Synthesis and in Silico Modelling of the Potential Dual Mechanistic Activity of Small Cationic Peptides Potentiating the Antibiotic Novobiocin against Susceptible and Multi-Drug Resistant Escherichia coli


 and
and
Abstract
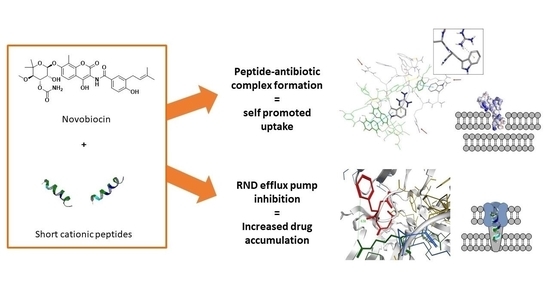
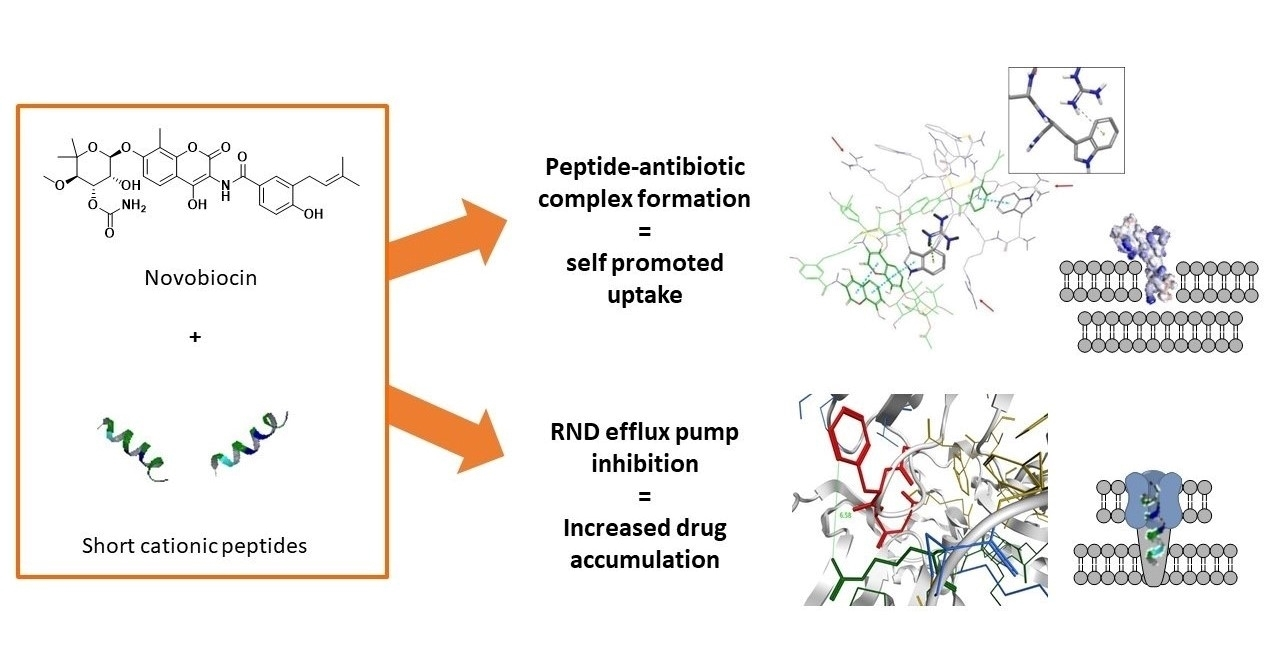
1. Introduction
2. Results and Discussion
2.1. Design and Synthesis of Peptides
2.2. Antimicrobial and Potentiating Properties
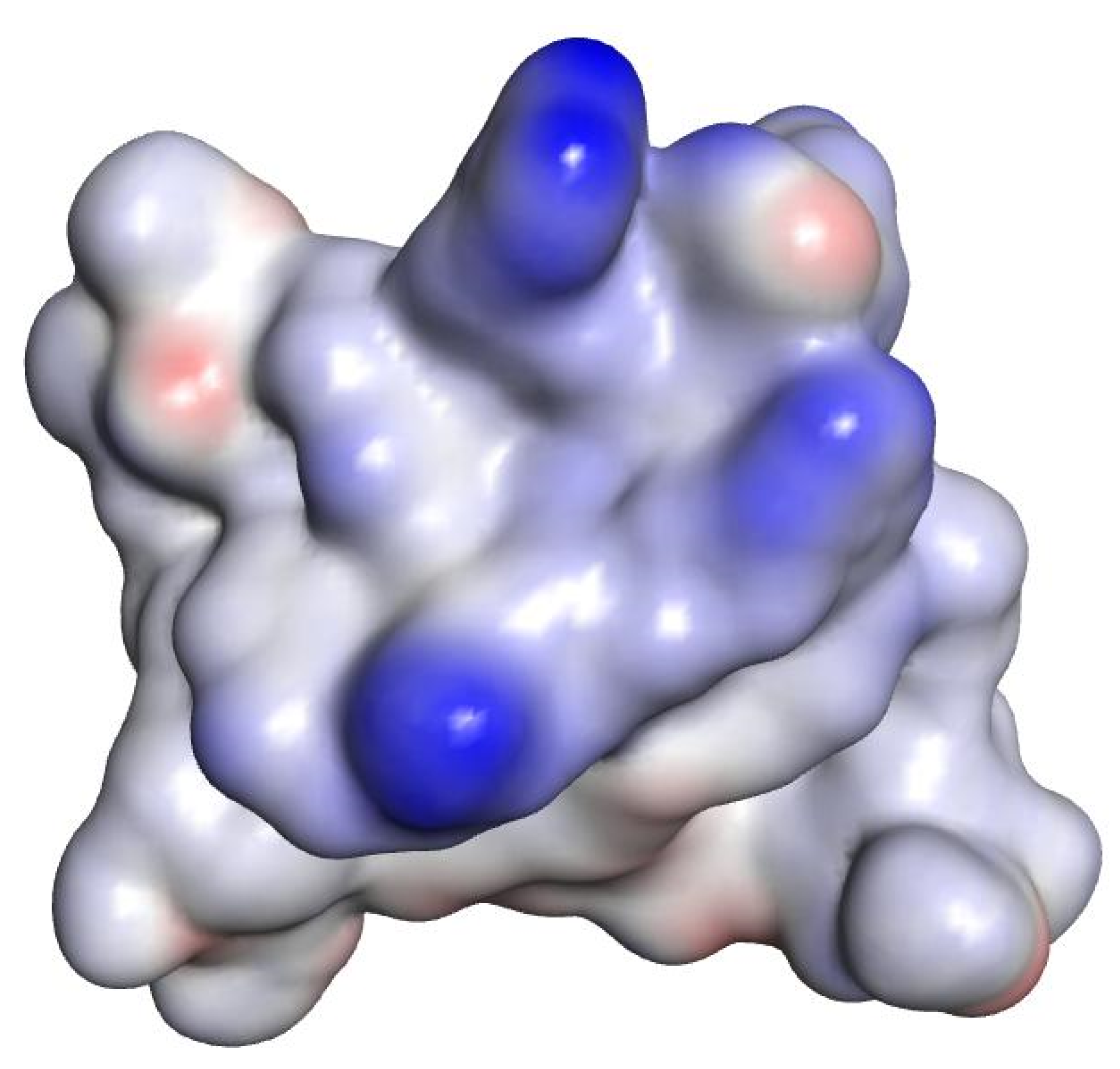
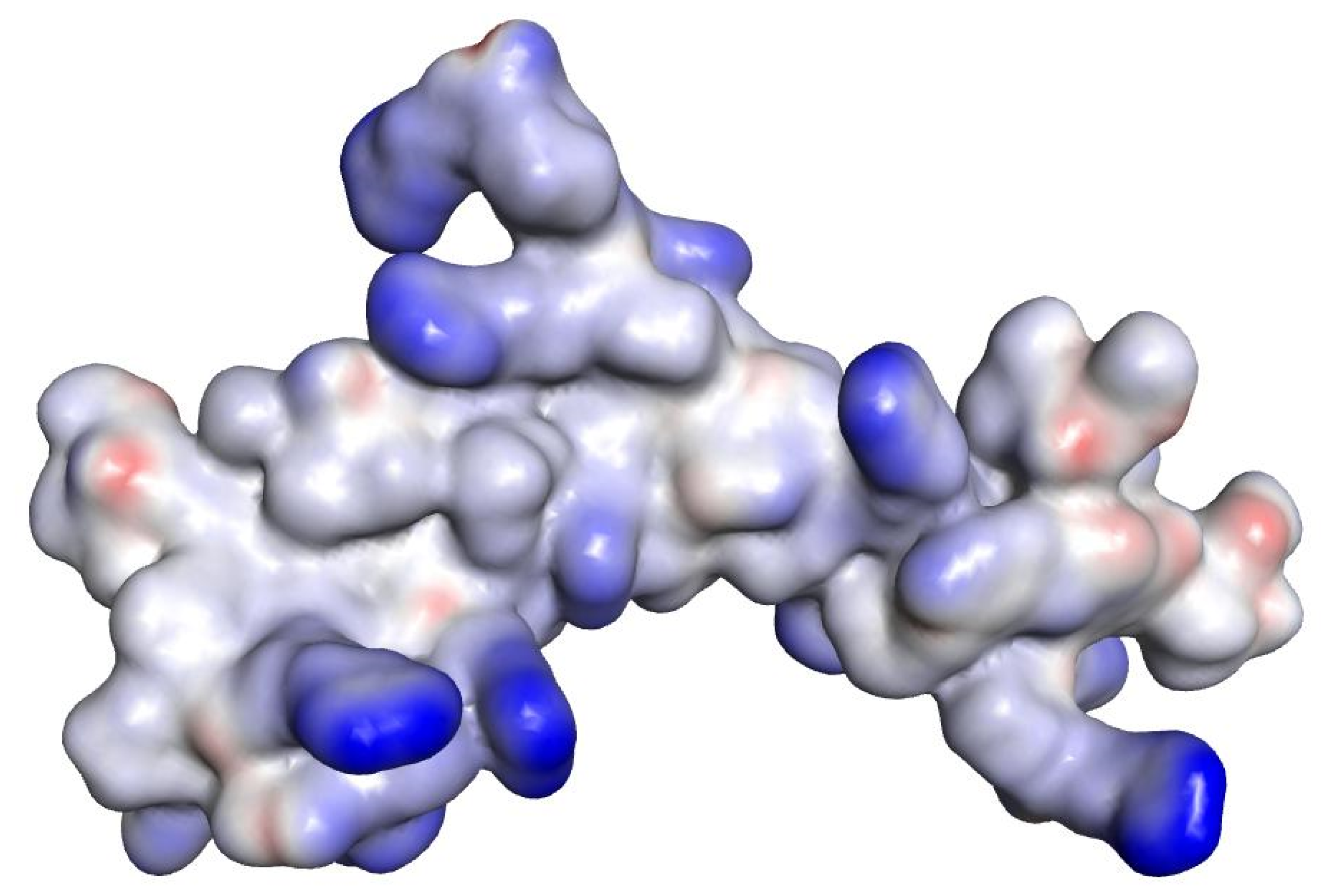
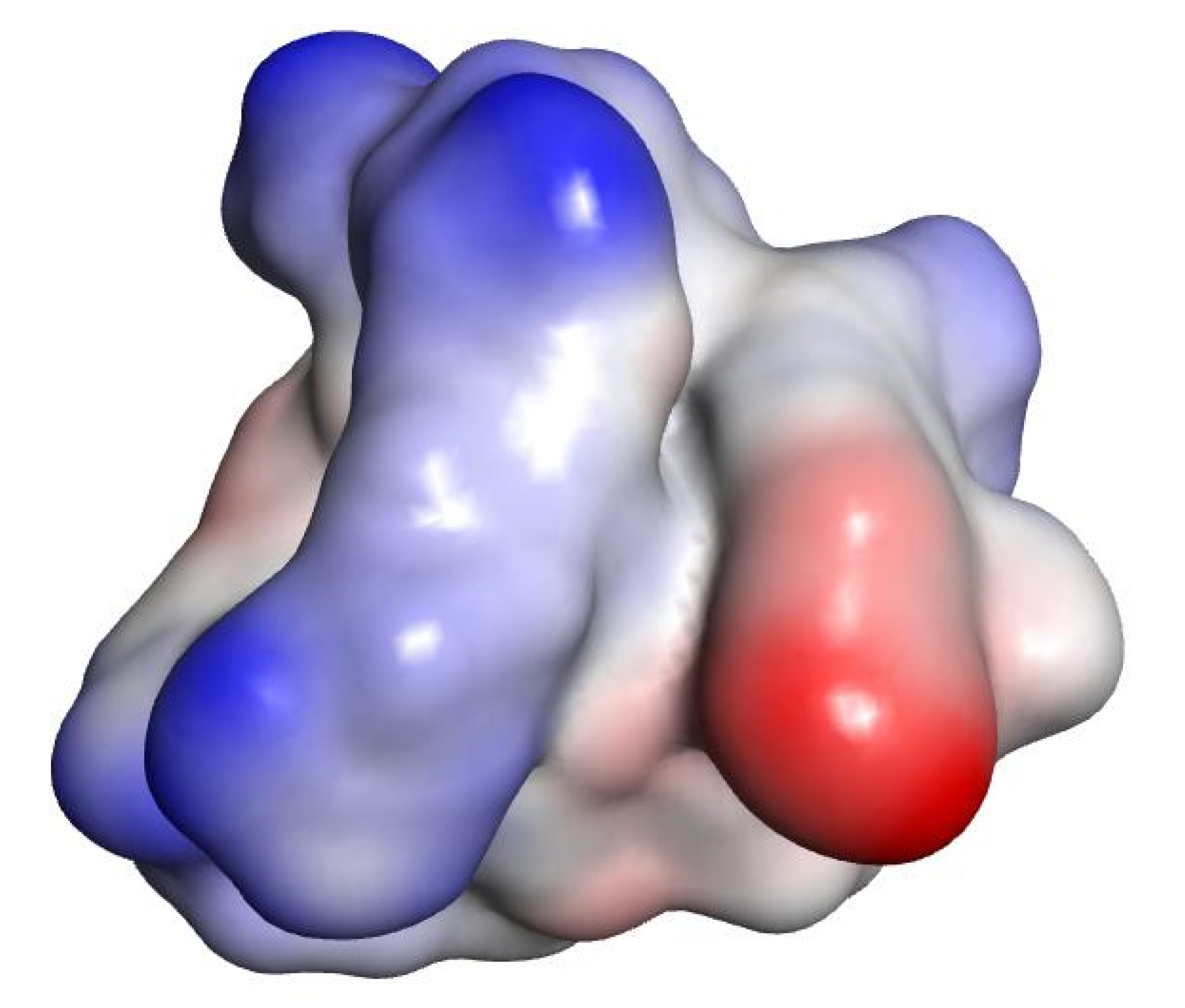
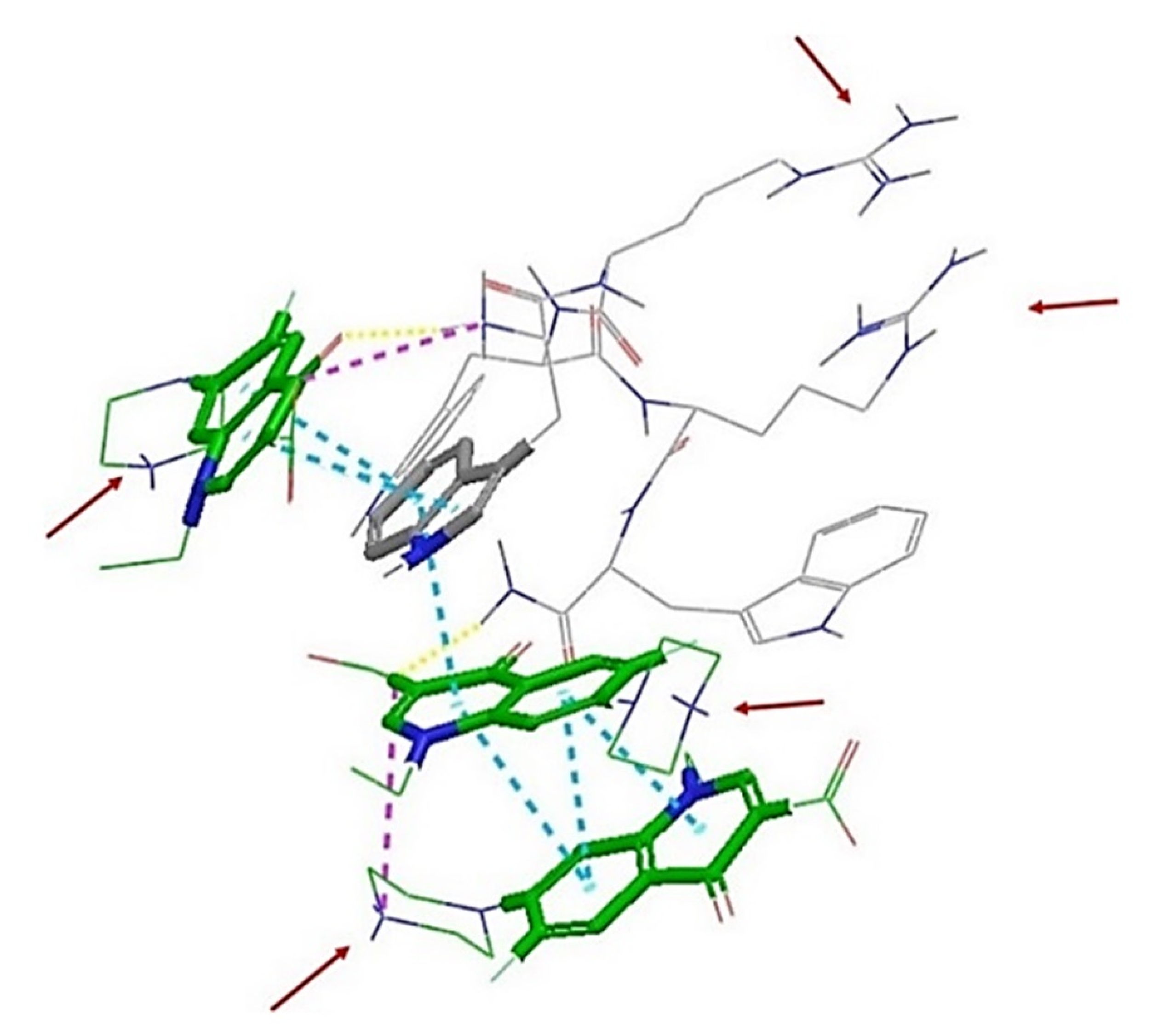
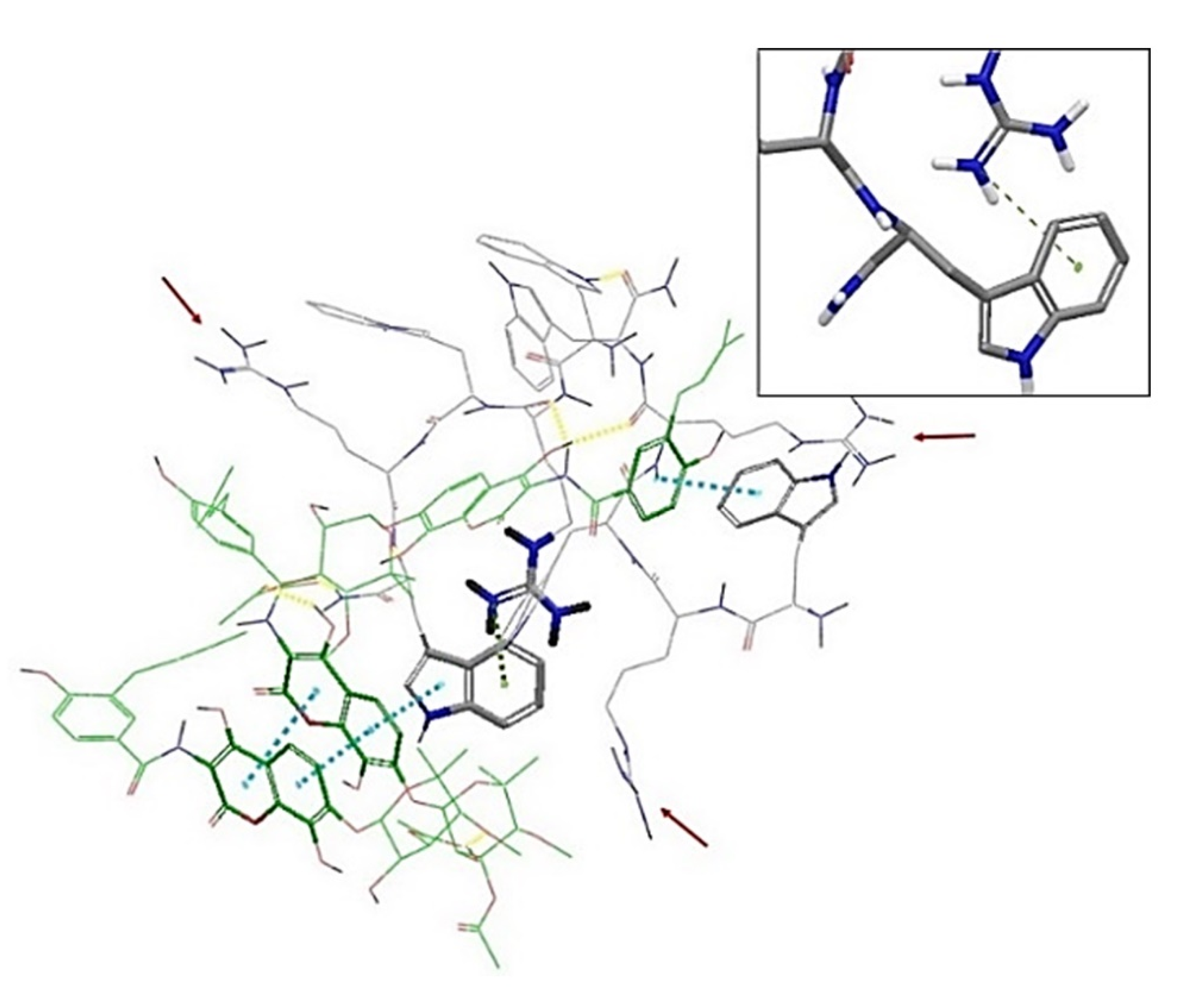
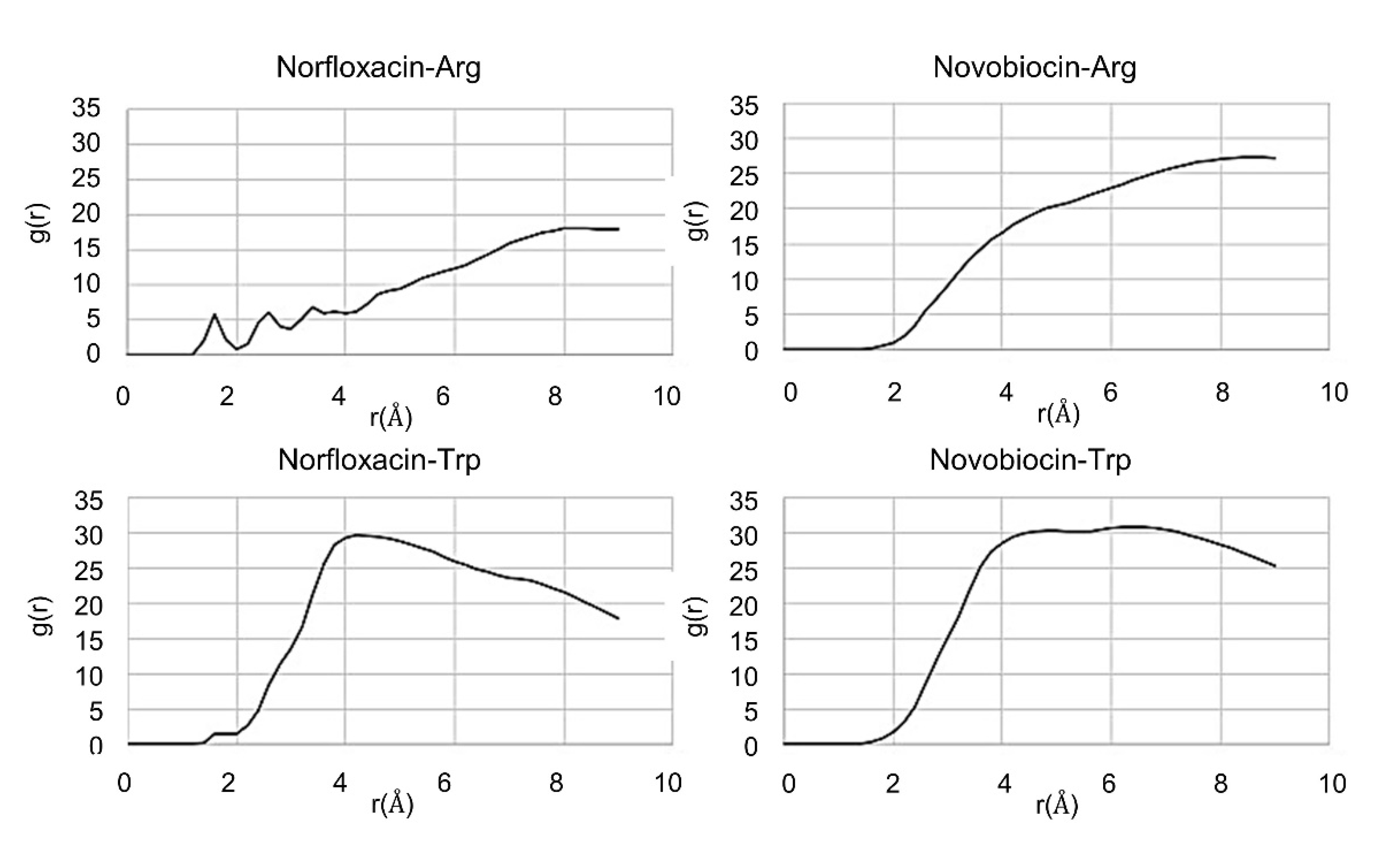
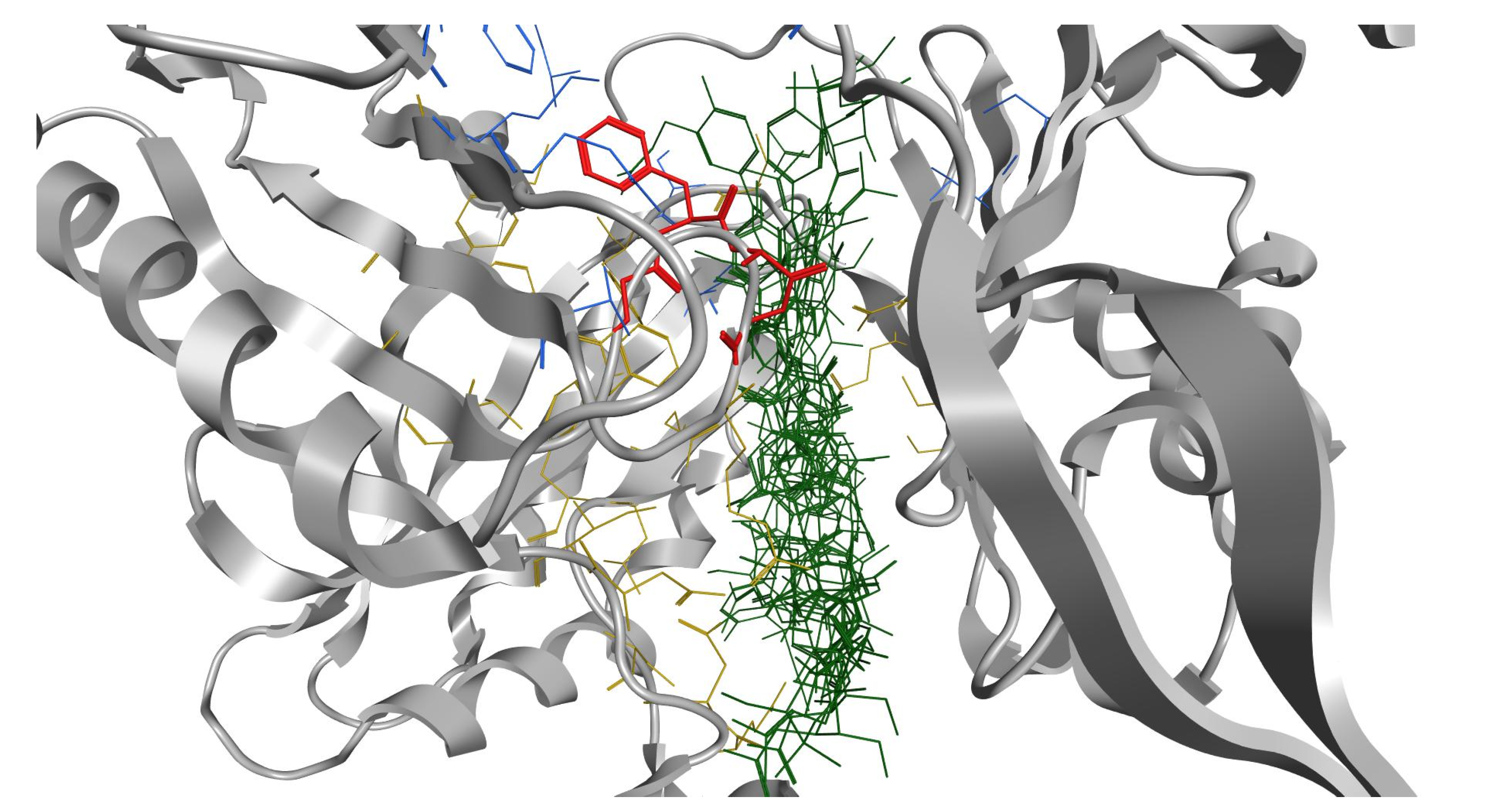
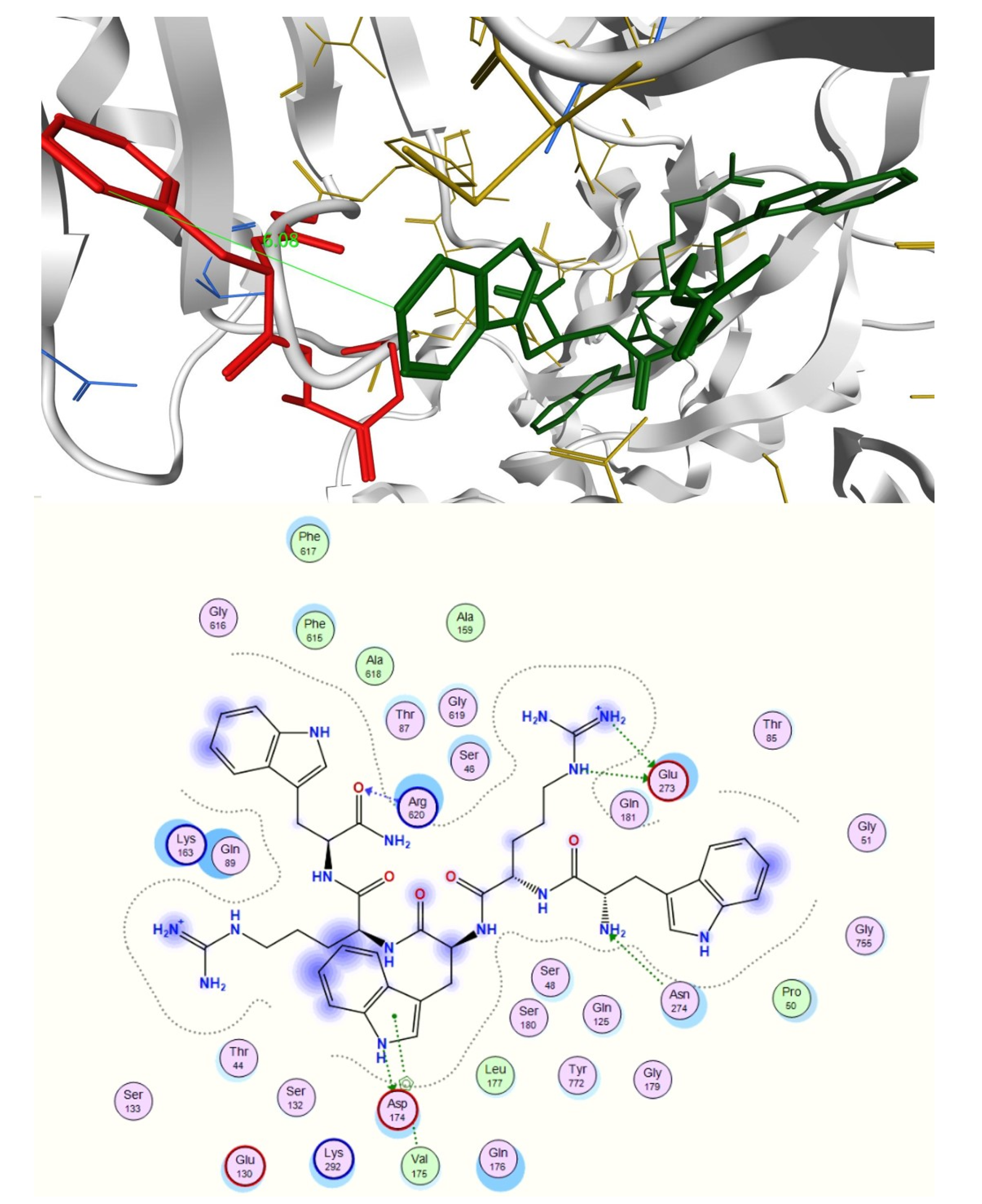
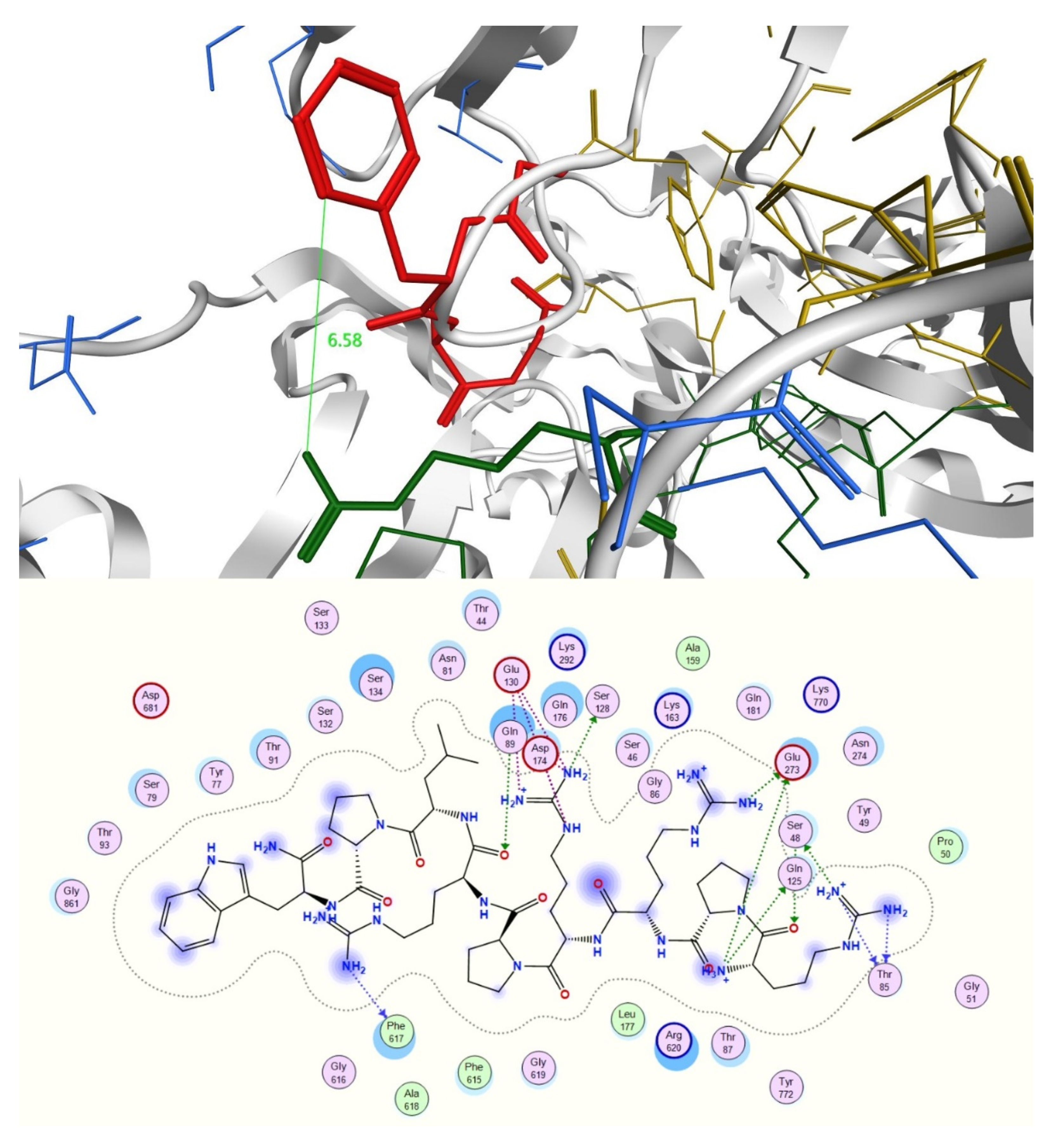
2.3. In Silico Studies
3. Materials and Methods
3.1. Design of Proline-Rich Cationic Peptides
- RPRPRPL (22): alternating Arg and Pro residues are maintained from the from parent structure. Peptide 22 did not show any significant potentiation of the MIC of novobiocin. This was not surprising as it does not contain any aromatic residues.
- RPWPPR (23): contains the last six residues of the parent structure but one Arg is substituted with a Trp to include aromaticity. Peptide 23 shows potentiation, but only against the susceptible strain.
- WKPLPPR (24): the aromatic residue is moved at the N terminal to better mimic PAβN. A leucine is also included to make the peptide slightly longer and include the chance of establishing interactions. It was also investigated whether substituting Lys for Arg could improve potentiation but no synergism was observed.
- FKPLPPH (25): the terminal residues were substituted with different amino acids but with the same electronic properties (Trp1 with Phe and Arg7 with Hys respectively). Potentiation was restored but again only against susceptible strain.
- The terminal portion of the parent structure was once again considered (RPPR) and this moiety repeated twice to increase length. Additionally, two Arg residues were substituted with Trp to include aromaticity, but not at the N terminus (peptide 24 did not show any improvement). This yielded peptide RPPWRPPW (26), however no potentiation was seen against the multidrug resistant strain.
- Pro2 and 3 were therefore separated with two Arg residues to add flexibility. Pro6 was also substituted with neutral leucine to further increase flexibility. Additionally, bulky Trp4 was eliminated to see if activity could be obtained with only one aromatic residue but without making the peptide too long. This led to RPPWRPPW (27), which shows an exciting profile.
3.2. Solid Phase Peptide Synthesis
Purification and Characterization
3.3. Biological Assays
3.4. Computational Studies
3.4.1. Clustering of MD Trajectories
3.4.2. Docking Studies of Peptides with RND Efflux Pump 4DX5
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AMP | Antimicrobial peptide |
| CFU | Colony-forming unit |
| DMSO | Dimethyl sulfoxide |
| Fmoc | Fluorenylmethyloxycarbonyl |
| HBTU | 2-(1H-Benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate |
| MLP | Molecular lipophilicity potential |
| PAβN | Phenylalanine-arginine-β-naphthylamide |
| PDB | Protein Data Bank |
| RMSD | Root mean square deviation |
| RND | Resistance nodulation division |
| RP-HPLC | Reverse phase high performance liquid chromatography |
| SPC | Statistical process control |
References
- Passarini, I.; Rossiter, S.; Malkinson, J.; Zloh, M. In Silico Structural Evaluation of Short Cationic Antimicrobial Peptides. Pharmaceutics 2018, 10, 72. [Google Scholar] [CrossRef] [PubMed]
- Magana, M.; Pushpanathan, M.; Santos, A.L.; Leanse, L.; Fernandez, M.; Ioannidis, A.; Giulianotti, M.A.; Apidianakis, Y.; Bradfute, S.; Ferguson, A.L. The value of antimicrobial peptides in the age of resistance. Lancet Infect. Dis. 2020, 20, e216–e230. [Google Scholar] [CrossRef]
- Koo, H.B.; Seo, J. Antimicrobial peptides under clinical investigation. Pept. Sci. 2019, 111, e24122. [Google Scholar] [CrossRef]
- Renau, T.E.; Leger, R.; Flamme, E.M.; Sanglang, J.; She, M.W.; Yen, R.; Gannon, C.L.; Griffith, D.; Chamberland, S.; Lomovskaya, O.; et al. Inhibitors of efflux pumps in Pseudomonas aeruginosa potentiate the activity of the fluoquinolone antibacterial levofloxacin. J. Med. Chem. 1999, 42, 4928–4931. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E. Peptide antibiotics. Lancet 1997, 349, 418–422. [Google Scholar] [CrossRef]
- Strøm, M.B.; Haug, B.E.; Skar, M.L.; Stensen, W.; Stiberg, T.; Svendsen, J.S. The Pharmacophore of Short Cationic Antibacterial Peptides. J. Med. Chem. 2003, 46, 1567–1570. [Google Scholar] [CrossRef]
- Scocchi, M.; Tossi, A.; Gennaro, R. Proline-rich antimicrobial peptides: Converging to a non-lytic mechanism of action. Cell. Mol. Life Sci. 2011, 68, 2317–2330. [Google Scholar] [CrossRef]
- Vitali, A. Proline-Rich Peptides: Multifunctional Bioactive Molecules as New Potential Therapeutic Drugs. Curr. Protein Pept. Sci. 2015, 16, 147–162. [Google Scholar] [CrossRef]
- Bechinger, B.; Gorr, S.-U. Antimicrobial Peptides: Mechanisms of Action and Resistance. J. Dent. Res. 2017, 96, 254–260. [Google Scholar] [CrossRef]
- Hof, W.V.; Veerman, E.C.; Helmerhorst, E.J.; Amerongen, A.V.N. Antimicrobial Peptides: Properties and Applicability. Biol. Chem. 2001, 382, 597–619. [Google Scholar] [CrossRef]
- Piers, K.L.; Hancock, R.E. The interaction of a recombinant cecropin/melittin hybrid peptide with the outer membrane ofPseudomonas aeruginosa. Mol. Microbiol. 1994, 12, 951–958. [Google Scholar] [CrossRef]
- Stapleton, P.; Shannon, K.P.; French, G.L. Carbapenem Resistance in Escherichia coli Associated with Plasmid-Determined CMY-4 β-Lactamase Production and Loss of an Outer Membrane Protein. Antimicrob. Agents Chemother. 1999, 43, 1206–1210. [Google Scholar] [CrossRef]
- Chevalier, J.; Malléa, M.; Pagès, J.M. Comparative aspects of the diffusion of norfloxacin, cefepime and spermine through the F porin channel of Enterobacter cloacae. Biochem. J. 2000, 348, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Giacometti, A.; Cirioni, O.; Barchiesi, F.; Fortuna, M.; Scalise, G. In-vitro activity of cationic peptides alone and in combination with clinically used antimicrobial agents against Pseudomonas aeruginosa. J. Antimicrob. Chemother. 1999, 44, 641–645. [Google Scholar] [CrossRef]
- Vaara, M.; Porro, M. Group of peptides that act synergistically with hydrophobic antibiotics against gram-negative enteric bacteria. Antimicrob. Agents Chemother. 1996, 40, 1801–1805. [Google Scholar] [CrossRef]
- Zloh, M.; Gibbons, S. The Role of Small Molecule; small Molecule Interactions in Overcoming Biological Barriers for Antibacterial Drug Action. Theor. Chem. Accounts 2006, 117, 231–238. [Google Scholar] [CrossRef]
- Lamers, R.P.; Cavallari, J.F.; Burrows, L.L. The Efflux Inhibitor Phenylalanine-Arginine Beta-Naphthylamide (PAβN) Permeabilizes the Outer Membrane of Gram-Negative Bacteria. PLoS ONE 2013, 8, e60666. [Google Scholar] [CrossRef] [PubMed]
- Lomovskaya, O.; Warren, M.S.; Lee, A.; Galazzo, J.; Fronko, R.; Lee, M.; Blais, J.; Cho, D.; Chamberland, S.; Renau, T.; et al. Identification and Characterization of Inhibitors of Multidrug Resistance Efflux Pumps in Pseudomonas aeruginosa: Novel Agents for Combination Therapy. Antimicrob. Agents Chemother. 2001, 45, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Sonnet, P.; Izard, D.; Mullié, C. Prevalence of efflux-mediated ciprofloxacin and levofloxacin resistance in recent clinical isolates of Pseudomonas aeruginosa and its reversal by the efflux pump inhibitors 1-(1-naphthylmethyl)-piperazine and phenylalanine-arginine-β -naphthylamide. Int. J. Antimicrob. Agents 2012, 39, 77–80. [Google Scholar] [CrossRef]
- Vargiu, A.V.; Nikaido, H. Multidrug binding properties of the AcrB efflux pump characterized by molecular dynamics simulations. Proc. Natl. Acad. Sci. USA 2012, 109, 20637–20642. [Google Scholar] [CrossRef] [PubMed]
- Eicher, T.; Cha, H.-J.; Seeger, M.A.; Brandstätter, L.; El-Delik, J.; Bohnert, J.A.; Kern, W.V.; Verrey, F.; Grütter, M.G.; Diederichs, K. Transport of drugs by the multidrug transporter AcrB involves an access and a deep binding pocket that are separated by a switch-loop. Proc. Natl. Acad. Sci. USA 2012, 109, 5687–5692. [Google Scholar] [CrossRef] [PubMed]
- Jamshidi, S.; Sutton, J.M.; Rahman, K.M. Computational Study Reveals the Molecular Mechanism of the Interaction between the Efflux Inhibitor PAβN and the AdeB Transporter from Acinetobacter baumannii. ACS Omega 2017, 2, 3002–3016. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, M.G.; Roy, R.N.; Lomakin, I.B.; Florin, T.; Mankin, A.S.; Steitz, T.A. Structures of proline-rich peptides bound to the ribosome reveal a common mechanism of protein synthesis inhibition. Nucleic Acids Res. 2016, 44, 2439–2450. [Google Scholar] [CrossRef] [PubMed]
- Andrews, J.M. Determination of minimum inhibitory concentrations. J. Antimicrob. Chemother. 2001, 48, 5–16. [Google Scholar] [CrossRef]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. Packmol: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef]
- Lemak, A.S.; Balabaev, N.K. On the Berendsen Thermostat. Mol. Simul. 1994, 13, 177–187. [Google Scholar] [CrossRef]
- Humphreys, D.D.; Friesner, R.A.; Berne, B.J. A Multiple-Time-Step Molecular Dynamics Algorithm for Macromolecules. J. Phys. Chem. 1994, 98, 6885–6892. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the 2006 ACM/IEEE Conference On Supercomputing, Tampa, FL, USA, 11–17 November 2006; Association for Computing Machinery: New York, NY, USA, 2006; p. 84-es. [Google Scholar] [CrossRef]
- Guo, Z.; Mohanty, U.; Noehre, J.; Sawyer, T.K.; Sherman, W.; Krilov, G. Probing the α-helical structural stability of stapled p53 peptides: Molecular dynamics simulations and analysis. Chem. Biol. Drug Des. 2010, 75, 348–359. [Google Scholar] [CrossRef]
- Pedretti, A.; Villa, L.; Vistoli, G. VEGA: A versatile program to convert, handle and visualize molecular structure on Windows-based PCs. J. Mol. Graph. Model. 2002, 21, 47–49. [Google Scholar] [CrossRef]
- Pedretti, A.; Villa, L.; Vistoli, G. VEGA: An open platform to develop chemo-bio-informatics applications, using plug-in architecture and script programming. J. Comput. Mol. Des. 2004, 18, 167–173. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Sequence-NH2 | MIC of Novobiocin (μg/mL) | |||
|---|---|---|---|---|---|
| E. coli NCTC 10418 | E. coli G69 | ||||
| Novobiocin | 32 | >128 | |||
| Added Peptide | +128 μg/mL | +32 μg/mL | +128 μg/mL | +32 μg/mL | |
| 1 | FRW | NP | NT | NP | NT |
| 2 | FWR | NP | NT | NP | NT |
| 3 | WRW | 8 | 64 | NP | NT |
| 4 | WRWR | 2 | 32 | NP | NT |
| 5 | RWRW | 2 | 64 | 16 | 64 |
| 6 | WRWRW | 0.125 | 8 | 16 | 8 |
| 7 | FRF | NP | NT | NP | NT |
| 8 | FRFR | NP | NT | 8 | NP |
| 9 | RFRF | 2 | NP | 8 | 128 |
| 10 | FRFRF | 2 | NT | 4 | 8 |
| 11 | RRFRF | 64 | NT | 64 | NT |
| 12 | WKW | 16 | 64 | NP | NT |
| 13 | WKWK | NP | NT | NP | NT |
| 14 | KWKW | NP | NT | NP | NT |
| 15 | WKWKW | 4 | 8 | 8 | 128 |
| 16 | FKF | NP | NT | NP | NT |
| 17 | FKFK | NP | NT | NP | NT |
| 18 | KFKF | NP | NT | NP | NT |
| 19 | FKFKF | 4 | 128 | 8 | 32 |
| 20 | WRRQRW | 4 | 16 | 32 | NP |
| 21 | FRRQRF | NP † | NT | NP | NT |
| 22 | RPRPRPL | NP | NT | NP | NT |
| 23 | RPWPPR | 4 | NT | NP | NT |
| 24 | WKPLPPR | NP | NT | NP | NT |
| 25 | FKPLPPH | 8 | NT | NP | NT |
| 26 | RPPWRPPW | 8 | 64 | NP | NT |
| 27 | RPRRPRLPW | 0.125 | 8 | 16 | 16 |
| +64 μg/mL PAβN ‡ | 8 | 2 | |||
| +32 μg/mL PAβN | 16 | 8 | |||
| Complex | Surface Area (Å2) | Broto log P | Broto Lipole | Virtual log P |
|---|---|---|---|---|
| Novobiocin | 968.9 (ds 17.6 Å) | 2.3840 | 1.4807 | 3.5449 |
| Norfloxacin | 556.1 (ds 13.3 Å) | −5.8350 | 0.4173 | −3.0671 |
| WRWRW-NH2 | 1280.5 (ds 20.2 Å) | −12.7310 | 6.0423 | −6.2840 |
| RPRRPRLPW-NH2 | 1861.8 (ds 24.3 Å) | −24.6980 | 5.0993 | −12.2239 |
| Novobiocin—WRWRW-NH2 A | 2353.1 (ds 27.4 Å) | −5.2310 | 2.6575 | −1.4918 |
| Novobiocin—WRWRW-NH2 B | 2219.3 (ds 26.6 Å) | −22.3820 | 1.8365 | −9.9088 |
| Norfloxacin—WRWRW-NH2 | 1648.9 (ds 22.9 Å) | −29.8880 | 1.4519 | −14.9976 |
| Novobiocin—RPRRPRLPW-NH2 | 6088.6 (ds 44.0 Å) | −59.7900 | 1.8135 | −24.2018 |
| Compound | Docking Score |
|---|---|
| PAβN | −7.3054 |
| Norfloxacin | −5.8704 |
| Novobiocin | −8.7469 |
| RPRRPRLPW (27) | −14.6003 |
| RPWPPR (23) | −11.3160 |
| WRWRW (6) | −12.2049 |
| WRW (3) | −8.1518 |
| Compound | Sequence-NH2 | Molar % Yield | Compound | Sequence-NH2 | Molar % Yield |
|---|---|---|---|---|---|
| 1 | FRW | 38 | 15 | WKWKW | 44 |
| 2 | FWR | 42 | 16 | FKF | 53 |
| 3 | WRW | 45 | 17 | FKFK | 56 |
| 4 | WRWR | 49 | 18 | KFKF | 55 |
| 5 | RWRW | 52 | 19 | FKFKF | 62 |
| 6 | WRWRW | 51 | 20 | WRRQRW | * |
| 7 | FRF | 55 | 21 | FRRQRF | * |
| 8 | FRFR | 58 | 22 | RPRPRPL | 52 |
| 9 | RFRF | 56 | 23 | RPWPPR | 56 |
| 10 | FRFRF | 60 | 24 | WKPLPPR | 48 |
| 11 | RRFRF | 48 | 25 | FKPLPPH | * |
| 12 | WKW | 36 | 26 | RPPWRPPW | 39 |
| 13 | WKWK | 39 | 27 | RPRRPRLPW | 45 |
| 14 | KWKW | 41 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Passarini, I.; Resende, P.E.d.; Soares, S.; Tahmasi, T.; Stapleton, P.; Malkinson, J.; Zloh, M.; Rossiter, S. Synthesis and in Silico Modelling of the Potential Dual Mechanistic Activity of Small Cationic Peptides Potentiating the Antibiotic Novobiocin against Susceptible and Multi-Drug Resistant Escherichia coli. Int. J. Mol. Sci. 2020, 21, 9134. https://doi.org/10.3390/ijms21239134
Passarini I, Resende PEd, Soares S, Tahmasi T, Stapleton P, Malkinson J, Zloh M, Rossiter S. Synthesis and in Silico Modelling of the Potential Dual Mechanistic Activity of Small Cationic Peptides Potentiating the Antibiotic Novobiocin against Susceptible and Multi-Drug Resistant Escherichia coli. International Journal of Molecular Sciences. 2020; 21(23):9134. https://doi.org/10.3390/ijms21239134
Chicago/Turabian StylePassarini, Ilaria, Pedro Ernesto de Resende, Sarah Soares, Tadeh Tahmasi, Paul Stapleton, John Malkinson, Mire Zloh, and Sharon Rossiter. 2020. "Synthesis and in Silico Modelling of the Potential Dual Mechanistic Activity of Small Cationic Peptides Potentiating the Antibiotic Novobiocin against Susceptible and Multi-Drug Resistant Escherichia coli" International Journal of Molecular Sciences 21, no. 23: 9134. https://doi.org/10.3390/ijms21239134
APA StylePassarini, I., Resende, P. E. d., Soares, S., Tahmasi, T., Stapleton, P., Malkinson, J., Zloh, M., & Rossiter, S. (2020). Synthesis and in Silico Modelling of the Potential Dual Mechanistic Activity of Small Cationic Peptides Potentiating the Antibiotic Novobiocin against Susceptible and Multi-Drug Resistant Escherichia coli. International Journal of Molecular Sciences, 21(23), 9134. https://doi.org/10.3390/ijms21239134