Cancer Stem Cell-Associated Pathways in the Metabolic Reprogramming of Breast Cancer



Abstract
1. Breast Cancer and CSCs
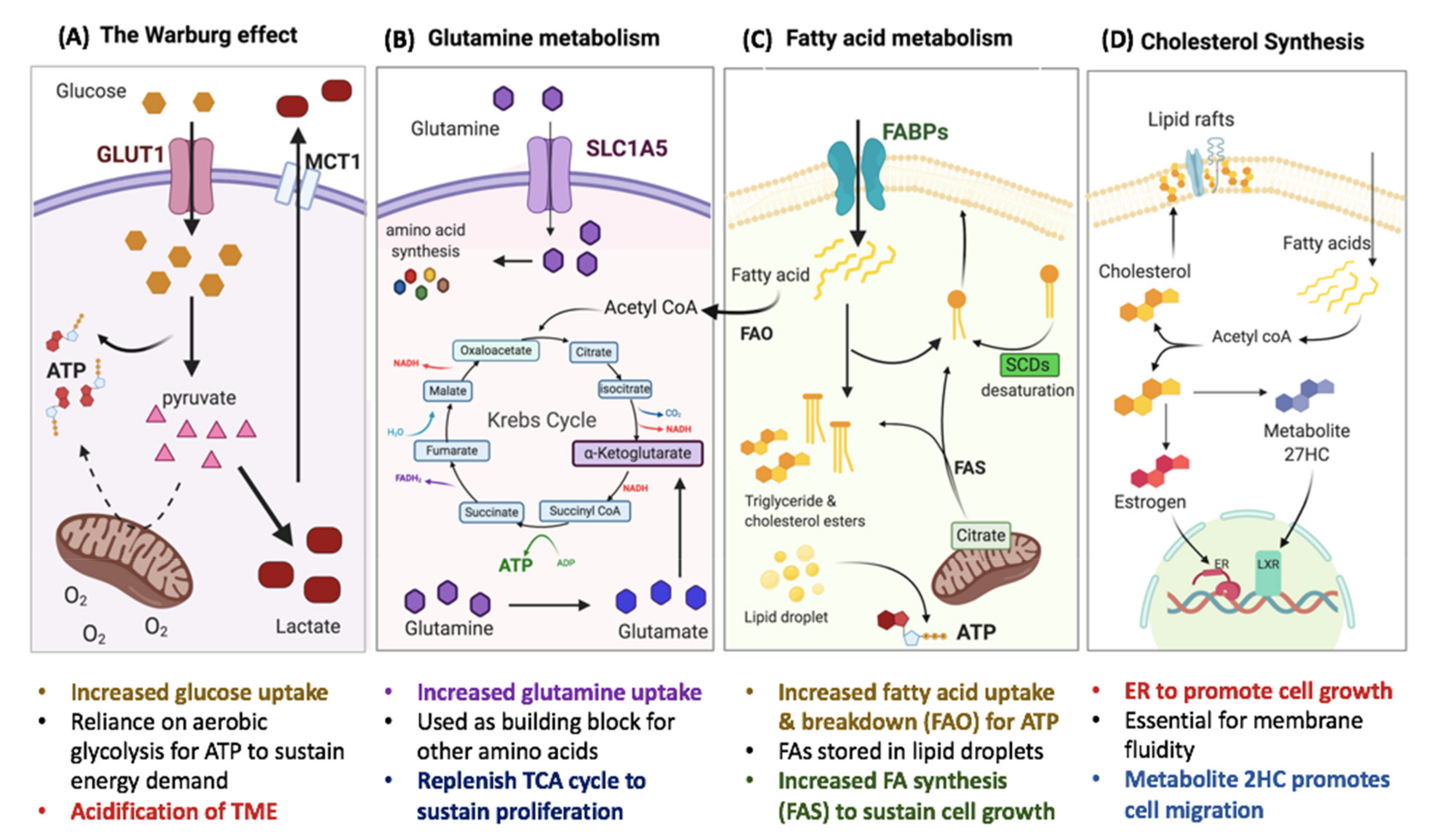
2. Metabolic Reprogramming in Breast Cancer and CSCs
3. YAP/TAZ Signaling
3.1. YAP in Breast Cancer Metabolism
3.2. Therapeutic Strategies for Targeting YAP-Mediated Cancer Metabolism
4. The JAK/STAT Pathway
4.1. STAT3 in Breast Cancer Metabolism
4.2. Therapeutic Strategies Targeting STAT3
5. PGC-1α Signaling Pathway
5.1. PGC-1α as a Regulator of Breast Cancer Metabolic Flexibility
5.2. Therapeutic Targeting of PGC-1a in Tumorigenesis
6. HIF-1α Signaling
6.1. HIF-1α in Breast Cancer Metabolism
6.2. Therapeutic Strategies Targeting HIF-1α
7. The NF-κB Pathway
7.1. NF-kB in Breast Cancer Metabolism
7.2. Therapeutic Strategies Targeting NF-κB Elements
8. The Wnt/β-Catenin Pathway
8.1. Wnt in Breast Cancer Metabolism
8.2. Therapeutic Targeting of Wnt Elements in Breast Cancer
9. Pathway Crosstalk in Metabolic Reprogramming
10. Concluding Remarks and Potential Future Avenues
Funding
Acknowledgments
Conflicts of Interest
References
- DeSantis, C.; Ma, J.; Bryan, L.; Jemal, A. Breast cancer statistics, 2013. CA Cancer J. Clin. 2014, 64, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Hon, J.D.; Singh, B.; Sahin, A.; Du, G.; Wang, J.; Wang, V.Y.; Deng, F.M.; Zhang, D.Y.; Monaco, M.E.; Lee, P. Breast cancer molecular subtypes: From TNBC to QNBC. Am. J. Cancer Res. 2016, 6, 1864–1872. [Google Scholar]
- Barnard, M.E.; Boeke, C.E.; Tamimi, R.M. Established breast cancer risk factors and risk of intrinsic tumor subtypes. Biochim. Biophys. Acta 2015, 1856, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Cheang, M.C.; Chia, S.K.; Voduc, D.; Gao, D.; Leung, S.; Snider, J.; Watson, M.; Davies, S.; Bernard, P.S.; Parker, J.S.; et al. Ki67 index, HER2 status, and prognosis of patients with luminal B breast cancer. J. Natl. Cancer Inst. 2009, 101, 736–750. [Google Scholar] [CrossRef] [PubMed]
- Molina, M.A.; Codony-Servat, J.; Albanell, J.; Rojo, F.; Arribas, J.; Baselga, J. Trastuzumab (herceptin), a humanized anti-Her2 receptor monoclonal antibody, inhibits basal and activated Her2 ectodomain cleavage in breast cancer cells. Cancer Res. 2001, 61, 4744–4749. [Google Scholar] [PubMed]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef]
- Saeg, F.; Anbalagan, M. Breast cancer stem cells and the challenges of eradication: A review of novel therapies. Stem Cell Investig. 2018, 5, 39. [Google Scholar] [CrossRef]
- Scioli, M.G.; Storti, G.; D’Amico, F.; Gentile, P.; Fabbri, G.; Cervelli, V.; Orlandi, A. The Role of Breast Cancer Stem Cells as a Prognostic Marker and a Target to Improve the Efficacy of Breast Cancer Therapy. Cancers 2019, 11, 1021. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Saunier, E.; Antonio, S.; Regazzetti, A.; Auzeil, N.; Laprevote, O.; Shay, J.W.; Coumoul, X.; Barouki, R.; Benelli, C.; Huc, L.; et al. Resveratrol reverses the Warburg effect by targeting the pyruvate dehydrogenase complex in colon cancer cells. Sci. Rep. 2017, 7, 6945. [Google Scholar] [CrossRef]
- O’Neill, S.; Porter, R.K.; McNamee, N.; Martinez, V.G.; O’Driscoll, L. 2-Deoxy-D-Glucose inhibits aggressive triple-negative breast cancer cells by targeting glycolysis and the cancer stem cell phenotype. Sci. Rep. 2019, 9, 3788. [Google Scholar] [CrossRef] [PubMed]
- Zacksenhaus, E.; Shrestha, M.; Liu, J.C.; Vorobieva, I.; Chung, P.E.D.; Ju, Y.; Nir, U.; Jiang, Z. Mitochondrial OXPHOS Induced by RB1 Deficiency in Breast Cancer: Implications for Anabolic Metabolism, Stemness, and Metastasis. Trends Cancer 2017, 3, 768–779. [Google Scholar] [CrossRef] [PubMed]
- Kinlaw, W.B.; Baures, P.W.; Lupien, L.E.; Davis, W.L.; Kuemmerle, N.B. Fatty Acids and Breast Cancer: Make Them on Site or Have Them Delivered. J. Cell. Physiol. 2016, 231, 2128–2141. [Google Scholar] [CrossRef] [PubMed]
- Giudetti, A.M.; De Domenico, S.; Ragusa, A.; Lunetti, P.; Gaballo, A.; Franck, J.; Simeone, P.; Nicolardi, G.; De Nuccio, F.; Santino, A.; et al. A specific lipid metabolic profile is associated with the epithelial mesenchymal transition program. Biochim. Biophys. Acta Mol. Cell. Biol. Lipids 2019, 1864, 344–357. [Google Scholar] [CrossRef]
- Hershey, B.J.; Vazzana, R.; Joppi, D.L.; Havas, K.M. Lipid Droplets Define a Sub-Population of Breast Cancer Stem Cells. J. Clin. Med. 2020, 9, 87. [Google Scholar] [CrossRef]
- Han, S.; Wei, R.; Zhang, X.; Jiang, N.; Fan, M.; Huang, J.H.; Xie, B.; Zhang, L.; Miao, W.; Butler, A.C.; et al. CPT1A/2-Mediated FAO Enhancement-A Metabolic Target in Radioresistant Breast Cancer. Front. Oncol. 2019, 9, 1201. [Google Scholar] [CrossRef]
- Chimento, A.; Casaburi, I.; Avena, P.; Trotta, F.; De Luca, A.; Rago, V.; Pezzi, V.; Sirianni, R. Cholesterol and Its Metabolites in Tumor Growth: Therapeutic Potential of Statins in Cancer Treatment. Front. Endocrinol. 2019, 9, 807. [Google Scholar] [CrossRef]
- Ehmsen, S.; Pedersen, M.H.; Wang, G.; Terp, M.G.; Arslanagic, A.; Hood, B.L.; Conrads, T.P.; Leth-Larsen, R.; Ditzel, H.J. Increased Cholesterol Biosynthesis Is a Key Characteristic of Breast Cancer Stem Cells Influencing Patient Outcome. Cell Rep. 2019, 27, 3927–3938.e6. [Google Scholar] [CrossRef]
- Lu, H.; Samanta, D.; Xiang, L.; Zhang, H.; Hu, H.; Chen, I.; Bullen, J.W.; Semenza, G.L. Chemotherapy triggers HIF-1-dependent glutathione synthesis and copper chelation that induces the breast cancer stem cell phenotype. Proc. Natl. Acad. Sci. USA 2015, 112, E4600–E4609. [Google Scholar] [CrossRef]
- Miran, T.; Vogg, A.T.J.; Drude, N.; Mottaghy, F.M.; Morgenroth, A. Modulation of glutathione promotes apoptosis in triple-negative breast cancer cells. FASEB J. 2018, 32, 2803–2813. [Google Scholar] [CrossRef]
- Dupuy, F.; Tabaries, S.; Andrzejewski, S.; Dong, Z.; Blagih, J.; Annis, M.G.; Omeroglu, A.; Gao, D.; Leung, S.; Amir, E.; et al. PDK1-Dependent Metabolic Reprogramming Dictates Metastatic Potential in Breast Cancer. Cell Metab. 2015, 22, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.X.; Zhao, B.; Guan, K.L. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Tumaneng, K.; Guan, K.L. The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat. Cell Biol. 2011, 13, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Chen, Y.; Luo, J.; Zheng, J.; Shao, G. YAP1 overexpression is associated with poor prognosis of breast cancer patients and induces breast cancer cell growth by inhibiting PTEN. FEBS Open Bio 2019, 9, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Shin, J.E.; Park, H.W. The Role of Hippo Pathway in Cancer Stem Cell Biology. Mol. Cells 2018, 41, 83–92. [Google Scholar] [CrossRef]
- Li, Y.W.; Xu, J.; Zhu, G.Y.; Huang, Z.J.; Lu, Y.; Li, X.Q.; Wang, N.; Zhang, F.X. Apigenin suppresses the stem cell-like properties of triple-negative breast cancer cells by inhibiting YAP/TAZ activity. Cell Death Discov. 2018, 4, 105. [Google Scholar] [CrossRef]
- Sulaiman, A.; McGarry, S.; Li, L.; Jia, D.; Ooi, S.; Addison, C.; Dimitroulakos, J.; Arnaout, A.; Nessim, C.; Yao, Z.; et al. Dual inhibition of Wnt and Yes-associated protein signaling retards the growth of triple-negative breast cancer in both mesenchymal and epithelial states. Mol. Oncol. 2018, 12, 423–440. [Google Scholar] [CrossRef]
- Shen, Y.; Zhao, S.; Wang, S.; Pan, X.; Zhang, Y.; Xu, J.; Jiang, Y.; Li, H.; Zhang, Q.; Gao, J.; et al. S1P/S1PR3 axis promotes aerobic glycolysis by YAP/c-MYC/PGAM1 axis in osteosarcoma. EBioMedicine 2019, 40, 210–223. [Google Scholar] [CrossRef]
- Kashihara, T.; Zablocki, D.; Sadoshima, J. YAP-dependent metabolic remodeling in local lymph node boosts the function of cancer cells. Biotarget 2019, 3. [Google Scholar] [CrossRef]
- Enzo, E.; Santinon, G.; Pocaterra, A.; Aragona, M.; Bresolin, S.; Forcato, M.; Grifoni, D.; Pession, A.; Zanconato, F.; Guzzo, G.; et al. Aerobic glycolysis tunes YAP/TAZ transcriptional activity. EMBO J. 2015, 34, 1349–1370. [Google Scholar] [CrossRef]
- Nokin, M.J.; Durieux, F.; Peixoto, P.; Chiavarina, B.; Peulen, O.; Blomme, A.; Turtoi, A.; Costanza, B.; Smargiasso, N.; Baiwir, D.; et al. Methylglyoxal, a glycolysis side-product, induces Hsp90 glycation and YAP-mediated tumor growth and metastasis. Elife 2016, 5. [Google Scholar] [CrossRef]
- Wang, W.; Xiao, Z.D.; Li, X.; Aziz, K.E.; Gan, B.; Johnson, R.L.; Chen, J. AMPK modulates Hippo pathway activity to regulate energy homeostasis. Nat. Cell Biol. 2015, 17, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Plouffe, S.W.; Meng, Z.; Lin, K.C.; Lin, B.; Hong, A.W.; Chun, J.V.; Guan, K.L. Characterization of Hippo Pathway Components by Gene Inactivation. Mol. Cell 2016, 64, 993–1008. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.S.; Stampouloglou, E.; Kingston, N.M.; Zhang, L.; Monti, S.; Varelas, X. Glutamine-utilizing transaminases are a metabolic vulnerability of TAZ/YAP-activated cancer cells. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef]
- Yi, M.; Li, J.; Chen, S.; Cai, J.; Ban, Y.; Peng, Q.; Zhou, Y.; Zeng, Z.; Peng, S.; Li, X.; et al. Emerging role of lipid metabolism alterations in Cancer stem cells. J. Exp. Clin. Cancer Res. 2018, 37, 118. [Google Scholar] [CrossRef]
- Noto, A.; De Vitis, C.; Pisanu, M.E.; Roscilli, G.; Ricci, G.; Catizone, A.; Sorrentino, G.; Chianese, G.; Taglialatela-Scafati, O.; Trisciuoglio, D.; et al. Stearoyl-CoA-desaturase 1 regulates lung cancer stemness via stabilization and nuclear localization of YAP/TAZ. Oncogene 2017, 36, 4573–4584. [Google Scholar] [CrossRef]
- Mukherjee, A.; Kenny, H.A.; Lengyel, E. Unsaturated Fatty Acids Maintain Cancer Cell Stemness. Cell Stem Cell 2017, 20, 291–292. [Google Scholar] [CrossRef]
- Yuan, L.; Mao, Y.; Luo, W.; Wu, W.; Xu, H.; Wang, X.L.; Shen, Y.H. Palmitic acid dysregulates the Hippo-YAP pathway and inhibits angiogenesis by inducing mitochondrial damage and activating the cytosolic DNA sensor cGAS-STING-IRF3 signaling mechanism. J. Biol. Chem. 2017, 292, 15002–15015. [Google Scholar] [CrossRef]
- Lee, C.K.; Jeong, S.H.; Jang, C.; Bae, H.; Kim, Y.H.; Park, I.; Kim, S.K.; Koh, G.Y. Tumor metastasis to lymph nodes requires YAP-dependent metabolic adaptation. Science 2019, 363, 644–649. [Google Scholar] [CrossRef]
- Dihge, L.; Vallon-Christersson, J.; Hegardt, C.; Saal, L.H.; Hakkinen, J.; Larsson, C.; Ehinger, A.; Loman, N.; Malmberg, M.; Bendahl, P.O.; et al. Prediction of Lymph Node Metastasis in Breast Cancer by Gene Expression and Clinicopathological Models: Development and Validation within a Population-Based Cohort. Clin. Cancer Res. 2019, 25, 6368–6381. [Google Scholar] [CrossRef]
- Shu, Z.; Gao, Y.; Zhang, G.; Zhou, Y.; Cao, J.; Wan, D.; Zhu, X.; Xiong, W. A functional interaction between Hippo-YAP signalling and SREBPs mediates hepatic steatosis in diabetic mice. J. Cell. Mol. Med. 2019, 23, 3616–3628. [Google Scholar] [CrossRef] [PubMed]
- Zalba, S.; Ten Hagen, T.L. Cell membrane modulation as adjuvant in cancer therapy. Cancer Treat. Rev. 2017, 52, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, G.; Ruggeri, N.; Specchia, V.; Cordenonsi, M.; Mano, M.; Dupont, S.; Manfrin, A.; Ingallina, E.; Sommaggio, R.; Piazza, S.; et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat. Cell Biol. 2014, 16, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wu, Y.; Wang, H.; Zhang, Y.; Mei, L.; Fang, X.; Zhang, X.; Zhang, F.; Chen, H.; Liu, Y.; et al. Interplay of mevalonate and Hippo pathways regulates RHAMM transcription via YAP to modulate breast cancer cell motility. Proc. Natl. Acad. Sci. USA 2014, 111, E89–E98. [Google Scholar] [CrossRef]
- Oku, Y.; Nishiya, N.; Shito, T.; Yamamoto, R.; Yamamoto, Y.; Oyama, C.; Uehara, Y. Small molecules inhibiting the nuclear localization of YAP/TAZ for chemotherapeutics and chemosensitizers against breast cancers. FEBS Open Bio 2015, 5, 542–549. [Google Scholar] [CrossRef]
- Di Agostino, S.; Sorrentino, G.; Ingallina, E.; Valenti, F.; Ferraiuolo, M.; Bicciato, S.; Piazza, S.; Strano, S.; Del Sal, G.; Blandino, G. YAP enhances the pro-proliferative transcriptional activity of mutant p53 proteins. EMBO Rep. 2016, 17, 188–201. [Google Scholar] [CrossRef]
- Borgquist, S.; Bjarnadottir, O.; Kimbung, S.; Ahern, T.P. Statins: A role in breast cancer therapy? J. Intern. Med. 2018, 284, 346–357. [Google Scholar] [CrossRef]
- Ma, J.; Fan, Z.; Tang, Q.; Xia, H.; Zhang, T.; Bi, F. Aspirin attenuates YAP and beta-catenin expression by promoting beta-TrCP to overcome docetaxel and vinorelbine resistance in triple-negative breast cancer. Cell Death Dis. 2020, 11, 530. [Google Scholar] [CrossRef]
- Galoczova, M.; Coates, P.; Vojtesek, B. STAT3, stem cells, cancer stem cells and p63. Cell. Mol. Biol. Lett. 2018, 23, 12. [Google Scholar] [CrossRef]
- Wu, P.; Wu, D.; Zhao, L.; Huang, L.; Shen, G.; Huang, J.; Chai, Y. Prognostic role of STAT3 in solid tumors: A systematic review and meta-analysis. Oncotarget 2016, 7, 19863–19883. [Google Scholar] [CrossRef]
- Wehde, B.L.; Radler, P.D.; Shrestha, H.; Johnson, S.J.; Triplett, A.A.; Wagner, K.U. Janus Kinase 1 Plays a Critical Role in Mammary Cancer Progression. Cell Rep. 2018, 25, 2192–2207.e2195. [Google Scholar] [CrossRef] [PubMed]
- Demaria, M.; Giorgi, C.; Lebiedzinska, M.; Esposito, G.; D’Angeli, L.; Bartoli, A.; Gough, D.J.; Turkson, J.; Levy, D.E.; Watson, C.J.; et al. A STAT3-mediated metabolic switch is involved in tumour transformation and STAT3 addiction. Aging 2010, 2, 823–842. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Raje, V.; Yakovlev, V.A.; Yacoub, A.; Szczepanek, K.; Meier, J.; Derecka, M.; Chen, Q.; Hu, Y.; Sisler, J.; et al. Mitochondrial localized Stat3 promotes breast cancer growth via phosphorylation of serine 727. J. Biol. Chem. 2013, 288, 31280–31288. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Fahrmann, J.F.; Lee, H.; Li, Y.J.; Tripathi, S.C.; Yue, C.; Zhang, C.; Lifshitz, V.; Song, J.; Yuan, Y.; et al. JAK/STAT3-Regulated Fatty Acid beta-Oxidation Is Critical for Breast Cancer Stem Cell Self-Renewal and Chemoresistance. Cell Metab. 2018, 27, 1357. [Google Scholar] [CrossRef]
- Cacace, A.; Sboarina, M.; Vazeille, T.; Sonveaux, P. Glutamine activates STAT3 to control cancer cell proliferation independently of glutamine metabolism. Oncogene 2017, 36, 2074–2084. [Google Scholar] [CrossRef]
- Turkson, J.; Zhang, S.; Mora, L.B.; Burns, A.; Sebti, S.; Jove, R. A novel platinum compound inhibits constitutive Stat3 signaling and induces cell cycle arrest and apoptosis of malignant cells. J. Biol. Chem. 2005, 280, 32979–32988. [Google Scholar] [CrossRef]
- Kim, J.W.; Gautam, J.; Kim, J.E.; Kim, J.A.; Kang, K.W. Inhibition of tumor growth and angiogenesis of tamoxifen-resistant breast cancer cells by ruxolitinib, a selective JAK2 inhibitor. Oncol. Lett. 2019, 17, 3981–3989. [Google Scholar] [CrossRef]
- Ventura-Clapier, R.; Garnier, A.; Veksler, V. Transcriptional control of mitochondrial biogenesis: The central role of PGC-1alpha. Cardiovasc. Res. 2008, 79, 208–217. [Google Scholar] [CrossRef]
- Im, Y.K.; Najyb, O.; Gravel, S.P.; McGuirk, S.; Ahn, R.; Avizonis, D.Z.; Chenard, V.; Sabourin, V.; Hudson, J.; Pawson, T.; et al. Interplay between ShcA Signaling and PGC-1alpha Triggers Targetable Metabolic Vulnerabilities in Breast Cancer. Cancer Res. 2018, 78, 4826–4838. [Google Scholar] [CrossRef]
- Andrzejewski, S.; Klimcakova, E.; Johnson, R.M.; Tabaries, S.; Annis, M.G.; McGuirk, S.; Northey, J.J.; Chenard, V.; Sriram, U.; Papadopoli, D.J.; et al. PGC-1alpha Promotes Breast Cancer Metastasis and Confers Bioenergetic Flexibility against Metabolic Drugs. Cell Metab. 2017, 26, 778–787.e5. [Google Scholar] [CrossRef]
- Jiang, W.G.; Douglas-Jones, A.; Mansel, R.E. Expression of peroxisome-proliferator activated receptor-gamma (PPARgamma) and the PPARgamma co-activator, PGC-1, in human breast cancer correlates with clinical outcomes. Int. J. Cancer 2003, 106, 752–757. [Google Scholar] [CrossRef] [PubMed]
- Cai, F.F.; Xu, C.; Pan, X.; Cai, L.; Lin, X.Y.; Chen, S.; Biskup, E. Prognostic value of plasma levels of HIF-1a and PGC-1a in breast cancer. Oncotarget 2016, 7, 77793–77806. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Huo, L.; Liu, Y.; Deng, P.; Szymanski, J.; Li, J.; Luo, X.; Hong, C.; Lin, J.; Wang, C.Y. PGC-1alpha Controls Skeletal Stem Cell Fate and Bone-Fat Balance in Osteoporosis and Skeletal Aging by Inducing TAZ. Cell Stem Cell 2018, 23, 193–209.e5. [Google Scholar] [CrossRef] [PubMed]
- Mahato, B.; Home, P.; Rajendran, G.; Paul, A.; Saha, B.; Ganguly, A.; Ray, S.; Roy, N.; Swerdlow, R.H.; Paul, S. Regulation of mitochondrial function and cellular energy metabolism by protein kinase C-lambda/iota: A novel mode of balancing pluripotency. Stem Cells 2014, 32, 2880–2892. [Google Scholar] [CrossRef] [PubMed]
- Sancho, P.; Burgos-Ramos, E.; Tavera, A.; Bou Kheir, T.; Jagust, P.; Schoenhals, M.; Barneda, D.; Sellers, K.; Campos-Olivas, R.; Grana, O.; et al. MYC/PGC-1alpha Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab. 2015, 22, 590–605. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; O’Connell, J.T.; Gonzalez Herrera, K.N.; Wikman, H.; Pantel, K.; Haigis, M.C.; de Carvalho, F.M.; Damascena, A.; Domingos Chinen, L.T.; Rocha, R.M.; et al. PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 2014, 16, 992–1003, 1001–1015. [Google Scholar] [CrossRef]
- McGuirk, S.; Gravel, S.P.; Deblois, G.; Papadopoli, D.J.; Faubert, B.; Wegner, A.; Hiller, K.; Avizonis, D.; Akavia, U.D.; Jones, R.G.; et al. PGC-1alpha supports glutamine metabolism in breast cancer. Cancer Metab. 2013, 1, 22. [Google Scholar] [CrossRef]
- Supruniuk, E.; Miklosz, A.; Chabowski, A. The Implication of PGC-1alpha on Fatty Acid Transport across Plasma and Mitochondrial Membranes in the Insulin Sensitive Tissues. Front. Physiol. 2017, 8, 923. [Google Scholar] [CrossRef]
- Wu, Q.; Li, B.; Li, Z.; Li, J.; Sun, S. Cancer-associated adipocytes: Key players in breast cancer progression. J. Hematol. Oncol. 2019, 12, 95. [Google Scholar] [CrossRef]
- Kim, B.; Jung, J.W.; Jung, J.; Han, Y.; Suh, D.H.; Kim, H.S.; Dhanasekaran, D.N.; Song, Y.S. PGC1alpha induced by reactive oxygen species contributes to chemoresistance of ovarian cancer cells. Oncotarget 2017, 8, 60299–60311. [Google Scholar] [CrossRef]
- Yun, C.W.; Han, Y.S.; Lee, S.H. PGC-1alpha Controls Mitochondrial Biogenesis in Drug-Resistant Colorectal Cancer Cells by Regulating Endoplasmic Reticulum Stress. Int. J. Mol. Sci. 2019, 20, 1707. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Bermudez, A.; Laza-Briviesca, R.; Vicente-Blanco, R.J.; Garcia-Grande, A.; Coronado, M.J.; Laine-Menendez, S.; Palacios-Zambrano, S.; Moreno-Villa, M.R.; Ruiz-Valdepenas, A.M.; Lendinez, C.; et al. Cisplatin resistance involves a metabolic reprogramming through ROS and PGC-1alpha in NSCLC which can be overcome by OXPHOS inhibition. Free Radic. Biol. Med. 2019, 135, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.K.; Jang, M.; Song, M.J.; Kim, D.; Kim, Y.; Jang, H.H. Redox-Mediated Mechanism of Chemoresistance in Cancer Cells. Antioxidants 2019, 8, 471. [Google Scholar] [CrossRef] [PubMed]
- Gabrielson, M.; Bjorklund, M.; Carlson, J.; Shoshan, M. Expression of mitochondrial regulators PGC1alpha and TFAM as putative markers of subtype and chemoresistance in epithelial ovarian carcinoma. PLoS ONE 2014, 9, e107109. [Google Scholar] [CrossRef]
- Polvani, S.; Tarocchi, M.; Tempesti, S.; Bencini, L.; Galli, A. Peroxisome proliferator activated receptors at the crossroad of obesity, diabetes, and pancreatic cancer. World J. Gastroenterol. 2016, 22, 2441–2459. [Google Scholar] [CrossRef]
- Park, S.; Chang, C.Y.; Safi, R.; Liu, X.; Baldi, R.; Jasper, J.S.; Anderson, G.R.; Liu, T.; Rathmell, J.C.; Dewhirst, M.W.; et al. ERRalpha-Regulated Lactate Metabolism Contributes to Resistance to Targeted Therapies in Breast Cancer. Cell Rep. 2016, 15, 323–335. [Google Scholar] [CrossRef]
- Wu, T.; Harder, B.G.; Wong, P.K.; Lang, J.E.; Zhang, D.D. Oxidative stress, mammospheres and Nrf2-new implication for breast cancer therapy? Mol. Carcinog. 2015, 54, 1494–1502. [Google Scholar] [CrossRef]
- Park, S.; Safi, R.; Liu, X.; Baldi, R.; Liu, W.; Liu, J.; Locasale, J.W.; Chang, C.Y.; McDonnell, D.P. Inhibition of ERRalpha Prevents Mitochondrial Pyruvate Uptake Exposing NADPH-Generating Pathways as Targetable Vulnerabilities in Breast Cancer. Cell Rep. 2019, 27, 3587–3601.e3584. [Google Scholar] [CrossRef]
- Yeo, E.J. Hypoxia and aging. Exp. Mol. Med. 2019, 51, 1–15. [Google Scholar] [CrossRef]
- Gruber, G.; Greiner, R.H.; Hlushchuk, R.; Aebersold, D.M.; Altermatt, H.J.; Berclaz, G.; Djonov, V. Hypoxia-inducible factor 1 alpha in high-risk breast cancer: An independent prognostic parameter? Breast Cancer Res. 2004, 6, R191–R198. [Google Scholar] [CrossRef]
- Nie, C.; Lv, H.; Bie, L.; Hou, H.; Chen, X. Hypoxia-inducible factor 1-alpha expression correlates with response to neoadjuvant chemotherapy in women with breast cancer. Medicine 2018, 97, e13551. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; AlTahan, A.; Jones, D.T.; Buffa, F.M.; Bridges, E.; Interiano, R.B.; Qu, C.; Vogt, N.; Li, J.L.; Baban, D.; et al. Estrogen receptor-alpha directly regulates the hypoxia-inducible factor 1 pathway associated with antiestrogen response in breast cancer. Proc. Natl. Acad. Sci. USA 2015, 112, 15172–15177. [Google Scholar] [CrossRef] [PubMed]
- Yehia, L.; Boulos, F.; Jabbour, M.; Mahfoud, Z.; Fakhruddin, N.; El-Sabban, M. Expression of HIF-1alpha and Markers of Angiogenesis Are Not Significantly Different in Triple Negative Breast Cancer Compared to Other Breast Cancer Molecular Subtypes: Implications for Future Therapy. PLoS ONE 2015, 10, e0129356. [Google Scholar] [CrossRef] [PubMed]
- Schwab, L.P.; Peacock, D.L.; Majumdar, D.; Ingels, J.F.; Jensen, L.C.; Smith, K.D.; Cushing, R.C.; Seagroves, T.N. Hypoxia-inducible factor 1alpha promotes primary tumor growth and tumor-initiating cell activity in breast cancer. Breast Cancer Res. 2012, 14, R6. [Google Scholar] [CrossRef]
- Gilkes, D.M.; Semenza, G.L. Role of hypoxia-inducible factors in breast cancer metastasis. Future Oncol. 2015, 9, 1623–1636. [Google Scholar] [CrossRef]
- Samanta, D.; Gilkes, D.M.; Chaturvedi, P.; Xiang, L.; Semenza, G.L. Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, E5429–E5438. [Google Scholar] [CrossRef]
- Munoz-Najar, U.M.; Neurath, K.M.; Vumbaca, F.; Claffey, K.P. Hypoxia stimulates breast carcinoma cell invasion through MT1-MMP and MMP-2 activation. Oncogene 2006, 25, 2379–2392. [Google Scholar] [CrossRef]
- Robey, I.F.; Lien, A.D.; Welsh, S.J.; Baggett, B.K.; Gillies, R.J. Hypoxia-inducible factor-1alpha and the glycolytic phenotype in tumors. Neoplasia 2005, 7, 324–330. [Google Scholar] [CrossRef]
- Prigione, A.; Rohwer, N.; Hoffmann, S.; Mlody, B.; Drews, K.; Bukowiecki, R.; Blumlein, K.; Wanker, E.E.; Ralser, M.; Cramer, T.; et al. HIF1alpha modulates cell fate reprogramming through early glycolytic shift and upregulation of PDK1-3 and PKM2. Stem Cells 2014, 32, 364–376. [Google Scholar] [CrossRef]
- Peng, F.; Wang, J.H.; Fan, W.J.; Meng, Y.T.; Li, M.M.; Li, T.T.; Cui, B.; Wang, H.F.; Zhao, Y.; An, F.; et al. Glycolysis gatekeeper PDK1 reprograms breast cancer stem cells under hypoxia. Oncogene 2018, 37, 1062–1074. [Google Scholar] [CrossRef]
- Dai, X.; Yan, X.; Wintergerst, K.A.; Cai, L.; Keller, B.B.; Tan, Y. Nrf2: Redox and Metabolic Regulator of Stem Cell State and Function. Trends Mol. Med. 2020, 26, 185–200. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Hallis, S.P.; Jung, K.A.; Ryu, D.; Kwak, M.K. Impairment of HIF-1alpha-mediated metabolic adaption by NRF2-silencing in breast cancer cells. Redox Biol. 2019, 24, 101210. [Google Scholar] [CrossRef] [PubMed]
- Bharti, S.K.; Mironchik, Y.; Wildes, F.; Penet, M.F.; Goggins, E.; Krishnamachary, B.; Bhujwalla, Z.M. Metabolic consequences of HIF silencing in a triple negative human breast cancer xenograft. Oncotarget 2018, 9, 15326–15339. [Google Scholar] [CrossRef] [PubMed]
- Briggs, K.J.; Koivunen, P.; Cao, S.; Backus, K.M.; Olenchock, B.A.; Patel, H.; Zhang, Q.; Signoretti, S.; Gerfen, G.J.; Richardson, A.L.; et al. Paracrine Induction of HIF by Glutamate in Breast Cancer: EglN1 Senses Cysteine. Cell 2016, 166, 126–139. [Google Scholar] [CrossRef]
- Liu, R.Z.; Graham, K.; Glubrecht, D.D.; Lai, R.; Mackey, J.R.; Godbout, R. A fatty acid-binding protein 7/RXRbeta pathway enhances survival and proliferation in triple-negative breast cancer. J. Pathol. 2012, 228, 310–321. [Google Scholar] [CrossRef]
- Kawashima, M.; Bensaad, K.; Zois, C.E.; Barberis, A.; Bridges, E.; Wigfield, S.; Lagerholm, C.; Dmitriev, R.I.; Tokiwa, M.; Toi, M.; et al. Disruption of hypoxia-inducible fatty acid binding protein 7 induces beige fat-like differentiation and thermogenesis in breast cancer cells. Cancer Metab. 2020, 8, 13. [Google Scholar] [CrossRef]
- Bensaad, K.; Favaro, E.; Lewis, C.A.; Peck, B.; Lord, S.; Collins, J.M.; Pinnick, K.E.; Wigfield, S.; Buffa, F.M.; Li, J.L.; et al. Fatty acid uptake and lipid storage induced by HIF-1alpha contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep. 2014, 9, 349–365. [Google Scholar] [CrossRef]
- Zhang, C.; Yang, C.; Feldman, M.J.; Wang, H.; Pang, Y.; Maggio, D.M.; Zhu, D.; Nesvick, C.L.; Dmitriev, P.; Bullova, P.; et al. Vorinostat suppresses hypoxia signaling by modulating nuclear translocation of hypoxia inducible factor 1 alpha. Oncotarget 2017, 8, 56110–56125. [Google Scholar] [CrossRef]
- Gartner, E.M.; Silverman, P.; Simon, M.; Flaherty, L.; Abrams, J.; Ivy, P.; Lorusso, P.M. A phase II study of 17-allylamino-17-demethoxygeldanamycin in metastatic or locally advanced, unresectable breast cancer. Breast Cancer Res. Treat. 2011, 131, 933–937. [Google Scholar] [CrossRef]
- Harrison, M.R.; Hahn, N.M.; Pili, R.; Oh, W.K.; Hammers, H.; Sweeney, C.; Kim, K.; Perlman, S.; Arnott, J.; Sidor, C.; et al. A phase II study of 2-methoxyestradiol (2ME2) NanoCrystal(R) dispersion (NCD) in patients with taxane-refractory, metastatic castrate-resistant prostate cancer (CRPC). Investig. New Drugs 2011, 29, 1465–1474. [Google Scholar] [CrossRef]
- Munster, P.N.; Thurn, K.T.; Thomas, S.; Raha, P.; Lacevic, M.; Miller, A.; Melisko, M.; Ismail-Khan, R.; Rugo, H.; Moasser, M.; et al. A phase II study of the histone deacetylase inhibitor vorinostat combined with tamoxifen for the treatment of patients with hormone therapy-resistant breast cancer. Br. J. Cancer 2011, 104, 1828–1835. [Google Scholar] [CrossRef] [PubMed]
- Campbell, E.J.; Dachs, G.U.; Morrin, H.R.; Davey, V.C.; Robinson, B.A.; Vissers, M.C.M. Activation of the hypoxia pathway in breast cancer tissue and patient survival are inversely associated with tumor ascorbate levels. BMC Cancer 2019, 19, 307. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Zhang, H.; Dinavahi, R.; Li, F.; Xiang, Y.; Raman, V.; Bhujwalla, Z.M.; Felsher, D.W.; Cheng, L.; Pevsner, J.; et al. HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer Cell 2007, 12, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Lacher, S.E.; Levings, D.C.; Freeman, S.; Slattery, M. Identification of a functional antioxidant response element at the HIF1A locus. Redox Biol. 2018, 19, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Uetaki, M.; Tabata, S.; Nakasuka, F.; Soga, T.; Tomita, M. Metabolomic alterations in human cancer cells by vitamin C-induced oxidative stress. Sci. Rep. 2015, 5, 13896. [Google Scholar] [CrossRef] [PubMed]
- Biswas, D.K.; Shi, Q.; Baily, S.; Strickland, I.; Ghosh, S.; Pardee, A.B.; Iglehart, J.D. NF-kappa B activation in human breast cancer specimens and its role in cell proliferation and apoptosis. Proc. Natl. Acad. Sci. USA 2004, 101, 10137–10142. [Google Scholar] [CrossRef]
- Bennett, L.; Mallon, E.A.; Horgan, P.G.; Paul, A.; McMillan, D.C.; Edwards, J. The relationship between members of the canonical NF-kappaB pathway, components of tumour microenvironment and survival in patients with invasive ductal breast cancer. Oncotarget 2017, 8, 33002–33013. [Google Scholar] [CrossRef]
- Van Laere, S.J.; Van der Auwera, I.; Van den Eynden, G.G.; van Dam, P.; Van Marck, E.A.; Vermeulen, P.B.; Dirix, L.Y. NF-kappaB activation in inflammatory breast cancer is associated with oestrogen receptor downregulation, secondary to EGFR and/or ErbB2 overexpression and MAPK hyperactivation. Br. J. Cancer 2007, 97, 659–669. [Google Scholar] [CrossRef]
- Yamaguchi, N.; Ito, T.; Azuma, S.; Ito, E.; Honma, R.; Yanagisawa, Y.; Nishikawa, A.; Kawamura, M.; Imai, J.; Watanabe, S.; et al. Constitutive activation of nuclear factor-kappaB is preferentially involved in the proliferation of basal-like subtype breast cancer cell lines. Cancer Sci. 2009, 100, 1668–1674. [Google Scholar] [CrossRef]
- Shostak, K.; Chariot, A. NF-kappaB, stem cells and breast cancer: The links get stronger. Breast Cancer Res. 2011, 13, 214. [Google Scholar] [CrossRef]
- Smith, S.M.; Lyu, Y.L.; Cai, L. NF-kappaB affects proliferation and invasiveness of breast cancer cells by regulating CD44 expression. PLoS ONE 2014, 9, e106966. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Santillan, K.; Melendez-Zajgla, J.; Jimenez-Hernandez, L.E.; Gaytan-Cervantes, J.; Munoz-Galindo, L.; Pina-Sanchez, P.; Martinez-Ruiz, G.; Torres, J.; Garcia-Lopez, P.; Gonzalez-Torres, C.; et al. NF-kappaBeta-inducing kinase regulates stem cell phenotype in breast cancer. Sci. Rep. 2016, 6, 37340. [Google Scholar] [CrossRef] [PubMed]
- deGraffenried, L.A.; Chandrasekar, B.; Friedrichs, W.E.; Donzis, E.; Silva, J.; Hidalgo, M.; Freeman, J.W.; Weiss, G.R. NF-kappa B inhibition markedly enhances sensitivity of resistant breast cancer tumor cells to tamoxifen. Ann. Oncol. 2004, 15, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Li, L.; Andrew, S.; Allan, D.; Li, X.; Lee, J.; Ji, G.; Yao, Z.; Gadde, S.; Figeys, D.; et al. An autocrine inflammatory forward-feedback loop after chemotherapy withdrawal facilitates the repopulation of drug-resistant breast cancer cells. Cell Death Dis. 2017, 8, e2932. [Google Scholar] [CrossRef] [PubMed]
- Londhe, P.; Yu, P.Y.; Ijiri, Y.; Ladner, K.J.; Fenger, J.M.; London, C.; Houghton, P.J.; Guttridge, D.C. Classical NF-kappaB Metabolically Reprograms Sarcoma Cells Through Regulation of Hexokinase 2. Front. Oncol. 2018, 8, 104. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, C.; Wu, R.; Lin, M.; Liang, Y.; Wang, X.; Yang, B.; Feng, Z. RRAD inhibits the Warburg effect through negative regulation of the NF-kappaB signaling. Oncotarget 2015, 6, 14982–14992. [Google Scholar] [CrossRef][Green Version]
- Chen, Z.; Li, S.; Shen, L.; Wei, X.; Zhu, H.; Wang, X.; Yang, M.; Zheng, X. NF-kappa B interacting long noncoding RNA enhances the Warburg effect and angiogenesis and is associated with decreased survival of patients with gliomas. Cell Death Dis. 2020, 11, 323. [Google Scholar] [CrossRef]
- Zha, X.; Hu, Z.; Ji, S.; Jin, F.; Jiang, K.; Li, C.; Zhao, P.; Tu, Z.; Chen, X.; Di, L.; et al. NFkappaB up-regulation of glucose transporter 3 is essential for hyperactive mammalian target of rapamycin-induced aerobic glycolysis and tumor growth. Cancer Lett. 2015, 359, 97–106. [Google Scholar] [CrossRef]
- Yang, W.; Xia, Y.; Cao, Y.; Zheng, Y.; Bu, W.; Zhang, L.; You, M.J.; Koh, M.Y.; Cote, G.; Aldape, K.; et al. EGFR-induced and PKCepsilon monoubiquitylation-dependent NF-kappaB activation upregulates PKM2 expression and promotes tumorigenesis. Mol. Cell 2012, 48, 771–784. [Google Scholar] [CrossRef]
- Johnson, R.F.; Witzel, I.I.; Perkins, N.D. p53-dependent regulation of mitochondrial energy production by the RelA subunit of NF-kappaB. Cancer Res. 2011, 71, 5588–5597. [Google Scholar] [CrossRef]
- Guido, C.; Whitaker-Menezes, D.; Lin, Z.; Pestell, R.G.; Howell, A.; Zimmers, T.A.; Casimiro, M.C.; Aquila, S.; Ando, S.; Martinez-Outschoorn, U.E.; et al. Mitochondrial fission induces glycolytic reprogramming in cancer-associated myofibroblasts, driving stromal lactate production, and early tumor growth. Oncotarget 2012, 3, 798–810. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.J.; Ratnam, N.M.; Byrd, J.C.; Guttridge, D.C. NF-kappaB functions in tumor initiation by suppressing the surveillance of both innate and adaptive immune cells. Cell Rep. 2014, 9, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Huber, V.; Camisaschi, C.; Berzi, A.; Ferro, S.; Lugini, L.; Triulzi, T.; Tuccitto, A.; Tagliabue, E.; Castelli, C.; Rivoltini, L. Cancer acidity: An ultimate frontier of tumor immune escape and a novel target of immunomodulation. Semin. Cancer Biol. 2017, 43, 74–89. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Sohn, K.H.; Rhee, S.H.; Hwang, D. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J. Biol. Chem. 2001, 276, 16683–16689. [Google Scholar] [CrossRef] [PubMed]
- Senga, S.; Kobayashi, N.; Kawaguchi, K.; Ando, A.; Fujii, H. Fatty acid-binding protein 5 (FABP5) promotes lipolysis of lipid droplets, de novo fatty acid (FA) synthesis and activation of nuclear factor-kappa B (NF-kappaB) signaling in cancer cells. Biochim. Biophys. Acta Mol. Cell. Biol. Lipids 2018, 1863, 1057–1067. [Google Scholar] [CrossRef]
- Li, J.; Condello, S.; Thomes-Pepin, J.; Ma, X.; Xia, Y.; Hurley, T.D.; Matei, D.; Cheng, J.X. Lipid Desaturation Is a Metabolic Marker and Therapeutic Target of Ovarian Cancer Stem Cells. Cell Stem Cell 2017, 20, 303–314.e305. [Google Scholar] [CrossRef]
- Qie, S.; Chu, C.; Li, W.; Wang, C.; Sang, N. ErbB2 activation upregulates glutaminase 1 expression which promotes breast cancer cell proliferation. J. Cell. Biochem. 2014, 115, 498–509. [Google Scholar] [CrossRef]
- Reid, M.A.; Lowman, X.H.; Pan, M.; Tran, T.Q.; Warmoes, M.O.; Ishak Gabra, M.B.; Yang, Y.; Locasale, J.W.; Kong, M. IKKbeta promotes metabolic adaptation to glutamine deprivation via phosphorylation and inhibition of PFKFB3. Genes Dev. 2016, 30, 1837–1851. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Shishodia, S.; Takada, Y.; Banerjee, S.; Newman, R.A.; Bueso-Ramos, C.E.; Price, J.E. Curcumin suppresses the paclitaxel-induced nuclear factor-kappaB pathway in breast cancer cells and inhibits lung metastasis of human breast cancer in nude mice. Clin. Cancer Res. 2005, 11, 7490–7498. [Google Scholar] [CrossRef]
- Marquardt, J.U.; Gomez-Quiroz, L.; Arreguin Camacho, L.O.; Pinna, F.; Lee, Y.H.; Kitade, M.; Dominguez, M.P.; Castven, D.; Breuhahn, K.; Conner, E.A.; et al. Curcumin effectively inhibits oncogenic NF-kappaB signaling and restrains stemness features in liver cancer. J. Hepatol. 2015, 63, 661–669. [Google Scholar] [CrossRef]
- Ghasemi, F.; Shafiee, M.; Banikazemi, Z.; Pourhanifeh, M.H.; Khanbabaei, H.; Shamshirian, A.; Amiri Moghadam, S.; ArefNezhad, R.; Sahebkar, A.; Avan, A.; et al. Curcumin inhibits NF-kB and Wnt/beta-catenin pathways in cervical cancer cells. Pathol. Res. Pract. 2019, 215, 152556. [Google Scholar] [CrossRef] [PubMed]
- Jiao, L.; Wang, S.; Zheng, Y.; Wang, N.; Yang, B.; Wang, D.; Yang, D.; Mei, W.; Zhao, Z.; Wang, Z. Betulinic acid suppresses breast cancer aerobic glycolysis via caveolin-1/NF-kappaB/c-Myc pathway. Biochem. Pharmacol. 2019, 161, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Yin, P.; Wang, W.; Zhang, Z.; Bai, Y.; Gao, J.; Zhao, C. Wnt signaling in human and mouse breast cancer: Focusing on Wnt ligands, receptors and antagonists. Cancer Sci. 2018, 109, 3368–3375. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.S.; Park, J.I. Wnt signaling in cancer: Therapeutic targeting of Wnt signaling beyond beta-catenin and the destruction complex. Exp. Mol. Med. 2020, 52, 183–191. [Google Scholar] [CrossRef]
- Pohl, S.G.; Brook, N.; Agostino, M.; Arfuso, F.; Kumar, A.P.; Dharmarajan, A. Wnt signaling in triple-negative breast cancer. Oncogenesis 2017, 6, e310. [Google Scholar] [CrossRef]
- Lamb, R.; Ablett, M.P.; Spence, K.; Landberg, G.; Sims, A.H.; Clarke, R.B. Wnt pathway activity in breast cancer sub-types and stem-like cells. PLoS ONE 2013, 8, e67811. [Google Scholar] [CrossRef]
- Domenici, G.; Aurrekoetxea-Rodriguez, I.; Simoes, B.M.; Rabano, M.; Lee, S.Y.; Millan, J.S.; Comaills, V.; Oliemuller, E.; Lopez-Ruiz, J.A.; Zabalza, I.; et al. A Sox2-Sox9 signalling axis maintains human breast luminal progenitor and breast cancer stem cells. Oncogene 2019, 38, 3151–3169. [Google Scholar] [CrossRef]
- Jang, G.B.; Hong, I.S.; Kim, R.J.; Lee, S.Y.; Park, S.J.; Lee, E.S.; Park, J.H.; Yun, C.H.; Chung, J.U.; Lee, K.J.; et al. Wnt/beta-Catenin Small-Molecule Inhibitor CWP232228 Preferentially Inhibits the Growth of Breast Cancer Stem-like Cells. Cancer Res. 2015, 75, 1691–1702. [Google Scholar] [CrossRef]
- Jang, G.B.; Kim, J.Y.; Cho, S.D.; Park, K.S.; Jung, J.Y.; Lee, H.Y.; Hong, I.S.; Nam, J.S. Blockade of Wnt/beta-catenin signaling suppresses breast cancer metastasis by inhibiting CSC-like phenotype. Sci. Rep. 2015, 5, 12465. [Google Scholar] [CrossRef]
- Yang, L.; Perez, A.A.; Fujie, S.; Warden, C.; Li, J.; Wang, Y.; Yung, B.; Chen, Y.R.; Liu, X.; Zhang, H.; et al. Wnt modulates MCL1 to control cell survival in triple negative breast cancer. BMC Cancer 2014, 14, 124. [Google Scholar] [CrossRef]
- Lee, S.Y.; Jeon, H.M.; Ju, M.K.; Kim, C.H.; Yoon, G.; Han, S.I.; Park, H.G.; Kang, H.S. Wnt/Snail signaling regulates cytochrome C oxidase and glucose metabolism. Cancer Res. 2012, 72, 3607–3617. [Google Scholar] [CrossRef] [PubMed]
- Vergara, D.; Stanca, E.; Guerra, F.; Priore, P.; Gaballo, A.; Franck, J.; Simeone, P.; Trerotola, M.; De Domenico, S.; Fournier, I.; et al. beta-Catenin Knockdown Affects Mitochondrial Biogenesis and Lipid Metabolism in Breast Cancer Cells. Front. Physiol. 2017, 8, 544. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Chen, Y.; Huo, D.; Khramtsov, A.; Khramtsova, G.; Zhang, C.; Goss, K.H.; Olopade, O.I. beta-catenin regulates c-Myc and CDKN1A expression in breast cancer cells. Mol. Carcinog. 2016, 55, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Tateishi, K.; Iafrate, A.J.; Ho, Q.; Curry, W.T.; Batchelor, T.T.; Flaherty, K.T.; Onozato, M.L.; Lelic, N.; Sundaram, S.; Cahill, D.P.; et al. Myc-Driven Glycolysis Is a Therapeutic Target in Glioblastoma. Clin. Cancer Res. 2016, 22, 4452–4465. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, N.; Zheng, Y.; Yang, B.; Liu, P.; Zhang, F.; Li, M.; Song, J.; Chang, X.; Wang, Z. Caveolin-1 inhibits breast cancer stem cells via c-Myc-mediated metabolic reprogramming. Cell Death Dis. 2020, 11, 450. [Google Scholar] [CrossRef]
- Bott, A.J.; Peng, I.C.; Fan, Y.; Faubert, B.; Zhao, L.; Li, J.; Neidler, S.; Sun, Y.; Jaber, N.; Krokowski, D.; et al. Oncogenic Myc Induces Expression of Glutamine Synthetase through Promoter Demethylation. Cell Metab. 2015, 22, 1068–1077. [Google Scholar] [CrossRef]
- Liao, J.; Liu, P.P.; Hou, G.; Shao, J.; Yang, J.; Liu, K.; Lu, W.; Wen, S.; Hu, Y.; Huang, P. Regulation of stem-like cancer cells by glutamine through beta-catenin pathway mediated by redox signaling. Mol. Cancer 2017, 16, 51. [Google Scholar] [CrossRef]
- Ishikawa, T.; Hosaka, Y.Z.; Beckwitt, C.; Wells, A.; Oltvai, Z.N.; Warita, K. Concomitant attenuation of HMG-CoA reductase expression potentiates the cancer cell growth-inhibitory effect of statins and expands their efficacy in tumor cells with epithelial characteristics. Oncotarget 2018, 9, 29304–29315. [Google Scholar] [CrossRef]
- Wang, P.; Gong, Y.; Guo, T.; Li, M.; Fang, L.; Yin, S.; Kamran, M.; Liu, Y.; Xu, J.; Xu, L.; et al. Activation of Aurora A kinase increases YAP stability via blockage of autophagy. Cell Death Dis. 2019, 10, 432. [Google Scholar] [CrossRef]
- Ouyang, X.; Han, S.N.; Zhang, J.Y.; Dioletis, E.; Nemeth, B.T.; Pacher, P.; Feng, D.; Bataller, R.; Cabezas, J.; Starkel, P.; et al. Digoxin Suppresses Pyruvate Kinase M2-Promoted HIF-1alpha Transactivation in Steatohepatitis. Cell Metab. 2019, 27, 339–350.e3. [Google Scholar] [CrossRef]
- Guerin, E.; Raffelsberger, W.; Pencreach, E.; Maier, A.; Neuville, A.; Schneider, A.; Bachellier, P.; Rohr, S.; Petitprez, A.; Poch, O.; et al. In vivo topoisomerase I inhibition attenuates the expression of hypoxia-inducible factor 1alpha target genes and decreases tumor angiogenesis. Mol. Med. 2012, 18, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Warfel, N.A. Targeting CDK4/6 to oppose hypoxia-mediated therapeutic resistance. Cell Cycle 2017, 16, 1241–1242. [Google Scholar] [CrossRef] [PubMed]
- Feldinger, K.; Kong, A. Profile of neratinib and its potential in the treatment of breast cancer. Breast Cancer (Dove Med. Press) 2015, 7, 147–162. [Google Scholar] [CrossRef] [PubMed]
- Barry, J.B.; Giguere, V. Epidermal growth factor-induced signaling in breast cancer cells results in selective target gene activation by orphan nuclear receptor estrogen-related receptor alpha. Cancer Res. 2005, 65, 6120–6129. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wu, T.; Wang, G.; Chen, W.; Zhu, Z.; Liu, Y.; Huang, Z.; Huang, Y.; Du, P.; Yang, Y.; Liu, C.Y.; et al. Co-inhibition of BET proteins and NF-kappaB as a potential therapy for colorectal cancer through synergistic inhibiting MYC and FOXM1 expressions. Cell Death Dis. 2018, 9, 315. [Google Scholar] [CrossRef]
- Liu, J.L.; Pan, Y.Y.; Chen, O.; Luan, Y.; Xue, X.; Zhao, J.J.; Liu, L.; Jia, H.Y. Curcumin inhibits MCF-7 cells by modulating the NF-kappaB signaling pathway. Oncol. Lett. 2017, 14, 5581–5584. [Google Scholar] [CrossRef]
- Kretzmann, N.A.; Chiela, E.; Matte, U.; Marroni, N.; Marroni, C.A. N-acetylcysteine improves antitumoural response of Interferon alpha by NF-kB downregulation in liver cancer cells. Comp. Hepatol. 2012, 11, 4. [Google Scholar] [CrossRef]
- Pantano, F.; Rossi, E.; Iuliani, M.; Facchinetti, A.; Simonetti, S.; Ribelli, G.; Zoccoli, A.; Vincenzi, B.; Tonini, G.; Zamarchi, R.; et al. Dynamic changes of Receptor activator of nuclear factor-kappaB expression in Circulating Tumor Cells during Denosumab predict treatment effectiveness in Metastatic Breast Cancer. Sci. Rep. 2020, 10, 1288. [Google Scholar] [CrossRef]
- Madan, B.; Ke, Z.; Harmston, N.; Ho, S.Y.; Frois, A.O.; Alam, J.; Jeyaraj, D.A.; Pendharkar, V.; Ghosh, K.; Virshup, I.H.; et al. Wnt addiction of genetically defined cancers reversed by PORCN inhibition. Oncogene 2016, 35, 2197–2207. [Google Scholar] [CrossRef]
- Furuya, K.; Sasaki, A.; Tsunoda, Y.; Tsuji, M.; Udaka, Y.; Oyamada, H.; Tsuchiya, H.; Oguchi, K. Eribulin upregulates miR-195 expression and downregulates Wnt3a expression in non-basal-like type of triple-negative breast cancer cell MDA-MB-231. Hum. Cell 2015, 29, 76–82. [Google Scholar] [CrossRef]
- Liu, J.; Pan, S.; Hsieh, M.H.; Ng, N.; Sun, F.; Wang, T.; Kasibhatla, S.; Schuller, A.G.; Li, A.G.; Cheng, D.; et al. Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc. Natl. Acad. Sci. USA 2013, 110, 20224–20229. [Google Scholar] [CrossRef] [PubMed]
- Tam, B.Y.; Chiu, K.; Chung, H.; Bossard, C.; Nguyen, J.D.; Creger, E.; Eastman, B.W.; Mak, C.C.; Ibanez, M.; Ghias, A.; et al. The CLK inhibitor SM08502 induces anti-tumor activity and reduces Wnt pathway gene expression in gastrointestinal cancer models. Cancer Lett. 2020, 473, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M. Antibody-drug conjugate targeting protein tyrosine kinase 7, a receptor tyrosine kinase-like molecule involved in WNT and vascular endothelial growth factor signaling: Effects on cancer stem cells, tumor microenvironment and whole-body homeostasis. Ann. Transl. Med. 2017, 5, 462. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.M.; Cancilla, B.; Yeung, V.P.; Cattaruzza, F.; Chartier, C.; Murriel, C.L.; Cain, J.; Tam, R.; Cheng, C.Y.; Evans, J.W.; et al. WNT antagonists exhibit unique combinatorial antitumor activity with taxanes by potentiating mitotic cell death. Sci. Adv. 2017, 3, e1700090. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.F.; Tai, W.T.; Hsu, C.Y.; Huang, J.W.; Liu, C.Y.; Chen, P.J.; Kim, I.; Shiau, C.W. Blockade of STAT3 activation by sorafenib derivatives through enhancing SHP-1 phosphatase activity. Eur. J. Med. Chem. 2012, 55, 220–227. [Google Scholar] [CrossRef]
- Hahn, Y.I.; Kim, S.J.; Choi, B.Y.; Cho, K.C.; Bandu, R.; Kim, K.P.; Kim, D.H.; Kim, W.; Park, J.S.; Han, B.W.; et al. Curcumin interacts directly with the Cysteine 259 residue of STAT3 and induces apoptosis in H-Ras transformed human mammary epithelial cells. Sci. Rep. 2018, 8, 6409. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, Y.; Dong, Y.; Chen, Z.; Eckols, T.K.; Kasembeli, M.M.; Tweardy, D.J.; Mitch, W.E. Pharmacokinetics and pharmacodynamics of TTI-101, a STAT3 inhibitor that blocks muscle proteolysis in rats with chronic kidney disease. Am. J. Physiol. Renal. Physiol. 2020, 319, F84–F92. [Google Scholar] [CrossRef]
- Stover, D.G.; Gil Del Alcazar, C.R.; Brock, J.; Guo, H.; Overmoyer, B.; Balko, J.; Xu, Q.; Bardia, A.; Tolaney, S.M.; Gelman, R.; et al. Phase II study of ruxolitinib, a selective JAK1/2 inhibitor, in patients with metastatic triple-negative breast cancer. NPJ Breast Cancer 2018, 4, 10. [Google Scholar] [CrossRef]
- Srirangam, A.; Milani, M.; Mitra, R.; Guo, Z.; Rodriguez, M.; Kathuria, H.; Fukuda, S.; Rizzardi, A.; Schmechel, S.; Skalnik, D.G.; et al. The human immunodeficiency virus protease inhibitor ritonavir inhibits lung cancer cells, in part, by inhibition of survivin. J. Thorac. Oncol. 2011, 6, 661–670. [Google Scholar] [CrossRef]
- Soleymani Abyaneh, H.; Gupta, N.; Alshareef, A.; Gopal, K.; Lavasanifar, A.; Lai, R. Hypoxia Induces the Acquisition of Cancer Stem-like Phenotype Via Upregulation and Activation of Signal Transducer and Activator of Transcription-3 (STAT3) in MDA-MB-231, a Triple Negative Breast Cancer Cell Line. Cancer Microenviron. 2018, 11, 141–152. [Google Scholar] [CrossRef]
- Gao, X.; Wang, H.; Yang, J.J.; Liu, X.; Liu, Z.R. Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase. Mol. Cell 2012, 45, 598–609. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Hu, H.; Chang, R.; Zhong, J.; Knabel, M.; O’Meally, R.; Cole, R.N.; Pandey, A.; Semenza, G.L. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell 2011, 145, 732–744. [Google Scholar] [CrossRef] [PubMed]
- Azoitei, N.; Becher, A.; Steinestel, K.; Rouhi, A.; Diepold, K.; Genze, F.; Simmet, T.; Seufferlein, T. PKM2 promotes tumor angiogenesis by regulating HIF-1alpha through NF-kappaB activation. Mol. Cancer 2016, 15, 3. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.P.; Fako, V.; Dang, H.; Dominguez, D.A.; Khatib, S.; Ma, L.; Wang, H.; Zheng, W.; Wang, X.W. PKM2 inhibition may reverse therapeutic resistance to transarterial chemoembolization in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2020, 39, 99. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zhu, Y.; Hu, J.; Jiang, L.; Li, L.; Jia, S.; Zen, K. Shikonin Inhibits Tumor Growth in Mice by Suppressing Pyruvate Kinase M2-mediated Aerobic Glycolysis. Sci. Rep. 2018, 8, 14517. [Google Scholar] [CrossRef]
- Kumar, S.; Donti, T.R.; Agnihotri, N.; Mehta, K. Transglutaminase 2 reprogramming of glucose metabolism in mammary epithelial cells via activation of inflammatory signaling pathways. Int. J. Cancer 2014, 134, 2798–2807. [Google Scholar] [CrossRef]
- Katt, W.P.; Blobel, N.J.; Komarova, S.; Antonyak, M.A.; Nakano, I.; Cerione, R.A. A small molecule regulator of tissue transglutaminase conformation inhibits the malignant phenotype of cancer cells. Oncotarget 2018, 9, 34379–34397. [Google Scholar] [CrossRef][Green Version]
- Deng, X.; Wang, Q.; Cheng, M.; Chen, Y.; Yan, X.; Guo, R.; Sun, L.; Li, Y.; Liu, Y. Pyruvate dehydrogenase kinase 1 interferes with glucose metabolism reprogramming and mitochondrial quality control to aggravate stress damage in cancer. J. Cancer 2020, 11, 962–973. [Google Scholar] [CrossRef]
- Yang, Z.; Zhang, S.L.; Hu, X.; Tam, K.Y. Inhibition of pyruvate dehydrogenase kinase 1 enhances the anti-cancer effect of EGFR tyrosine kinase inhibitors in non-small cell lung cancer. Eur. J. Pharmacol. 2018, 838, 41–52. [Google Scholar] [CrossRef]
- Qin, W.; Tian, Y.; Zhang, J.; Liu, W.; Zhou, Q.; Hu, S.; Yang, F.; Lu, L.; Lu, H.; Cui, S.; et al. The double inhibition of PDK1 and STAT3-Y705 prevents liver metastasis in colorectal cancer. Sci. Rep. 2019, 9, 12973. [Google Scholar] [CrossRef]
- Kennedy, B.E.; Murphy, J.P.; Clements, D.R.; Konda, P.; Holay, N.; Kim, Y.; Pathak, G.P.; Giacomantonio, M.A.; Hiani, Y.E.; Gujar, S. Inhibition of Pyruvate Dehydrogenase Kinase Enhances the Antitumor Efficacy of Oncolytic Reovirus. Cancer Res. 2019, 79, 3824–3836. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Hottiger, M.O. Crosstalk between Wnt/beta-Catenin and NF-kappaB Signaling Pathway during Inflammation. Front. Immunol. 2016, 7, 378. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, M.L.; Ortolano, N.A.; Romero-Morales, A.I.; Gama, V. Wnt Signaling and Its Impact on Mitochondrial and Cell Cycle Dynamics in Pluripotent Stem Cells. Genes 2018, 9, 109. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Ma, L.; Li, J.; Yu, Y.; Zhang, D.; Wei, J.; Jin, H.; Xu, D.; Gao, J.; Huang, C. NF-kappaB1 inhibits c-Myc protein degradation through suppression of FBW7 expression. Oncotarget 2014, 5, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Mammoto, A.; Muyleart, M.; Kadlec, A.; Gutterman, D.; Mammoto, T. YAP1-TEAD1 signaling controls angiogenesis and mitochondrial biogenesis through PGC1alpha. Microvasc. Res. 2018, 119, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Petty, A.J.; Li, A.; Wang, X.; Dai, R.; Heyman, B.; Hsu, D.; Huang, X.; Yang, Y. Hedgehog signaling promotes tumor-associated macrophage polarization to suppress intratumoral CD8+ T cell recruitment. J. Clin. Investig. 2019, 129, 5151–5162. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Pathway | Therapy | Subtype | Mechanism of Action | Clinical Trial ID |
|---|---|---|---|---|
| YAP/TAZ | Zoledronate | TNBC | Inhibits the mevalonate pathway via enzyme farnesyl diphosphate synthase [43]. | NCT03358017 |
| Statin | Invasive BC | Inhibits the mevalonate pathway via HMG-coA Reductase [148]. | NCT03971019 | |
| Dasatnib | TNBC | Inhibits the nuclear translocation of YAP via its phosphorylation and degradation [45]. | NCT02720185 | |
| Alisertib | ER+ HER-2- | Inhibits Aurora A kinase, decreasing YAP stability [149]. | NCT02187991 | |
| HIF-1α | Digoxin | Invasive BC | Inhibits PKM2-mediated transcription, indirectly reducing HIF-1α [150]. | NCT01763931 |
| Irinotecan | Metastatic BC | Inhibits Topoisomerase1, shown to decrease HIF-1α expression [151]. | NCT03562390 | |
| Vorinostat | HER-2+ BC | Reduces nuclear accumulation of HIF-1α via HDAC inhibition [98]. | NCT00574587 | |
| Palbociclib | ER+ HER-2- | Disrupts HIF-1α stabilization via CDK inhibition [152]. | NCT04247633 | |
| PGC-1α | TPST-1120 | TNBC | Inhibits PPARα as a small molecule selective antagonist. | NCT03829436 |
| Neratinib | ER+ HER2- | Reduces ERRα-mediated gene expression via EGFR inhibition [153,154]. | NCT04460430 | |
| NF-kB | Bortezomib | TNBC | Prevents nuclear translocation of NF-kB elements by proteosome inhibition [155]. | NCT04265872 |
| IMX-110 | TNBC | Decreases NF-kB signaling via nanoparticle encapsulation of curcumin [156]. | NCT03382340 | |
| Reparixin | TNBC | Attenuates NF-kB signaling by inhibition of upstream IL8 receptor CXCR1/2 [114]. | NCT02370238 | |
| N-acetylcysteine | Stage I BC | Reduces NF-kB activity [157]. | NCT01878695 | |
| Denosumab | Early BC | Inhibits receptor activator of NF-kB ligand (RANKL) [158]. | NCT03324932 | |
| Wnt signaling | ETC-1922159 | Advanced solid tumors | Prevents the processing of Wnt proteins as a porcupine inhibitor [159]. | NCT02521844 |
| Eribulin | ER+/PR+ BC | Increases miR-195 expression, in turn decreasing expression of Wnt3a [160]. | NCT03795012 | |
| LGK974 | TNBC | Targets porcupine inhibiting Wnt signaling [161]. | NCT01351103 | |
| SM08502 | Advanced tumors | Reduces Wnt-mediated gene expression by interfering with alternative splicing [162]. | NCT03355066 | |
| PTK7-ADC | TNBC | Disrupts Wnt signaling by targeting protein tyrosine kinase 7 [163]. | NCT03243331 | |
| Vantictumab | Metastatic breast cancer | Inhibits Wnt-mediated signaling by targeting the frizzled receptors [164]. | NCT01973309 | |
| STAT3 | Sorafenib | Recurrent BC | Inhibits phosphorylation of STAT3 by enhancing phosphatase activity [165]. | NCT00499525 |
| IMX-110 | BC | Decreases STAT3 activity via nanoparticle encapsulation of curcumin [166]. | NCT03382340 | |
| TTI-101 | Advanced BC | Specifically targets and binds to STAT3 preventing activation [167]. | NCT03195699 | |
| Ruxolitinib | TNBC | Inhibits STAT3 phosphorylation and downstream target genes [168]. | NCT02876302 | |
| Ritonavir | BC (all types) | Inhibits phosphorylation of STAT3 via HIV protease inhibition [169]. | NCT01009437 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Sahli, S.; Wang, L. Cancer Stem Cell-Associated Pathways in the Metabolic Reprogramming of Breast Cancer. Int. J. Mol. Sci. 2020, 21, 9125. https://doi.org/10.3390/ijms21239125
El-Sahli S, Wang L. Cancer Stem Cell-Associated Pathways in the Metabolic Reprogramming of Breast Cancer. International Journal of Molecular Sciences. 2020; 21(23):9125. https://doi.org/10.3390/ijms21239125
Chicago/Turabian StyleEl-Sahli, Sara, and Lisheng Wang. 2020. "Cancer Stem Cell-Associated Pathways in the Metabolic Reprogramming of Breast Cancer" International Journal of Molecular Sciences 21, no. 23: 9125. https://doi.org/10.3390/ijms21239125
APA StyleEl-Sahli, S., & Wang, L. (2020). Cancer Stem Cell-Associated Pathways in the Metabolic Reprogramming of Breast Cancer. International Journal of Molecular Sciences, 21(23), 9125. https://doi.org/10.3390/ijms21239125