The Therapeutic Potential of Neuronal K-Cl Co-Transporter KCC2 in Huntington’s Disease and Its Comorbidities


Abstract

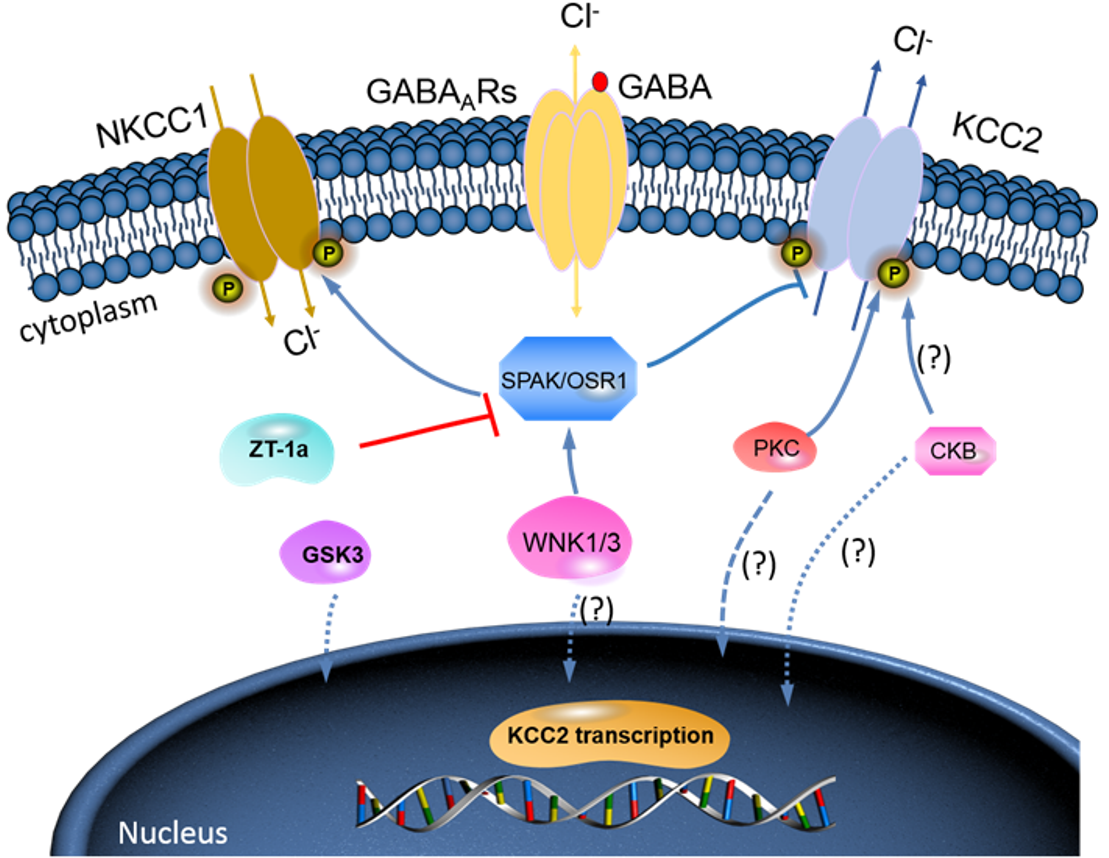{kind=link}
{kind=link}
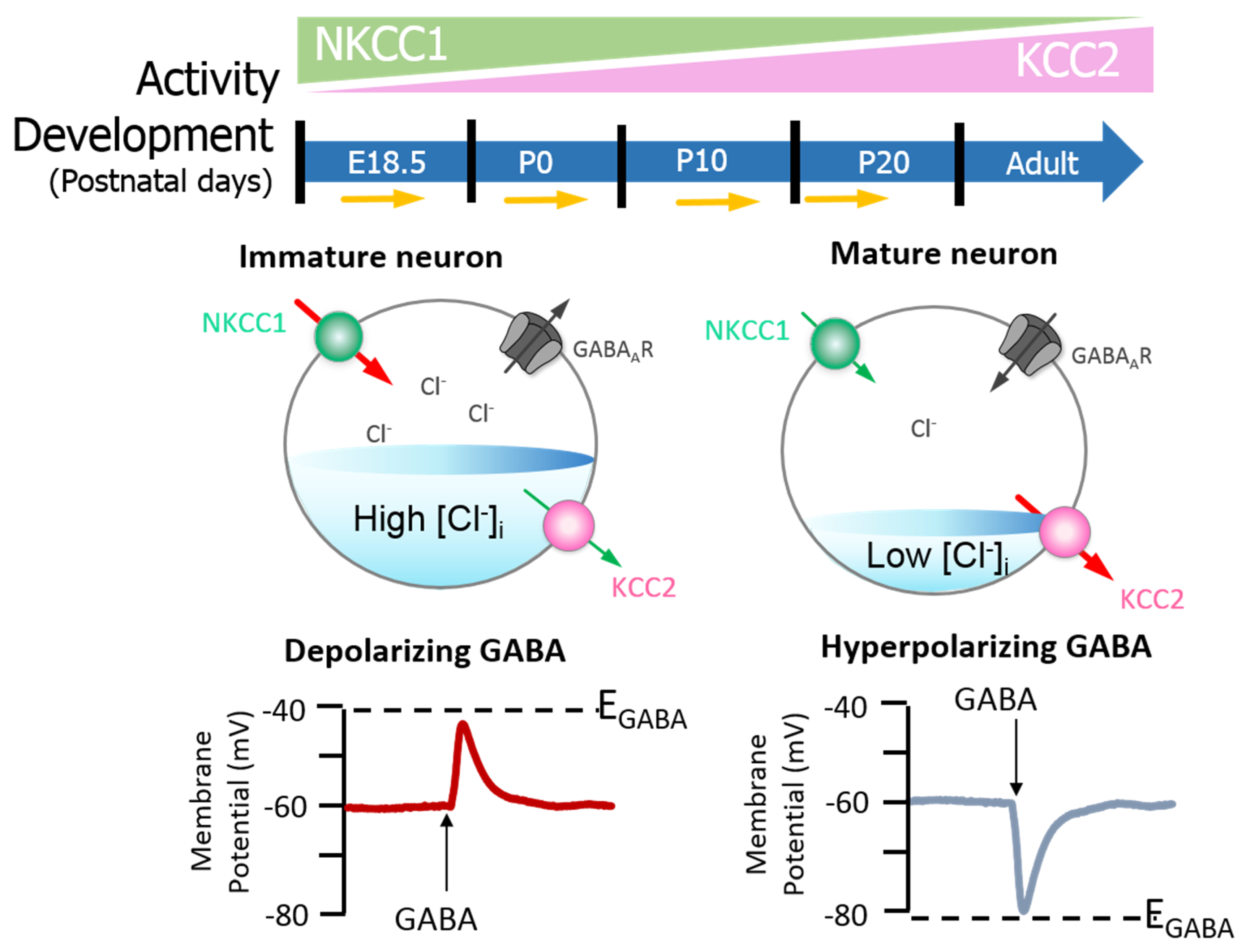{kind=link}
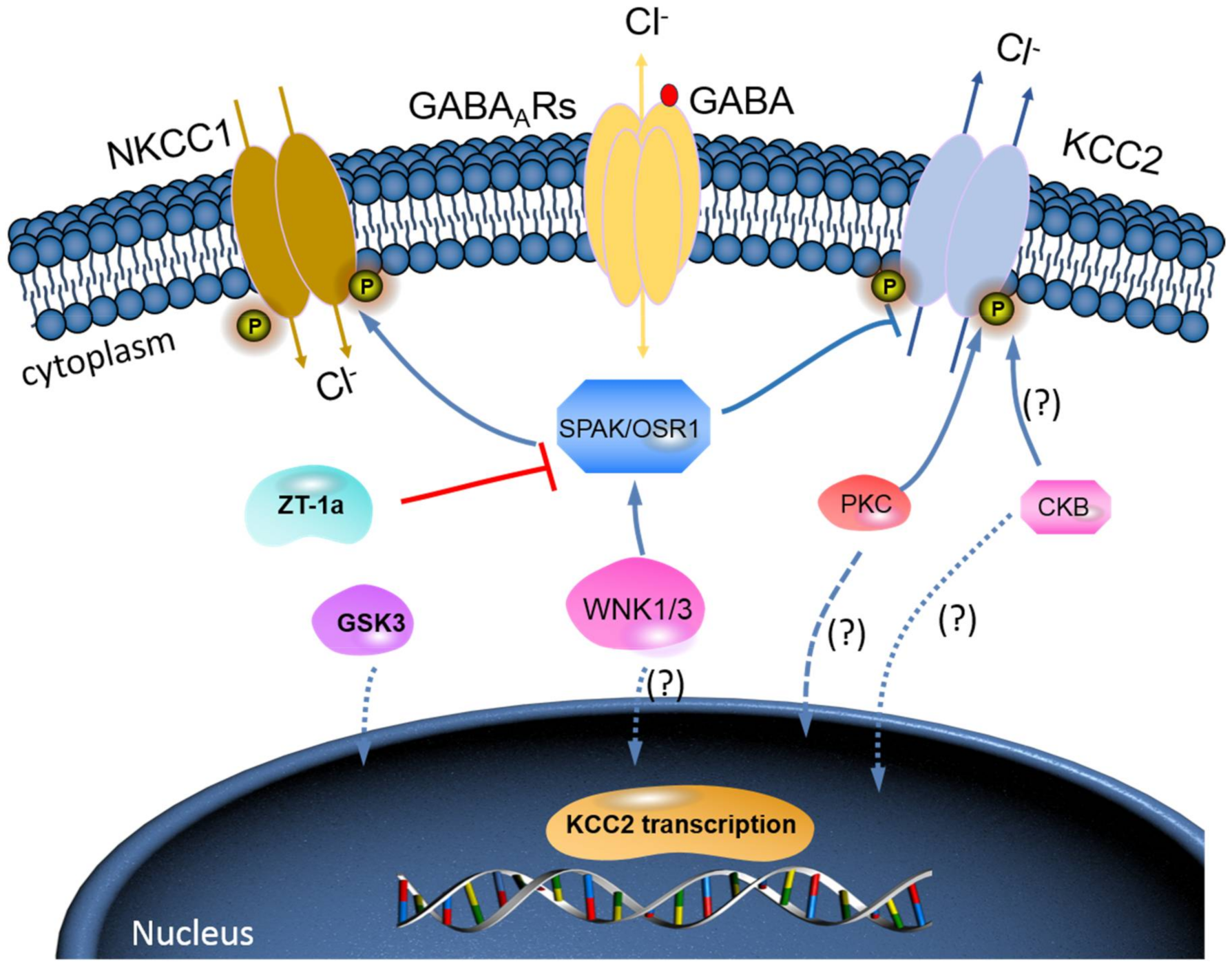{kind=link}
1. Introduction
2. KCC2 Regulation and Function in the Healthy Brain
Phosphorylation Regulation of KCC2 by Protein Kinase Signalling Pathways
3. KCC2 Regulation and Function in the HD Brain
3.1. Mechanisms of Reduced KCC2 Function in HD
3.2. Sleep Disorders in Huntington’s Disease
3.3. Sleep Disorders Treatment
3.4. Hypothalamic Changes in the HD: Implications for Sleep and Circadian Rhythmicity
3.5. KCC2 and GABA Involvement
3.6. Drug Development for KCC2 Activation
4. Conclusions and Future Prospective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Payne, J.A. Functional characterization of the neuronal-specific K-Cl cotransporter: Implications for [K+] oregulation. Am. J. Physiol. Cell Physiol. 1997, 273, C1516–C1525. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.T.; Chang, Y.G.; Chern, Y. Insights into GABAAergic system alteration in Huntington′s disease. Open Biol. 2018, 8. [Google Scholar] [CrossRef]
- Jimenez-Sanchez, M.; Licitra, F.; Underwood, B.R.; Rubinsztein, D.C. Huntington′s Disease: Mechanisms of Pathogenesis and Therapeutic Strategies. Cold Spring Harb. Perspect. Med. 2017, 7. [Google Scholar] [CrossRef]
- Rosenblatt, A.; Brinkman, R.R.; Liang, K.Y.; Almqvist, E.W.; Margolis, R.L.; Huang, C.Y.; Sherr, M.; Franz, M.L.; Abbott, M.H.; Hayden, M.R.; et al. Familial influence on age of onset among siblings with Huntington disease. Am. J. Med. Genet. 2001, 105, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Genetic Modifiers of Huntington’s Disease Consortium. Identification of Genetic Factors that Modify Clinical Onset of Huntington’s Disease. Cell 2015, 162, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Moss, D.J.H.; Pardiñas, A.F.; Langbehn, D.; Lo, K.; Leavitt, B.R.; Roos, R.; Durr, A.; Mead, S.; Holmans, P.; Jones, L.; et al. Identification of genetic variants associated with Huntington’s disease progression: A genome-wide association study. Lancet Neurol. 2017, 16, 701–711. [Google Scholar] [CrossRef]
- Georgiou, N.; Bradshaw, J.L.; Chiu, E.; Tudor, A.; O′Gorman, L.; Phillips, J.G. Differential clinical and motor control function in a pair of monozygotic twins with Huntington’s disease. Mov. Disord. 1999, 14, 320–325. [Google Scholar] [CrossRef]
- Walker, F.O. Huntington’s disease. Lancet 2007, 369, 218–228. [Google Scholar] [CrossRef]
- Zuccato, C.; Valenza, M.; Cattaneo, E. Molecular mechanisms and potential therapeutical targets in Huntington’s disease. Physiol. Rev. 2010, 90, 905–981. [Google Scholar] [CrossRef]
- Goodman, A.O.; Rogers, L.; Pilsworth, S.; McAllister, C.J.; Shneerson, J.M.; Morton, A.J.; Barker, R.A. Asymptomatic sleep abnormalities are a common early feature in patients with Huntington’s disease. Curr. Neurol. Neurosci. Rep. 2011, 11, 211–217. [Google Scholar] [CrossRef]
- Pellegrino, C.M.; Rybicki, A.C.; Musto, S.; Nagel, R.L.; Schwartz, R.S. Molecular identification and expression of erythroid K: Cl cotransporter in human and mouse erythroleukemic cells. Blood Cells Mol. Dis. 1998, 24, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Russell, J.M. Sodium-potassium-chloride cotransport. Physiol. Rev. 2000, 80, 211–276. [Google Scholar] [CrossRef] [PubMed]
- Schulte, J.T.; Wierenga, C.J.; Bruining, H. Chloride transporters and GABA polarity in developmental, neurological and psychiatric conditions. Neurosci. Biobehav. Rev. 2018, 90, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Shulga, A.; Magalhães, A.C.; Autio, H.; Plantman, S.; di Lieto, A.; Nykjær, A.; Carlstedt, T.; Risling, M.; Arumäe, U.; Castrén, E. The loop diuretic bumetanide blocks posttraumatic p75NTR upregulation and rescues injured neurons. J. Neurosci. 2012, 32, 1757–1770. [Google Scholar] [CrossRef] [PubMed]
- Kravitz, A.V.; Kreitzer, A.C. Striatal mechanisms underlying movement, reinforcement, and punishment. Physiology (Bethesda) 2012, 27, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Simpson, E.H.; Kellendonk, C.; Kandel, E. A possible role for the striatum in the pathogenesis of the cognitive symptoms of schizophrenia. Neuron 2010, 65, 585–596. [Google Scholar] [CrossRef]
- Lemiere, J.; Decruyenaere, M.; Evers-Kiebooms, G.; Vandenbussche, E.; Dom, R. Cognitive changes in patients with Huntington’s disease (HD) and asymptomatic carriers of the HD mutation—A longitudinal follow-up study. J. Neurol. 2004, 251, 935–942. [Google Scholar] [CrossRef]
- Begeti, F.; Schwab, L.C.; Mason, S.L.; Barker, R.A. Hippocampal dysfunction defines disease onset in Huntington’s disease. J. Neurol. Neurosurg. Psychiatry 2016, 87, 975–981. [Google Scholar] [CrossRef]
- Paulsen, J.S. Cognitive impairment in Huntington disease: Diagnosis and treatment. Curr. Neurol. Neurosci. Rep. 2011, 11, 474–483. [Google Scholar] [CrossRef]
- Petersen, A.; Gabery, S. Hypothalamic and Limbic System Changes in Huntington’s Disease. J. Huntingt. Dis. 2012, 1, 5–16. [Google Scholar] [CrossRef]
- Arnulf, I.; Nielsen, J.; Lohmann, E.; Schieffer, J.; Wild, E.; Jennum, P.; Konofal, E.; Walker, M.; Oudiette, D.; Tabrizi, S.; et al. Rapid Eye Movement Sleep Disturbances in Huntington Disease. Arch. Neurol. 2008, 65, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Horta, S.; Perez-Perez, J.; van Duijn, E.; Fernandez-Bobadilla, R.; Carceller, M.; Pagonabarraga, J.; Pascual-Sedano, B.; Campolongo, A.; Ruiz-Idiago, J.; Sampedro, F.; et al. Neuropsychiatric symptoms are very common in premanifest and early stage Huntington’s Disease. Parkinsonism Relat. Disord. 2016, 25, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Orth, M.; Schippling, S.; Schneider, S.A.; Bhatia, K.P.; Talelli, P.; Tabrizi, S.J.; Rothwell, J.C. Abnormal motor cortex plasticity in premanifest and very early manifest Huntington disease. J. Neurol. Neurosurg. Psychiatry 2010, 81, 267–270. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, J.S.; Langbehn, D.R.; Stout, J.C.; Aylward, E.; Ross, C.A.; Nance, M.; Guttman, M.; Johnson, S.; MacDonald, M.; Beglinger, L.J.; et al. Detection of Huntington’s disease decades before diagnosis: The Predict-HD study. J. Neurol. Neurosurg. Psychiatry 2008, 79, 874–880. [Google Scholar] [CrossRef]
- Bellosta Diago, E.; Perez Perez, J.; Santos Lasaosa, S.; Viloria Alebesque, A.; Martinez Horta, S.; Kulisevsky, J.; Lopez Del Val, J. Circadian rhythm and autonomic dysfunction in presymptomatic and early Huntington’s disease. Parkinsonism Relat. Disord. 2017, 44, 95–100. [Google Scholar] [CrossRef]
- Vonsattel, J.-P.; Myers, R.H.; Stevens, T.J.; Ferrante, R.J.; Bird, E.D.; Richardson, E.P., Jr. Neuropathological Classification of Huntington’s Disease. J. Neuropathol. Exp. Neurol. 1985, 44, 559–577. [Google Scholar] [CrossRef]
- Li, J.Y.; Plomann, M.; Brundin, P. Huntington’s disease: A synaptopathy? Trends Mol. Med. 2003, 9, 414–420. [Google Scholar] [CrossRef]
- Tyebji, S.; Hannan, A.J. Synaptopathic mechanisms of neurodegeneration and dementia: Insights from Huntington’s disease. Prog. Neurobiol. 2017, 153, 18–45. [Google Scholar] [CrossRef]
- Garret, M.; Du, Z.; Chazalon, M.; Cho, Y.H.; Baufreton, J. Alteration of GABAergic neurotransmission in Huntington’s disease. CNS Neurosci. Ther. 2018, 24, 292–300. [Google Scholar] [CrossRef]
- Sardini, A.; Amey, J.S.; Weylandt, K.H.; Nobles, M.; Valverde, M.A.; Higgins, C.F. Cell volume regulation and swelling-activated chloride channels. Biochim. Biophys. Acta 2003, 1618, 153–162. [Google Scholar] [CrossRef]
- Wilson, C.S.; Mongin, A.A. The signaling role for chloride in the bidirectional communication between neurons and astrocytes. Neurosci. Lett. 2019, 689, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Duy, P.Q.; David, W.B.; Kahle, K.T. Identification of KCC2 Mutations in Human Epilepsy Suggests Strategies for Therapeutic Transporter Modulation. Front. Cell Neurosci. 2019, 13, 515. [Google Scholar] [CrossRef] [PubMed]
- Elorza-Vidal, X.; Gaitan-Penas, H.; Estevez, R. Chloride Channels in Astrocytes: Structure, Roles in Brain Homeostasis and Implications in Disease. Int. J. Mol. Sci. 2019, 20, 1034. [Google Scholar] [CrossRef] [PubMed]
- Deeb, T.Z.; Lee, H.H.; Walker, J.A.; Davies, P.A.; Moss, S.J. Hyperpolarizing GABAergic transmission depends on KCC2 function and membrane potential. Channels (Austin) 2011, 5, 475–481. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hübner, C.A.; Stein, V.; Hermans-Borgmeyer, I.; Meyer, T.; Ballanyi, K.; Jentsch, T.J. Disruption of KCC2 Reveals an Essential Role of K-Cl Cotransport Already in Early Synaptic Inhibition. Neuron 2001, 30, 515–524. [Google Scholar] [CrossRef]
- Huebner, C.A.; Holthoff, K. Anion transport and GABA signaling. Front. Cell. Neurosci. 2013, 7, 177. [Google Scholar] [CrossRef]
- Staley, K.J.; Soldo, B.L.; Proctor, W.R. Ionic mechanisms of neuronal excitation by inhibitory GABAA receptors. Science 1995, 269, 977–981. [Google Scholar] [CrossRef]
- Dallwig, R.; Deitmer, J.W.; Backus, K.H. On the mechanism of GABA-induced currents in cultured rat cortical neurons. Pflügers Arch. 1999, 437, 289–297. [Google Scholar] [CrossRef]
- Kim, D.-Y.; Fenoglio, K.A.; Kerrigan, J.F.; Rho, J.M. Bicarbonate contributes to GABAA receptor-mediated neuronal excitation in surgically resected human hypothalamic hamartomas. Epilepsy Res. 2009, 83, 89–93. [Google Scholar] [CrossRef]
- Ma, B.F.; Xie, M.J.; Zhou, M. Bicarbonate efflux via GABAA receptors depolarizes membrane potential and inhibits two-pore domain potassium channels of astrocytes in rat hippocampal slices. Glia 2012, 60, 1761–1772. [Google Scholar] [CrossRef]
- Lombardi, A.; Jedlicka, P.; Luhmann, H.J.; Kilb, W. Interactions between membrane resistance, GABA-A receptor properties, bicarbonate dynamics and Cl−-transport shape activity-dependent changes of intracellular Cl− concentration. Int. J. Mol. Sci. 2019, 20, 1416. [Google Scholar] [CrossRef] [PubMed]
- Sibbe, M.; Kulik, A. GABAergic regulation of adult hippocampal neurogenesis. Mol. Neurobiol. 2017, 54, 5497–5510. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Jun, I.; Shin, D.H.; Yoon, J.G.; Piao, H.; Jung, J.; Park, H.W.; Cheng, M.H.; Bahar, I.; Whitcomb, D.C.; et al. Regulation of CFTR Bicarbonate Channel Activity by WNK1: Implications for Pancreatitis and CFTR-related disorders. Cell Mol. Gastroenterol. Hepatol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.Q.; Barberis, A.; Higley, M.J. Preserving the balance: Diverse forms of long-term GABAergic synaptic plasticity. Nat. Rev. Neurosci. 2019, 20, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Rivera, C.; Voipio, J.; Payne, J.A.; Ruusuvuori, E.; Lahtinen, H.; Lamsa, K.; Pirvola, U.; Saarma, M.; Kaila, K. The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 1999, 397, 251–255. [Google Scholar] [CrossRef]
- Medina, I.; Friedel, P.; Rivera, C.; Kahle, K.T.; Kourdougli, N.; Uvarov, P.; Pellegrino, C. Current view on the functional regulation of the neuronal K+-Cl− cotransporter KCC2. Front. Cell Neurosci. 2014, 8, 27. [Google Scholar] [CrossRef] [PubMed]
- Singh Jaggi, A.; Kaur, A.; Bali, A.; Singh, N. Expanding spectrum of sodium potassium chloride co-transporters in the pathophysiology of diseases. Curr. Neuropharmacol. 2015, 13, 369–388. [Google Scholar] [CrossRef]
- Smith, K.R.; Muir, J.; Rao, Y.; Browarski, M.; Gruenig, M.C.; Sheehan, D.F.; Haucke, V.; Kittler, J.T. Stabilization of GABAA receptors at endocytic zones is mediated by an AP2 binding motif within the GABAA receptor β3 subunit. J. Neurosci. 2012, 32, 2485–2498. [Google Scholar] [CrossRef]
- Plotkin, M.D.; Snyder, E.Y.; Hebert, S.C.; Delpire, E. Expression of the Na-K-2Cl cotransporter is developmentally regulated in postnatal rat brains: A possible mechanism underlying GABA′s excitatory role in immature brain. J. Neurobiol. 1997, 33, 781–795. [Google Scholar] [CrossRef]
- Yamada, J.; Okabe, A.; Toyoda, H.; Kilb, W.; Luhmann, H.J.; Fukuda, A. Cl− uptake promoting depolarizing GABA actions in immature rat neocortical neurones is mediated by NKCC1. J. Physiol. 2004, 557, 829–841. [Google Scholar] [CrossRef]
- Deng, X.; Dzamko, N.; Prescott, A.; Davies, P.; Liu, Q.; Yang, Q.; Lee, J.D.; Patricelli, M.P.; Nomanbhoy, T.K.; Alessi, D.R.; et al. Characterization of a selective inhibitor of the Parkinson’s disease kinase LRRK2. Nat. Chem. Biol. 2011, 7, 203–205. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Song, S.; Banerjee, S.; Jiang, T.; Zhang, J.; Kahle, K.T.; Sun, D.; Zhang, Z. The WNK-SPAK/OSR1 Kinases and the Cation-Chloride Cotransporters as Therapeutic Targets for Neurological Diseases. Aging Dis. 2019, 10, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Alessi, D.R.; Zhang, J.; Khanna, A.; Hochdorfer, T.; Shang, Y.; Kahle, K.T. The WNK-SPAK/OSR1 pathway: Master regulator of cation-chloride cotransporters. Sci. Signal. 2014, 7, re3. [Google Scholar] [CrossRef] [PubMed]
- Gamba, G. Molecular physiology and pathophysiology of electroneutral cation-chloride cotransporters. Physiol. Rev. 2005, 85, 423–493. [Google Scholar] [CrossRef] [PubMed]
- Orlov, S.N.; Koltsova, S.V.; Kapilevich, L.V.; Gusakova, S.V.; Dulin, N.O. NKCC1 and NKCC2: The pathogenetic role of cation-chloride cotransporters in hypertension. Genes Dis. 2015, 2, 186–196. [Google Scholar] [CrossRef]
- Nezu, A.; Parvin, M.N.; Turner, R.J. A conserved hydrophobic tetrad near the C terminus of the secretory Na+-K+-2Cl− cotransporter (NKCC1) is required for its correct intracellular processing. J. Biol. Chem. 2009, 284, 6869–6876. [Google Scholar] [CrossRef]
- Chew, T.A.; Orlando, B.J.; Zhang, J.; Latorraca, N.R.; Wang, A.; Hollingsworth, S.A.; Chen, D.-H.; Dror, R.O.; Liao, M.; Feng, L. Structure and mechanism of the cation–chloride cotransporter NKCC1. Nature 2019, 572, 488–492. [Google Scholar] [CrossRef]
- Hsu, Y.T.; Chang, Y.G.; Liu, Y.C.; Wang, K.Y.; Chen, H.M.; Lee, D.J.; Yang, S.S.; Tsai, C.H.; Lien, C.C.; Chern, Y. Enhanced Na+-K+-2Cl− 1 underlies motor dysfunction in huntington’s disease. Mov. Disord. 2019, 34, 845–857. [Google Scholar] [CrossRef]
- Dargaei, Z.; Bang, J.Y.; Mahadevan, V.; Khademullah, C.S.; Bedard, S.; Parfitt, G.M.; Kim, J.C.; Woodin, M.A. Restoring GABAergic inhibition rescues memory deficits in a Huntington’s disease mouse model. Proc. Natl. Acad. Sci. USA 2018, 115, E1618–E1626. [Google Scholar] [CrossRef]
- Tang, B.L. The Expanding Therapeutic Potential of Neuronal KCC2. Cells 2020, 9, 240. [Google Scholar] [CrossRef]
- Moore, Y.E.; Kelley, M.R.; Brandon, N.J.; Deeb, T.Z.; Moss, S.J. Seizing Control of KCC2: A New Therapeutic Target for Epilepsy. Trends Neurosci. 2017, 40, 555–571. [Google Scholar] [CrossRef] [PubMed]
- Kahle, K.T.; Khanna, A.R.; Alper, S.L.; Adragna, N.C.; Lauf, P.K.; Sun, D.; Delpire, E. K-Cl cotransporters, cell volume homeostasis, and neurological disease. Trends Mol. Med. 2015, 21, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Tillman, L.; Zhang, J. Crossing the Chloride Channel: The Current and Potential Therapeutic Value of the Neuronal K+–Cl− Cotransporter KCC2. Biomed. Res. Int. 2019, 2019, 8941046. [Google Scholar] [CrossRef] [PubMed]
- de Los Heros, P.; Alessi, D.R.; Gourlay, R.; Campbell, D.G.; Deak, M.; Macartney, T.J.; Kahle, K.T.; Zhang, J. The WNK-regulated SPAK/OSR1 kinases directly phosphorylate and inhibit the K+–Cl− co-transporters. Biochem. J. 2014, 458, 559–573. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.C.-Y.; Hannemann, A.; Wadud, R.; Rees, D.C.; Brewin, J.N.; Low, P.S.; Gibson, J.S. The role of WNK in modulation of KCl cotransport activity in red cells from normal individuals and patients with sickle cell anaemia. Pflug. Arch. Eur. J. Phy. 2019, 471, 1539–1549. [Google Scholar] [CrossRef] [PubMed]
- Shekarabi, M.; Zhang, J.; Khanna, A.R.; Ellison, D.H.; Delpire, E.; Kahle, K.T. WNK Kinase Signaling in Ion Homeostasis and Human Disease. Cell Metab. 2017, 25, 285–299. [Google Scholar] [CrossRef]
- Heubl, M.; Zhang, J.; Pressey, J.C.; Al Awabdh, S.; Renner, M.; Gomez-Castro, F.; Moutkine, I.; Eugene, E.; Russeau, M.; Kahle, K.T.; et al. GABAA receptor dependent synaptic inhibition rapidly tunes KCC2 activity via the Cl−-sensitive WNK1 kinase. Nat. Commun. 2017, 8, 1776. [Google Scholar] [CrossRef]
- Brown, A.; Meor Azlan, N.F.; Wu, Z.; Zhang, J. WNK-SPAK/OSR1-NCC kinase signaling pathway as a novel target for the treatment of salt-sensitive hypertension. Acta Pharm. Sin. 2020. [Google Scholar] [CrossRef]
- Meor Azlan, N.F.; Zhang, J. Role of the Cation-chloride-cotransporters in Cardiovascular Disease. Cells 2020, 9, 2293. [Google Scholar] [CrossRef]
- Kahle, K.T.; Khanna, A.R.; Duan, J.; Staley, K.J.; Delpire, E.; Poduri, A. The KCC2 Cotransporter and Human Epilepsy: Getting Excited About Inhibition. Neuroscientist 2016, 22, 555–562. [Google Scholar] [CrossRef]
- Yeo, M.; Berglund, K.; Augustine, G.; Liedtke, W. Novel repression of Kcc2 transcription by REST-RE-1 controls developmental switch in neuronal chloride. J. Neurosci. 2009, 29, 14652–14662. [Google Scholar] [CrossRef]
- Smith-Dijak, A.I.; Sepers, M.D.; Raymond, L.A. Alterations in synaptic function and plasticity in Huntington disease. J. Neurochem. 2019, 150, 346–365. [Google Scholar] [CrossRef] [PubMed]
- Rinehart, J.; Maksimova, Y.D.; Tanis, J.E.; Stone, K.L.; Hodson, C.A.; Zhang, J.; Risinger, M.; Pan, W.; Wu, D.; Colangelo, C.M.; et al. Sites of regulated phosphorylation that control K-Cl cotransporter activity. Cell 2009, 138, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Friedel, P.; Kahle, K.T.; Zhang, J.; Hertz, N.; Pisella, L.I.; Buhler, E.; Schaller, F.; Duan, J.; Khanna, A.R.; Bishop, P.N.; et al. WNK1-regulated inhibitory phosphorylation of the KCC2 cotransporter maintains the depolarizing action of GABA in immature neurons. Sci. Signal. 2015, 8, ra65. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Zhang, J.; Mansuri, M.S.; Duan, J.; Karimy, J.K.; Delpire, E.; Alper, S.L.; Lifton, R.P.; Fukuda, A.; Kahle, K.T. Developmentally regulated KCC2 phosphorylation is essential for dynamic GABA-mediated inhibition and survival. Sci. Signal. 2019, 12, eaaw9315. [Google Scholar] [CrossRef] [PubMed]
- Moore, Y.E.; Conway, L.C.; Wobst, H.J.; Brandon, N.J.; Deeb, T.Z.; Moss, S.J. Developmental Regulation of KCC2 Phosphorylation Has Long-Term Impacts on Cognitive Function. Front. Mol. Neurosci. 2019, 12, 173. [Google Scholar] [CrossRef]
- Lee, H.H.; Deeb, T.Z.; Walker, J.A.; Davies, P.A.; Moss, S.J. NMDA receptor activity downregulates KCC2 resulting in depolarizing GABAA receptor-mediated currents. Nat. Neurosci. 2011, 14, 736–743. [Google Scholar] [CrossRef]
- Lee, H.H.; Walker, J.A.; Williams, J.R.; Goodier, R.J.; Payne, J.A.; Moss, S.J. Direct protein kinase C-dependent phosphorylation regulates the cell surface stability and activity of the potassium chloride cotransporter KCC2. J. Biol. Chem. 2007, 282, 29777–29784. [Google Scholar] [CrossRef]
- Zhang, S.; Hennessey, T.; Yang, L.; Starkova, N.; Beal, M.; Starkov, A. Impaired brain creatine kinase activity in Huntington’s disease. Neurodegener. Dis. 2011, 8, 194–201. [Google Scholar] [CrossRef]
- Inoue, K.; Ueno, S.; Fukuda, A. Interaction of neuron-specific K+–Cl− cotransporter, KCC2, with brain-type creatine kinase. FEBS Lett. 2004, 564, 131–135. [Google Scholar] [CrossRef]
- Hemmer, W.; Furter-Graves, E.M.; Frank, G.; Wallimann, T.; Furter, R. Autophosphorylation of creatine kinase: Characterization and identification of a specifically phosphorylated peptide. Biochim. Et Biophys. Acta-Protein Struct. Mol. Enzymol. 1995, 1251, 81–90. [Google Scholar] [CrossRef]
- Inoue, K.; Yamada, J.; Ueno, S.; Fukuda, A. Brain-type creatine kinase activates neuron-specific K+-Cl− co-transporter KCC2. J. Neurochem. 2006, 96, 598–608. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-S.; Chen, C.-M.; Soong, B.-w.; Wu, Y.-R.; Chen, H.-M.; Yeh, W.-Y.; Wu, D.-R.; Lin, Y.-J.; Poon, P.W.-F.; Cheng, M.-L. Dysregulated brain creatine kinase is associated with hearing impairment in mouse models of Huntington disease. J. Clin. Investig. 2011, 121, 1519–1523. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.S.; Cheng, T.H.; Chang, C.P.; Chen, H.M.; Chern, Y. Enhancement of brain-type creatine kinase activity ameliorates neuronal deficits in Huntington’s disease. Biochim. Biophys. Acta 2013, 1832, 742–753. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.T.; Chang, Y.G.; Chang, C.P.; Siew, J.J.; Chen, H.M.; Tsai, C.H.; Chern, Y. Altered behavioral responses to gamma—Aminobutyric acid pharmacological agents in a mouse model of Huntington’s disease. Mov. Disord. 2017, 32, 1600–1609. [Google Scholar] [CrossRef] [PubMed]
- Hinz, L.; Barrufet, J.T.; Heine, V.M. KCC2 expression levels are reduced in post mortem brain tissue of Rett syndrome patients. Acta Neuropathol. Commun. 2019, 7, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Bhuiyan, M.I.H.; Zhang, T.; Karimy, J.K.; Wu, Z.; Fiesler, V.M.; Zhang, J.; Huang, H.; Hasan, M.N.; Skrzypiec, A.E.; et al. Modulation of brain cation-Cl− cotransport via the SPAK kinase inhibitor ZT-1a. Nat. Commun. 2020, 11, 78. [Google Scholar] [CrossRef]
- Schmidt, T.; Ghaffarian, N.; Philippot, C.; Seifert, G.; Steinhauser, C.; Pape, H.C.; Blaesse, P. Differential regulation of chloride homeostasis and GABAergic transmission in the thalamus. Sci. Rep. 2018, 8, 13929. [Google Scholar] [CrossRef]
- Benraiss, A.; Wang, S.; Herrlinger, S.; Li, X.; Chandler-Militello, D.; Mauceri, J.; Burm, H.B.; Toner, M.; Osipovitch, M.; Xu, Q.J. Human glia can both induce and rescue aspects of disease phenotype in Huntington disease. Nat. Commun. 2016, 7, 1–13. [Google Scholar] [CrossRef]
- Dunah, A.W.; Jeong, H.; Griffin, A.; Kim, Y.-M.; Standaert, D.G.; Hersch, S.M.; Mouradian, M.M.; Young, A.B.; Tanese, N.; Krainc, D. Sp1 and TAFII130 Transcriptional Activity Disrupted in Early Huntington’s Disease. Science 2002, 296, 2238–2243. [Google Scholar] [CrossRef]
- Zuccato, C.; Tartari, M.; Crotti, A.; Goffredo, D.; Valenza, M.; Conti, L.; Cataudella, T.; Leavitt, B.R.; Hayden, M.R.; Timmusk, T.; et al. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat. Genet. 2003, 35, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, G.D.; Gokhan, S.; Molero, A.E.; Mehler, M.F. Selective roles of normal and mutant huntingtin in neural induction and early neurogenesis. PLoS ONE 2013, 8, e64368. [Google Scholar] [CrossRef] [PubMed]
- Uvarov, P.; Ludwig, A.; Markkanen, M.; Rivera, C.; Airaksinen, M.S. Upregulation of the neuron-specific K+/Cl− cotransporter expression by transcription factor early growth response 4. J. Neurosci. 2006, 26, 13463–13473. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Uvarov, P. Neuronal K-Cl Cotransporter: Transcriptional Mechanisms of KCC2 Gene Regulation. Helsingin yliopisto. Ph.D. Thesis, University of Helsinki, Helsinki, Finland, 2010. Available online: http://urn.fi/URN:ISBN:978-952-10-6300-8.
- Ludwig, A.; Uvarov, P.; Soni, S.; Thomas-Crusells, J.; Airaksinen, M.S.; Rivera, C. Early growth response 4 mediates BDNF induction of potassium chloride cotransporter 2 transcription. J. Neurosci. 2011, 31, 644–649. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, X.; Wang, W.; Lu, Y.-G.; Pan, Z.Z. Brain-Derived Neurotrophic Factor–Mediated Downregulation of Brainstem K+–Cl− Cotransporter and Cell-Type–Specific GABA Impairment for Activation of Descending Pain Facilitation. Mol. Pharmacol. 2013, 84, 511–520. [Google Scholar] [CrossRef]
- Rivera, C.; Li, H.; Thomas-Crusells, J.; Lahtinen, H.; Viitanen, T.; Nanobashvili, A.; Kokaia, Z.; Airaksinen, M.S.; Voipio, J.; Kaila, K.; et al. BDNF-induced TrkB activation down-regulates the K+–Cl− cotransporter KCC2 and impairs neuronal Cl− extrusion. J. Cell Biol. 2002, 159, 747–752. [Google Scholar] [CrossRef]
- Wardle, R.A.; Poo, M.-m. Brain-derived neurotrophic factor modulation of GABAergic synapses by postsynaptic regulation of chloride transport. J. Neurosci. 2003, 23, 8722–8732. [Google Scholar] [CrossRef]
- Coull, J.A.; Beggs, S.; Boudreau, D.; Boivin, D.; Tsuda, M.; Inoue, K.; Gravel, C.; Salter, M.W.; De Koninck, Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 2005, 438, 1017–1021. [Google Scholar] [CrossRef]
- Miletic, G.; Miletic, V. Loose ligation of the sciatic nerve is associated with TrkB receptor-dependent decreases in KCC2 protein levels in the ipsilateral spinal dorsal horn. Pain 2008, 137, 532–539. [Google Scholar] [CrossRef][Green Version]
- Zhang, W.; Liu, L.-Y.; Xu, T.-L. Reduced potassium-chloride co-transporter expression in spinal cord dorsal horn neurons contributes to inflammatory pain hypersensitivity in rats. Neuroscience 2008, 152, 502–510. [Google Scholar] [CrossRef]
- Ferrini, F.; Trang, T.; Mattioli, T.-A.M.; Laffray, S.; Del′Guidice, T.; Lorenzo, L.-E.; Castonguay, A.; Doyon, N.; Zhang, W.; Godin, A.G. Morphine hyperalgesia gated through microglia-mediated disruption of neuronal Cl− homeostasis. Nat. Neurosci. 2013, 16, 183–192. [Google Scholar] [CrossRef] [PubMed]
- McKinstry, S.U.; Karadeniz, Y.B.; Worthington, A.K.; Hayrapetyan, V.Y.; Ozlu, M.I.; Serafin-Molina, K.; Risher, W.C.; Ustunkaya, T.; Dragatsis, I.; Zeitlin, S.; et al. Huntingtin is required for normal excitatory synapse development in cortical and striatal circuits. J. Neurosci. 2014, 34, 9455–9472. [Google Scholar] [CrossRef] [PubMed]
- Li, S.H.; Li, X.J. Huntingtin-protein interactions and the pathogenesis of Huntington’s disease. Trends Genet. 2004, 20, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Lee, C.J.; Schroeder, A.; Kim, Y.S.; Jung, S.H.; Kim, J.S.; Kim, D.Y.; Son, E.J.; Han, H.C.; Hong, S.K.; et al. Excitatory actions of GABA in the suprachiasmatic nucleus. J. Neurosci. 2008, 28, 5450–5459. [Google Scholar] [CrossRef]
- Gauthier, L.R.; Charrin, B.C.; Borrell-Pages, M.; Dompierre, J.P.; Rangone, H.; Cordelieres, F.P.; De Mey, J.; MacDonald, M.E.; Lessmann, V.; Humbert, S.; et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 2004, 118, 127–138. [Google Scholar] [CrossRef]
- Farajnia, S.; van Westering, T.L.; Meijer, J.H.; Michel, S. Seasonal induction of GABAergic excitation in the central mammalian clock. Proc. Natl. Acad. Sci. USA 2014, 111, 9627–9632. [Google Scholar] [CrossRef]
- Morton, A.J.; Wood, N.I.; Hastings, M.H.; Hurelbrink, C.; Barker, R.A.; Maywood, E.S. Disintegration of the sleep-wake cycle and circadian timing in Huntington’s disease. J. Neurosci. 2005, 25, 157–163. [Google Scholar] [CrossRef]
- Morton, A.J.; Rudiger, S.R.; Wood, N.I.; Sawiak, S.J.; Brown, G.C.; McLaughlan, C.J.; Kuchel, T.R.; Snell, R.G.; Faull, R.L.; Bawden, C.S. Early and progressive circadian abnormalities in Huntington’s disease sheep are unmasked by social environment. Hum. Mol. Genet. 2014, 23, 3375–3383. [Google Scholar] [CrossRef]
- Kreutzmann, J.C.; Havekes, R.; Abel, T.; Meerlo, P. Sleep deprivation and hippocampal vulnerability: Changes in neuronal plasticity, neurogenesis and cognitive function. Neuroscience 2015, 309, 173–190. [Google Scholar] [CrossRef]
- Kim, D.J.; Lee, H.P.; Kim, M.S.; Park, Y.J.; Go, H.J.; Kim, K.S.; Lee, S.P.; Chae, J.H.; Lee, C.T. The effect of total sleep deprivation on cognitive functions in normal adult male subjects. Int. J. Neurosci. 2001, 109, 127–137. [Google Scholar] [CrossRef]
- Wichniak, A.; Wierzbicka, A.; Walecka, M.; Jernajczyk, W. Effects of Antidepressants on Sleep. Curr. Psychiatry Rep. 2017, 19, 63. [Google Scholar] [CrossRef] [PubMed]
- Lazar, A.S.; Panin, F.; Goodman, A.O.; Lazic, S.E.; Lazar, Z.I.; Mason, S.L.; Rogers, L.; Murgatroyd, P.R.; Watson, L.P.; Singh, P.; et al. Sleep deficits but no metabolic deficits in premanifest Huntington’s disease. Ann. Neurol. 2015, 78, 630–648. [Google Scholar] [CrossRef]
- Diago, E.B.; Martinez-Horta, S.; Lasaosa, S.S.; Alebesque, A.V.; Perez-Perez, J.; Kulisevsky, J.; Del Val, J.L. Circadian Rhythm, Cognition, and Mood Disorders in Huntington′s Disease. J. Huntingt. Dis. 2018, 7, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Piano, C.; Della Marca, G.; Losurdo, A.; Imperatori, C.; Solito, M.; Calandra-Buonaura, G.; Provini, F.; Cortelli, P.; Bentivoglio, A.R. Subjective Assessment of Sleep in Huntington Disease: Reliability of Sleep Questionnaires Compared to Polysomnography. Neurodegener. Dis. 2017, 17, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Jha, M.; Kamble, N.; Lenka, A.; Yadav, R.; Purushottam, M.; Jain, S.; Pal, P. Sleep disturbances in patients with Huntington’s disease: A questionnaire-based study. Ann. Mov. Disord. 2019, 2, 9–14. [Google Scholar] [CrossRef]
- Solomon, A.C.; Stout, J.C.; Weaver, M.; Queller, S.; Tomusk, A.; Whitlock, K.B.; Hui, S.L.; Marshall, J.; Jackson, J.G.; Siemers, E.R.; et al. Ten-year rate of longitudinal change in neurocognitive and motor function in prediagnosis Huntington disease. Mov. Disord. 2008, 23, 1830–1836. [Google Scholar] [CrossRef] [PubMed]
- Rupp, J.; Blekher, T.; Jackson, J.; Beristain, X.; Marshall, J.; Hui, S.; Wojcieszek, J.; Foroud, T. Progression in prediagnostic Huntington disease. J. Neurol. Neurosurg. Psychiatry 2010, 81, 379–384. [Google Scholar] [CrossRef][Green Version]
- Paulsen, J.S. Early Detection of Huntington Disease. Future Neurol. 2010, 5. [Google Scholar] [CrossRef]
- Townhill, J.; Hughes, A.C.; Thomas, B.; Busse, M.E.; Price, K.; Dunnett, S.B.; Hastings, M.H.; Rosser, A.E. Using Actiwatch to monitor circadian rhythm disturbance in Huntington’ disease: A cautionary note. J. Neurosci. Methods 2016, 265, 13–18. [Google Scholar] [CrossRef]
- Anderson, K.E.; van Duijn, E.; Craufurd, D.; Drazinic, C.; Edmondson, M.; Goodman, N.; van Kammen, D.; Loy, C.; Priller, J.; Goodman, L.V. Clinical Management of Neuropsychiatric Symptoms of Huntington Disease: Expert-Based Consensus Guidelines on Agitation, Anxiety, Apathy, Psychosis and Sleep Disorders. J. Huntingt. Dis. 2018, 7, 355–366. [Google Scholar] [CrossRef]
- Franzen, P.L.; Buysse, D.J. Sleep disturbances and depression: Risk relationships for subsequent depression and therapeutic implications. Dialogues Clin. Neurosci. 2008, 10, 473–481. [Google Scholar] [PubMed]
- Aziz, N.A.; Anguelova, G.V.; Marinus, J.; Lammers, G.J.; Roos, R.A. Sleep and circadian rhythm alterations correlate with depression and cognitive impairment in Huntington’s disease. Parkinsonism Relat. Disord. 2010, 16, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Pallier, P.N.; Morton, A.J. Management of sleep/wake cycles improves cognitive function in a transgenic mouse model of Huntington’s disease. Brain Res. 2009, 1279, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Raymer, B.K. Sleep modulating agents. Bioorg. Med. Chem. Lett. 2019, 29, 2025–2033. [Google Scholar] [CrossRef]
- Politis, M.; Pavese, N.; Tai, Y.F.; Tabrizi, S.J.; Barker, R.A.; Piccini, P. Hypothalamic involvement in Huntington’s disease: An in vivo PET study. Brain 2008, 131, 2860–2869. [Google Scholar] [CrossRef]
- Kassubek, J.; Juengling, F.D.; Kioschies, T.; Henkel, K.; Karitzky, J.; Kramer, B.; Ecker, D.; Andrich, J.; Saft, C.; Kraus, P.; et al. Topography of cerebral atrophy in early Huntington’s disease: A voxel based morphometric MRI study. J. Neurol. Neurosurg. Psychiatry 2004, 75, 213–220. [Google Scholar]
- Douaud, G.; Gaura, V.; Ribeiro, M.J.; Lethimonnier, F.; Maroy, R.; Verny, C.; Krystkowiak, P.; Damier, P.; Bachoud-Levi, A.C.; Hantraye, P.; et al. Distribution of grey matter atrophy in Huntington’s disease patients: A combined ROI-based and voxel-based morphometric study. Neuroimage 2006, 32, 1562–1575. [Google Scholar] [CrossRef]
- Aziz, N.A.; Anguelova, G.V.; Marinus, J.; van Dijk, J.G.; Roos, R.A. Autonomic symptoms in patients and pre-manifest mutation carriers of Huntington’s disease. Eur. J. Neurol. 2010, 17, 1068–1074. [Google Scholar] [CrossRef]
- Ono, D.; Honma, K.I.; Yanagawa, Y.; Yamanaka, A.; Honma, S. Role of GABA in the regulation of the central circadian clock of the suprachiasmatic nucleus. J. Physiol. Sci. 2018, 68, 333–343. [Google Scholar] [CrossRef]
- McNeill, J.K.t.; Walton, J.C.; Albers, H.E. Functional Significance of the Excitatory Effects of GABA in the Suprachiasmatic Nucleus. J. Biol. Rhythm. 2018, 33, 376–387. [Google Scholar] [CrossRef]
- Rohr, K.E.; Pancholi, H.; Haider, S.; Karow, C.; Modert, D.; Raddatz, N.J.; Evans, J. Seasonal plasticity in GABAA signaling is necessary for restoring phase synchrony in the master circadian clock network. eLife 2019, 8. [Google Scholar] [CrossRef]
- Olde Engberink, A.H.O.; Meijer, J.H.; Michel, S. Chloride cotransporter KCC2 is essential for GABAergic inhibition in the SCN. Neuropharmacology 2018, 138, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Garbazza, C.; Benedetti, F. Genetic Factors Affecting Seasonality, Mood, and the Circadian Clock. Front. Endocrinol. (Lausanne) 2018, 9, 481. [Google Scholar] [CrossRef] [PubMed]
- Albers, H.E.; Walton, J.C.; Gamble, K.L.; McNeill, J.K.t.; Hummer, D.L. The dynamics of GABA signaling: Revelations from the circadian pacemaker in the suprachiasmatic nucleus. Front. Neuroendocr. 2017, 44, 35–82. [Google Scholar] [CrossRef]
- DeWoskin, D.; Myung, J.; Belle, M.D.; Piggins, H.D.; Takumi, T.; Forger, D.B. Distinct roles for GABA across multiple timescales in mammalian circadian timekeeping. Proc. Natl. Acad. Sci. USA 2015, 112, E3911–3919. [Google Scholar] [CrossRef]
- Belenky, M.A.; Yarom, Y.; Pickard, G.E. Heterogeneous expression of gamma-aminobutyric acid and gamma-aminobutyric acid-associated receptors and transporters in the rat suprachiasmatic nucleus. J. Comp. Neurol. 2008, 506, 708–732. [Google Scholar] [CrossRef]
- Song, S.; Luo, L.; Sun, B.; Sun, D. Roles of glial ion transporters in brain diseases. Glia 2020, 68, 472–494. [Google Scholar] [CrossRef]
- Mintz, E.M.; Jasnow, A.M.; Gillespie, C.F.; Huhman, K.L.; Albers, H.E. GABA interacts with photic signaling in the suprachiasmatic nucleus to regulate circadian phase shifts. Neuroscience 2002, 109, 773–778. [Google Scholar] [CrossRef]
- Kaila, K.; Price, T.J.; Payne, J.A.; Puskarjov, M.; Voipio, J. Cation-chloride cotransporters in neuronal development, plasticity and disease. Nat. Rev. Neurosci. 2014, 15, 637–654. [Google Scholar] [CrossRef]
- Jeu, M.D.; Pennartz, C. Circadian Modulation of GABA Function in the Rat Suprachiasmatic Nucleus: Excitatory Effects During the Night Phase. J. Neurophysiol. 2002, 87, 834–844. [Google Scholar] [CrossRef][Green Version]
- Belenky, M.A.; Sollars, P.J.; Mount, D.B.; Alper, S.L.; Yarom, Y.; Pickard, G.E. Cell-type specific distribution of chloride transporters in the rat suprachiasmatic nucleus. Neuroscience 2010, 165, 1519–1537. [Google Scholar] [CrossRef] [PubMed]
- Klett, N.J.; Allen, C.N. Intracellular Chloride Regulation in AVP+ and VIP+ Neurons of the Suprachiasmatic Nucleus. Sci. Rep. 2017, 7, 10226. [Google Scholar] [CrossRef] [PubMed]
- Kahle, K.T.; Delpire, E. Kinase-KCC2 coupling: Cl− rheostasis, disease susceptibility, therapeutic target. J. Neurophysiol. 2016, 115, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Myung, J.; Hong, S.; DeWoskin, D.; De Schutter, E.; Forger, D.B.; Takumi, T. GABA-mediated repulsive coupling between circadian clock neurons in the SCN encodes seasonal time. Proc. Natl. Acad. Sci. USA 2015, 112, E3920–3929. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Park, H.M.; Rigel, D.F.; DiPetrillo, K.; Whalen, E.J.; Anisowicz, A.; Beil, M.; Berstler, J.; Brocklehurst, C.E.; Burdick, D.A.; et al. Small-molecule WNK inhibition regulates cardiovascular and renal function. Nat. Chem. Biol. 2016, 12, 896–898. [Google Scholar] [CrossRef] [PubMed]
- Apsel, B.; Blair, J.A.; Gonzalez, B.; Nazif, T.M.; Feldman, M.E.; Aizenstein, B.; Hoffman, R.; Williams, R.L.; Shokat, K.M.; Knight, Z.A. Targeted polypharmacology: Discovery of dual inhibitors of tyrosine and phosphoinositide kinases. Nat. Chem. Biol. 2008, 4, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, E.; Mori, T.; Zeniya, M.; Isobe, K.; Ishigami-Yuasa, M.; Fujii, S.; Kagechika, H.; Ishihara, T.; Mizushima, T.; Sasaki, S.; et al. Discovery of Novel SPAK Inhibitors That Block WNK Kinase Signaling to Cation Chloride Transporters. J. Am. Soc. Nephrol. 2015, 26, 1525–1536. [Google Scholar] [CrossRef]
- AlAmri, M.A.; Kadri, H.; Alderwick, L.J.; Simpkins, N.S.; Mehellou, Y. Rafoxanide and Closantel Inhibit SPAK and OSR1 Kinases by Binding to a Highly Conserved Allosteric Site on Their C-terminal Domains. ChemMedChem 2017, 12, 639–645. [Google Scholar] [CrossRef]
- AlAmri, M.A.; Kadri, H.; Alderwick, L.J.; Jeeves, M.; Mehellou, Y. The Photosensitising Clinical Agent Verteporfin Is an Inhibitor of SPAK and OSR1 Kinases. Chembiochem 2018, 19, 2072–2080. [Google Scholar] [CrossRef]
- Mori, T.; Kikuchi, E.; Watanabe, Y.; Fujii, S.; Ishigami-Yuasa, M.; Kagechika, H.; Sohara, E.; Rai, T.; Sasaki, S.; Uchida, S. Chemical library screening for WNK signalling inhibitors using fluorescence correlation spectroscopy. Biochem. J. 2013, 455, 339–345. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andrews, K.; Josiah, S.S.; Zhang, J. The Therapeutic Potential of Neuronal K-Cl Co-Transporter KCC2 in Huntington’s Disease and Its Comorbidities. Int. J. Mol. Sci. 2020, 21, 9142. https://doi.org/10.3390/ijms21239142
Andrews K, Josiah SS, Zhang J. The Therapeutic Potential of Neuronal K-Cl Co-Transporter KCC2 in Huntington’s Disease and Its Comorbidities. International Journal of Molecular Sciences. 2020; 21(23):9142. https://doi.org/10.3390/ijms21239142
Chicago/Turabian StyleAndrews, Katie, Sunday Solomon Josiah, and Jinwei Zhang. 2020. "The Therapeutic Potential of Neuronal K-Cl Co-Transporter KCC2 in Huntington’s Disease and Its Comorbidities" International Journal of Molecular Sciences 21, no. 23: 9142. https://doi.org/10.3390/ijms21239142
APA StyleAndrews, K., Josiah, S. S., & Zhang, J. (2020). The Therapeutic Potential of Neuronal K-Cl Co-Transporter KCC2 in Huntington’s Disease and Its Comorbidities. International Journal of Molecular Sciences, 21(23), 9142. https://doi.org/10.3390/ijms21239142