Rethinking Intellectual Disability from Neuro- to Astro-Pathology



{kind=link}
{kind=link}
Abstract
1. Introduction
2. Astrocyte Function in the Healthy Brain
3. Astrocyte Involvement in Learning and Memory in the Healthy Brain
4. Astrocyte Dysfunction in Neurodevelopmental Disorders
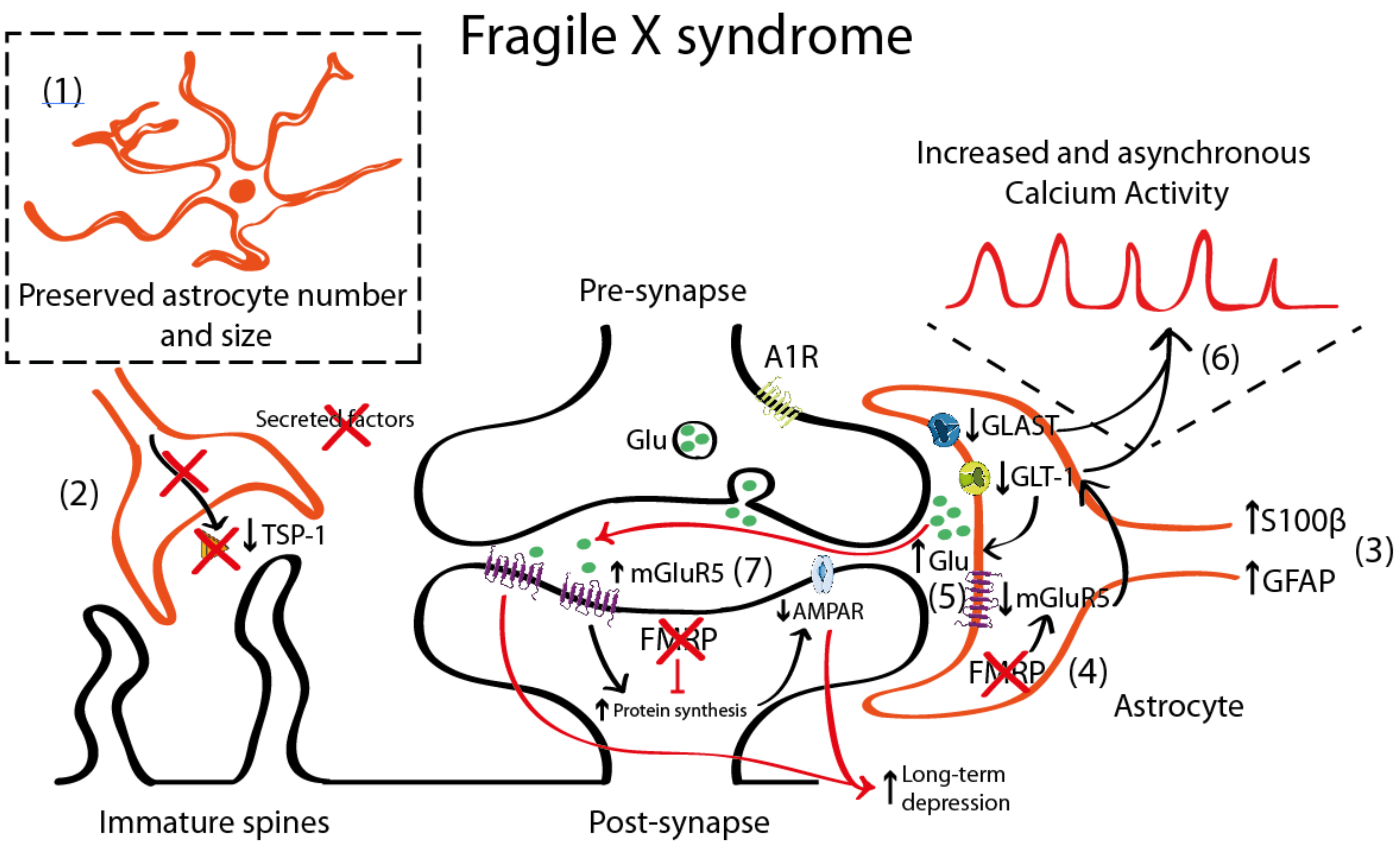
4.1. Astrocyte Pathology in Fragile X Syndrome (FXS)
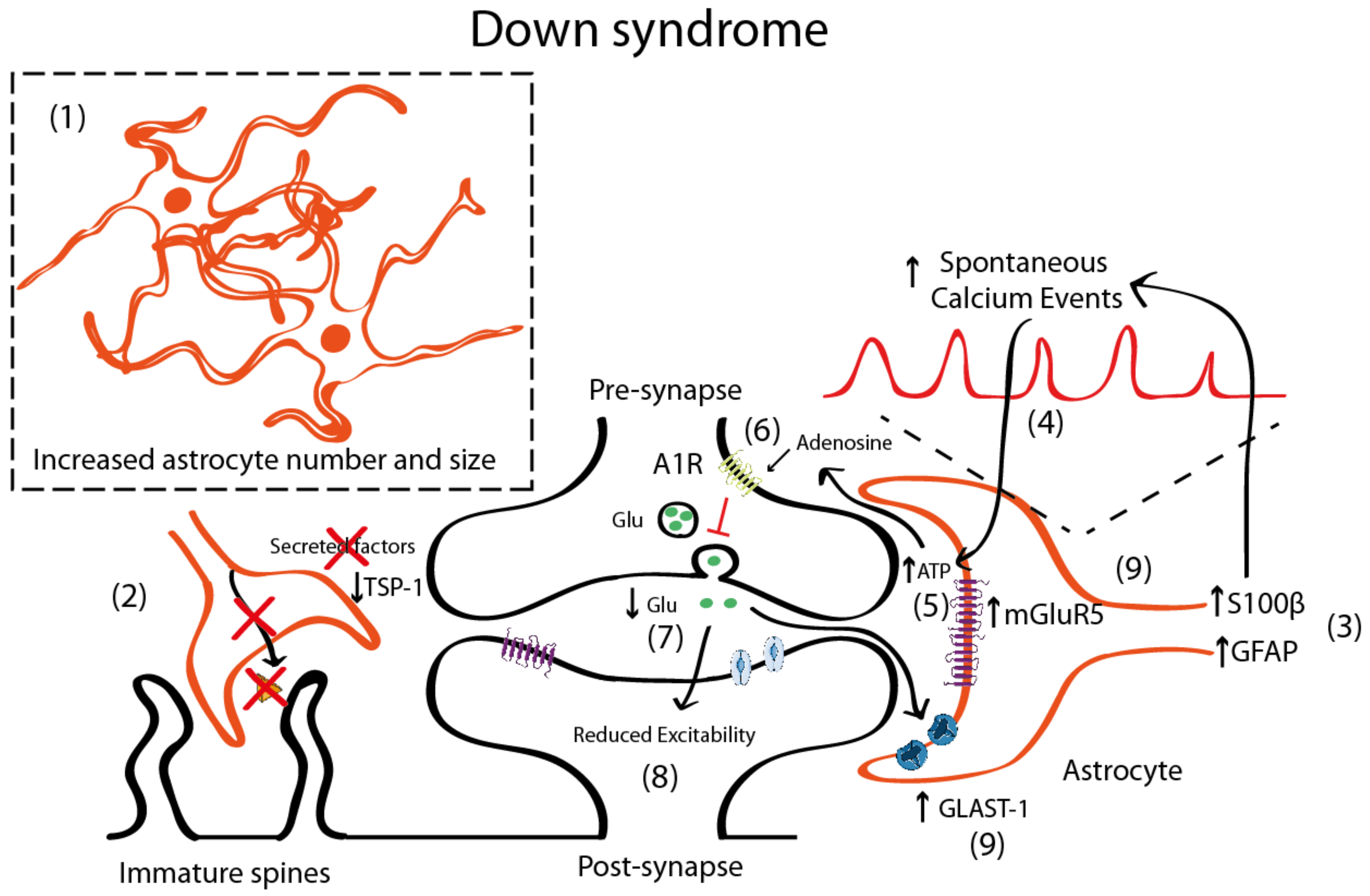
4.2. Astrocyte Pathology in Down Syndrome
5. Astrocytic Phenotypes in DS and FXS: Same Players for Different Phenotypes?
6. Astrocyte Involvement in Memory Pathology in Neurodevelopmental Disorders: A Look into the Future
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DS | Down syndrome |
| FXS | Fragile X syndrome |
| S100β | S100 calcium-binding protein β |
| HSA21 | Homo Sapiens Autosome 21 |
| GFAP | Glial Fibrillary Acidic Protein |
| GABA | Gamma aminobutyric acid |
| ATP | Adenosine triphosphate |
| PAP | Perisynaptic astrocytic process |
| LTP | Long-term potentiation |
| LTD | Long-term depression |
| TeNT | Tetanus Neurotoxin |
| hM3Dq | human Gq-coupled M3 muscarinic receptor |
| CA1 | Cornu Ammonis 1 |
| CA3 | Cornu Ammonis 3 |
| ACC | Anterior Cingulate Cortex |
| FMR1 | fragile X mental retardation 1 |
| FMRP | Fragile X Mental Retardation Protein |
| CA4 | Cornu Ammonis 4 |
| KO | Knock-out |
| mGluR5 | Metabotropic Glutamate Receptor 5 |
| GLT-1 | Glutamate Transporter 1 |
| GLAST-1 | Glutamate Aspartate Transporter 1 |
| TSP-1 | Thrombospondin-1 |
| iPSC | Induced pluripotent stem cells |
| YRK1A | Dual-specificity tyrosine phosphorylation-regulated kinase-1 |
| STAT | Signal Transducer and Activator of Transcription |
| NPC | Neural Progenitor Cell |
| A1R | Adenosine 1 Receptor |
| DPCPX | 8-Cyclopentyl-1,3-dipropyl xanthine |
References
- Dierssen, M. Top ten discoveries of the year: Neurodevelopmental disorders. Free Neuropathol. 2020, 1, 13. Available online: https://www.uni-muenster.de/Ejournals/index.php/fnp/article/view/2672 (accessed on 26 October 2020).
- Papazoglou, A.; Jacobson, L.A.; McCabe, M.; Kaufmann, W.; Zabel, T.A. To ID or Not to ID? Changes in Classification Rates of Intellectual Disability Using DSM-5. Intellect. Dev. Disabil. 2014, 52, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Ardiles, A.O.; Grabrucker, A.M.; Scholl, F.G.; Rudenko, G.; Borsello, T. Molecular and Cellular Mechanisms of Synaptopathies. Neural Plast. 2017, 2017, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Norris, R.; Gordon, S.; Nithianantharajah, J. Neurodevelopmental synaptopathies: Insights from behaviour in rodent models of synapse gene mutations. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2018, 84, 424–439. [Google Scholar] [CrossRef]
- Dierssen, M.; Ramakers, G.J.A. Dendritic pathology in mental retardation: From molecular genetics to neurobiology. Genes Brain Behav. 2006, 5, 48–60. [Google Scholar] [CrossRef]
- Irwin, S.A.; Galvez, R.; Greenough, W.T. Dendritic Spine Structural Anomalies in Fragile-X Mental Retardation Syndrome. Cereb. Cortex 2000, 10, 1038–1044. [Google Scholar] [CrossRef]
- Dierssen, M.; Benavides-Piccione, R.; Martínez-Cué, C.; Estivill, X.; Flórez, J.; Elston, G.; DeFelipe, J. Alterations of neocortical pyramidal cell phenotype in the Ts65Dn mouse model of Down syndrome: Effects of environmental enrichment. Cereb. Cortex 2003, 13, 758–764. [Google Scholar] [CrossRef]
- Belichenko, P.V.; Masliah, E.; Kleschevnikov, A.M.; Villar, A.J.; Epstein, C.J.; Salehi, A.; Mobley, W.C. Synaptic structural abnormalities in the Ts65Dn mouse model of down syndrome. J. Comp. Neurol. 2004, 480, 281–298. [Google Scholar] [CrossRef]
- Catuara-Solarz, S.; Espinosa-Carrasco, J.; Erb, I.; Langohr, K.; Gonzalez, J.R.; Notredame, C.; Dierssen, M. Combined Treatment with Environmental Enrichment and (-)-Epigallocatechin-3-Gallate Ameliorates Learning Deficits and Hippocampal Alterations in a Mouse Model of Down Syndrome. eNeuro 2016, 3. [Google Scholar] [CrossRef]
- Comery, T.A.; Harris, J.B.; Willems, P.J.; Oostra, B.A.; Irwin, S.A.; Weiler, I.J.; Greenough, W.T. Abnormal dendritic spines in fragile X knockout mice: Maturation and pruning deficits. Proc. Natl. Acad. Sci. USA 1997, 94, 5401–5404. [Google Scholar] [CrossRef]
- Hinton, V.J.; Brown, W.T.; Wisniewski, K.; Rudelli, R.D. Analysis of neocortex in three males with the fragile X syndrome. Am. J. Med. Genet. 1991, 41, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Miller, E.C.; Pozzo-Miller, L. Dendritic spine dysgenesis in Rett syndrome. Front. Neuroanat. 2014, 8. [Google Scholar] [CrossRef] [PubMed]
- Landi, S.; Putignano, E.; Boggio, E.M.; Giustetto, M.; Pizzorusso, T.; Ratto, G.M. The short-time structural plasticity of dendritic spines is altered in a model of Rett syndrome. Sci. Rep. 2011, 1, 45. [Google Scholar] [CrossRef] [PubMed]
- Garcia, O.; Torres, M.; Helguera, P.; Coskun, P.; Busciglio, J. A Role for Thrombospondin-1 Deficits in Astrocyte-Mediated Spine and Synaptic Pathology in Down’s Syndrome. PLoS ONE 2010, 5, e14200. [Google Scholar] [CrossRef] [PubMed]
- Stagni, F.; Salvalai, M.E.; Giacomini, A.; Emili, M.; Uguagliati, B.; Xia, E.; Grilli, M.; Bartesaghi, R.; Bartesaghi, R. Neonatal treatment with cyclosporine A restores neurogenesis and spinogenesis in the Ts65Dn model of Down syndrome. Neurobiol. Dis. 2019, 129, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.; Doering, L.C. Astrocytes Prevent Abnormal Neuronal Development in the Fragile X Mouse. J. Neurosci. 2010, 30, 4508–4514. [Google Scholar] [CrossRef]
- Fukuda, T.; Itoh, M.; Ichikawa, T.; Washiyama, K.; Goto, Y.-I. Delayed Maturation of Neuronal Architecture and Synaptogenesis in Cerebral Cortex ofMecp2-Deficient Mice. J. Neuropathol. Exp. Neurol. 2005, 64, 537–544. [Google Scholar] [CrossRef]
- Kleschevnikov, A.M.; Belichenko, P.V.; Villar, A.J.; Epstein, C.J.; Malenka, R.C.; Mobley, W.C. Hippocampal Long-Term Potentiation Suppressed by Increased Inhibition in the Ts65Dn Mouse, a Genetic Model of Down Syndrome. J. Neurosci. 2004, 24, 8153–8160. [Google Scholar] [CrossRef]
- Zhao, M.-G.; Toyoda, H.; Ko, S.W.; Ding, H.-K.; Wu, L.-J.; Zhuo, M. Deficits in Trace Fear Memory and Long-Term Potentiation in a Mouse Model for Fragile X Syndrome. J. Neurosci. 2005, 25, 7385–7392. [Google Scholar] [CrossRef]
- Martin, H.G.S.; Lassalle, O.; Brown, J.T.; Manzoni, O.J. Age-Dependent Long-Term Potentiation Deficits in the Prefrontal Cortex of theFmr1Knockout Mouse Model of Fragile X Syndrome. Cereb. Cortex 2015, 26, 2084–2092. [Google Scholar] [CrossRef]
- Weng, S.-M.; McLeod, F.; Bailey, M.E.S.; Cobb, S.R. Synaptic plasticity deficits in an experimental model of rett syndrome: Long-term potentiation saturation and its pharmacological reversal. Neuroscience 2011, 180, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Fred Attneave, M.B.; Hebb, D.O. The Organization of Behavior; A Neuropsychological Theory. Am. J. Psychol. 1950, 63, 633. [Google Scholar] [CrossRef]
- Liu, X.; Ramirez, S.; Pang, P.T.; Puryear, C.B.; Govindarajan, A.; Deisseroth, K.; Tonegawa, S. Optogenetic stimulation of a hippocampal engram activates fear memory recall. Nat. Cell Biol. 2012, 484, 381–385. [Google Scholar] [CrossRef] [PubMed]
- De La Torre, R.; De Sola, S.; Pons, M.; Duchon, A.; De Lagran, M.M.; Farré, M.; Fitó, M.; Benejam, B.; Langohr, K.; Rodriguez, J.; et al. Epigallocatechin-3-gallate, a DYRK1A inhibitor, rescues cognitive deficits in Down syndrome mouse models and in humans. Mol. Nutr. Food Res. 2014, 58, 278–288. [Google Scholar] [CrossRef] [PubMed]
- De La Torre, R.; De Sola, S.; Hernandez, G.; Farré, M.; Pujol, J.; Rodriguez, J.; Espadaler, J.M.; Langohr, K.; Cuenca-Royo, A.; Principe, A.; et al. Safety and efficacy of cognitive training plus epigallocatechin-3-gallate in young adults with Down’s syndrome (TESDAD): A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2016, 15, 801–810. [Google Scholar] [CrossRef]
- Jacquemont, S.; Berry-Kravis, E.; Hagerman, R.; Von Raison, F.; Gasparini, F.; Apostol, G.; Ufer, M.; Portes, V.D.; Gomez-Mancilla, B. The challenges of clinical trials in fragile X syndrome. Psychopharmacology 2014, 231, 1237–1250. [Google Scholar] [CrossRef]
- Perea, G.; Araque, A. Astrocytes Potentiate Transmitter Release at Single Hippocampal Synapses. Science 2007, 317, 1083–1086. [Google Scholar] [CrossRef]
- Adamsky, A.; Kol, A.; Kreisel, T.; Doron, A.; Ozeri-Engelhard, N.; Melcer, T.; Refaeli, R.; Horn, H.; Regev, L.; Groysman, M.; et al. Astrocytic Activation Generates De Novo Neuronal Potentiation and Memory Enhancement. Cell 2018, 174, 59–71. [Google Scholar] [CrossRef]
- Kol, A.; Adamsky, A.; Groysman, M.; Kreisel, T.; London, M.; Goshen, I. Astrocytes contribute to remote memory formation by modulating hippocampal–cortical communication during learning. Nat. Neurosci. 2020, 23, 1229–1239. [Google Scholar] [CrossRef]
- Lee, H.S.; Ghetti, A.; Pinto-Duarte, A.; Wang, X.; Dziewczapolski, G.; Galimi, F.; Huitron-Resendiz, S.; Piña-Crespo, J.C.; Roberts, A.J.; Verma, I.M.; et al. Astrocytes contribute to gamma oscillations and recognition memory. Proc. Natl. Acad. Sci. USA 2014, 111, E3343–E3352. [Google Scholar] [CrossRef]
- Suzuki, A.; Stern, S.A.; Bozdagi, O.; Huntley, G.W.; Walker, R.H.; Magistretti, P.J.; Alberini, C.M. Astrocyte-Neuron Lactate Transport Is Required for Long-Term Memory Formation. Cell 2011, 144, 810–823. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Lau, S.K.M.; Doering, L.C. Astrocyte-secreted thrombospondin-1 modulates synapse and spine defects in the fragile X mouse model. Mol. Brain 2016, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Hodges, J.L.; Yu, X.; Gilmore, A.; Bennett, H.; Tjia, M.; Perna, J.F.; Chen, C.-C.; Li, X.; Lu, J.; Zuo, Y. Astrocytic Contributions to Synaptic and Learning Abnormalities in a Mouse Model of Fragile X Syndrome. Biol. Psychiatry 2017, 82, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Torres, M.D.; Garcia, O.; Tang, C.; Busciglio, J. Dendritic spine pathology and thrombospondin-1 deficits in Down syndrome. Free. Radic. Biol. Med. 2017, 114, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Dossi, E.; Vasile, F.; Rouach, N. Human astrocytes in the diseased brain. Brain Res. Bull. 2018, 136, 139–156. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.; Ince, P.; Lace, G.; Forster, G.; Shaw, P.; Matthews, F.; Savva, G.; Brayne, C.; Wharton, S.B. Astrocyte phenotype in relation to Alzheimer-type pathology in the ageing brain. Neurobiol. Aging 2010, 31, 578–590. [Google Scholar] [CrossRef]
- Kuchibhotla, K.V.; Lattarulo, C.R.; Hyman, B.T.; Bacskai, B.J. Synchronous Hyperactivity and Intercellular Calcium Waves in Astrocytes in Alzheimer Mice. Science 2009, 323, 1211–1215. [Google Scholar] [CrossRef]
- Santello, M.; Toni, N.; Volterra, A. Astrocyte function from information processing to cognition and cognitive impairment. Nat. Neurosci. 2019, 22, 154–166. [Google Scholar] [CrossRef]
- Blanco-Suárez, E.; Caldwell, A.L.M.; Allen, N.J. Role of astrocyte-synapse interactions in CNS disorders. J. Physiol. 2017, 595, 1903–1916. [Google Scholar] [CrossRef]
- Bally, B.P.; Farmer, W.T.; Jones, E.V.; Jessa, S.; Kacerovsky, J.B.; Mayran, A.; Peng, H.; Lefebvre, J.L.; Drouin, J.; Hayer, A.; et al. Human iPSC-derived Down syndrome astrocytes display genome-wide perturbations in gene expression, an altered adhesion profile, and increased cellular dynamics. Hum. Mol. Genet. 2020, 29, 785–802. [Google Scholar] [CrossRef]
- Mizuno, G.O.; Wang, Y.; Shi, G.; Wang, Y.; Sun, J.; Papadopoulos, S.; Broussard, G.J.; Unger, E.K.; Deng, W.; Weick, J.; et al. Aberrant Calcium Signaling in Astrocytes Inhibits Neuronal Excitability in a Human Down Syndrome Stem Cell Model. Cell Rep. 2018, 24, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Mito, T.; Becker, L.E. Developmental Changes of S-100 Protein and Glial Fibrillary Acidic Protein in the Brain in Down Syndrome. Exp. Neurol. 1993, 120, 170–176. [Google Scholar] [CrossRef] [PubMed]
- J∅Rgensen, O.S.; Brooksbank, B.W.; Balazs, R. Neuronal plasticity and astrocytic reaction in Down syndrome and Alzheimer disease. J. Neurol. Sci. 1990, 98, 63–79. [Google Scholar] [CrossRef]
- Quinlan, R.A.; Brenner, M.; Goldman, J.E.; Messing, A. GFAP and its role in Alexander disease. Exp. Cell Res. 2007, 313, 2077–2087. [Google Scholar] [CrossRef] [PubMed]
- Laurence, J.A.; Fatemi, S.H. Glial fibrillary acidic protein is elevated in superior frontal, parietal and cerebellar cortices of autistic subjects. Cerebellum 2005, 4, 206–210. [Google Scholar] [CrossRef]
- Wu, Z.; Guo, Z.; Gearing, M.; Chen, G. Tonic inhibition in dentate gyrus impairs long-term potentiation and memory in an Alzheimer’s disease model. Nat. Commun. 2014, 5, 1–13. [Google Scholar] [CrossRef]
- Jo, S.; Yarishkin, O.; Hwang, Y.J.; Chun, Y.E.; Park, M.; Woo, D.H.; Bae, J.Y.; Kim, T.; Lee, J.; Chun, H.; et al. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat. Med. 2014, 20, 886–896. [Google Scholar] [CrossRef]
- Gerlai, R.; Wojtowicz, J.M.; Marks, A.; Roder, J. Overexpression of a calcium-binding protein, S100 beta, in astrocytes alters synaptic plasticity and impairs spatial learning in transgenic mice. Learn. Mem. 1995, 2, 26–39. [Google Scholar] [CrossRef]
- Durkee, C.A.; Araque, A. Diversity and Specificity of Astrocyte–neuron Communication. Neuroscience 2019, 396, 73–78. [Google Scholar] [CrossRef]
- Panatier, A.; Robitaille, R. Astrocytic mGluR5 and the tripartite synapse. Neuroscience 2016, 323, 29–34. [Google Scholar] [CrossRef]
- Haydon, P.G. Glia: Listening and talking to the synapse. Nat. Rev. Neurosci. 2001, 2, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Araque, A.; Carmignoto, G.; Haydon, P.G.; Oliet, S.H.R.; Robitaille, R.; Volterra, A. Gliotransmitters Travel in Time and Space. Neuron 2014, 81, 728–739. [Google Scholar] [CrossRef] [PubMed]
- Ben Achour, S.; Pont-Lezica, L.; Béchade, C.; Pascual, O. Is astrocyte calcium signaling relevant for synaptic plasticity? Neuron Glia Biol. 2010, 6, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Mederos, S.; Perea, G. GABAergic-astrocyte signaling: A refinement of inhibitory brain networks. Glia 2019, 67, 1842–1851. [Google Scholar] [CrossRef]
- Cavaccini, A.; Durkee, C.; Kofuji, P.; Tonini, R.; Araque, A. Astrocyte Signaling Gates Long-Term Depression at Corticostriatal Synapses of the Direct Pathway. J. Neurosci. 2020, 40, 5757–5768. [Google Scholar] [CrossRef]
- Araque, A.; Parpura, V.; Sanzgiri, R.P.; Haydon, P.G. Tripartite synapses: Glia, the unacknowledged partner. Trends Neurosci. 1999, 22, 208–215. [Google Scholar] [CrossRef]
- Volterra, A.; Liaudet, N.; Savtchouk, I. Astrocyte Ca2+ signalling: An unexpected complexity. Nat. Rev. Neurosci. 2014, 15, 327–335. [Google Scholar] [CrossRef]
- Savtchouk, I.; Volterra, A. Gliotransmission: Beyond Black-and-White. J. Neurosci. 2018, 38, 14–25. [Google Scholar] [CrossRef]
- Ventura, R.; Harris, K.M. Three-Dimensional Relationships between Hippocampal Synapses and Astrocytes. J. Neurosci. 1999, 19, 6897–6906. [Google Scholar] [CrossRef]
- Bushong, E.A.; Martone, M.E.; Jones, Y.Z.; Ellisman, M.H. Protoplasmic Astrocytes in CA1 Stratum Radiatum Occupy Separate Anatomical Domains. J. Neurosci. 2002, 22, 183–192. [Google Scholar] [CrossRef]
- Theodosis, D.T. Oxytocin-Secreting Neurons: A Physiological Model of Morphological Neuronal and Glial Plasticity in the Adult Hypothalamus. Front. Neuroendocr. 2002, 23, 101–135. [Google Scholar] [CrossRef] [PubMed]
- Nishida, H.; Okabe, S. Direct Astrocytic Contacts Regulate Local Maturation of Dendritic Spines. J. Neurosci. 2007, 27, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Hirrlinger, J.; Hülsmann, S.; Kirchhoff, F. Astroglial processes show spontaneous motility at active synaptic terminals in situ. Eur. J. Neurosci. 2004, 20, 2235–2239. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.-S.; Allen, N.J.; Eroglu, C. Astrocytes Control Synapse Formation, Function, and Elimination. Cold Spring Harb. Perspect. Biol. 2015, 7, a020370. [Google Scholar] [CrossRef]
- Van Horn, M.R.; Ruthazer, E.S. Glial regulation of synapse maturation and stabilization in the developing nervous system. Curr. Opin. Neurobiol. 2019, 54, 113–119. [Google Scholar] [CrossRef]
- Christopherson, K.S.; Ullian, E.M.; Stokes, C.C.; Mullowney, C.E.; Hell, J.W.; Agah, A.; Lawler, J.; Mosher, D.F.; Bornstein, P.; Barres, B.A. Thrombospondins are Astrocyte-Secreted Proteins that Promote CNS Synaptogenesis. Cell 2005, 120, 421–433. [Google Scholar] [CrossRef]
- Nishiyama, H.; Knöpfel, T.; Endo, S.; Itohara, S. Glial protein S100B modulates long-term neuronal synaptic plasticity. Proc. Natl. Acad. Sci. USA 2002, 99, 4037–4042. [Google Scholar] [CrossRef]
- Morquette, P.; Verdier, D.; Kadala, A.; Féthière, J.; Philippe, A.G.; Robitaille, R.; Kolta, A. An astrocyte-dependent mechanism for neuronal rhythmogenesis. Nat. Neurosci. 2015, 18, 844–854. [Google Scholar] [CrossRef]
- Ahlemeyer, B.; Beier, H.; Semkova, I.; Schaper, C.; Krieglstein, J. S-100β protects cultured neurons against glutamate- and staurosporine-induced damage and is involved in the antiapoptotic action of the 5 HT1A-receptor agonist, Bay x 3702. Brain Res. 2000, 858, 121–128. [Google Scholar] [CrossRef]
- Mori, T.; Tan, J.; Arendash, G.W.; Koyama, N.; Nojima, Y.; Town, T. Overexpression of Human S100B Exacerbates Brain Damage and Periinfarct Gliosis after Permanent Focal Ischemia. Stroke 2008, 39, 2114–2121. [Google Scholar] [CrossRef]
- Villarreal, A.; Avilés-Reyes, R.; Angelo, M.F.; Reines, A.G.; Ramos, A.J. S100B alters neuronal survival and dendrite extension via RAGE?mediated NF??B signaling. J. Neurochem. 2011, 117, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Benchenane, K.; Tiesinga, P.H.; Battaglia, F.P. Oscillations in the prefrontal cortex: A gateway to memory and attention. Curr. Opin. Neurobiol. 2011, 21, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Escuela, D.O.B.; Carlsson, J.; Ambrogini, P.; Narváez, M.; Wydra, K.; Tarakanov, A.O.; Li, X.; Millón, C.; Ferraro, L.; Cuppini, R.; et al. Understanding the Role of GPCR Heteroreceptor Complexes in Modulating the Brain Networks in Health and Disease. Front. Cell. Neurosci. 2017, 11. [Google Scholar] [CrossRef]
- Durkee, C.A.; Covelo, A.; Lines, J.; Kofuji, P.; Aguilar, J.; Araque, A. G i/o protein-coupled receptors inhibit neurons but activate astrocytes and stimulate gliotransmission. Glia 2019, 67, 1076–1093. [Google Scholar] [CrossRef] [PubMed]
- Doron, A.; Goshen, I. Investigating the transition from recent to remote memory using advanced tools. Brain Res. Bull. 2018, 141, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Frankland, P.W.; Bontempi, B. The organization of recent and remote memories. Nat. Rev. Neurosci. 2005, 6, 119–130. [Google Scholar] [CrossRef]
- Moscovitch, M.; Cabeza, R.; Winocur, G.; Nadel, L. Episodic Memory and Beyond: The Hippocampus and Neocortex in Transformation. Annu. Rev. Psychol. 2016, 67, 105–134. [Google Scholar] [CrossRef]
- Araujo, B.H.S.; Kaid, C.; De Souza, J.S.; Da Silva, S.G.; Goulart, E.; Caires, L.C.J.; Musso, C.M.; Torres, L.B.; Ferrasa, A.; Herai, R.; et al. Down Syndrome iPSC-Derived Astrocytes Impair Neuronal Synaptogenesis and the mTOR Pathway In Vitro. Mol. Neurobiol. 2017, 55, 5962–5975. [Google Scholar] [CrossRef]
- Murphy, G.M.; Ellis, W.G.; Lee, Y.-L.; Stultz, K.E.; Shrivastava, R.; Tinklenberg, J.R.; Eng, L.F. Chapter 40: Astrocytic Gliosis in the Amygdala in Down’s Syndrome and Alzheimer’s Disease; Elsevier: Amsterdam, The Netherlands, 1992; Volume 94, pp. 475–483. [Google Scholar]
- Guttenplan, K.A.; Stafford, B.K.; El-Danaf, R.N.; Adler, D.I.; Münch, A.E.; Weigel, M.K.; Huberman, A.D.; Liddelow, S.A. Neurotoxic Reactive Astrocytes Drive Neuronal Death after Retinal Injury. Cell Rep. 2020, 31, 107776. [Google Scholar] [CrossRef]
- Kia, A.; McAvoy, K.; Krishnamurthy, K.; Trotti, D.; Pasinelli, P. Astrocytes expressing ALS-linked mutant FUS induce motor neuron death through release of tumor necrosis factor-alpha. Glia 2018, 66, 1016–1033. [Google Scholar] [CrossRef]
- Sofroniew, M. Astrogliosis. Cold Spring Harb. Perspect. Biol. 2015, 7, a020420. [Google Scholar] [CrossRef] [PubMed]
- Pekny, M.; Pekna, M. Astrocyte Reactivity and Reactive Astrogliosis: Costs and Benefits. Physiol. Rev. 2014, 94, 1077–1098. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009, 32, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Griffin, W.; Sheng, J.G.; McKenzie, J.E.; Royston, M.C.; Gentleman, S.M.; Brumback, R.A.; Cork, L.C.; Del Bigio, M.R.; Roberts, G.W.; Mrak, R.E. Life-long Overexpression of S100β in Down’s Syndrome: Implications for Alzheimer Pathogenesis. Neurobiol. Aging 1999, 19, 401–405. [Google Scholar] [CrossRef]
- Banerjee, A.; Ifrim, M.F.; Valdez, A.N.; Raj, N.; Bassell, G.J. Aberrant RNA translation in fragile X syndrome: From FMRP mechanisms to emerging therapeutic strategies. Brain Res. 2018, 1693, 24–36. [Google Scholar] [CrossRef]
- Dockendorff, T.C.; Labrador, M. The Fragile X Protein and Genome Function. Mol. Neurobiol. 2018, 56, 711–721. [Google Scholar] [CrossRef]
- Reiss, A.L.; Aylward, E.; Freund, L.S.; Joshi, P.K.; Bryan, R.N. Neuroanatomy of fragile X syndrome: The posterior fossa. Ann. Neurol. 1991, 29, 26–32. [Google Scholar] [CrossRef]
- Sabaratnam, M. Pathological and neuropathological findings in two males with fragile-X syndrome. J. Intellect. Disabil. Res. 2000, 44, 81–85. [Google Scholar] [CrossRef]
- The Dutch-Belgian Fragile X Consorthrum. Fmr1 knockout mice: A model to study fragile X mental retardation. Cell 1994, 78. [CrossRef]
- Higashimori, H.; Morel, L.; Huth, J.; Lindemann, L.; Dulla, C.; Taylor, A.; Freeman, M.; Yang, Y. Astroglial FMRP-dependent translational down-regulation of mGluR5 underlies glutamate transporter GLT1 dysregulation in the fragile X mouse. Hum. Mol. Genet. 2013, 22, 2041–2054. [Google Scholar] [CrossRef]
- Bear, M.F.; Huber, K.M.; Warren, S.T. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004, 27, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Dölen, G.; Osterweil, E.; Rao, B.S.S.; Smith, G.B.; Auerbach, B.D.; Chattarji, S.; Bear, M.F. Correction of Fragile X Syndrome in Mice. Neuron 2007, 56, 955–962. [Google Scholar] [CrossRef] [PubMed]
- Veloz, M.F.V.; Buijsen, R.A.; Willemsen, R.; Cupido, A.; Bosman, L.W.; Koekkoek, S.K.E.; Potters, J.W.; Oostra, B.A.; De Zeeuw, C.I. The effect of an mGluR5 inhibitor on procedural memory and avoidance discrimination impairments in Fmr1 KO mice. Genes Brain Behav. 2012, 11, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Pop, A.S.; Levenga, J.; De Esch, C.E.F.; Buijsen, R.A.; Nieuwenhuizen, I.M.; Li, T.; Isaacs, A.; Gasparini, F.; Oostra, B.A.; Willemsen, R. (Rob) Rescue of dendritic spine phenotype in Fmr1 KO mice with the mGluR5 antagonist AFQ056/Mavoglurant. Psychopharmacology 2012, 231, 1227–1235. [Google Scholar] [CrossRef]
- Aloisi, E.; Le Corf, K.; Dupuis, J.; Zhang, P.; Ginger, M.; Labrousse, V.; Spatuzza, M.; Haberl, M.G.; Costa, L.; Shigemoto, R.; et al. Altered surface mGluR5 dynamics provoke synaptic NMDAR dysfunction and cognitive defects in Fmr1 knockout mice. Nat. Commun. 2017, 8, 1103. [Google Scholar] [CrossRef]
- Carroll, R.C.; Lissin, D.V.; Von Zastrow, M.; Nicoll, R.A.; Malenka, R.C. Rapid redistribution of glutamate receptors contributes to long-term depression in hippocampal cultures. Nat. Neurosci. 1999, 2, 454–460. [Google Scholar] [CrossRef]
- Snyder, E.M.; Philpot, B.D.; Huber, K.M.; Dong, X.; Fallon, J.R.; Bear, M.F. Internalization of ionotropic glutamate receptors in response to mGluR activation. Nat. Neurosci. 2001, 4, 1079–1085. [Google Scholar] [CrossRef]
- Zakharenko, S.S.; Zablow, L.; Siegelbaum, S.A. Altered presynaptic vesicle release and cycling during mGluR-dependent LTD. Neuron 2002, 35, 1099–1110. [Google Scholar] [CrossRef]
- Huber, K.M.; Gallagher, S.M.; Warren, S.T.; Bear, M.F. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc. Natl. Acad. Sci. USA 2002, 99, 7746–7750. [Google Scholar] [CrossRef]
- Yuskaitis, C.J.; Beurel, E.; Jope, R.S. Evidence of reactive astrocytes but not peripheral immune system activation in a mouse model of Fragile X syndrome. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2010, 1802, 1006–1012. [Google Scholar] [CrossRef]
- Pacey, L.K.K.; Guan, S.; Tharmalingam, S.; Thomsen, C.; Hampson, D.R. Persistent astrocyte activation in the fragile X mouse cerebellum. Brain Behav. 2015, 5, e00400. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Hulsizer, S.; Cui, Y.; Pretto, D.L.; Kim, K.H.; Hagerman, P.J.; Tassone, F.; Pessah, I.N. Enhanced Asynchronous Ca2+Oscillations Associated with Impaired Glutamate Transport in Cortical Astrocytes ExpressingFmr1Gene Premutation Expansion. J. Biol. Chem. 2013, 288, 13831–13841. [Google Scholar] [CrossRef] [PubMed]
- Asch, A.S.; Leung, L.L.; Shapiro, J.; Nachman, R.L. Human brain glial cells synthesize thrombospondin. Proc. Natl. Acad. Sci. USA 1986, 83, 2904–2908. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Ge, J.; Summers, J.B.; Li, F.; Liu, X.; Ma, P.; Kaminski, J.; Zhuang, J. TSP-1 Secreted by Bone Marrow Stromal Cells Contributes to Retinal Ganglion Cell Neurite Outgrowth and Survival. PLoS ONE 2008, 3, e2470. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.C.; Tucker, R.P. The thrombospondin type 1 repeat (TSR) superfamily: Diverse proteins with related roles in neuronal development. Dev. Dyn. 2000. [Google Scholar] [CrossRef]
- Lu, Z.; Kipnis, J. Thrombospondin 1—A key astrocyte-derived neurogenic factor. FASEB J. 2010, 24, 1925–1934. [Google Scholar] [CrossRef]
- Pinter, J.D.; Eliez, S.; Schmitt, J.E.; Capone, G.T.; Reiss, A.L. Neuroanatomy of Down’s Syndrome: A High-Resolution MRI Study. Am. J. Psychiatry 2001, 158, 1659–1665. [Google Scholar] [CrossRef]
- Raz, N.; Torres, I.J.; Briggs, S.D.; Spencer, W.D.; Thornton, A.E.; Loken, W.J.; Gunning, F.M.; McQuain, J.D.; Driesen, N.R.; Acker, J.D. Selective neuroanatornic abnormalities in Down’s syndrome and their cognitive correlates: Evidence from MRI morphometry. Neurology 1995, 45, 356–366. [Google Scholar] [CrossRef]
- De Lagran, M.M.; Benavides-Piccione, R.; Ballesteros-Yanez, I.; Calvo, M.; Morales, M.; Fillat, C.; DeFelipe, J.; Ramakers, G.J.A.; Dierssen, M. Dyrk1A Influences Neuronal Morphogenesis Through Regulation of Cytoskeletal Dynamics in Mammalian Cortical Neurons. Cereb. Cortex 2012, 22, 2867–2877. [Google Scholar] [CrossRef]
- Lott, I.; Dierssen, M. Cognitive deficits and associated neurological complications in individuals with Down’s syndrome. Lancet Neurol. 2010, 9, 623–633. [Google Scholar] [CrossRef]
- Griffin, W.S.; Stanley, L.C.; Ling, C.; White, L.; MacLeod, V.; Perrot, L.J.; White, C.L.; Araoz, C. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc. Natl. Acad. Sci. USA 1989, 86, 7611–7615. [Google Scholar] [CrossRef] [PubMed]
- Lockrow, J.P.; Fortress, A.M.; Granholm, A.-C.E. Age-Related Neurodegeneration and Memory Loss in Down Syndrome. Curr. Gerontol. Geriatr. Res. 2012, 2012, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Goodison, K.L.; Parhad, I.M.; White, C.L.; Sima, A.A.F.; Clark, A.W. Neuronal and Glial Gene Expression in Neocortex of Downʼs Syndrome and Alzheimerʼs Disease. J. Neuropathol. Exp. Neurol. 1993, 52, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Kanaumi, T.; Milenkovic, I.; Adle-Biassette, H.; Aronica, E.; Kovacs, G.G. Non-neuronal cell responses differ between normal and Down syndrome developing brains. Int. J. Dev. Neurosci. 2013, 31, 796–803. [Google Scholar] [CrossRef]
- Guidi, S.; Bonasoni, P.; Ceccarelli, C.; Santini, D.; Gualtieri, F.; Ciani, E.; Bartesaghi, R. RESEARCH ARTICLE: Neurogenesis Impairment and Increased Cell Death Reduce Total Neuron Number in the Hippocampal Region of Fetuses with Down Syndrome. Brain Pathol. 2007, 18, 180–197. [Google Scholar] [CrossRef]
- Hibaoui, Y.; Grad, I.; Letourneau, A.; Sailani, M.R.; Dahoun, S.; Santoni, F.A.; Gimelli, S.; Guipponi, M.; Pelte, M.F.; Bena, F.S.; et al. Modelling and rescuing neurodevelopmental defect of D own syndrome using induced pluripotent stem cells from monozygotic twins discordant for trisomy 21. EMBO Mol. Med. 2013, 6, 259–277. [Google Scholar] [CrossRef]
- Kurabayashi, N.; Nguyen, M.D.; Sanada, K. DYRK 1A overexpression enhances STAT activity and astrogliogenesis in a Down syndrome mouse model. EMBO Rep. 2015, 16, 1548–1562. [Google Scholar] [CrossRef]
- Lorenzi, H.A.; Reeves, R.H. Hippocampal hypocellularity in the Ts65Dn mouse originates early in development. Brain Res. 2006, 1104, 153–159. [Google Scholar] [CrossRef]
- Contestabile, A.; Fíla, T.; Cappellini, A.; Bartesaghi, R.; Ciani, E. Widespread impairment of cell proliferation in the neonate Ts65Dn mouse, a model for Down syndrome. Cell Prolif. 2009, 42, 171–181. [Google Scholar] [CrossRef]
- Anderson, A.J.; Stoltzner, S.; Lai, F.; Su, J.; Nixon, R.A. Morphological and biochemical assessment of DNA damage and apoptosis in Down syndrome and Alzheimer disease, and effect of postmortem tissue archival on TUNEL. Neurobiol. Aging 2000, 21, 511–524. [Google Scholar] [CrossRef]
- Halassa, M.M.; Fellin, T.; Takano, H.; Dong, J.-H.; Haydon, P.G. Synaptic Islands Defined by the Territory of a Single Astrocyte. J. Neurosci. 2007, 27, 6473–6477. [Google Scholar] [CrossRef]
- Colombo, J.A.; Reisin, H.D.; Jones, M.; Bentham, C. Development of interlaminar astroglial processes in the cerebral cortex of control and Down’s syndrome human cases. Exp. Neurol. 2005, 193, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-M.; Wang, H.-K.; Ye, C.-Q.; Ge, W.; Chen, Y.; Jiang, Z.-L.; Wu, C.-P.; Poo, M.-M.; Duan, S. ATP Released by Astrocytes Mediates Glutamatergic Activity-Dependent Heterosynaptic Suppression. Neuron 2003, 40, 971–982. [Google Scholar] [CrossRef]
- Pascual, O.; Casper, K.B.; Kubera, C.; Zhang, J.; Revilla-Sanchez, R.; Sul, J.-Y.; Takano, H.; Moss, S.J.; McCarthy, K.; Haydon, P.G. Astrocytic Purinergic Signaling Coordinates Synaptic Networks. Science 2005, 310, 113–116. [Google Scholar] [CrossRef] [PubMed]
- Dunwiddie, T.V.; Masino, S.A. The Role and Regulation of Adenosine in the Central Nervous System. Annu. Rev. Neurosci. 2001, 24, 31–55. [Google Scholar] [CrossRef] [PubMed]
- Lindquist, B.E.; Shuttleworth, C.W. Adenosine receptor activation is responsible for prolonged depression of synaptic transmission after spreading depolarization in brain slices. Neuroscience 2012, 223, 365–376. [Google Scholar] [CrossRef]
- Risser, D.; Lubec, G.; Cairns, N.; Herrera-Marschitz, M. Excitatory amino acids and monoamines in parahippocampal gyrus and frontal cortical pole of adults with down syndrome. Life Sci. 1997, 60, 1231–1237. [Google Scholar] [CrossRef]
- Reynolds, G.P.; Warner, C.E. Amino acid neurotransmitter deficits in adult Down’s syndrome brain tissue. Neurosci. Lett. 1988, 94, 224–227. [Google Scholar] [CrossRef]
- Begni, B.; Brighina, L.; Fumagalli, L.; Andreoni, S.; Castelli, E.; Francesconi, C.; Del Bo, R.; Bresolin, N.; Ferrarese, C. Altered glutamate uptake in peripheral tissues from Down Syndrome patients. Neurosci. Lett. 2003, 343, 73–76. [Google Scholar] [CrossRef]
- Chen, C.; Jiang, P.; Xue, H.; Peterson, S.E.; Tran, H.T.; McCann, A.E.; Parast, M.M.; Li, S.; Pleasure, D.E.; Laurent, L.C.; et al. Role of astroglia in Down’s syndrome revealed by patient-derived human-induced pluripotent stem cells. Nat. Commun. 2014, 5, 4430. [Google Scholar] [CrossRef]
- Belichenko, P.V.; Kleschevnikov, A.M.; Masliah, E.; Wu, C.; Takimoto-Kimura, R.; Salehi, A.; Mobley, W.C. Excitatory-inhibitory relationship in the fascia dentata in the Ts65Dn mouse model of down syndrome. J. Comp. Neurol. 2009, 512, 453–466. [Google Scholar] [CrossRef] [PubMed]
- Harashima, C.; Jacobowitz, D.M.; Stoffel, M.; Chakrabarti, L.; Haydar, T.F.; Siarey, R.J.; Galdzicki, Z. Elevated Expression of the G-Protein-Activated Inwardly Rectifying Potassium Channel 2 (GIRK2) in Cerebellar Unipolar Brush Cells of a Down Syndrome Mouse Model. Cell. Mol. Neurobiol. 2006, 26, 717–732. [Google Scholar] [CrossRef] [PubMed]
- Best, T.K.; Cramer, N.P.; Chakrabarti, L.; Haydar, T.F.; Galdzicki, Z. Dysfunctional hippocampal inhibition in the Ts65Dn mouse model of Down syndrome. Exp. Neurol. 2011, 233, 749–757. [Google Scholar] [CrossRef]
- Costa, A.C.; Grybko, M.J. Deficits in hippocampal CA1 LTP induced by TBS but not HFS in the Ts65Dn mouse: A model of Down syndrome. Neurosci. Lett. 2005, 382, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Kurt, M.; Davies, D.C.; Kidd, M.; Dierssen, M.; Flórez, J. Synaptic deficit in the temporal cortex of partial trisomy 16 (Ts65Dn) mice. Brain Res. 2000, 858, 191–197. [Google Scholar] [CrossRef]
- Oka, A.; Takashima, S. The up-regulation of metabotropic glutamate receptor 5 (mGluR5) in Down’s syndrome brains. Acta Neuropathol. 1999, 97, 275–278. [Google Scholar] [CrossRef]
- Gibson, J.R.; Bartley, A.F.; Hays, S.A.; Huber, K.M. Imbalance of Neocortical Excitation and Inhibition and Altered UP States Reflect Network Hyperexcitability in the Mouse Model of Fragile X Syndrome. J. Neurophysiol. 2008, 100, 2615–2626. [Google Scholar] [CrossRef]
- Olmos-Serrano, J.L.; Paluszkiewicz, S.M.; Martin, B.S.; Kaufmann, W.E.; Corbin, J.G.; Huntsman, M.M. Defective GABAergic Neurotransmission and Pharmacological Rescue of Neuronal Hyperexcitability in the Amygdala in a Mouse Model of Fragile X Syndrome. J. Neurosci. 2010, 30, 9929–9938. [Google Scholar] [CrossRef]
- Ethridge, L.E.; White, S.P.; Mosconi, M.W.; Wang, J.; Byerly, M.J.; Sweeney, J.A. Reduced habituation of auditory evoked potentials indicate cortical hyper-excitability in Fragile X Syndrome. Transl. Psychiatry 2016, 6, e787. [Google Scholar] [CrossRef]
- Iyer, A.M.; Van Scheppingen, J.; Milenkovic, I.; Anink, J.J.; Lim, D.; Genazzani, A.A.; Adle-Biassette, H.; Kovacs, G.G.; Aronica, E. Metabotropic Glutamate Receptor 5 in Down’s Syndrome Hippocampus During Development: Increased Expression in Astrocytes. Curr. Alzheimer Res. 2014, 11, 694–705. [Google Scholar] [CrossRef]
- Piers, T.M.; Kim, D.H.; Kim, B.C.; Regan, P.; Whitcomb, D.J.; Cho, K. Translational Concepts of mGluR5 in Synaptic Diseases of the Brain. Front. Pharm. 2012, 3, 199. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi-Yamaguchi, Y.; Furuichi, T. The Homer family proteins. Genome Biol. 2007, 8, 206. [Google Scholar] [CrossRef] [PubMed]
- Won, H.; Lee, H.-R.; Gee, H.Y.; Mah, W.; Kim, J.-I.; Lee, J.; Ha, S.; Chung, C.; Jung, E.S.; Cho, Y.S.; et al. Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nat. Cell Biol. 2012, 486, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Gregory, K.J.; Dong, E.N.; Meiler, J.; Conn, P.J. Allosteric modulation of metabotropic glutamate receptors: Structural insights and therapeutic potential. Neuropharmacology 2011, 60, 66–81. [Google Scholar] [CrossRef] [PubMed]
- Huber, K.M. Role for Rapid Dendritic Protein Synthesis in Hippocampal mGluR-Dependent Long-Term Depression. Science 2000, 288, 1254–1256. [Google Scholar] [CrossRef]
- Lepannetier, S.; Gualdani, R.; Tempesta, S.; Schakman, O.; Seghers, F.; Kreis, A.; Yerna, X.; Slimi, A.; De Clippele, M.; Tajeddine, N.; et al. Activation of TRPC1 Channel by Metabotropic Glutamate Receptor mGluR5 Modulates Synaptic Plasticity and Spatial Working Memory. Front. Cell. Neurosci. 2018, 12, 318. [Google Scholar] [CrossRef]
- Sun, Y.; Lipton, J.O.; Boyle, L.M.; Madsen, J.R.; Goldenberg, M.C.; Pascual-Leone, A.; Sahin, M.; Rotenberg, A. Direct current stimulation induces mGluR5-dependent neocortical plasticity. Ann. Neurol. 2016, 80, 233–246. [Google Scholar] [CrossRef]
- Mor-Shaked, H.; Eiges, R. Reevaluation of FMR1 Hypermethylation Timing in Fragile X Syndrome. Front. Mol. Neurosci. 2018, 11, 31. [Google Scholar] [CrossRef]
- Liu, X.S.; Wu, H.; Krzisch, M.; Wu, X.; Graef, J.; Muffat, J.; Hnisz, D.; Li, C.H.; Yuan, B.; Xu, C.; et al. Rescue of Fragile X Syndrome Neurons by DNA Methylation Editing of the FMR1 Gene. Cell 2018, 172, 979–992.e6. [Google Scholar] [CrossRef]
- Lu, J.; McCarter, M.; Lian, G.; Esposito, G.; Capoccia, E.; Delli-Bovi, L.C.; Hecht, J.; Sheen, V. Global hypermethylation in fetal cortex of Down syndrome due to DNMT3L overexpression. Hum. Mol. Genet. 2016, 25, 1714–1727. [Google Scholar] [CrossRef]
- Jin, S.; Lee, Y.K.; Lim, Y.C.; Zheng, Z.; Lin, X.M.; Ng, D.P.Y.; Holbrook, J.D.; Law, H.Y.; Kwek, K.Y.C.; Yeo, G.S.H.; et al. Global DNA Hypermethylation in Down Syndrome Placenta. PLoS Genet. 2013, 9, e1003515. [Google Scholar] [CrossRef] [PubMed]
- Laufer, B.I.; Hwang, H.; Ciernia, A.V.; Mordaunt, C.E.; LaSalle, J.M. Whole genome bisulfite sequencing of Down syndrome brain reveals regional DNA hypermethylation and novel disorder insights. Epigenetics 2019, 14, 672–684. [Google Scholar] [CrossRef] [PubMed]
- De Toma, I.; Ortega, M.; Catuara-Solarz, S.; Sierra, C.; Sabidó, E.; Dierssen, M. Re-establishment of the epigenetic state and rescue of kinome deregulation in Ts65Dn mice upon treatment with green tea extract and environmental enrichment. Sci. Rep. 2020, 10, 1–18. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández-Blanco, Á.; Dierssen, M. Rethinking Intellectual Disability from Neuro- to Astro-Pathology. Int. J. Mol. Sci. 2020, 21, 9039. https://doi.org/10.3390/ijms21239039
Fernández-Blanco Á, Dierssen M. Rethinking Intellectual Disability from Neuro- to Astro-Pathology. International Journal of Molecular Sciences. 2020; 21(23):9039. https://doi.org/10.3390/ijms21239039
Chicago/Turabian StyleFernández-Blanco, Álvaro, and Mara Dierssen. 2020. "Rethinking Intellectual Disability from Neuro- to Astro-Pathology" International Journal of Molecular Sciences 21, no. 23: 9039. https://doi.org/10.3390/ijms21239039
APA StyleFernández-Blanco, Á., & Dierssen, M. (2020). Rethinking Intellectual Disability from Neuro- to Astro-Pathology. International Journal of Molecular Sciences, 21(23), 9039. https://doi.org/10.3390/ijms21239039