Loss. Investig. Ophthalmol. Vis. Sci. 2015, 56, 7545–7550. [Google Scholar] [CrossRef]
- Sakai, A.; Sugawara, Y.; Kuroishi, T.; Sasano, T.; Sugawara, S. Identification of IL-18 and Th17 cells in salivary glands of patients with Sjogren’s syndrome, and amplification of IL-17-mediated secretion of inflammatory cytokines from salivary gland cells by IL-18. J. Immunol. 2008, 181, 2898–2906. [Google Scholar] [CrossRef]
- Manoussakis, M.N.; Boiu, S.; Korkolopoulou, P.; Kapsogeorgou, E.K.; Kavantzas, N.; Ziakas, P.; Patsouris, E.; Moutsopoulos, H.M. Rates of infiltration by macrophages and dendritic cells and expression of interleukin-18 and interleukin-12 in the chronic inflammatory lesions of Sjogren’s syndrome: Correlation with certain features of immune hyperactivity and factors associated with high risk of lymphoma development. Arthritis Rheum. 2007, 56, 3977–3988. [Google Scholar] [CrossRef]
- Delaleu, N.; Immervoll, H.; Cornelius, J.; Jonsson, R. Biomarker profiles in serum and saliva of experimental Sjogren’s syndrome: Associations with specific autoimmune manifestations. Arthritis Res. Ther. 2008, 10, R22. [Google Scholar] [CrossRef]
- Brayer, J.B.; Cha, S.; Nagashima, H.; Yasunari, U.; Lindberg, A.; Diggs, S.; Martinez, J.; Goa, J.; Humphreys-Beher, M.G.; Peck, A.B. IL-4-dependent effector phase in autoimmune exocrinopathy as defined by the NOD.IL-4-gene knockout mouse model of Sjogren’s syndrome. Scand. J. Immunol. 2001, 54, 133–140. [Google Scholar] [CrossRef]
- Gao, J.; Killedar, S.; Cornelius, J.G.; Nguyen, C.; Cha, S.; Peck, A.B. Sjogren’s syndrome in the NOD mouse model is an interleukin-4 time-dependent, antibody isotype-specific autoimmune disease. J. Autoimmun. 2006, 26, 90–103. [Google Scholar] [CrossRef]
- Veldhoen, M.; Hocking, R.J.; Atkins, C.J.; Locksley, R.M.; Stockinger, B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 2006, 24, 179–189. [Google Scholar] [CrossRef]
- Jin, W.; Dong, C. IL-17 cytokines in immunity and inflammation. Emerg. Microbes. Infect. 2013, 2, e60. [Google Scholar] [CrossRef]
- Fei, Y.; Zhang, W.; Lin, D.; Wu, C.; Li, M.; Zhao, Y.; Zeng, X.; Zhang, F. Clinical parameter and Th17 related to lymphocytes infiltrating degree of labial salivary gland in primary Sjögren’s syndrome. Clin. Rheumatol. 2014, 33, 523–529. [Google Scholar] [CrossRef]
- Nguyen, C.Q.; Hu, M.H.; Li, Y.; Stewart, C.; Peck, A.B. Salivary gland tissue expression of interleukin-23 and interleukin-17 in Sjogren’s syndrome: Findings in humans and mice. Arthritis Rheum. 2008, 58, 734–743. [Google Scholar] [CrossRef]
- Katsifis, G.E.; Rekka, S.; Moutsopoulos, N.M.; Pillemer, S.; Wahl, S.M. Systemic and local interleukin-17 and linked cytokines associated with Sjogren’s syndrome immunopathogenesis. Am. J. Pathol. 2009, 175, 1167–1177. [Google Scholar] [CrossRef]
- Matsui, K.; Sano, H. T Helper 17 Cells in Primary Sjögren’s Syndrome. J. Clin. Med. 2017, 6, 65. [Google Scholar] [CrossRef]
- Korn, T.; Bettelli, E.; Gao, W.; Awasthi, A.; Jager, A.; Strom, T.B.; Oukka, M.; Kuchroo, V.K. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature 2007, 448, 484–487. [Google Scholar] [CrossRef]
- Huber, M.; Brustle, A.; Reinhard, K.; Guralnik, A.; Walter, G.; Mahiny, A.; von Low, E.; Lohoff, M. IRF4 is essential for IL-21-mediated induction, amplification, and stabilization of the Th17 phenotype. Proc. Natl. Acad. Sci. USA 2008, 105, 20846–20851. [Google Scholar] [CrossRef]
- Chen, Q.; Yang, W.; Gupta, S.; Biswas, P.; Smith, P.; Bhagat, G.; Pernis, A.B. IRF-4-binding protein inhibits interleukin-17 and interleukin-21 production by controlling the activity of IRF-4 transcription factor. Immunity 2008, 29, 899–911. [Google Scholar] [CrossRef]
- Kuchen, S.; Robbins, R.; Sims, G.P.; Sheng, C.; Phillips, T.M.; Lipsky, P.E.; Ettinger, R. Essential role of IL-21 in B cell activation, expansion, and plasma cell generation during CD4+ T cell-B cell collaboration. J. Immunol. 2007, 179, 5886–5896. [Google Scholar] [CrossRef]
- Pene, J.; Gauchat, J.F.; Lecart, S.; Drouet, E.; Guglielmi, P.; Boulay, V.; Delwail, A.; Foster, D.; Lecron, J.C.; Yssel, H. Cutting edge: IL-21 is a switch factor for the production of IgG1 and IgG3 by human B cells. J. Immunol. 2004, 172, 5154–5157. [Google Scholar] [CrossRef]
- Ettinger, R.; Kuchen, S.; Lipsky, P.E. Interleukin 21 as a target of intervention in autoimmune disease. Ann. Rheum. Dis. 2008, 67 (Suppl. 3), iii83–iii86. [Google Scholar] [CrossRef]
- Maehara, T.; Moriyama, M.; Nakashima, H.; Miyake, K.; Hayashida, J.N.; Tanaka, A.; Shinozaki, S.; Kubo, Y.; Nakamura, S. Interleukin-21 contributes to germinal centre formation and immunoglobulin G4 production in IgG4-related dacryoadenitis and sialoadenitis, so-called Mikulicz’s disease. Ann. Rheum. Dis. 2012, 71, 2011–2019. [Google Scholar] [CrossRef]
- Kang, K.Y.; Kim, H.O.; Kwok, S.K.; Ju, J.H.; Park, K.S.; Sun, D.I.; Jhun, J.Y.; Oh, H.J.; Park, S.H.; Kim, H.Y. Impact of interleukin-21 in the pathogenesis of primary Sjogren’s syndrome: Increased serum levels of interleukin-21 and its expression in the labial salivary glands. Arthritis Res. Ther. 2011, 13, R179. [Google Scholar] [CrossRef]
- Lavoie, T.N.; Stewart, C.M.; Berg, K.M.; Li, Y.; Nguyen, C.Q. Expression of interleukin-22 in Sjogren’s syndrome: Significant correlation with disease parameters. Scand. J. Immunol. 2011, 74, 377–382. [Google Scholar] [CrossRef]
- Verstappen, G.M.; Corneth, O.B.J.; Bootsma, H.; Kroese, F.G.M. Th17 cells in primary Sjogren’s syndrome: Pathogenicity and plasticity. J. Autoimmun. 2018, 87, 16–25. [Google Scholar] [CrossRef]
- Voigt, A.; Esfandiary, L.; Wanchoo, A.; Glenton, P.; Donate, A.; Craft, W.F.; Craft, S.L.; Nguyen, C.Q. Sexual dimorphic function of IL-17 in salivary gland dysfunction of the C57BL/6.NOD-Aec1Aec2 model of Sjogren’s syndrome. Sci. Rep. 2016, 6, 38717. [Google Scholar] [CrossRef]
- Fontenot, J.D.; Rasmussen, J.P.; Williams, L.M.; Dooley, J.L.; Farr, A.G.; Rudensky, A.Y. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity 2005, 22, 329–341. [Google Scholar] [CrossRef]
- Miyara, M.; Chader, D.; Sage, E.; Sugiyama, D.; Nishikawa, H.; Bouvry, D.; Claer, L.; Hingorani, R.; Balderas, R.; Rohrer, J.; et al. Sialyl Lewis x (CD15s) identifies highly differentiated and most suppressive FOXP3high regulatory T cells in humans. Proc. Natl. Acad. Sci. USA 2015, 112, 7225–7230. [Google Scholar] [CrossRef]
- Deenick, E.K.; Ma, C.S. The regulation and role of T follicular helper cells in immunity. Immunology 2011, 134, 361–367. [Google Scholar] [CrossRef]
- Moens, L.; Tangye, S.G. Cytokine-Mediated Regulation of Plasma Cell Generation: IL-21 Takes Center Stage. Front. Immunol. 2014, 5, 65. [Google Scholar] [CrossRef]
- Crotty, S. T follicular helper cell differentiation, function, and roles in disease. Immunity 2014, 41, 529–542. [Google Scholar] [CrossRef]
- Simpson, N.; Gatenby, P.A.; Wilson, A.; Malik, S.; Fulcher, D.A.; Tangye, S.G.; Manku, H.; Vyse, T.J.; Roncador, G.; Huttley, G.A.; et al. Expansion of circulating T cells resembling follicular helper T cells is a fixed phenotype that identifies a subset of severe systemic lupus erythematosus. Arthritis Rheum. 2010, 62, 234–244. [Google Scholar] [CrossRef]
- Szabo, K.; Papp, G.; Barath, S.; Gyimesi, E.; Szanto, A.; Zeher, M. Follicular helper T cells may play an important role in the severity of primary Sjogren’s syndrome. Clin. Immunol. 2013, 147, 95–104. [Google Scholar] [CrossRef]
- Zhao, Y.; Lutalo, P.M.; Thomas, J.E.; Sangle, S.; Choong, L.M.; Tyler, J.R.; Tree, T.; Spencer, J.; D’Cruz, D.P. Circulating T follicular helper cell and regulatory T cell frequencies are influenced by B cell depletion in patients with granulomatosis with polyangiitis. Rheumatology (Oxford) 2014, 53, 621–630. [Google Scholar] [CrossRef][Green Version]
- Brokstad, K.A.; Fredriksen, M.; Zhou, F.; Bergum, B.; Brun, J.G.; Cox, R.J.; Skarstein, K. T follicular-like helper cells in the peripheral blood of patients with primary Sjogren’s syndrome. Scand. J. Immunol. 2018, 88, e12679. [Google Scholar] [CrossRef]
- Gong, Y.Z.; Nititham, J.; Taylor, K.; Miceli-Richard, C.; Sordet, C.; Wachsmann, D.; Bahram, S.; Georgel, P.; Criswell, L.A.; Sibilia, J.; et al. Differentiation of follicular helper T cells by salivary gland epithelial cells in primary Sjogren’s syndrome. J. Autoimmun. 2014, 51, 57–66. [Google Scholar] [CrossRef]
- Wikenheiser, D.J.; Stumhofer, J.S. ICOS Co-Stimulation: Friend or Foe? Front. Immunol. 2016, 7. [Google Scholar] [CrossRef]
- Sage, P.T.; Sharpe, A.H. T follicular regulatory cells in the regulation of B cell responses. Trends Immunol. 2015, 36, 410–418. [Google Scholar] [CrossRef]
- Verstappen, G.M.; Nakshbandi, U.; Mossel, E.; Haacke, E.A.; van der Vegt, B.; Vissink, A.; Bootsma, H.; Kroese, F.G.M. Is the T Follicular Regulatory: Follicular Helper T Cell Ratio in Blood a Biomarker for Ectopic Lymphoid Structure Formation in Sjogren’s Syndrome? Comment on the Article by Fonseca et al. Arthritis Rheumatol. 2018, 70, 1354–1355. [Google Scholar] [CrossRef]
- Fonseca, V.R.; Romao, V.C.; Agua-Doce, A.; Santos, M.; Lopez-Presa, D.; Ferreira, A.C.; Fonseca, J.E.; Graca, L. The Ratio of Blood T Follicular Regulatory Cells to T Follicular Helper Cells Marks Ectopic Lymphoid Structure Formation While Activated Follicular Helper T Cells Indicate Disease Activity in Primary Sjogren’s Syndrome. Arthritis Rheumatol. 2018, 70, 774–784. [Google Scholar] [CrossRef]
- Skopouli, F.N.; Dafni, U.; Ioannidis, J.P.A.; Moutsopoulos, H.M. Clinical evolution, and morbidity and mortality of primary Sjogren’s syndrome. Semin. Arthritis Rheu. 2000, 29, 296–304. [Google Scholar] [CrossRef]
- Vogelsang, P.; Jonsson, M.V.; Dalvin, S.T.; Appel, S. Role of Dendritic Cells in Sjogren’s Syndrome. Scand. J. Immunol. 2006, 64, 219–226. [Google Scholar] [CrossRef]
- Vogelsang, P.; Brun, J.G.; Oijordsbakken, G.; Skarstein, K.; Jonsson, R.; Appel, S. Levels of plasmacytoid dendritic cells and type-2 myeloid dendritic cells are reduced in peripheral blood of patients with primary Sjogren’s syndrome. Ann. Rheum. Dis. 2010, 69, 1235–1238. [Google Scholar] [CrossRef]
- Bjordal, O.; Norheim, K.B.; Rodahl, E.; Jonsson, R.; Omdal, R. Primary Sjogren’s syndrome and the eye. Surv. Ophthalmol. 2020, 65, 119–132. [Google Scholar] [CrossRef]
- Gottenberg, J.E.; Cagnard, N.; Lucchesi, C.; Letourneur, F.; Mistou, S.; Lazure, T.; Jacques, S.; Ba, N.; Ittah, M.; Lepajolec, C.; et al. Activation of IFN pathways and plasmacytoid dendritic cell recruitment in target organs of primary Sjogren’s syndrome. Proc. Natl. Acad. Sci. USA 2006, 103, 2770–2775. [Google Scholar] [CrossRef]
- Colonna, M.; Trinchieri, G.; Liu, Y.J. Plasmacytoid dendritic cells in immunity. Nat. Immunol. 2004, 5, 1219–1226. [Google Scholar] [CrossRef]
- Gilliet, M.; Cao, W.; Liu, Y.J. Plasmacytoid dendritic cells: Sensing nucleic acids in viral infection and autoimmune diseases. Nat. Rev. Immunol. 2008, 8, 594–606. [Google Scholar] [CrossRef]
- Swiecki, M.; Colonna, M. Unraveling the functions of plasmacytoid dendritic cells during viral infections, autoimmunity, and tolerance. Immunol. Rev. 2010, 234, 142–162. [Google Scholar] [CrossRef] [PubMed]
- Bave, U.; Nordmark, G.; Lovgren, T.; Ronnelid, J.; Cajander, S.; Eloranta, M.L.; Alm, G.V.; Ronnblom, L. Activation of the type I interferon system in primary Sjogren’s syndrome: A possible etiopathogenic mechanism. Arthritis Rheum. 2005, 52, 1185–1195. [Google Scholar] [CrossRef]
- Vakaloglou, K.M.; Mavragani, C.P. Activation of the type I interferon pathway in primary Sjogren’s syndrome: An update. Curr. Opin. Rheumatol. 2011, 23, 459–464. [Google Scholar] [CrossRef]
- Rusakiewicz, S.; Nocturne, G.; Lazure, T.; Semeraro, M.; Flament, C.; Caillat-Zucman, S.; Sene, D.; Delahaye, N.; Vivier, E.; Chaba, K.; et al. NCR3/NKp30 contributes to pathogenesis in primary Sjogren’s syndrome. Sci. Transl. Med. 2013, 5, 195ra196. [Google Scholar] [CrossRef]
- Izumi, Y.; Ida, H.; Huang, M.; Iwanaga, N.; Tanaka, F.; Aratake, K.; Arima, K.; Tamai, M.; Kamachi, M.; Nakamura, H.; et al. Characterization of peripheral natural killer cells in primary Sjogren’s syndrome: Impaired NK cell activity and low NK cell number. J. Lab. Clin. Med. 2006, 147, 242–249. [Google Scholar] [CrossRef]
- Chen, X.; Aqrawi, L.A.; Utheim, T.P.; Tashbayev, B.; Utheim, Ø.A.; Reppe, S.; Hove, L.H.; Herlofson, B.B.; Singh, P.B.; Palm, Ø.; et al. Elevated cytokine levels in tears and saliva of patients with primary Sjögren’s syndrome correlate with clinical ocular and oral manifestations. Sci. Rep. 2019, 9, 7319. [Google Scholar] [CrossRef]
- Zhou, D.; Chen, Y.T.; Chen, F.; Gallup, M.; Vijmasi, T.; Bahrami, A.F.; Noble, L.B.; van Rooijen, N.; McNamara, N.A. Critical involvement of macrophage infiltration in the development of Sjogren’s syndrome-associated dry eye. Am. J. Pathol. 2012, 181, 753–760. [Google Scholar] [CrossRef]
- Ushio, A.; Arakaki, R.; Yamada, A.; Saito, M.; Tsunematsu, T.; Kudo, Y.; Ishimaru, N. Crucial roles of macrophages in the pathogenesis of autoimmune disease. World. J. Immunol. 2017, 7, 1–8. [Google Scholar] [CrossRef]
- Ushio, A.; Arakaki, R.; Otsuka, K.; Yamada, A.; Tsunematsu, T.; Kudo, Y.; Aota, K.; Azuma, M.; Ishimaru, N. CCL22-Producing Resident Macrophages Enhance T Cell Response in Sjogren’s Syndrome. Front. Immunol. 2018, 9, 2594. [Google Scholar] [CrossRef]
- Stefanski, A.L.; Tomiak, C.; Pleyer, U.; Dietrich, T.; Burmester, G.R.; Dorner, T. The Diagnosis and Treatment of Sjogren’s Syndrome. Dtsch. Arztebl. Int. 2017, 114, 354–361. [Google Scholar] [CrossRef]
- Felten, R.; Scher, F.; Sibilia, J.; Gottenberg, J.E.; Arnaud, L. The pipeline of targeted therapies under clinical development for primary Sjogren’s syndrome: A systematic review of trials. Autoimmun. Rev. 2019, 18, 576–582. [Google Scholar] [CrossRef]
- Fox, R.I.; Fox, C.M.; Gottenberg, J.E.; Dorner, T. Treatment of Sjogren’s syndrome: Current therapy and future directions. Rheumatology (Oxford) 2019. (ahead of print). [Google Scholar] [CrossRef]
- Fasano, S.; Isenberg, D.A. Present and novel biologic drugs in primary Sjogren’s syndrome. Clin. Exp. Rheumatol. 2019, 37 (Suppl. 118), 167–174. [Google Scholar]
- Johr, C.R.; Vivino, F.B. Biologic Therapy in the Treatment of Sjögren’s Syndrome: A Clinical Perspective. Curr. Treat. Options. Rheumatol. 2018, 4, 85–98. [Google Scholar] [CrossRef]
- Sada, P.R.; Isenberg, D.; Ciurtin, C. Biologic treatment in Sjogren’s syndrome. Rheumatology (Oxford) 2015, 54, 219–230. [Google Scholar] [CrossRef]
- Chen, X.; Wu, H.; Wei, W. Advances in the diagnosis and treatment of Sjogren’s syndrome. Clin. Rheumatol. 2018, 37, 1743–1749. [Google Scholar] [CrossRef]
- Shiboski, C.H.; Shiboski, S.C.; Seror, R.; Criswell, L.A.; Labetoulle, M.; Lietman, T.M.; Rasmussen, A.; Scofield, H.; Vitali, C.; Bowman, S.J.; et al. 2016 American College of Rheumatology/European League Against Rheumatism Classification Criteria for Primary Sjogren’s Syndrome: A Consensus and Data-Driven Methodology Involving Three International Patient Cohorts. Arthritis Rheumatol. 2017, 69, 35–45. [Google Scholar] [CrossRef]
- Seror, R.; Bowman, S.J.; Brito-Zeron, P.; Theander, E.; Bootsma, H.; Tzioufas, A.; Gottenberg, J.E.; Ramos-Casals, M.; Dorner, T.; Ravaud, P.; et al. EULAR Sjogren’s syndrome disease activity index (ESSDAI): A user guide. RMD Open 2015, 1, e000022. [Google Scholar] [CrossRef]
- Seror, R.; Ravaud, P.; Mariette, X.; Bootsma, H.; Theander, E.; Hansen, A.; Ramos-Casals, M.; Dorner, T.; Bombardieri, S.; Hachulla, E.; et al. EULAR Sjogren’s Syndrome Patient Reported Index (ESSPRI): Development of a consensus patient index for primary Sjogren’s syndrome. Ann. Rheum. Dis. 2011, 70, 968–972. [Google Scholar] [CrossRef]
- Brito-Zeron, P.; Baldini, C.; Bootsma, H.; Bowman, S.J.; Jonsson, R.; Mariette, X.; Sivils, K.; Theander, E.; Tzioufas, A.; Ramos-Casals, M. Sjogren syndrome. Nat. Rev. Dis. Primers. 2016, 2, 16047. [Google Scholar] [CrossRef]
- Voulgarelis, M.; Tzioufas, A.G. Pathogenetic mechanisms in the initiation and perpetuation of Sjögren’s syndrome. Nat. Rev. Rheumatol. 2010, 6, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, L.; Szodoray, P.; Kiss, E. Secondary tumours in Sjogren’s syndrome. Autoimmun. Rev. 2010, 9, 203–206. [Google Scholar] [CrossRef]
- Dass, S.; Bowman, S.J.; Vital, E.M.; Ikeda, K.; Pease, C.T.; Hamburger, J.; Richards, A.; Rauz, S.; Emery, P. Reduction of fatigue in Sjogren syndrome with rituximab: Results of a randomised, double-blind, placebo-controlled pilot study. Ann. Rheum. Dis. 2008, 67, 1541–1544. [Google Scholar] [CrossRef] [PubMed]
- Meijer, J.M.; Meiners, P.M.; Vissink, A.; Spijkervet, F.K.; Abdulahad, W.; Kamminga, N.; Brouwer, E.; Kallenberg, C.G.; Bootsma, H. Effectiveness of rituximab treatment in primary Sjogren’s syndrome: A randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2010, 62, 960–968. [Google Scholar] [CrossRef]
- Gottenberg, J.E.; Cinquetti, G.; Larroche, C.; Combe, B.; Hachulla, E.; Meyer, O.; Pertuiset, E.; Kaplanski, G.; Chiche, L.; Berthelot, J.M.; et al. Efficacy of rituximab in systemic manifestations of primary Sjogren’s syndrome: Results in 78 patients of the AutoImmune and Rituximab registry. Ann. Rheum. Dis. 2013, 72, 1026–1031. [Google Scholar] [CrossRef]
- Carubbi, F.; Cipriani, P.; Marrelli, A.; Benedetto, P.; Ruscitti, P.; Berardicurti, O.; Pantano, I.; Liakouli, V.; Alvaro, S.; Alunno, A.; et al. Efficacy and safety of rituximab treatment in early primary Sjogren’s syndrome: A prospective, multi-center, follow-up study. Arthritis Res. Ther. 2013, 15, R172. [Google Scholar] [CrossRef]
- Meiners, P.M.; Arends, S.; Brouwer, E.; Spijkervet, F.K.; Vissink, A.; Bootsma, H. Responsiveness of disease activity indices ESSPRI and ESSDAI in patients with primary Sjogren’s syndrome treated with rituximab. Ann. Rheum. Dis. 2012, 71, 1297–1302. [Google Scholar] [CrossRef]
- Devauchelle-Pensec, V.; Mariette, X.; Jousse-Joulin, S.; Berthelot, J.M.; Perdriger, A.; Puechal, X.; Le Guern, V.; Sibilia, J.; Gottenberg, J.E.; Chiche, L.; et al. Treatment of primary Sjogren syndrome with rituximab: A randomized trial. Ann. Intern. Med. 2014, 160, 233–242. [Google Scholar] [CrossRef]
- Brown, S.; Navarro Coy, N.; Pitzalis, C.; Emery, P.; Pavitt, S.; Gray, J.; Hulme, C.; Hall, F.; Busch, R.; Smith, P.; et al. The TRACTISS protocol: A randomised double blind placebo controlled clinical trial of anti-B-cell therapy in patients with primary Sjogren’s Syndrome. BMC Musculoskelet. Disord. 2014, 15, 21. [Google Scholar] [CrossRef]
- Bowman, S.J.; Everett, C.C.; O’Dwyer, J.L.; Emery, P.; Pitzalis, C.; Ng, W.F.; Pease, C.T.; Price, E.J.; Sutcliffe, N.; Gendi, N.S.T.; et al. Randomized Controlled Trial of Rituximab and Cost-Effectiveness Analysis in Treating Fatigue and Oral Dryness in Primary Sjogren’s Syndrome. Arthritis Rheumatol. 2017, 69, 1440–1450. [Google Scholar] [CrossRef]
- Steinfeld, S.D.; Tant, L.; Burmester, G.R.; Teoh, N.K.; Wegener, W.A.; Goldenberg, D.M.; Pradier, O. Epratuzumab (humanised anti-CD22 antibody) in primary Sjogren’s syndrome: An open-label phase I/II study. Arthritis Res. Ther. 2006, 8, R129. [Google Scholar] [CrossRef]
- Gottenberg, J.E.; Dorner, T.; Bootsma, H.; Devauchelle-Pensec, V.; Bowman, S.J.; Mariette, X.; Bartz, H.; Oortgiesen, M.; Shock, A.; Koetse, W.; et al. Efficacy of Epratuzumab, an Anti-CD22 Monoclonal IgG Antibody, in Systemic Lupus Erythematosus Patients With Associated Sjogren’s Syndrome: Post Hoc Analyses From the EMBODY Trials. Arthritis Rheumatol. 2018, 70, 763–773. [Google Scholar] [CrossRef]
- Mariette, X.; Seror, R.; Quartuccio, L.; Baron, G.; Salvin, S.; Fabris, M.; Desmoulins, F.; Nocturne, G.; Ravaud, P.; De Vita, S. Efficacy and safety of belimumab in primary Sjogren’s syndrome: Results of the BELISS open-label phase II study. Ann. Rheum. Dis. 2015, 74, 526–531. [Google Scholar] [CrossRef]
- Dorner, T.; Posch, M.G.; Li, Y.; Petricoul, O.; Cabanski, M.; Milojevic, J.M.; Kamphausen, E.; Valentin, M.A.; Simonett, C.; Mooney, L.; et al. Treatment of primary Sjogren’s syndrome with ianalumab (VAY736) targeting B cells by BAFF receptor blockade coupled with enhanced, antibody-dependent cellular cytotoxicity. Ann. Rheum. Dis. 2019, 78, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Dörner, T.; Bowman, S.J.; Fox, R.; Mariette, X.; Papas, A.; Grader-Beck, T.; Fisher, B.A.; Barcelos, F.; De Vita, S.; Schulze-Koops, H.; et al. OP0302 IANALUMAB (VAY736), a dual mode of action biologic combining baff receptor inhibition with b cell depletion, reaches primary endpoint for treatment of primary sjogren’s syndrome. Ann. Rheum. Dis. 2020, 79 (Suppl. 1), 187. [Google Scholar] [CrossRef]
- St Clair, E.W.; Baer, A.N.; Wei, C.W.; Noaiseh, G.; Parke, A.; Coca, A.; Utset, T.O.; Genovese, M.C.; Wallace, D.J.; McNamara, J.; et al. Clinical Efficacy and Safety of Baminercept, a Lymphotoxin beta Receptor Fusion Protein, in Primary Sjogren’s Syndrome Results from a Phase II Randomized, Double-Blind, Placebo-Controlled Trial. Arthritis Rheumatol. 2018, 70, 1470–1480. [Google Scholar] [CrossRef]
- Pescovitz, M.D. Rituximab, an anti-cd20 monoclonal antibody: History and mechanism of action. Am. J. Transpl. 2006, 6 Pt 1, 859–866. [Google Scholar] [CrossRef]
- Carubbi, F.; Alunno, A.; Cipriani, P.; Berardicurti, O.; Ruscitti, P.; Liakouli, V.; Ciccia, F.; Triolo, G.; Gerli, R.; Giacomelli, R. Use of Rituximab in the Management of Sjögren’s Syndrome. Curr. Treat. Options Rheumatol. 2015, 1, 277–291. [Google Scholar] [CrossRef]
- Perosa, F.; Prete, M.; Racanelli, V.; Dammacco, F. CD20-depleting therapy in autoimmune diseases: From basic research to the clinic. J. Intern. Med. 2010, 267, 260–277. [Google Scholar] [CrossRef]
- Ciccia, F.; Giardina, A.; Rizzo, A.; Guggino, G.; Cipriani, P.; Carubbi, F.; Giacomelli, R.; Triolo, G. Rituximab modulates the expression of IL-22 in the salivary glands of patients with primary Sjogren’s syndrome. Ann. Rheum. Dis. 2013, 72, 782–783. [Google Scholar] [CrossRef]
- Heinz, C.; Merz, H.; Nieschalk, M.; Mueller-Miny, H.; Koch, P.; Heiligenhaus, A. Rituximab for the treatment of extranodal marginal zone B-cell lymphoma of the lacrimal gland. Br. J. Ophthalmol. 2007, 91, 1563–1564. [Google Scholar] [CrossRef]
- Verstappen, G.M.; van Nimwegen, J.F.; Vissink, A.; Kroese, F.G.M.; Bootsma, H. The value of rituximab treatment in primary Sjogren’s syndrome. Clin. Immunol. 2017, 182, 62–71. [Google Scholar] [CrossRef]
- Cornec, D.; Devauchelle-Pensec, V.; Mariette, X.; Jousse-Joulin, S.; Berthelot, J.M.; Perdriger, A.; Puechal, X.; Le Guern, V.; Sibilia, J.; Gottenberg, J.E.; et al. Development of the Sjogren’s Syndrome Responder Index, a data-driven composite endpoint for assessing treatment efficacy. Rheumatology (Oxford) 2015, 54, 1699–1708. [Google Scholar] [CrossRef]
- Payandeh, Z.; Bahrami, A.A.; Hoseinpoor, R.; Mortazavi, Y.; Rajabibazl, M.; Rahimpour, A.; Taromchi, A.H.; Khalil, S. The applications of anti-CD20 antibodies to treat various B cells disorders. Biomed. Pharm. 2019, 109, 2415–2426. [Google Scholar] [CrossRef]
- Clark, E.A.; Giltiay, N.V. CD22: A Regulator of Innate and Adaptive B Cell Responses and Autoimmunity. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Dorner, T.; Goldenberg, D.M. Targeting CD22 as a strategy for treating systemic autoimmune diseases. Ther. Clin. Risk. Manag. 2007, 3, 953–959. [Google Scholar]
- Leonard, J.P.; Coleman, M.; Ketas, J.C.; Chadburn, A.; Furman, R.; Schuster, M.W.; Feldman, E.J.; Ashe, M.; Schuster, S.J.; Wegener, W.A.; et al. Epratuzumab, a Humanized Anti-CD22 Antibody, in Aggressive Non-Hodgkin’s Lymphoma. Phase. I/Ii Clin. Trial. Results 2004, 10, 5327–5334. [Google Scholar] [CrossRef]
- Haas, K.M.; Sen, S.; Sanford, I.G.; Miller, A.S.; Poe, J.C.; Tedder, T.F. CD22 ligand binding regulates normal and malignant B lymphocyte survival in vivo. J. Immunol. 2006, 177, 3063–3073. [Google Scholar] [CrossRef]
- Adler, S.; Korner, M.; Forger, F.; Huscher, D.; Caversaccio, M.D.; Villiger, P.M. Evaluation of histologic, serologic, and clinical changes in response to abatacept treatment of primary Sjogren’s syndrome: A pilot study. Arthritis Care. Res. (Hoboken) 2013, 65, 1862–1868. [Google Scholar] [CrossRef] [PubMed]
- Meiners, P.M.; Vissink, A.; Kroese, F.G.; Spijkervet, F.K.; Smitt-Kamminga, N.S.; Abdulahad, W.H.; Bulthuis-Kuiper, J.; Brouwer, E.; Arends, S.; Bootsma, H. Abatacept treatment reduces disease activity in early primary Sjogren’s syndrome (open-label proof of concept ASAP study). Ann. Rheum. Dis. 2014, 73, 1393–1396. [Google Scholar] [CrossRef]
- Verstappen, G.M.; Meiners, P.M.; Corneth, O.B.J.; Visser, A.; Arends, S.; Abdulahad, W.H.; Hendriks, R.W.; Vissink, A.; Kroese, F.G.M.; Bootsma, H. Attenuation of Follicular Helper T Cell-Dependent B Cell Hyperactivity by Abatacept Treatment in Primary Sjogren’s Syndrome. Arthritis Rheumatol. 2017, 69, 1850–1861. [Google Scholar] [CrossRef]
- Van Nimwegen, J.F.; Mossel, E.; van Zuiden, G.S.; Wijnsma, R.F.; Delli, K.; Stel, A.J.; van der Vegt, B.; Haacke, E.A.; Olie, L.; Los, L.I.; et al. Abatacept treatment for patients with early active primary Sjögren’s syndrome: A single-centre, randomised, double-blind, placebo-controlled, phase 3 trial (ASAP-III study). Lancet. Rheumatol. 2020, 2, e153–e163. [Google Scholar] [CrossRef]
- Rigby, M.R.; DiMeglio, L.A.; Rendell, M.S.; Felner, E.I.; Dostou, J.M.; Gitelman, S.E.; Patel, C.M.; Griffin, K.J.; Tsalikian, E.; Gottlieb, P.A.; et al. Targeting of memory T cells with alefacept in new-onset type 1 diabetes (T1DAL study): 12 month results of a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Diabetes Endocrinol. 2013, 1, 284–294. [Google Scholar] [CrossRef]
- Cancro, M.P. The BLyS/BAFF family of ligands and receptors: Key targets in the therapy and understanding of autoimmunity. Ann. Rheum. Dis. 2006, 65 (Suppl. 3), iii34–iii36. [Google Scholar] [CrossRef]
- Ramos-Casals, M. The B-lymphocyte stimulator connection in Sjogren’s syndrome. Rheumatology (Oxford) 2013, 52, 223–225. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lavie, F.; Miceli-Richard, C.; Ittah, M.; Sellam, J.; Gottenberg, J.E.; Mariette, X. B-cell activating factor of the tumour necrosis factor family expression in blood monocytes and T cells from patients with primary Sjogren’s syndrome. Scand. J. Immunol. 2008, 67, 185–192. [Google Scholar] [CrossRef]
- Groom, J.; Kalled, S.L.; Cutler, A.H.; Olson, C.; Woodcock, S.A.; Schneider, P.; Tschopp, J.; Cachero, T.G.; Batten, M.; Wheway, J.; et al. Association of BAFF/BLyS overexpression and altered B cell differentiation with Sjogren’s syndrome. J. Clin. Investig. 2002, 109, 59–68. [Google Scholar] [CrossRef]
- Felten, R.; Sibilia, J.; Gottenberg, J.E. Chapter 17—Outcome Measures in Sjögren’s Syndrome and Perspectives in Clinical Trial Design. In Sjögren’s Syndrome; Gerli, R., Bartoloni, E., Alunno, A., Eds.; Academic Press: Cambridge, MA, USA, 2016; pp. 263–271. [Google Scholar]
- De Vita, S.; Quartuccio, L.; Salvin, S.; Picco, L.; Scott, C.A.; Rupolo, M.; Fabris, M. Sequential therapy with belimumab followed by rituximab in Sjogren’s syndrome associated with B-cell lymphoproliferation and overexpression of BAFF: Evidence for long-term efficacy. Clin. Exp. Rheumatol. 2014, 32, 490–494. [Google Scholar]
- Kamal, A.; Khamashta, M. The efficacy of novel B cell biologics as the future of SLE treatment: A review. Autoimmun. Rev. 2014, 13, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Wiestner, A. The role of B-cell receptor inhibitors in the treatment of patients with chronic lymphocytic leukemia. Haematologica 2015, 100, 1495–1507. [Google Scholar] [CrossRef] [PubMed]
- Mielle, J.; Tison, A.; Cornec, D.; Le Pottier, L.; Daien, C.; Pers, J.-O. B cells in Sjögren’s syndrome: From pathophysiology to therapeutic target. Rheumatology 2019. (ahead of print). [Google Scholar] [CrossRef]
- Nayar, S.; Campos, J.; Smith, C.G.; Iannizzotto, V.; Gardner, D.H.; Colafrancesco, S.; Pipi, E.; Kollert, F.; Hunter, K.J.; Brewer, C.; et al. Phosphatidylinositol 3-kinase delta pathway: A novel therapeutic target for Sjogren’s syndrome. Ann. Rheum. Dis. 2019, 78, 249–260. [Google Scholar] [CrossRef]
- Corneth, O.B.J.; Verstappen, G.M.P.; Paulissen, S.M.J.; de Bruijn, M.J.W.; Rip, J.; Lukkes, M.; van Hamburg, J.P.; Lubberts, E.; Bootsma, H.; Kroese, F.G.M.; et al. Enhanced Bruton’s Tyrosine Kinase Activity in Peripheral Blood B Lymphocytes From Patients With Autoimmune Disease. Arthritis Rheumatol. 2017, 69, 1313–1324. [Google Scholar] [CrossRef]
- Remouchamps, C.; Boutaffala, L.; Ganeff, C.; Dejardin, E. Biology and signal transduction pathways of the Lymphotoxin-αβ/LTβR system. Cytokine. Growth. Factor. Rev. 2011, 22, 301–310. [Google Scholar] [CrossRef]
- Browning, J.L. Inhibition of the lymphotoxin pathway as a therapy for autoimmune disease. Immunol. Rev. 2008, 223, 202–220. [Google Scholar] [CrossRef]
- Fava, R.A.; Kennedy, S.M.; Wood, S.G.; Bolstad, A.I.; Bienkowska, J.; Papandile, A.; Kelly, J.A.; Mavragani, C.P.; Gatumu, M.; Skarstein, K.; et al. Lymphotoxin-beta receptor blockade reduces CXCL13 in lacrimal glands and improves corneal integrity in the NOD model of Sjogren’s syndrome. Arthritis Res. Ther. 2011, 13, R182. [Google Scholar] [CrossRef]
- Singh, N.; Cohen, P.L. The T cell in Sjogren’s syndrome: Force majeure, not spectateur. J. Autoimmun. 2012, 39, 229–233. [Google Scholar] [CrossRef]
- Linsley, P.S.; Nadler, S.G. The clinical utility of inhibiting CD28-mediated costimulation. Immunol. Rev. 2009, 229, 307–321. [Google Scholar] [CrossRef]
- Tsuboi, H.; Matsumoto, I.; Hagiwara, S.; Hirota, T.; Takahashi, H.; Ebe, H.; Yokosawa, M.; Yagishita, M.; Takahashi, H.; Kurata, I.; et al. Effectiveness of abatacept for patients with Sjogren’s syndrome associated with rheumatoid arthritis. An open label, multicenter, one-year, prospective study: ROSE (Rheumatoid Arthritis with Orencia Trial toward Sjogren’s syndrome Endocrinopathy) trial. Mod. Rheumatol. 2016, 26, 891–899. [Google Scholar] [CrossRef]
- Haacke, E.A.; van der Vegt, B.; Meiners, P.M.; Vissink, A.; Spijkervet, F.K.; Bootsma, H.; Kroese, F.G. Abatacept treatment of patients with primary Sjogren’s syndrome results in a decrease of germinal centres in salivary gland tissue. Clin. Exp. Rheumatol. 2017, 35, 317–320. [Google Scholar]
- Ramos-Casals, M.; Brito-Zerón, P. Emerging biological therapies in primary Sjögren’s syndrome. Rheumatology 2007, 46, 1389–1396. [Google Scholar] [CrossRef]
- Papp, K.A.; Henninger, E. Evaluation of efalizumab using safe psoriasis control. BMC Derm. 2006, 6, 8. [Google Scholar] [CrossRef]
- Prater, E.F.; Day, A.; Patel, M.; Menter, A. A retrospective analysis of 72 patients on prior efalizumab subsequent to the time of voluntary market withdrawal in 2009. J. Drugs. Derm. 2014, 13, 712–718. [Google Scholar]
- Crow, J.M. Therapeutics: Silencing psoriasis. Nature 2012, 492, S58–S59. [Google Scholar] [CrossRef]
- Papp, K.A. The long-term efficacy and safety of new biological therapies for psoriasis. Arch. Derm. Res. 2006, 298, 7–15. [Google Scholar] [CrossRef]
- Yu, X.; Riemekasten, G.; Petersen, F. Autoantibodies against muscarinic acetylcholine receptor M3 in Sjogren’s syndrome and corresponding mouse models. Front. Biosci. (Landmark Ed.) 2018, 23, 2053–2064. [Google Scholar] [CrossRef]
- Sumida, T.; Iizuka, M.; Asashima, H.; Tsuboi, H.; Matsumoto, I. Pathogenic role of anti-M3 muscarinic acetylcholine receptor immune response in Sjögren’s syndrome. Press. Med. 2012, 41, e461–e466. [Google Scholar] [CrossRef]
- Bacman, S.; Berra, A.; Sterin-Borda, L.; Borda, E. Muscarinic acetylcholine receptor antibodies as a new marker of dry eye Sjogren syndrome. Investig. Ophthalmol. Vis. Sci. 2001, 42, 321–327. [Google Scholar]
- Asashima, H.; Tsuboi, H.; Takahashi, H.; Hirota, T.; Iizuka, M.; Kondo, Y.; Matsui, M.; Matsumoto, I.; Sumida, T. The anergy induction of M3 muscarinic acetylcholine receptor-reactive CD4+ T cells suppresses experimental sialadenitis-like Sjogren’s syndrome. Arthritis Rheumatol. 2015, 67, 2213–2225. [Google Scholar] [CrossRef]
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Moorman, M.A.; Simonetti, D.W.; Craig, S.; Marshak, D.R. Multilineage potential of adult human mesenchymal stem cells. Science 1999, 284, 143–147. [Google Scholar] [CrossRef]
- Munir, H.; McGettrick, H.M. Mesenchymal Stem Cell Therapy for Autoimmune Disease: Risks and Rewards. Stem. Cells. Dev. 2015, 24, 2091–2100. [Google Scholar] [CrossRef]
- Liu, R.; Su, D.; Zhou, M.; Feng, X.; Li, X.; Sun, L. Umbilical cord mesenchymal stem cells inhibit the differentiation of circulating T follicular helper cells in patients with primary Sjogren’s syndrome through the secretion of indoleamine 2,3-dioxygenase. Rheumatology (Oxford) 2015, 54, 332–342. [Google Scholar] [CrossRef]
- Xu, J.; Wang, D.; Liu, D.; Fan, Z.; Zhang, H.; Liu, O.; Ding, G.; Gao, R.; Zhang, C.; Ding, Y.; et al. Allogeneic mesenchymal stem cell treatment alleviates experimental and clinical Sjogren syndrome. Blood 2012, 120, 3142–3151. [Google Scholar] [CrossRef]
- Alunno, A.; Montanucci, P.; Bistoni, O.; Basta, G.; Caterbi, S.; Pescara, T.; Pennoni, I.; Bini, V.; Bartoloni, E.; Gerli, R.; et al. In vitro immunomodulatory effects of microencapsulated umbilical cord Wharton jelly-derived mesenchymal stem cells in primary Sjogren’s syndrome. Rheumatology (Oxford) 2015, 54, 163–168. [Google Scholar] [CrossRef]
- Shi, B.; Qi, J.; Yao, G.; Feng, R.; Zhang, Z.; Wang, D.; Chen, C.; Tang, X.; Lu, L.; Chen, W.; et al. Mesenchymal stem cell transplantation ameliorates Sjogren’s syndrome via suppressing IL-12 production by dendritic cells. Stem. Cell. Res. Ther. 2018, 9, 308. [Google Scholar] [CrossRef]
- Maria, N.I.; van Helden-Meeuwsen, C.G.; Brkic, Z.; Paulissen, S.M.; Steenwijk, E.C.; Dalm, V.A.; van Daele, P.L.; Martin van Hagen, P.; Kroese, F.G.; van Roon, J.A.; et al. Association of Increased Treg Cell Levels With Elevated Indoleamine 2,3-Dioxygenase Activity and an Imbalanced Kynurenine Pathway in Interferon-Positive Primary Sjögren’s Syndrome. Arthritis Rheumatol. 2016, 68, 1688–1699. [Google Scholar] [CrossRef]
- Liu, Y.; Li, C.; Wang, S.; Guo, J.; Guo, J.; Fu, J.; Ren, L.; An, Y.; He, J.; Li, Z. Human umbilical cord mesenchymal stem cells confer potent immunosuppressive effects in Sjögren’s syndrome by inducing regulatory T cells. Mod. Rheumatol. 2020, 1–11, (ahead of print). [Google Scholar] [CrossRef]
- Maria, O.M.; Tran, S.D. Human mesenchymal stem cells cultured with salivary gland biopsies adopt an epithelial phenotype. Stem. Cells. Dev. 2011, 20, 959–967. [Google Scholar] [CrossRef]
- Park, Y.J.; Koh, J.; Gauna, A.E.; Chen, S.; Cha, S. Identification of regulatory factors for mesenchymal stem cell-derived salivary epithelial cells in a co-culture system. PLoS ONE 2014, 9, e112158. [Google Scholar] [CrossRef]
- Aluri, H.S.; Samizadeh, M.; Edman, M.C.; Hawley, D.R.; Armaos, H.L.; Janga, S.R.; Meng, Z.; Sendra, V.G.; Hamrah, P.; Kublin, C.L.; et al. Delivery of Bone Marrow-Derived Mesenchymal Stem Cells Improves Tear Production in a Mouse Model of Sjogren’s Syndrome. Stem. Cells. Int. 2017, 2017, 3134543. [Google Scholar] [CrossRef]
- Dietrich, J.; Ott, L.; Roth, M.; Witt, J.; Geerling, G.; Mertsch, S.; Schrader, S. MSC Transplantation Improves Lacrimal Gland Regeneration after Surgically Induced Dry Eye Disease in Mice. Sci. Rep. 2019, 9, 18299. [Google Scholar] [CrossRef]
- Dietrich, J.; Schrader, S. Towards Lacrimal Gland Regeneration: Current Concepts and Experimental Approaches. Curr. Eye Res. 2020, 45, 230–240. [Google Scholar] [CrossRef]
- Liang, J.; Zhang, H.; Kong, W.; Deng, W.; Wang, D.; Feng, X.; Zhao, C.; Hua, B.; Wang, H.; Sun, L. Safety analysis in patients with autoimmune disease receiving allogeneic mesenchymal stem cells infusion: A long-term retrospective study. Stem. Cell. Res. Ther. 2018, 9, 312. [Google Scholar] [CrossRef]
- Santamaria, P. Cytokines and chemokines in autoimmune disease: An overview. Adv. Exp. Med. Biol. 2003, 520, 1–7. [Google Scholar] [CrossRef]
- Retamozo, S.; Flores-Chavez, A.; Consuegra-Fernandez, M.; Lozano, F.; Ramos-Casals, M.; Brito-Zeron, P. Cytokines as therapeutic targets in primary Sjogren syndrome. Pharm. Ther. 2018, 184, 81–97. [Google Scholar] [CrossRef]
- Steinfeld, S.D.; Demols, P.; Salmon, I.; Kiss, R.; Appelboom, T. Infliximab in patients with primary Sjogren’s syndrome: A pilot study. Arthritis Rheum. 2001, 44, 2371–2375. [Google Scholar] [CrossRef]
- Mariette, X.; Ravaud, P.; Steinfeld, S.; Baron, G.; Goetz, J.; Hachulla, E.; Combe, B.; Puechal, X.; Pennec, Y.; Sauvezie, B.; et al. Inefficacy of infliximab in primary Sjogren’s syndrome: Results of the randomized, controlled Trial of Remicade in Primary Sjogren’s Syndrome (TRIPSS). Arthritis Rheum. 2004, 50, 1270–1276. [Google Scholar] [CrossRef]
- Sankar, V.; Brennan, M.T.; Kok, M.R.; Leakan, R.A.; Smith, J.A.; Manny, J.; Baum, B.J.; Pillemer, S.R. Etanercept in Sjogren’s syndrome: A twelve-week randomized, double-blind, placebo-controlled pilot clinical trial. Arthritis Rheum. 2004, 50, 2240–2245. [Google Scholar] [CrossRef]
- Zandbelt, M.M.; de Wilde, P.; van Damme, P.; Hoyng, C.B.; van de Putte, L.; van den Hoogen, F. Etanercept in the treatment of patients with primary Sjogren’s syndrome: A pilot study. J. Rheumatol. 2004, 31, 96–101. [Google Scholar]
- Khurshudian, A.V. A pilot study to test the efficacy of oral administration of interferon-alpha lozenges to patients with Sjogren’s syndrome. Oral. Surg. Oral. Med. Oral. Pathol. Oral. Radiol. Endod. 2003, 95, 38–44. [Google Scholar] [CrossRef]
- Cummins, M.J.; Papas, A.; Kammer, G.M.; Fox, P.C. Treatment of primary Sjogren’s syndrome with low-dose human interferon alfa administered by the oromucosal route: Combined phase III results. Arthritis Rheum. 2003, 49, 585–593. [Google Scholar] [CrossRef]
- Liew, S.H.; Nichols, K.K.; Klamerus, K.J.; Li, J.Z.; Zhang, M.; Foulks, G.N. Tofacitinib (CP-690,550), a Janus kinase inhibitor for dry eye disease: Results from a phase 1/2 trial. Ophthalmology 2012, 119, 1328–1335. [Google Scholar] [CrossRef]
- Norheim, K.B.; Harboe, E.; Goransson, L.G.; Omdal, R. Interleukin-1 inhibition and fatigue in primary Sjogren’s syndrome--a double blind, randomised clinical trial. PLoS ONE 2012, 7, e30123. [Google Scholar] [CrossRef]
- Justet, A.; Ottaviani, S.; Dieude, P.; Taille, C. Tocilizumab for refractory organising pneumonia associated with Sjogren’s disease. BMJ Case Rep. 2015, 2015. [Google Scholar] [CrossRef]
- Le Page, C.; Génin, P.; Baines, M.G.; Hiscott, J. Interferon activation and innate immunity. Rev. Immunogenet. 2000, 2, 374–386. [Google Scholar]
- Platanias, L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef]
- Ambrosi, A.; Wahren-Herlenius, M. Update on the immunobiology of Sjogren’s syndrome. Curr. Opin. Rheumatol. 2015, 27, 468–475. [Google Scholar] [CrossRef]
- Yao, Y.; Liu, Z.; Jallal, B.; Shen, N.; Ronnblom, L. Type I interferons in Sjogren’s syndrome. Autoimmun. Rev. 2013, 12, 558–566. [Google Scholar] [CrossRef]
- Ha, Y.J.; Choi, Y.S.; Kang, E.H.; Chung, J.H.; Cha, S.; Song, Y.W.; Lee, Y.J. Increased expression of interferon-lambda in minor salivary glands of patients with primary Sjogren’s syndrome and its synergic effect with interferon-alpha on salivary gland epithelial cells. Clin. Exp. Rheumatol. 2018, 36 (Suppl. 112), 31–40. [Google Scholar] [PubMed]
- Sjostrand, M.; Johansson, A.; Aqrawi, L.; Olsson, T.; Wahren-Herlenius, M.; Espinosa, A. The Expression of BAFF Is Controlled by IRF Transcription Factors. J. Immunol. 2016, 196, 91–96. [Google Scholar] [CrossRef]
- Mathian, A.; Hie, M.; Cohen-Aubart, F.; Amoura, Z. Targeting interferons in systemic lupus erythematosus: Current and future prospects. Drugs 2015, 75, 835–846. [Google Scholar] [CrossRef]
- Charras, A.; Arvaniti, P.; Le Dantec, C.; Arleevskaya, M.I.; Zachou, K.; Dalekos, G.N.; Bordon, A.; Renaudineau, Y. JAK Inhibitors Suppress Innate Epigenetic Reprogramming: A Promise for Patients with Sjogren’s Syndrome. Clin. Rev. Allergy Immunol. 2020, 58, 182–193. [Google Scholar] [CrossRef]
- Lee, J.; Lee, J.; Kwok, S.K.; Baek, S.; Jang, S.G.; Hong, S.M.; Min, J.W.; Choi, S.S.; Lee, J.; Cho, M.L.; et al. JAK-1 Inhibition Suppresses Interferon-Induced BAFF Production in Human Salivary Gland: Potential Therapeutic Strategy for Primary Sjogren’s Syndrome. Arthritis Rheumatol. 2018, 70, 2057–2066. [Google Scholar] [CrossRef]
- Khamashta, M.; Merrill, J.T.; Werth, V.P.; Furie, R.; Kalunian, K.; Illei, G.G.; Drappa, J.; Wang, L.; Greth, W.; Investigators, C.D. Sifalimumab, an anti-interferon-alpha monoclonal antibody, in moderate to severe systemic lupus erythematosus: A randomised, double-blind, placebo-controlled study. Ann. Rheum. Dis. 2016, 75, 1909–1916. [Google Scholar] [CrossRef]
- McBride, J.M.; Jiang, J.; Abbas, A.R.; Morimoto, A.; Li, J.; Maciuca, R.; Townsend, M.; Wallace, D.J.; Kennedy, W.P.; Drappa, J. Safety and pharmacodynamics of rontalizumab in patients with systemic lupus erythematosus: Results of a phase I, placebo-controlled, double-blind, dose-escalation study. Arthritis Rheum. 2012, 64, 3666–3676. [Google Scholar] [CrossRef]
- Furie, R.; Khamashta, M.; Merrill, J.T.; Werth, V.P.; Kalunian, K.; Brohawn, P.; Illei, G.G.; Drappa, J.; Wang, L.; Yoo, S.; et al. Anifrolumab, an Anti-Interferon-alpha Receptor Monoclonal Antibody, in Moderate-to-Severe Systemic Lupus Erythematosus. Arthritis Rheumatol. 2017, 69, 376–386. [Google Scholar] [CrossRef]
- Ogawa, Y.; Shimizu, E.; Tsubota, K. Interferons and Dry Eye in Sjögren’s Syndrome. Int. J. Mol. Sci. 2018, 19, 3548. [Google Scholar] [CrossRef]
- Coursey, T.G.; Bohat, R.; Barbosa, F.L.; Pflugfelder, S.C.; de Paiva, C.S. Desiccating stress-induced chemokine expression in the epithelium is dependent on upregulation of NKG2D/RAE-1 and release of IFN-γ in experimental dry eye. J. Immunol. 2014, 193, 5264–5272. [Google Scholar] [CrossRef]
- Willeke, P.; Schluter, B.; Schotte, H.; Domschke, W.; Gaubitz, M.; Becker, H. Interferon-gamma is increased in patients with primary Sjogren’s syndrome and Raynaud’s phenomenon. Semin. Arthritis Rheum. 2009, 39, 197–202. [Google Scholar] [CrossRef]
- Yamada, A.; Arakaki, R.; Kudo, Y.; Ishimaru, N. Targeting IL-1 in Sjogren’s syndrome. Expert. Opin. Targets 2013, 17, 393–401. [Google Scholar] [CrossRef]
- Chen, Y.T.; Nikulina, K.; Lazarev, S.; Bahrami, A.F.; Noble, L.B.; Gallup, M.; McNamara, N.A. Interleukin-1 as a phenotypic immunomodulator in keratinizing squamous metaplasia of the ocular surface in Sjogren’s syndrome. Am. J. Pathol. 2010, 177, 1333–1343. [Google Scholar] [CrossRef]
- Ye, C.; Brand, D.; Zheng, S.G. Targeting IL-2: An unexpected effect in treating immunological diseases. Signal. Transduct. Target. Ther. 2018, 3, 2. [Google Scholar] [CrossRef]
- Klatzmann, D.; Abbas, A.K. The promise of low-dose interleukin-2 therapy for autoimmune and inflammatory diseases. Nat. Rev. Immunol. 2015, 15, 283–294. [Google Scholar] [CrossRef]
- Dwyer, C.J.; Ward, N.C.; Pugliese, A.; Malek, T.R. Promoting Immune Regulation in Type 1 Diabetes Using Low-Dose Interleukin-2. Curr. Diab. Rep. 2016, 16, 46. [Google Scholar] [CrossRef]
- Humrich, J.Y.; Riemekasten, G. Restoring regulation—IL-2 therapy in systemic lupus erythematosus. Expert. Rev. Clin. Immunol. 2016, 12, 1153–1160. [Google Scholar] [CrossRef]
- Miao, M.; Hao, Z.; Guo, Y.; Zhang, X.; Zhang, S.; Luo, J.; Zhao, X.; Zhang, C.; Liu, X.; Wu, X.; et al. Short-term and low-dose IL-2 therapy restores the Th17/Treg balance in the peripheral blood of patients with primary Sjogren’s syndrome. Ann. Rheum. Dis. 2018, 77, 1838–1840. [Google Scholar] [CrossRef]
- Lim, S.A.; Nam, D.H.; Lee, J.H.; Kwok, S.K.; Park, S.H.; Chung, S.H. Association of IL-21 cytokine with severity of primary Sjogren syndrome dry eye. Cornea 2015, 34, 248–252. [Google Scholar] [CrossRef]
- Papp, G.; Gyimesi, E.; Szabo, K.; Zold, E.; Zeher, M. Increased IL-21 Expression Induces Granzyme B in Peripheral CD5(+) B Cells as a Potential Counter-Regulatory Effect in Primary Sjogren’s Syndrome. Mediat. Inflamm. 2016, 2016, 4328372. [Google Scholar] [CrossRef]
- Ma, C.S.; Deenick, E.K.; Batten, M.; Tangye, S.G. The origins, function, and regulation of T follicular helper cells. J. Exp. Med. 2012, 209, 1241–1253. [Google Scholar] [CrossRef]
- Komai, T.; Shoda, H.; Yamaguchi, K.; Sakurai, K.; Shibuya, M.; Kubo, K.; Takahashi, T.; Fujio, K.; Yamamoto, K. Neuromyelitis optica spectrum disorder complicated with Sjogren syndrome successfully treated with tocilizumab: A case report. Mod. Rheumatol. 2016, 26, 294–296. [Google Scholar] [CrossRef]
- Ciccia, F.; Guggino, G.; Rizzo, A.; Bombardieri, M.; Raimondo, S.; Carubbi, F.; Cannizzaro, A.; Sireci, G.; Dieli, F.; Campisi, G.; et al. Interleukin (IL)-22 receptor 1 is over-expressed in primary Sjogren’s syndrome and Sjogren-associated non-Hodgkin lymphomas and is regulated by IL-18. Clin. Exp. Immunol. 2015, 181, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Siebert, S.; Tsoukas, A.; Robertson, J.; McInnes, I. Cytokines as therapeutic targets in rheumatoid arthritis and other inflammatory diseases. Pharm. Rev. 2015, 67, 280–309. [Google Scholar] [CrossRef]
- Vosters, J.L.; Baum, B.J.; Tak, P.P.; Illei, G.G.; Chiorini, J.A. Developing Developing a gene therapy for Sjogren’s syndrome. Future Rheumatol. 2006, 1, 433–440. [Google Scholar] [CrossRef]
- Nguyen, C.Q.; Yin, H.; Lee, B.H.; Chiorini, J.A.; Peck, A.B. IL17: Potential therapeutic target in Sjogren’s syndrome using adenovirus-mediated gene transfer. Lab. Investig. 2011, 91, 54–62. [Google Scholar] [CrossRef]
- Lee, B.H.; Carcamo, W.C.; Chiorini, J.A.; Peck, A.B.; Nguyen, C.Q. Gene therapy using IL-27 ameliorates Sjögren’s syndrome-like autoimmune exocrinopathy. Arthritis Res. Ther. 2012, 14, R172. [Google Scholar] [CrossRef]
- Su, Y.; Gu, Y.; Wu, R.; Wang, H. Bone Morphogenetic Protein 6 Inhibits the Immunomodulatory Property of BMMSCs via Id1 in Sjogren’s Syndrome. Stem. Cells. Int. 2018, 2018, 9837035. [Google Scholar] [CrossRef]
- Yin, H.; Cabrera-Perez, J.; Lai, Z.; Michael, D.; Weller, M.; Swaim, W.D.; Liu, X.; Catalan, M.A.; Rocha, E.M.; Ismail, N.; et al. Association of bone morphogenetic protein 6 with exocrine gland dysfunction in patients with Sjogren’s syndrome and in mice. Arthritis Rheum. 2013, 65, 3228–3238. [Google Scholar] [CrossRef]
- Lai, Z.; Yin, H.; Cabrera-Pérez, J.; Guimaro, M.C.; Afione, S.; Michael, D.G.; Glenton, P.; Patel, A.; Swaim, W.D.; Zheng, C.; et al. Aquaporin gene therapy corrects Sjögren’s syndrome phenotype in mice. Proc. Natl. Acad. Sci. USA 2016, 113, 5694. [Google Scholar] [CrossRef]
- Baum, B.J.; Alevizos, I.; Zheng, C.; Cotrim, A.P.; Liu, S.; McCullagh, L.; Goldsmith, C.M.; Burbelo, P.D.; Citrin, D.E.; Mitchell, J.B.; et al. Early responses to adenoviral-mediated transfer of the aquaporin-1 cDNA for radiation-induced salivary hypofunction. Proc. Natl. Acad. Sci. USA 2012, 109, 19403–19407. [Google Scholar] [CrossRef]
- Carroll, D. A CRISPR approach to gene targeting. Mol. Ther. 2012, 20, 1658–1660. [Google Scholar] [CrossRef]
- Wang, L.; Yang, Y.; Breton, C.; Bell, P.; Li, M.; Zhang, J.; Che, Y.; Saveliev, A.; He, Z.; White, J.; et al. A mutation-independent CRISPR-Cas9-mediated gene targeting approach to treat a murine model of ornithine transcarbamylase deficiency. Sci. Adv. 2020, 6, eaax5701. [Google Scholar] [CrossRef]
- Cabral, T.; DiCarlo, J.E.; Justus, S.; Sengillo, J.D.; Xu, Y.; Tsang, S.H. CRISPR applications in ophthalmologic genome surgery. Curr. Opin. Ophthalmol. 2017, 28, 252–259. [Google Scholar] [CrossRef]








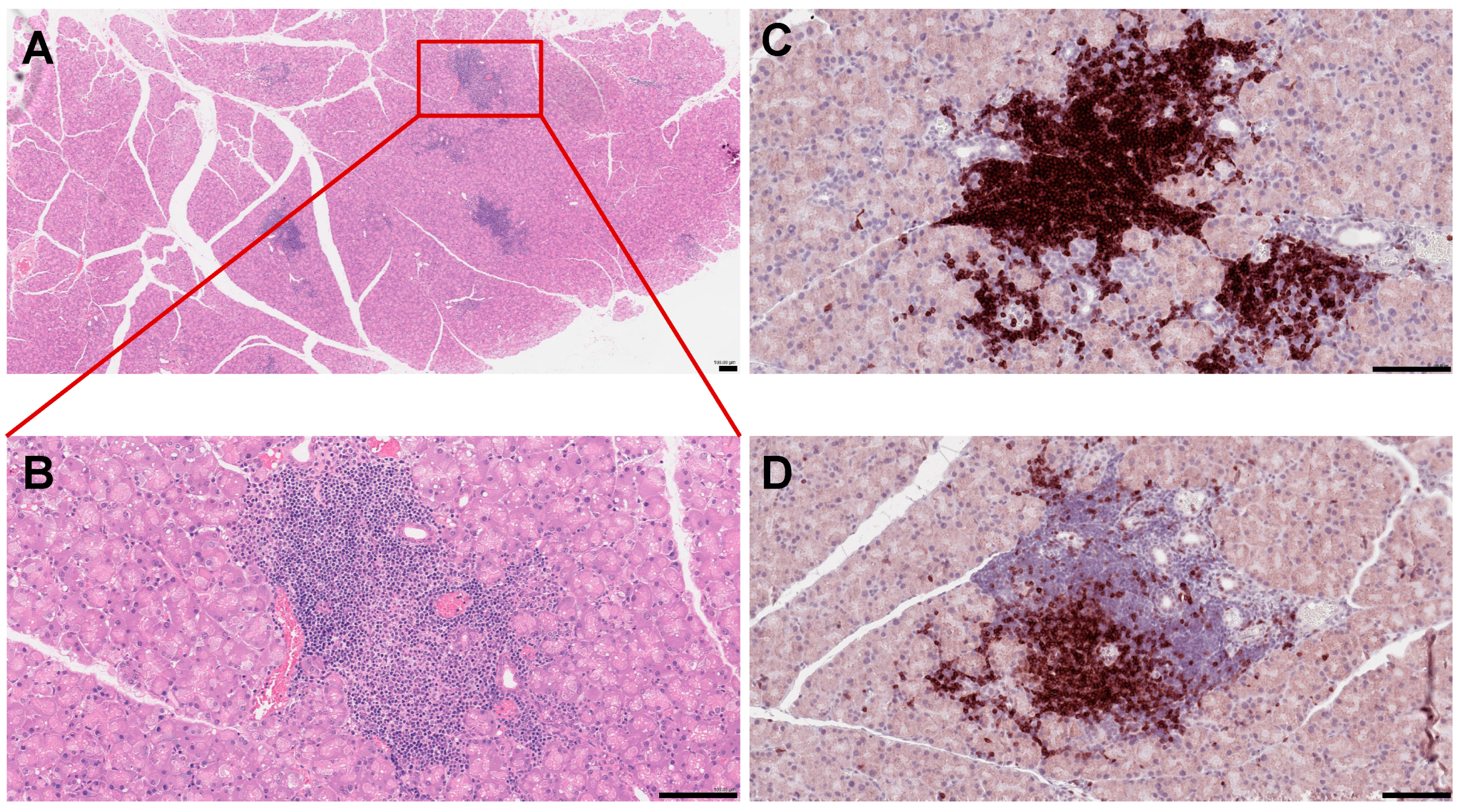{kind=link}
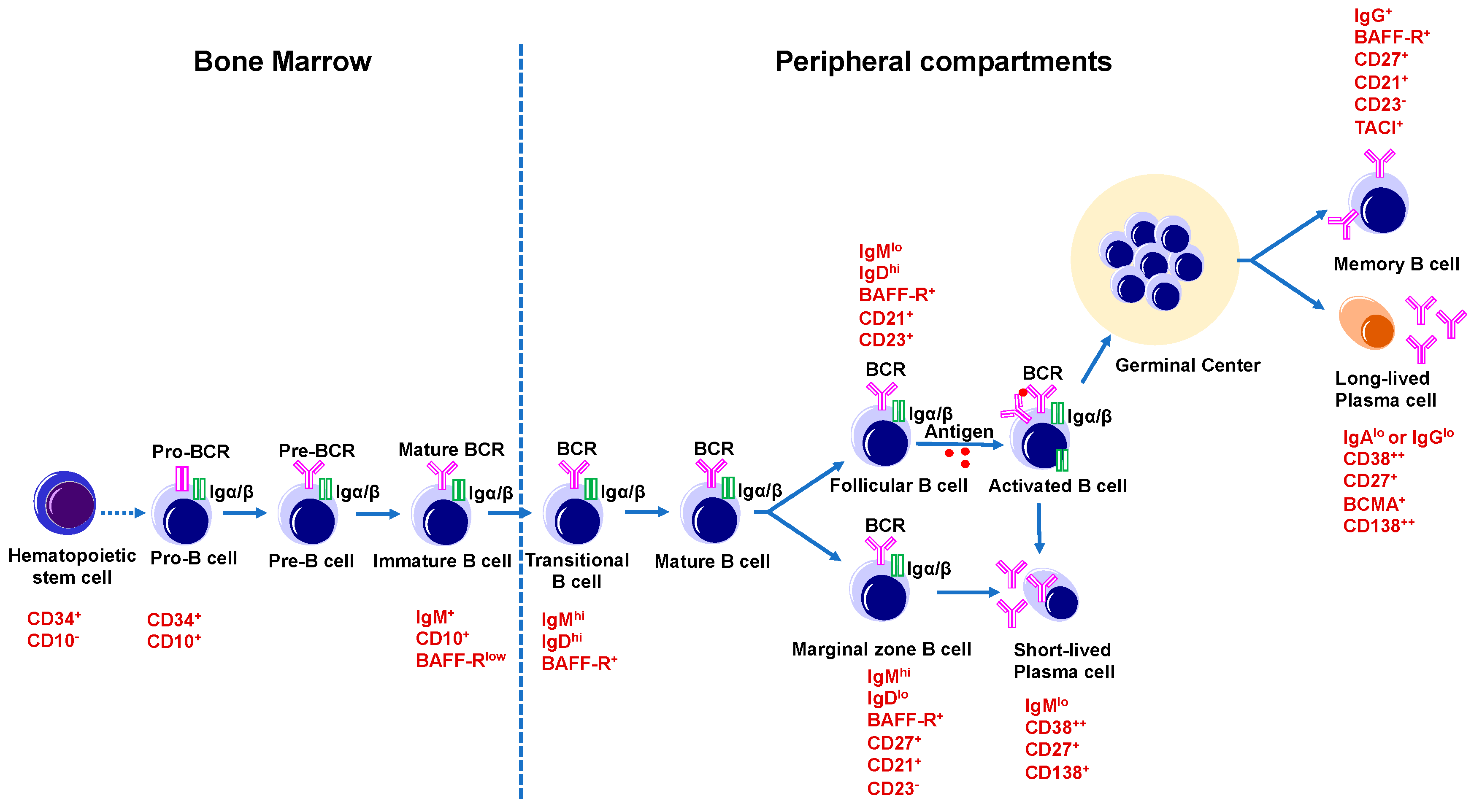{kind=link}
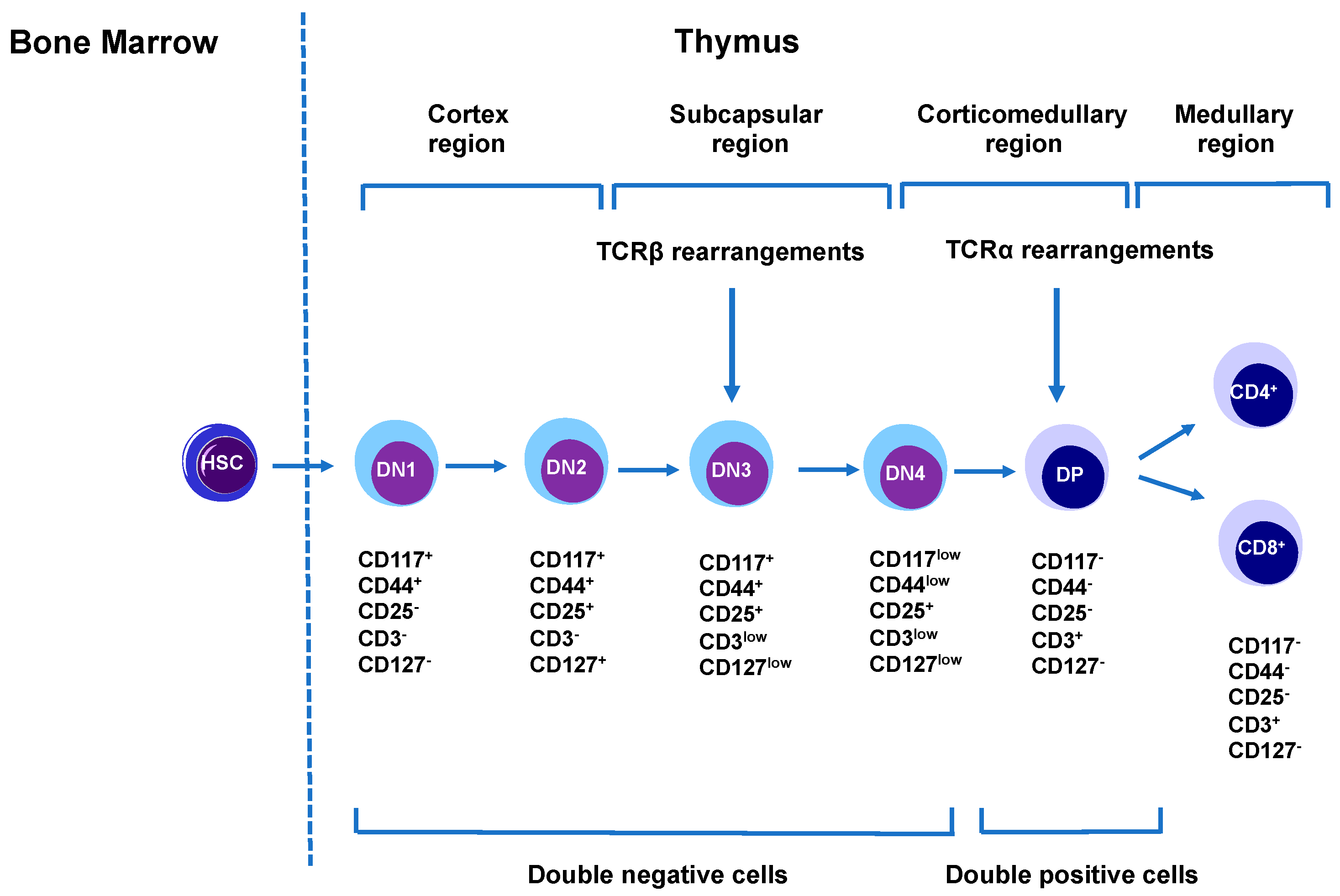{kind=link}
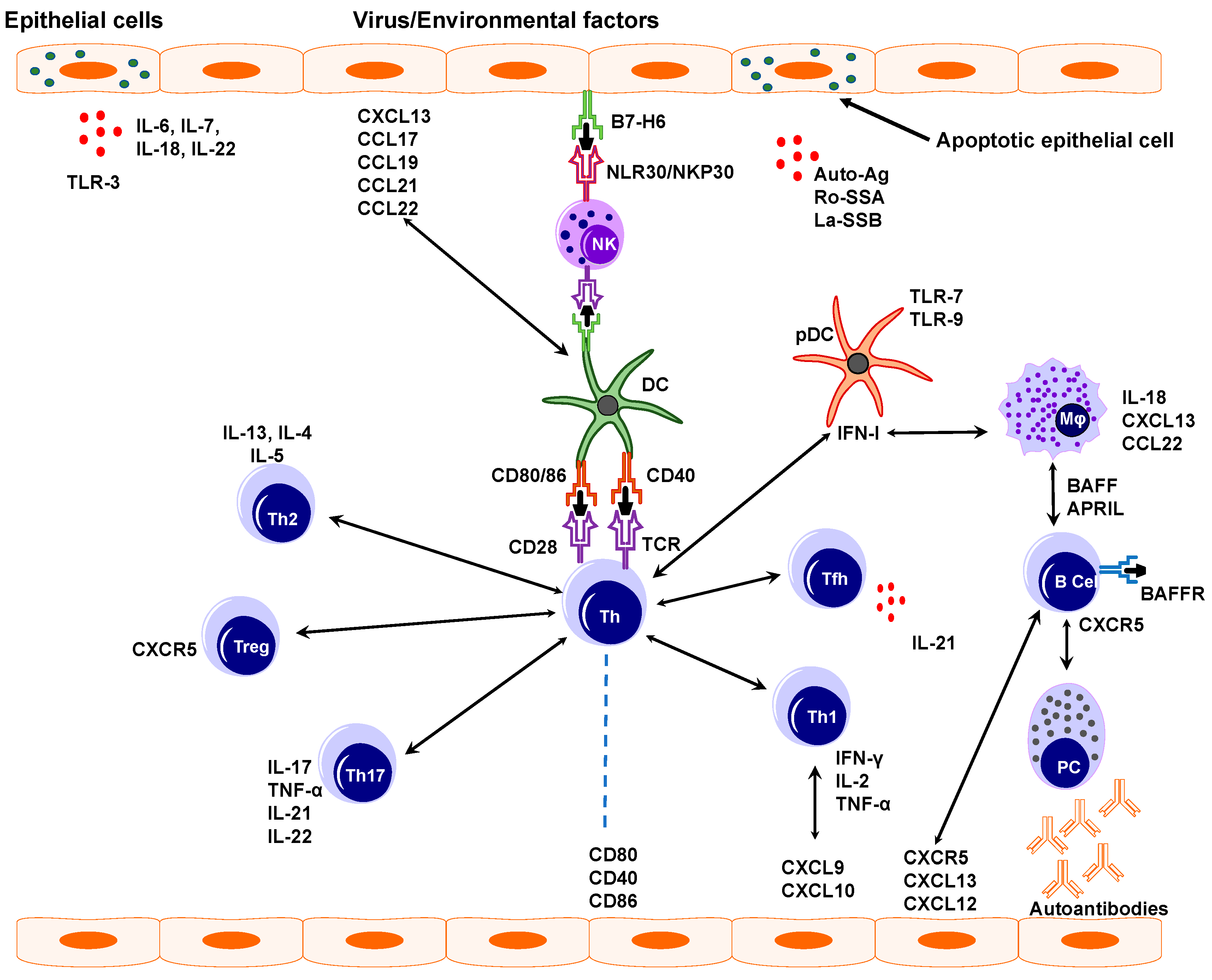{kind=link}
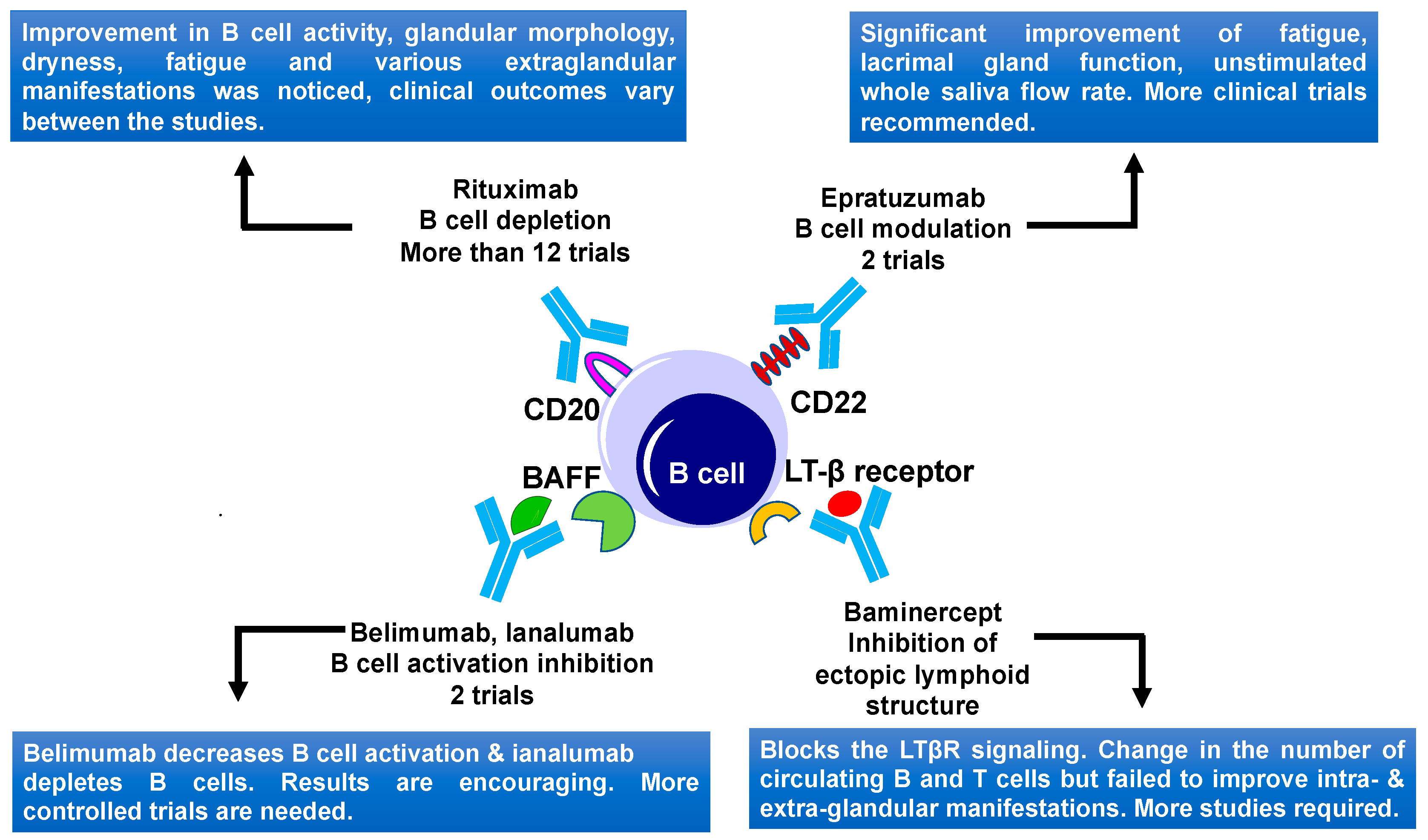{kind=link}
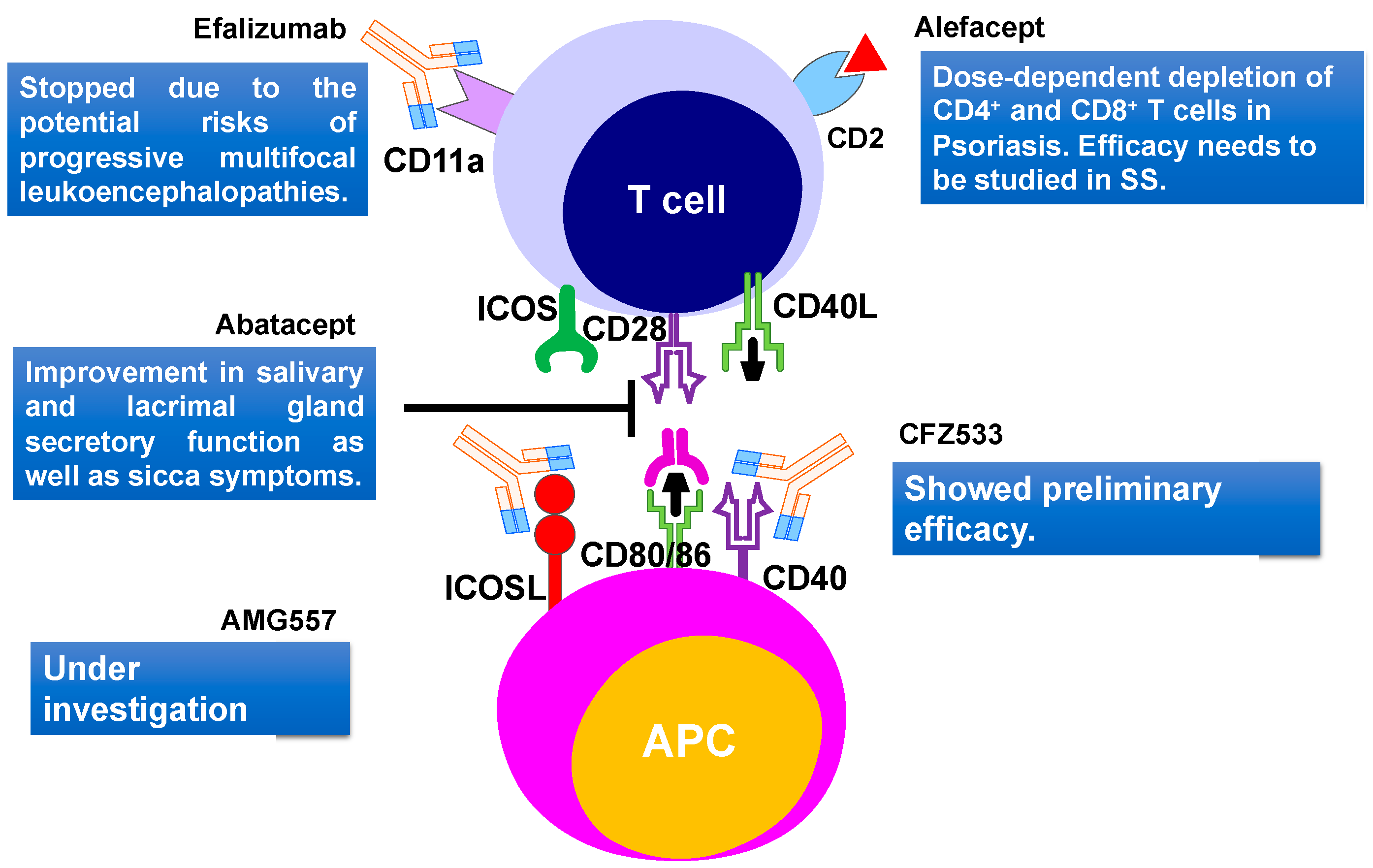{kind=link}
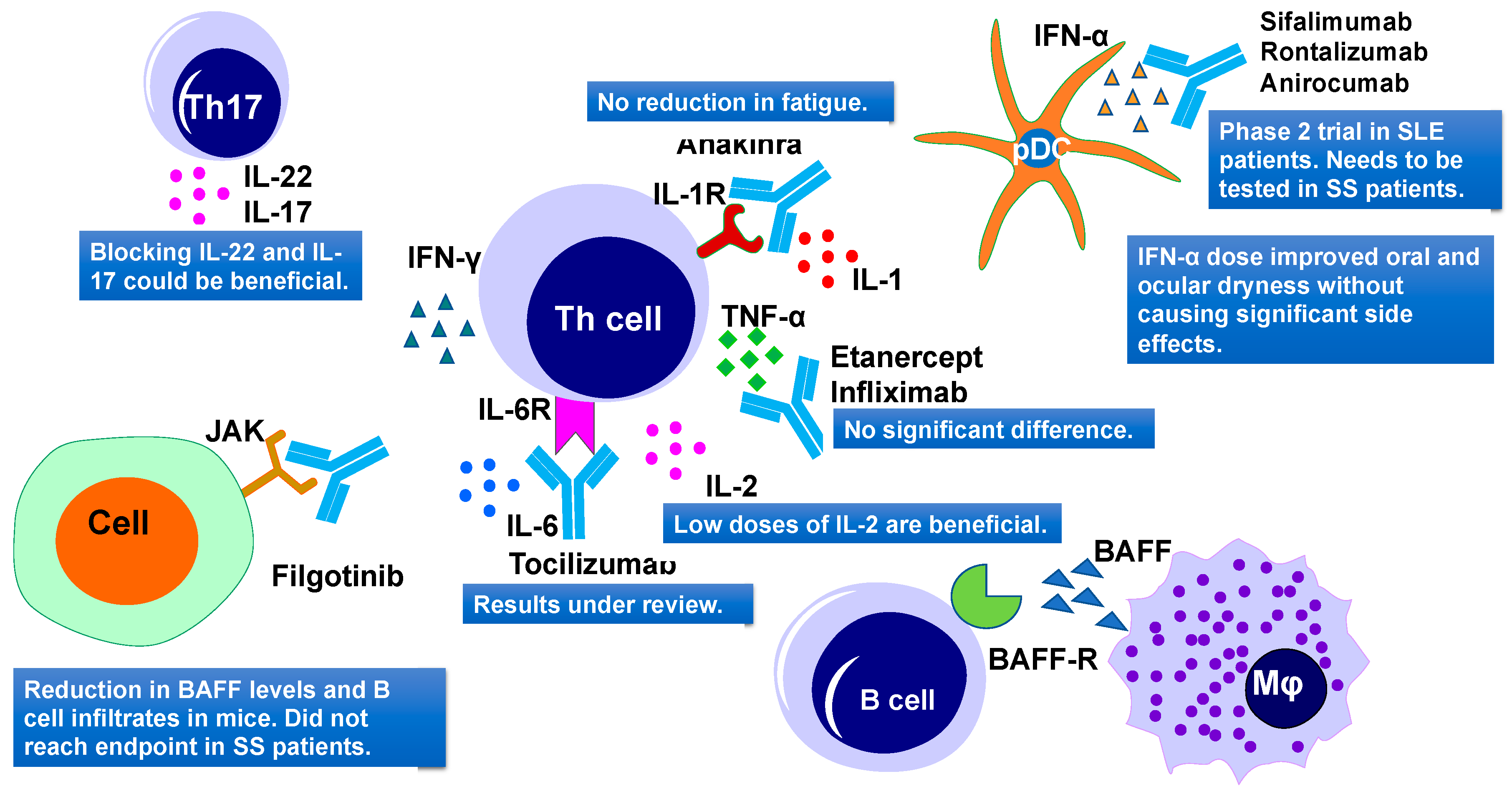{kind=link}
| Drug | Target | Dose | No. of Pats | Type of Study | Efficacy | Side Effects | Refs |
|---|---|---|---|---|---|---|---|
| Rituximab | Chimeric mAb against CD20 | Twice 1 g on days 1 and 15 | 17 | Randomized, double-blind, Placebo-controlled pilot study | Improvement after 6 months, sicca symptoms did not improve | IRR, SSR | [196] |
| 1 g with an interval of 2 weeks or placebo | 30 | Prospective, single center, randomized, double-blind, placebo-controlled trial | Stimulated saliva flow rate and lacrimal gland function improvement | SSR | [197] | ||
| 375 mg/m2/week for 4 weeks or 1 g on days 1 and 15 | 78 | Prospective study (AIR registry) | 1st cycle efficacy in 47 patients (60 %) After 6 m ESSDAI decrease | IRR, SSR | [198] | ||
| 1 g with an interval of 15 days. patients received 6 courses of therapy | 41 | Prospective, multicenter, follow-up study | ESSDAI decrease. Reduction of infiltrate and GCs after treatment | No adverse effects | [199] | ||
| Twice 1 g, 15 days apart | 28 | Prospective single-center study | ESSDAI and ESSPRI score improved. | Not reported | [200] | ||
| Twice 1 g, two weeks apart | 120 | Randomized, double-blind, Placebo-controlled, parallel-group trial (TEARS) | No significant difference | Few patients had IRR | [201] | ||
| two doses of rituximab (1 g) or placebo, two weeks apart | 110 | A randomized double-blind placebo-controlled clinical trial | No significant difference | Not reported | [202] | ||
| Two courses of rituximab (1 g) at weeks 0, 2, 24, and 26 or placebo. | 133 | A multicenter, randomized, double-blind, placebo-controlled, parallel-group trial | No significant improvement in any outcome except unstimulated saliva flow | Few serious adverse events were reported but there were no deaths | [203] | ||
| Epratuzumab | Humanized anti-CD22 monoclonal antibody | 4 infusions of 360 mg/m2 biweekly | 16 | An open-label phase I/II study | Improvements in fatigue. B-cell reduction, T cells did not change | Not reported | [204] |
| 600 mg every week, or epratuzumab 1200 mg every other week for 4 weeks | 1584 | Randomized, double-blind, placebo-controlled, multicenter studies | Disease activity in patients with SLE and associated SS showed improvements | Adverse events were comparable in the treated and placebo group | [205] | ||
| Belimumab | Human IgG1ʎ mAb targeting BAFF | 10 mg/kg, monthly dose | 30 | Phase II open-label | In 60% of patients improvement in dryness, fatigue, and musculoskeletal pain | One patient develops pneumococcal meningitis | [206] |
| Ianalumab (VAY736) | a B cell-depleting, BAFF-R blocking, monoclonal antibody | single infusion at either 3 mg/kg, 10 mg/kg or placebo. | 27 | Double-blind, placebo-controlled, phase II, single-center study | Both doses lead to depletion of B cells for a long time | Moderate infusion related side effects | [207] |
| BAFF-R | Monthly s.c. doses (5, 50, 300 mg) or placebo. | 190 | Phase 2b Study | Primary endpoint achieved, improvement for 300 mg dose | Safety profile looked good | [208] | |
| Baminercept | Lymphotoxin-β receptor Fusion protein, reduces B cell infiltration | s.c. injections of 100 mg of baminercept every week for 24 weeks or placebo | 52 | Phase II multicenter, randomized, double-blind, placebo-controlled trial | No significant difference in ESSDAI, no difference in salivary gland secretion and ocular dryness | Higher incidence of liver toxicity | [209] |
| Drug | Target | Dose | No. of Pats | Phase of Study | Efficacy | Side Effects | Refs |
|---|---|---|---|---|---|---|---|
| Abatacept | Anti-CD80/86, targets activation of T cells | 8 doses of 500/750 mg, 2 weeks apart | 11 | A pilot study | CTLA-4 Ig treatment significantly reduces salivary gland inflammation, increases saliva production | No serious adverse effects | [222] |
| 8 infusions, first 3 were 2 weeks apart, then 4 weeks apart | 15 | Open-label study | Improvement in disease symptoms and fatigue. | No serious side effects or infections were seen | [223] | ||
| ~10 mg/kg by i.v. infusion on days 1, 15, and 29 and every 4 weeks thereafter for 24 weeks | 15 | Open-label study | Reduction of circulating Tfh cells and ICOS expression on T cells was noticed | Not reported | [224] | ||
| 125 mg s.c. once a week for 24 weeks or placebo | 80 | Single center, randomized, double-blind, phase 3 trial | ESSDAI no significant difference | Few serious adverse events reported | [225] | ||
| Alefacept | Anti-CD2 dimeric fusion protein | Two 12-week courses of 15 mg i.m. per week with a two-week interval or a placebo | 73 | Phase 2, double-blind, placebo-controlled | Lowered insulin usage and reduced hypoglycemic events | A severe drop in CD4+ and CD8+ T cells pose a major concern | [226] |
| MSCs | Cell Number, Origin | Administration | Effect | Refs |
|---|---|---|---|---|
| UMSCs | 1 × 106 /Kg one dose | iv | Increase saliva flow, reduction in anti-SSA/Ro and anti-SSB/La antibodies | [257] |
| UMSCs | Human N/A | Coculture | Differentiation and proliferation of Tfh cells decreased | [256] |
| UMSCs microencapsulated | Human N/A | Coculture | Decrease in proliferation of T cells, and numbers of Th1, Th17; Treg increased | [258] |
| UMSCs | Human 1 × 106 /Kg | iv | Reduced IL-12, decrease inTh17 and Tfh cells; Treg increased | [259] |
| Drug | Cytokines | Target | Dose | No of Pats | Phase of Study | Efficacy | Side Effects | Refs |
|---|---|---|---|---|---|---|---|---|
| Infliximab | TNF family | TNF-α | 3 mg/kg two weeks apart, three infusions | 16 | Phase II | Improvement in the visual analog score, fatigue, and dryness | No significant adverse events were seen | [270] |
| infliximab | TNF family | TNF-α | 3 infusions of 5 mg/kg drug or placebo two weeks apart | 103 | Randomized, double-blind, placebo-controlled study | No significant differences | Severe adverse events reported in the infliximab group | [271] |
| Etanercept | TNF family | TNF-α | 25 mg s.c. twice per week for 12 weeks | 15 | Pilot study | No increase in salivary or lacrimal gland function | Injection-site reactions occurring in about one-third of patients | [272,273] |
| IFN-α | IFN-α | 150 IU of interferon-α 3 times a day for 24 weeks | 12 | Double-blind placebo-controlled | Improvement in symptoms of xerostomia and xerophthalmia | Well tolerated | [274] | |
| IFN-α | 150 IU of interferon-α 3 times a day for 24 weeks | 497 | 2 Phase III clinical trials | Majority of symptoms improved | No significant adverse effect noted | [275] | ||
| Tofacitinib | IFN | 0.0003–0.005% daily | 327 | Phase 1/2 prospective, randomized | Better patient-reported ocular tolerability | Well tolerated | [276] | |
| Anakinra, a non-glycosylated recombinant version of the human IL-1 receptor antagonist, IL-lRa | IL-1 | IL-1R blockade | 100 mg/day or a placebo for 4 weeks | 26 | A double-blind, placebo-controlled parallel-group study | No significant changes | Two serious adverse events (SAE) were observed | [277] |
| Tocilizumab | IL-6 | anti-IL-6 mAb | 8 mg/kg | 1 | Case study | EULAR SS activity Index was stabilized at 4, CT scan and pulmonary function normalized | Treatment was well tolerated | [278] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Srivastava, A.; Makarenkova, H.P. Innate Immunity and Biological Therapies for the Treatment of Sjögren’s Syndrome. Int. J. Mol. Sci. 2020, 21, 9172. https://doi.org/10.3390/ijms21239172
Srivastava A, Makarenkova HP. Innate Immunity and Biological Therapies for the Treatment of Sjögren’s Syndrome. International Journal of Molecular Sciences. 2020; 21(23):9172. https://doi.org/10.3390/ijms21239172
Chicago/Turabian StyleSrivastava, Amrita, and Helen P. Makarenkova. 2020. "Innate Immunity and Biological Therapies for the Treatment of Sjögren’s Syndrome" International Journal of Molecular Sciences 21, no. 23: 9172. https://doi.org/10.3390/ijms21239172
APA StyleSrivastava, A., & Makarenkova, H. P. (2020). Innate Immunity and Biological Therapies for the Treatment of Sjögren’s Syndrome. International Journal of Molecular Sciences, 21(23), 9172. https://doi.org/10.3390/ijms21239172