Role of PD-L1 in Gut Mucosa Tolerance and Chronic Inflammation

Abstract
1. Introduction
2. B7 Immunoglobulin Superfamily in Peripheral Immune Tolerance
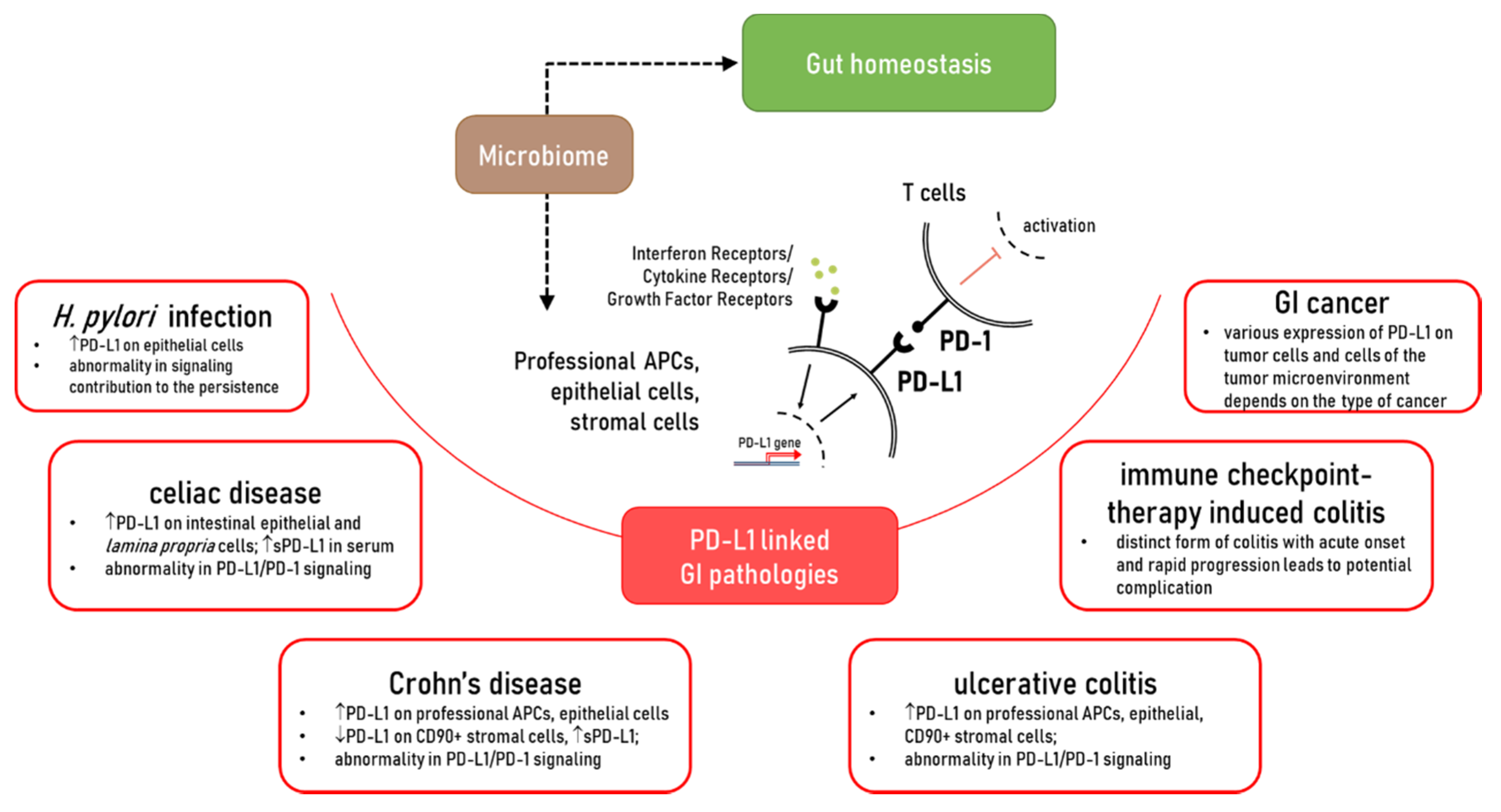
3. PD-L1 in Gut Homeostasis
4. The Role of PD-L1 in the Immunopathogenesis of Chronic Gastrointestinal Diseases
4.1. Alterations in the Expression of PD-L1 in Crohn’s Disease (CD) and Ulcerative Colitis (UC): Contribution to Chronic Inflammation
4.2. The Role of PD-L1 in IBD Associated Fibrosis
4.3. Regulation of PD-L1 Expression: Implication in IBD
5. Celiac Disease
6. PD-L1/PD-1 as a Potential Therapeutic Target in Gut Chronic Inflammatory Diseases: Lesson Learned from Immune Checkpoint Therapy of Solid Cancers
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Okumura, R.; Takeda, K. Roles of intestinal epithelial cells in the maintenance of gut homeostasis. Exp. Mol. Med. 2017, 49, 338. [Google Scholar] [CrossRef]
- Chassaing, B.; Kumar, M.; Baker, M.T.; Singh, V.; Vijay-Kumar, M. Mammalian gut immunity. Biomed. J. 2014, 37, 246–258. [Google Scholar]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and Its Ligands in Tolerance and Immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef]
- Okazaki, T.; Honjo, T. The PD-1-PD-L pathway in immunological tolerance. Trends Immunol. 2006, 27, 195–201. [Google Scholar] [CrossRef]
- Schnell, A.; Bod, L.; Madi, A.; Kuchroo, V.K. The yin and yang of co-inhibitory receptors: Toward anti-tumor immunity without autoimmunity. Cell Res. 2020, 30, 285–299. [Google Scholar] [CrossRef]
- Francisco, L.M.; Salinas, V.H.; Brown, K.E.; Vanguri, V.K.; Freeman, G.J.; Kuchroo, V.K.; Sharpe, A.H. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J. Exp. Med. 2009, 206, 3015–3029. [Google Scholar] [CrossRef]
- Zamani, M.R.; Aslani, S.; Salmaninejad, A.; Javan, M.R.; Rezaei, N. PD-1/PD-L and autoimmunity: A growing relationship. Cell. Immunol. 2016, 310, 27–41. [Google Scholar] [CrossRef]
- Nakazawa, A.; Dotan, I.; Brimnes, J.; Allez, M.; Shao, L.; Tsushima, F.; Azuma, M.; Mayer, L. The Expression and Function of Costimulatory Molecules B7h and B7-H1 on Colonic Epithelial Cells. Gastroenterology 2004, 126, 1347–1357. [Google Scholar] [CrossRef]
- Faleiro, R.; Liu, J.; Karunarathne, D.; Edmundson, A.; Winterford, C.; Nguyen, T.H.; Simms, L.A.; Radford-Smith, G.; Wykes, M. Crohn’s disease is facilitated by a disturbance of programmed death-1 ligand 2 on blood dendritic cells. Clin. Transl. Immunol. 2019, 8. [Google Scholar] [CrossRef]
- Cassol, C.A.; Owen, D.; Kendra, K.; Braga, J.R.; Frankel, W.L.; Arnold, C.A. Programmed cell death-1 (PD-1) and programmed death-ligand 1 (PD-L1) expression in PD-1 inhibitor-associated colitis and its mimics. Histopathology 2020, 77, 240–249. [Google Scholar] [CrossRef]
- Pinchuk, I.V.; Saada, J.I.; Beswick, E.J.; Boya, G.; Qiu, S.M.; Mifflin, R.C.; Raju, G.S.; Reyes, V.E.; Powell, D.W. PD-1 Ligand Expression by Human Colonic Myofibroblasts/Fibroblasts Regulates CD4+ T-Cell Activity. Gastroenterology 2008, 135, 1228–1237. [Google Scholar] [CrossRef]
- Beswick, E.J.; Grim, C.; Singh, A.; Aguirre, J.E.; Tafoya, M.; Qiu, S.; Rogler, G.; McKee, R.; Samedi, V.; Ma, T.Y.; et al. Expression of programmed death-ligand 1 by human colonic CD90+ stromal cells differs between ulcerative colitis and Crohn’s disease and determines their capacity to suppress Th1 cells. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Robertson, J.; Haas, C.T.; Pele, L.C.; Monie, T.P.; Charalambos, C.; Parkes, M.; Hewitt, R.E.; Powell, J.J. Intestinal APCs of the endogenous nanomineral pathway fail to express PD-L1 in Crohn’s disease. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Lina, T.T.; Alzahrani, S.; House, J.; Yamaoka, Y.; Sharpe, A.H.; Rampy, B.A.; Pinchuk, I.V.; Reyes, V.E. Helicobacter pylori cag pathogenicity island’s role in B7-H1 induction and immune evasion. PLoS ONE 2015, 10, e121841. [Google Scholar] [CrossRef]
- Beswick, E.J.; Pinchuk, I.V.; Das, S.; Powell, D.W.; Reyes, V.E. Expression of the programmed death ligand 1, B7-H1, on gastric epithelial cells after Helicobacter pylori exposure promotes development of CD4+ CD25+ FoxP3+ regulatory T cells. Infect. Immun. 2007, 75, 4334–4341. [Google Scholar] [CrossRef]
- Ponce de León, C.; Angel López-Casado, M.; Lorite, P.; Palomeque, T.; Isabel Torres, M. Dysregulation of the PD-1/PD-L1 pathway contributes to the pathogenesis of celiac disease. Cell. Mol. Immunol. 2019, 16, 777–779. [Google Scholar] [CrossRef]
- Shan, T.; Chen, S.; Wu, T.; Yang, Y.; Li, S.; Chen, X. PD-L1 expression in colon cancer and its relationship with clinical prognosis. Int. J. Clin. Exp. Pathol. 2019, 12, 1764–1769. [Google Scholar]
- Wyss, J.; Dislich, B.; Koelzer, V.H.; Galván, J.A.; Dawson, H.; Hädrich, M.; Inderbitzin, D.; Lugli, A.; Zlobec, I.; Berger, M.D. Stromal PD-1/PD-L1 Expression Predicts Outcome in Colon Cancer Patients. Clin. Colorectal Cancer 2019, 18, e20–e38. [Google Scholar] [CrossRef]
- Zhao, Q.; Hu, F.; Xiao, Z.; Li, M.; Wu, X.; Zhao, Y.; Wu, Y.; Yin, J.; Lin, L.; Zhang, H.; et al. Comprehensive molecular profiling of the B7 family in gastrointestinal cancer. Cell Prolif. 2018, 51, e12468. [Google Scholar] [CrossRef]
- Janakiram, M.; Chinai, J.M.; Fineberg, S.; Fiser, A.; Montagna, C.; Medavarapu, R.; Castano, E.; Jeon, H.; Ohaegbulam, K.C.; Zhao, R.; et al. Expression, clinical significance, and receptor identification of the newest B7 family member HHLA2 protein. Clin. Cancer Res. 2015, 21, 2359–2366. [Google Scholar] [CrossRef]
- Qin, W.; Hu, L.; Zhang, X.; Jiang, S.; Li, J.; Zhang, Z.; Wang, X. The Diverse Function of PD-1/PD-L Pathway Beyond Cancer. Front. Immunol. 2019, 10, 2298. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, M.W.; Bledsoe, J.R.; Morales-Oyarvide, V.; Huynh, T.G.; Mino-Kenudson, M. PD-L1 expression in colorectal cancer is associated with microsatellite instability, BRAF mutation, medullary morphology and cytotoxic tumor-infiltrating lymphocytes. Mod. Pathol. 2016, 29, 1104–1112. [Google Scholar] [CrossRef]
- Wang, D.Y.; Johnson, D.B.; Davis, E.J. Toxicities Associated with PD-1/PD-L1 Blockade. Cancer J. 2018, 24, 36–40. [Google Scholar] [CrossRef]
- Ramos-Casals, M.; Brahmer, J.R.; Callahan, M.K.; Flores-Chávez, A.; Keegan, N.; Khamashta, M.A.; Lambotte, O.; Mariette, X.; Prat, A.; Suárez-Almazor, M.E. Immune-related adverse events of checkpoint inhibitors. Nat. Rev. Dis. Prim. 2020, 6, 1–21. [Google Scholar]
- Uhlig, H.H.; Powrie, F. Translating Immunology into Therapeutic Concepts for Inflammatory Bowel Disease. Annu. Rev. Immunol. 2018, 36, 755–781. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zang, X. Structures of Immune Checkpoints: An Overview on the CD28-B7 Family. In Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2019; Volume 1172, pp. 63–78. [Google Scholar]
- Collins, M.; Ling, V.; Carreno, B.M. The B7 family of immune-regulatory ligands. Genome Biol. 2005, 6, 223. [Google Scholar] [CrossRef][Green Version]
- Janakiram, M.; Shah, U.A.; Liu, W.; Zhao, A.; Schoenberg, M.P.; Zang, X. The third group of the B7-CD28 immune checkpoint family: HHLA2, TMIGD2, B7x, and B7-H3. Immunol. Rev. 2017, 276, 26–39. [Google Scholar] [CrossRef]
- Sharpe, A.H.; Freeman, G.J. The B7-CD28 superfamily. Nat. Rev. Immunol. 2002, 2, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Zhu, H.X.; Yao, Y.; Bian, Z.H.; Zheng, Y.J.; Li, L.; Moutsopoulos, H.M.; Gershwin, M.E.; Lian, Z.X. Immune checkpoint molecules. Possible future therapeutic implications in autoimmune diseases. J. Autoimmun. 2019, 104, 102333. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zheng, Q.; Jin, L. The Role of B7 Family Molecules in Maternal–Fetal Immunity. Front. Immunol. 2020, 11, 458. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.; Aoyama, K.; Taylor, P.A.; Koehn, B.H.; Veenstra, R.G.; Panoskaltsis-Mortari, A.; Munn, D.H.; Murphy, W.J.; Azuma, M.; Yagita, H.; et al. Host programmed death ligand 1 is dominant over programmed death ligand 2 expression in regulating graft-versus-host disease lethality. Blood 2013, 122, 3062–3073. [Google Scholar] [CrossRef] [PubMed]
- Joosse, M.E.; Nederlof, I.; Walker, L.S.K.; Samsom, J.N. Tipping the balance: Inhibitory checkpoints in intestinal homeostasis. Mucosal Immunol. 2019, 12, 21–35. [Google Scholar] [CrossRef]
- Sharpe, A.H.; Wherry, E.J.; Ahmed, R.; Freeman, G.J. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat. Immunol. 2007, 8, 239–245. [Google Scholar] [CrossRef] [PubMed]
- O’Malley, G.; Treacy, O.; Lynch, K.; Naicker, S.D.; Leonard, N.A.; Lohan, P.; Dunne, P.D.; Ritter, T.; Egan, L.J.; Ryan, A.E. Stromal cell PD-L1 inhibits CD8+ T-cell antitumor immune responses and promotes colon cancer. Cancer Immunol. Res. 2018, 6, 1426–1441. [Google Scholar] [CrossRef] [PubMed]
- Beswick, E.J.; Johnson, J.R.; Saada, J.I.; Humen, M.; House, J.; Dann, S.; Qiu, S.; Brasier, A.R.; Powell, D.W.; Reyes, V.E.; et al. TLR4 Activation Enhances the PD-L1–Mediated Tolerogenic Capacity of Colonic CD90+ Stromal Cells. J. Immunol. 2014, 193, 2218–2229. [Google Scholar] [CrossRef]
- Aguirre, J.E.; Beswick, E.J.; Grim, C.; Uribe, G.; Tafoya, M.; Chacon Palma, G.; Samedi, V.; McKee, R.; Villeger, R.; Fofanov, Y.; et al. Matrix metalloproteinases cleave membrane-bound PD-L1 on CD90+ (myo-) fibroblasts in Crohn’s disease and regulate Th1/Th17 cell responses. Int. Immunol. 2020, 32, 57–68. [Google Scholar] [CrossRef]
- Mezache, L.; Magro, C.; Hofmeister, C.; Pichiorri, F.; Sborov, D.; Nuovo, G.J. Modulation of PD-L1 and CD8 Activity in Idiopathic and Infectious Chronic Inflammatory Conditions. Appl. Immunohistochem. Mol. Morphol. 2017, 25, 100–109. [Google Scholar] [CrossRef]
- Maruya, M.; Kawamoto, S.; Kato, L.M.; Fagarasan, S. Impaired selection of IgA and intestinal dysbiosis associated with PD-1-deficiency. Gut Microbes 2013, 4, 165–171. [Google Scholar] [CrossRef]
- Kawamoto, S.; Tran, T.H.; Maruya, M.; Suzuki, K.; Doi, Y.; Tsutsui, Y.; Kato, L.M.; Fagarasan, S. The inhibitory receptor PD-1 regulates IgA selection and bacterial composition in the gut. Science 2012, 336, 485–489. [Google Scholar] [CrossRef]
- Rabe, H.; Nordström, I.; Andersson, K.; Lundell, A.C.; Rudin, A. Staphylococcus aureus convert neonatal conventional CD4+ T cells into FOXP3+ CD25+ CD127low T cells via the PD-1/PD-L1 axis. Immunology 2014, 141, 467–481. [Google Scholar] [CrossRef]
- Scandiuzzi, L.; Ghosh, K.; Hofmeyer, K.A.; Abadi, Y.M.; Lázár-Molnár, E.; Lin, E.Y.; Liu, Q.; Jeon, H.; Almo, S.C.; Chen, L.; et al. Tissue-Expressed B7-H1 Critically Controls Intestinal Inflammation. Cell Rep. 2014, 6, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Mallett, G.; Laurence, A.; Amarnath, S. Programmed cell death-1 receptor (Pd-1)-mediated regulation of innate lymphoid cells. Int. J. Mol. Sci. 2019, 20, 2836. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, C.; Khan, A.R.; Floudas, A.; Saunders, S.P.; Hams, E.; Rodewald, H.R.; McKenzie, A.N.J.; Fallon, P.G. ILC2s regulate adaptive Th2 cell functions via PD-L1 checkpoint control. J. Exp. Med. 2017, 214, 2507–2521. [Google Scholar] [CrossRef] [PubMed]
- Reynoso, E.D.; Elpek, K.G.; Francisco, L.; Bronson, R.; Bellemare-Pelletier, A.; Sharpe, A.H.; Freeman, G.J.; Turley, S.J. Intestinal Tolerance Is Converted to Autoimmune Enteritis upon PD-1 Ligand Blockade. J. Immunol. 2009, 182, 2102–2112. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.A.; Dorfman, D.M.; Ma, F.-R.; Sullivan, E.L.; Munoz, O.; Wood, C.R.; Greenfield, E.A.; Freeman, G.J. Blockade of Programmed Death-1 Ligands on Dendritic Cells Enhances T Cell Activation and Cytokine Production. J. Immunol. 2003, 170, 1257–1266. [Google Scholar] [CrossRef] [PubMed]
- Kryczek, I.; Wei, S.; Gong, W.; Shu, X.; Szeliga, W.; Vatan, L.; Chen, L.; Wang, G.; Zou, W. Cutting Edge: IFN-γ Enables APC to Promote Memory Th17 and Abate Th1 Cell Development. J. Immunol. 2008, 181, 5842–5846. [Google Scholar] [CrossRef] [PubMed]
- Karakhanova, S.; Meisel, S.; Ring, S.; Mahnke, K.; Enk, A.H. ERK/p38 MAP-kinases and PI3K are involved in the differential regulation of B7-H1 expression in DC subsets. Eur. J. Immunol. 2009, 40, 254–266. [Google Scholar] [CrossRef]
- Zhao, Q.; Xiao, X.; Wu, Y.; Wei, Y.; Zhu, L.-Y.; Zhou, J.; Kuang, D.-M. Interleukin-17-educated monocytes suppress cytotoxic T-cell function through B7-H1 in hepatocellular carcinoma patients. Eur. J. Immunol. 2011, 41, 2314–2322. [Google Scholar] [CrossRef]
- Carol, M.; Lambrechts, A.; Van Gossum, A.; Libin, M.; Goldman, M.; Mascart-Lemone, F. Spontaneous secretion of interferon γ and interleukin 4 by human intraepithelial and lamina propria gut lymphocytes. Gut 1998, 42, 643–649. [Google Scholar] [CrossRef]
- Troncone, E.; Marafini, I.; Stolfi, C.; Monteleone, G. Transforming growth factor-β1/Smad7 in intestinal immunity, inflammation, and cancer. Front. Immunol. 2018, 9, 1407. [Google Scholar] [CrossRef]
- Garo, L.P.; Ajay, A.K.; Fujiwara, M.; Beynon, V.; Kuhn, C.; Gabriely, G.; Sadhukan, S.; Raheja, R.; Rubino, S.; Weiner, H.L.; et al. Smad7 Controls Immunoregulatory PDL2/1-PD1 Signaling in Intestinal Inflammation and Autoimmunity. Cell Rep. 2019, 28, 3353–3366. [Google Scholar] [CrossRef]
- Song, M.Y.; Hong, C.P.; Park, S.J.; Kim, J.H.; Yang, B.G.; Park, Y.; Kim, S.W.; Kim, K.S.; Lee, J.Y.; Lee, S.W.; et al. Protective effects of Fc-fused PD-L1 on two different animal models of colitis. Gut 2015, 64, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Ihan, A.; Pinchuk, I.V.; Beswick, E.J. Inflammation, Immunity, and Vaccines for Helicobacter pylori Infection. Helicobacter 2012, 17, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Alzahrani, S.; Lina, T.T.; Gonzalez, J.; Pinchuk, I.V.; Beswick, E.J.; Reyes, V.E. Effect of Helicobacter pylori on gastric epithelial cells. World J. Gastroenterol. 2014, 20, 12767–12780. [Google Scholar] [CrossRef] [PubMed]
- Lina, T.T.; Alzahrani, S.; Gonzalez, J.; Pinchuk, I.V.; Beswick, E.J.; Reyes, V.E. Immune evasion strategies used by Helicobacter pylori. World J. Gastroenterol. 2014, 20, 12753–12766. [Google Scholar] [CrossRef]
- Rajabian, Z.; Kalani, F.; Taghiloo, S.; Tehrani, M.; Rafiei, A.; Hosseini-Khah, Z.; Hosseini, V.; Ajami, A. Over-expression of immunosuppressive molecules, PD-L1 and PD-L2, in ulcerative colitis patients. Iran. J. Immunol. 2019, 16, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, R.E.; Pele, L.C.; Tremelling, M.; Metz, A.; Parkes, M.; Powell, J.J. Immuno-inhibitory PD-L1 can be induced by a Peptidoglycan/NOD2 mediated pathway in primary monocytic cells and is deficient in Crohn’s patients with homozygous NOD2 mutations. Clin. Immunol. 2012, 143, 162–169. [Google Scholar] [CrossRef] [PubMed]
- De Souza, H.S.P.P.; Fiocchi, C. Immunopathogenesis of IBD: Current state of the art. Nat. Rev. Gastroenterol. Hepatol. 2015, 13, 13–27. [Google Scholar] [CrossRef]
- Ahluwalia, B.; Moraes, L.; Magnusson, M.K.; Öhman, L. Immunopathogenesis of inflammatory bowel disease and mechanisms of biological therapies. Scand. J. Gastroenterol. 2018, 53, 379–389. [Google Scholar] [CrossRef]
- Neurath, M.F. Current and emerging therapeutic targets for IBD. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 269–278. [Google Scholar] [CrossRef]
- Hovhannisyan, Z.; Treatman, J.; Littman, D.R.; Mayer, L. Characterization of interleukin-17-producing regulatory T cells in inflamed intestinal mucosa from patients with inflammatory bowel diseases. Gastroenterology 2011, 140, 957–965. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ueno, A.; Iacucci, M.; Fort Gasia, M.; Jijon, H.B.; Panaccione, R.; Kaplan, G.G.; Beck, P.L.; Luider, J.; Barkema, H.W.; et al. Crossover Subsets of CD4+ T Lymphocytes in the Intestinal Lamina Propria of Patients with Crohn’s Disease and Ulcerative Colitis. Dig. Dis. Sci. 2017, 62, 2357–2368. [Google Scholar] [CrossRef] [PubMed]
- Pinchuk, I.V.; Beswick, E.J.; Saada, J.I.; Boya, G.; Schmitt, D.; Raju, G.S.; Brenmoehl, J.; Rogler, G.; Reyes, V.E.; Powell, D.W. Human colonic myofibroblasts promote expansion of CD4+ CD25high Foxp3+ regulatory T cells. Gastroenterology 2011, 140, 2019–2030. [Google Scholar] [CrossRef] [PubMed]
- Totsuka, T.; Kanai, T.; Nemoto, Y.; Tomita, T.; Tsuchiya, K.; Sakamoto, N.; Okamoto, R.; Watanabe, M. Immunosenescent colitogenic CD4+ T cells convert to regulatory cells and suppress colitis. Eur. J. Immunol. 2008, 38, 1275–1286. [Google Scholar] [CrossRef]
- Hornung, M.; Farkas, S.A.; Sattler, C.; Schlitt, H.J.; Geissler, E.K. DX5+NKT cells induce the death of colitis-associated cells: Involvement of programmed death ligand-1. Eur. J. Immunol. 2006, 36, 1210–1221. [Google Scholar] [CrossRef]
- Kanai, T.; Totsuka, T.; Uraushihara, K.; Makita, S.; Nakamura, T.; Koganei, K.; Fukushima, T.; Akiba, H.; Yagita, H.; Okumura, K.; et al. Blockade of B7-H1 Suppresses the Development of Chronic Intestinal Inflammation. J. Immunol. 2003, 171, 4156–4163. [Google Scholar] [CrossRef]
- Menzies, A.M.; Johnson, D.B.; Ramanujam, S.; Atkinson, V.G.; Wong, A.N.M.; Park, J.J.; McQuade, J.L.; Shoushtari, A.N.; Tsai, K.K.; Eroglu, Z.; et al. Anti-PD-1 therapy in patients with advanced melanoma and preexisting autoimmune disorders or major toxicity with ipilimumab. Ann. Oncol. 2017, 28, 368–376. [Google Scholar] [CrossRef]
- Saha, A.; O’Connor, R.S.; Thangavelu, G.; Lovitch, S.B.; Dandamudi, D.B.; Wilson, C.B.; Vincent, B.G.; Tkachev, V.; Pawlicki, J.M.; Furlan, S.N.; et al. Programmed death ligand-1 expression on donor T cells drives graft-versus-host disease lethality. J. Clin. Investig. 2016, 126, 2642–2660. [Google Scholar] [CrossRef]
- Munir, S.; Lundsager, M.T.; Jørgensen, M.A.; Hansen, M.; Petersen, T.H.; Bonefeld, C.M.; Friese, C.; Met, Ö.; Straten, P.T.; Andersen, M.H. Inflammation induced PD-L1-specific T cells. Cell Stress 2019, 3, 319–327. [Google Scholar] [CrossRef]
- Tang, L.; Ma, S.; Gong, H.; Wang, J.; Xu, Y.; Wu, D.; Sun, A. PD-L1 Ameliorates Murine Acute Graft-Versus-Host Disease by Suppressing Effector But Not Regulatory T Cells Function. Arch. Immunol. Ther. Exp. 2019, 67, 179–187. [Google Scholar] [CrossRef]
- Rieder, F. Managing Intestinal Fibrosis in Patients with Inflammatory Bowel Disease. Gastroenterol. Hepatol. 2018, 14, 120–122. [Google Scholar]
- Geng, Y.; Liu, X.; Liang, J.; Habiel, D.M.; Kulur, V.; Coelho, A.L.; Deng, N.; Xie, T.; Wang, Y.; Liu, N.; et al. PD-L1 on invasive fibroblasts drives fibrosis in a humanized model of idiopathic pulmonary fibrosis. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Celada, L.J.; Kropski, J.A.; Herazo-Maya, J.D.; Luo, W.; Creecy, A.; Abad, A.T.; Chioma, O.S.; Lee, G.; Hassell, N.E.; Shaginurova, G.I.; et al. PD-1 up-regulation on CD4+ T cells promotes pulmonary fibrosis through STAT3-mediated IL-17A and TGF-β1 production. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Ni, K.; Liu, M.; Zheng, J.; Wen, L.; Chen, Q.; Xiang, Z.; Lam, K.-T.; Liu, Y.; Chan, G.C.-F.; Lau, Y.-L.; et al. PD-1/PD-L1 Pathway Mediates the Alleviation of Pulmonary Fibrosis by Human Mesenchymal Stem Cells in Humanized Mice. Am. J. Respir. Cell Mol. Biol. 2018, 58, 684–695. [Google Scholar] [CrossRef] [PubMed]
- Ju, X.; Zhang, H.; Zhou, Z.; Wang, Q. Regulation of PD-L1 expression in cancer and clinical implications in immunotherapy. Am. J. Cancer Res. 2020, 10, 1–11. [Google Scholar] [PubMed]
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and Function of the PD-L1 Checkpoint. Immunity 2018, 48, 434–452. [Google Scholar] [CrossRef] [PubMed]
- Zerdes, I.; Matikas, A.; Bergh, J.; Rassidakis, G.Z.; Foukakis, T. Genetic, transcriptional and post-translational regulation of the programmed death protein ligand 1 in cancer: Biology and clinical correlations. Oncogene 2018, 37, 4639–4661. [Google Scholar] [CrossRef]
- Lastwika, K.J.; Wilson, W.; Li, Q.K.; Norris, J.; Xu, H.; Ghazarian, S.R.; Kitagawa, H.; Kawabata, S.; Taube, J.M.; Yao, S.; et al. Control of PD-L1 Expression by Oncogenic Activation of the AKT–mTOR Pathway in Non–Small Cell Lung Cancer. Cancer Res. 2016, 76, 227–238. [Google Scholar] [CrossRef]
- Loke, P.; Allison, J.P. PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proc. Natl. Acad. Sci. USA 2003, 100, 5336–5341. [Google Scholar] [CrossRef]
- Wang, J.-F.; Li, J.-B.; Zhao, Y.-J.; Yi, W.-J.; Bian, J.-J.; Wan, X.-J.; Zhu, K.-M.; Deng, X.-M. Up-regulation of Programmed Cell Death 1 Ligand 1 on Neutrophils May Be Involved in Sepsis-induced Immunosuppression. Anesthesiology 2015, 122, 852–863. [Google Scholar] [CrossRef]
- De Kleijn, S.; Langereis, J.D.; Leentjens, J.; Kox, M.; Netea, M.G.; Koenderman, L.; Ferwerda, G.; Pickkers, P.; Hermans, P.W.M. IFN-γ-Stimulated Neutrophils Suppress Lymphocyte Proliferation through Expression of PD-L1. PLoS ONE 2013, 8, e72249. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Crabill, G.A.; Pritchard, T.S.; McMiller, T.L.; Wei, P.; Pardoll, D.M.; Pan, F.; Topalian, S.L. Mechanisms regulating PD-L1 expression on tumor and immune cells. J. Immunother. Cancer 2019, 7, 305. [Google Scholar] [CrossRef] [PubMed]
- Neurath, M.F. Cytokines in inflammatory bowel disease. Nat. Rev. Immunol. 2014, 14, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Guan, Q.; Zhang, J. Recent Advances: The Imbalance of Cytokines in the Pathogenesis of Inflammatory Bowel Disease. Mediators Inflamm. 2017, 2017, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.K.; Seo, S.H.; Kim, B.S.; Kim, C.D.; Lee, J.H.; Kang, J.S.; Pil, J.M.; Lim, J.S. IFN-gamma regulates the expression of B7-H1 in dermal fibroblast cells. J. Dermatol. Sci. 2005, 40, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hamrouni, A.; Wolowiec, D.; Coiteux, V.; Kuliczkowski, K.; Hetuin, D.; Saudemont, A.; Quesnel, B. Plasma cells from multiple myeloma patients express B7-H1 (PD-L1) and increase expression after stimulation with IFN-γ and TLR ligands via a MyD88-, TRAF6-, and MEK-dependent pathway. Blood 2007, 110, 296–304. [Google Scholar] [CrossRef]
- Norén, E.; Almer, S.; Söderman, J. Genetic variation and expression levels of tight junction genes identifies association between MAGI3 and inflammatory bowel disease. BMC Gastroenterol. 2017, 17, 68. [Google Scholar] [CrossRef][Green Version]
- Long, S.H.; He, Y.; Chen, M.H.; Cao, K.; Chen, Y.J.; Chen, B.L.; Mao, R.; Zhang, S.H.; Zhu, Z.H.; Zeng, Z.R.; et al. Activation of PI3K/Akt/mTOR signaling pathway triggered by PTEN downregulation in the pathogenesis of Crohn’s disease. J. Dig. Dis. 2013, 14, 662–669. [Google Scholar] [CrossRef]
- Baas, M.; Besançon, A.; Goncalves, T.; Valette, F.; Yagita, H.; Sawitzki, B.; Volk, H.D.; Waeckel-Enée, E.; Rocha, B.; Chatenoud, L.; et al. TGFβ-dependent expression of PD-1 and PD-L1 controls CD8+ T cell anergy in transplant tolerance. Elife 2016, 5. [Google Scholar] [CrossRef]
- Ou, J.N.; Wiedeman, A.E.; Stevens, A.M. TNF-α and TGF-β counter-regulate PD-L1 expression on monocytes in systemic lupus erythematosus. Sci. Rep. 2012, 2. [Google Scholar] [CrossRef]
- David, J.M.; Dominguez, C.; McCampbell, K.K.; Gulley, J.L.; Schlom, J.; Palena, C. A novel bifunctional anti-PD-L1/TGF-β Trap fusion protein (M7824) efficiently reverts mesenchymalization of human lung cancer cells. Oncoimmunology 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Zhang, L.; Li, J.; Li, Y.; Wang, Y.; Xu, Z.X. Recent findings in the regulation of programmed death ligand 1 expression. Front. Immunol. 2019, 10, 1337. [Google Scholar] [CrossRef] [PubMed]
- Fantini, M.C.; Rizzo, A.; Fina, D.; Caruso, R.; Sarra, M.; Stolfi, C.; Becker, C.; MacDonald, T.T.; Pallone, F.; Neurath, M.F.; et al. Smad7 Controls Resistance of Colitogenic T Cells to Regulatory T Cell-Mediated Suppression. Gastroenterology 2009, 136, 1308–1316. [Google Scholar] [CrossRef] [PubMed]
- Marafini, I.; Zorzi, F.; Codazza, S.; Pallone, F.; Monteleone, G. TGF-Beta Signaling Manipulation as Potential Therapy for IBD. Curr. Drug Targets 2013, 14, 1400–1404. [Google Scholar] [CrossRef]
- Pulko, V.; Liu, X.; Krco, C.J.; Harris, K.J.; Frigola, X.; Kwon, E.D.; Dong, H. TLR3-Stimulated Dendritic Cells Up-regulate B7-H1 Expression and Influence the Magnitude of CD8 T Cell Responses to Tumor Vaccination. J. Immunol. 2009, 183, 3634–3641. [Google Scholar] [CrossRef]
- Groeger, S.; Denter, F.; Lochnit, G.; Schmitz, M.L.; Meyle, J. Porphyromonas gingivalis cell wall components induce programmed death ligand 1 (PD-L1) expression on human oral carcinoma cells by a receptor-interacting protein kinase 2 (RIP2)-dependent mechanism. Infect. Immun. 2020, 88. [Google Scholar] [CrossRef]
- Lu, Y.; Li, X.; Liu, S.; Zhang, Y.; Zhang, D. Toll-like receptors and inflammatory bowel disease. Front. Immunol. 2018, 9, 72. [Google Scholar] [CrossRef]
- Lauret, E.; Rodrigo, L. Celiac Disease and Autoimmune-Associated Conditions. Biomed Res. Int. 2013, 2013, 1–17. [Google Scholar] [CrossRef]
- Tursi, A.; Giorgetti, G.M.; Brandimarte, G.; Elisei, W. High Prevalence of Celiac Disease Among Patients Affected by Crohn’s Disease. Inflamm. Bowel Dis. 2005, 11, 662–666. [Google Scholar] [CrossRef]
- Pascual, V.; Dieli-Crimi, R.; López-Palacios, N.; Bodas, A.; Medrano, L.M.; Núñez, C. Inflammatory bowel disease and celiac disease: Overlaps and differences. World J. Gastroenterol. 2014, 20, 4846–4856. [Google Scholar] [CrossRef]
- Jacobs, J.; Smits, E.; Lardon, F.; Pauwels, P.; Deschoolmeester, V. Immune Checkpoint Modulation in Colorectal Cancer: What’s New and What to Expect. J. Immunol. Res. 2015, 2015, 158038. [Google Scholar] [CrossRef] [PubMed]
- Valentini, A.M.; Di Pinto, F.; Cariola, F.; Guerra, V.; Giannelli, G.; Caruso, M.L.; Pirrelli, M. PD-L1 expression in colorectal cancer defines three subsets of tumor immune microenvironments. Oncotarget 2018, 9, 8584–8596. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Gerber, D.E. Autoimmunity, checkpoint inhibitor therapy and immune-related adverse events: A review. Semin. Cancer Biol. 2020, 64, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Siakavellas, S.I.; Bamias, G. Checkpoint inhibitor colitis: A new model of inflammatory bowel disease? Curr. Opin. Gastroenterol. 2018, 34, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Som, A.; Mandaliya, R.; Alsaadi, D.; Farshidpour, M.; Charabaty, A.; Malhotra, N.; Mattar, M.C. Immune checkpoint inhibitor-induced colitis: A comprehensive review. World J. Clin. Cases 2019, 7, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Bellaguarda, E.; Hanauer, S. Checkpoint Inhibitor-Induced Colitis. Am. J. Gastroenterol. 2020, 115, 202–210. [Google Scholar] [CrossRef]
- Postow, M.A.; Sidlow, R.; Hellmann, M.D. Immune-related adverse events associated with immune checkpoint blockade. N. Engl. J. Med. 2018, 378, 158–168. [Google Scholar] [CrossRef]
- Ibraheim, H.; Perucha, E.; Powell, N. Pathology of immune-mediated tissue lesions following treatment with immune checkpoint inhibitors. Rheumatology 2019, 58, vii17–vii28. [Google Scholar] [CrossRef]
- Chen, J.H.; Pezhouh, M.K.; Lauwers, G.Y.; Masia, R. Histopathologic Features of Colitis Due to Immunotherapy with Anti-PD-1 Antibodies. Am. J. Surg. Pathol. 2017, 41, 643–654. [Google Scholar] [CrossRef]
- Lee, S.A.; Wang, Y.; Liu, F.; Riordan, S.M.; Liu, L.; Zhang, L. Escherichia coli K12 upregulates PD-L1 expression in IFN-γ sensitized intestinal epithelial cells via the NF-κB pathway. Infect. Immun. 2020. [Google Scholar] [CrossRef]
- Alpdundar Bulut, E.; Bayyurt Kocabas, B.; Yazar, V.; Aykut, G.; Guler, U.; Salih, B.; Surucu Yilmaz, N.; Ayanoglu, I.C.; Polat, M.M.; Akcali, K.C.; et al. Human Gut Commensal Membrane Vesicles Modulate Inflammation by Generating M2-like Macrophages and Myeloid-Derived Suppressor Cells. J. Immunol. 2020, 205, 2707–2718. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Liu, D.; Xie, Y.; Yao, X.; Li, Y. Bifidobacterium infantis Induces Protective Colonic PD-L1 and Foxp3 Regulatory T Cells in an Acute Murine Experimental Model of Inflammatory Bowel Disease. Gut Liver 2019, 13, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, L.C.; Bhatia, S.; Thompson, J.A.; Grivas, P. Preexisting autoimmune disease: Implications for immune checkpoint inhibitor therapy in solid tumors. J. Natl. Compr. Cancer Netw. 2019, 17, 750–757. [Google Scholar] [CrossRef] [PubMed]
- Abu-Sbeih, H.; Faleck, D.M.; Ricciuti, B.; Mendelsohn, R.B.; Naqash, A.R.; Cohen, J.V.; Sellers, M.C.; Balaji, A.; Ben-Betzalel, G.; Hajir, I.; et al. Immune Checkpoint Inhibitor Therapy in Patients With Preexisting Inflammatory Bowel Disease. J. Clin. Oncol. 2020, 38, 576–583. [Google Scholar] [CrossRef]


{kind=link}
| What Was Shown | Animal Model | PD-L1 Source and Regime | Mechanism of Action | Ref. |
|---|---|---|---|---|
| PD-L1-mediated protection | DSS- and TNBS-colitis in PD-L1−/−; Rag-1−/−; PD-1−/− C57BL/6 background mice | Knockout | PD-L1-mediated protection during intestinal inflammation through epithelial cells or myofibroblasts and pericytes. Adaptive immune response was not required for PD-L1 protection in this model. | [42] |
| PD-L1-Fc displayed protective effects | DSS-colitis in wild-type B6 | Adenovirus expressing PD-L1-Fc (Ad/PD-L1-Fc) at a dose of 1 × 108 plaque forming units (PFU) on days 0, 3, and 6, or PD-L1-Fc protein at a dose of 30 μg on day 2 | PD-L1-Fc displayed protective effects on both systemic and mucosal inflammation, due to the downregulation of IFN-γ and IL-17 production. PD-L1-Fc might exert a protective effect by targeting different types of immune cells driving inflammation, such as myeloid cells in the DSS model and CD4 Th cells in the T-cell transfer model. | [53] |
| T-cell-induced colitis model in Rag-1 KO mice | Three weeks after T-cell reconstitution, recombinant PD-L1-Fc protein was administered at 1-week intervals at a dose of 20 μg for 4 weeks | |||
| PD-L1-Fc displayed protective effects | DSS-colitis in C57BL/6J WT | PDL1-Fc treatment were given i.p. (100 mg per mouse every other day, four times) | This colitis mitigation in PDL2-Fc- or PDL1-Fc-treated mice is associated with increased colonic Foxp3+T cells. | [52] |
| Blocking PD-L1 is reduced ability of DX5+NKT to killing of colitogenic T cells | Chronic DSS-colitis in BALB/c SCID mice | Anti-PD-L1 antibody | In vitro blocking PD-L1 with specific antibody resulted in a reduced ability of DX5+NKT cells to induce the death of CD62L+CD4+ T cells and lymphocytes derived from intestinal lymph node tissue of mice with chronic DSS-mediated colitis. | [66] |
| Anti-PD-L1 mAb prevents the development of colitis | Adoptive transfer of CD4+CD45RBhigh T cells to SCID mice | Anti-PD-L1 (MIH6) mAb 250 μg/mouse three times per week starting on the day of T cell transfer and continuing up to 7 weeks | Decreased expansion of pathogenic T cells and the down-regulated Th1 cytokine production (i.e., IFN-γ, IL-2, and TNF-α) by LP CD4+ T cells. The endogenous PD-L1 might be required for the expansion and differentiation of adoptively transferred CD4+CD45Bhigh T cells in vivo. | [67] |
| PD-1−/− mice are resistant to colitis | DSS-induced colitis in specific-pathogen-free PD-1−/− mice in C57BL/6 (B6)-background | PD-1 KO mice | PD-1−/− mice are resistant to DSS-induced colitis; altered microbiota in PD-1−/− mice modulates gut inflammation, colon microbiota of PD-1−/− mice is less colitogenic than WT commensal bacteria. | [68] |
| PD-1 expression on CD4+ LP T cells associated with protection from colitis | Adoptive transfers of the colitic LP CD4+ T cells after developing CD4+CD45RBhigh T cell in Balb/c or colitic LP CD4+ T cell-transferred colitis in SCID mice | LP CD4+ T cells obtained from non-colitic mice after over seven or more transfers expressed significantly higher levels of PD-1. | [65] | |
| PD-L1 blockade leads autoimmune enteritis | Naive congenic CD45.1+ OT-I T cells were transferred i.v. into CD45.2+ C57BL/6 or iFABP-tOVA recipients | Mice were injected i.p. with 200 μg of anti-PD-L1 Ab on day –1, day 2, and every other day | Ab-mediated PD-L1 blockade leads to a considerable expansion of OVA-specific CD8+ T cells and their differentiation into effector cells capable of producing pro-inflammatory cytokines. It breaks the intestinal tolerance in iFABP-tOVA mice leading to CD8+T cell-mediated autoimmune enteritis. | [45] |
| decreased gut homing and cytokine production by Pd-L1–/– donor T cells in animal model of GVHD | Lethally irradiated BALB/c recipients were infused with 107 WT B6 BM cells alone or with 2 × 106 WT B6 or Pd-L1–/– purified T cells | PD-L1 KO donor | Upregulation of PD-L1 on GVHD-causing CD4 and CD8+ T cells is necessary for their survival, proliferation, and optimal T eff function. PD-L1–deficient T cells had reduced expression of gut homing receptors, diminished production of inflammatory cytokines, and enhanced rates of apoptosis. | [69] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chulkina, M.; Beswick, E.J.; Pinchuk, I.V. Role of PD-L1 in Gut Mucosa Tolerance and Chronic Inflammation. Int. J. Mol. Sci. 2020, 21, 9165. https://doi.org/10.3390/ijms21239165
Chulkina M, Beswick EJ, Pinchuk IV. Role of PD-L1 in Gut Mucosa Tolerance and Chronic Inflammation. International Journal of Molecular Sciences. 2020; 21(23):9165. https://doi.org/10.3390/ijms21239165
Chicago/Turabian StyleChulkina, Marina, Ellen J. Beswick, and Irina V. Pinchuk. 2020. "Role of PD-L1 in Gut Mucosa Tolerance and Chronic Inflammation" International Journal of Molecular Sciences 21, no. 23: 9165. https://doi.org/10.3390/ijms21239165
APA StyleChulkina, M., Beswick, E. J., & Pinchuk, I. V. (2020). Role of PD-L1 in Gut Mucosa Tolerance and Chronic Inflammation. International Journal of Molecular Sciences, 21(23), 9165. https://doi.org/10.3390/ijms21239165