Abstract
Alternative splicing (AS) is a critical post-transcriptional regulatory mechanism used by more than 95% of transcribed human genes and responsible for structural transcript variation and proteome diversity. In the past decade, genome-wide transcriptome sequencing has revealed that AS is tightly regulated in a tissue- and developmental stage-specific manner, and also frequently dysregulated in multiple human cancer types. It is currently recognized that splicing defects, including genetic alterations in the spliced gene, altered expression of both core components or regulators of the precursor messenger RNA (pre-mRNA) splicing machinery, or both, are major drivers of tumorigenesis. Hence, in this review we provide an overview of our current understanding of splicing alterations in cancer, and emphasize the need to further explore the cancer-specific splicing programs in order to obtain new insights in oncology. Furthermore, we also discuss the recent advances in the identification of dysregulated splicing signatures on a genome-wide scale and their potential use as biomarkers. Finally, we highlight the therapeutic opportunities arising from dysregulated splicing and summarize the current approaches to therapeutically target AS in cancer.
1. Introduction
In higher eukaryotes, the primary gene transcripts, also called precursor messenger RNAs (pre-mRNAs), undergo a finely tuned post-transcriptional regulatory process that removes the non-coding regions (introns) and splices together the coding sequences (exons), thus generating the mature mRNAs. This mechanism is designated as pre-mRNA splicing and is a critical step in gene expression. In addition, it is well known that the splicing patterns of a gene vary widely as result of the process of alternative splicing (AS) that differentially retains or excludes certain exons from the pre-mRNA transcript. Consequently, various combinations of exons from a single gene can produce a diversity of mRNA variants, which is determinant to structural transcript variation and proteome diversity [1] and can generate different protein isoforms with related, distinct, or even opposing functions [2,3]. Remarkably, AS is a widespread event affecting more than 95% of transcribed human genes, as suggested by data provided by whole transcriptome sequencing projects [2,4]. This complex and tightly regulated mechanism is shared across different tissues and developmental stages, and frequently dysregulated in various human diseases, including cancer [5]. This dysregulation was verified in various types of cancer through detection of aberrant splicing patterns in tumor tissues when compared to their normal counterparts by high-throughput sequencing techniques [6,7,8,9]. Additionally, accumulating evidence clearly supports that the aberrant splicing profiles found in cancer are contributing to neoplastic transformation, cancer progression, and therapy resistance [10,11]. Therefore, it is of utmost relevance to identify pathological splicing isoforms for the development of new effective biomarkers, as well as to clarify the mechanisms behind aberrant AS, thereby elucidating its impact on cancer and providing novel therapeutic strategies.
Hence, this review summarizes our current understanding of splicing alterations in cancer and emphasizes the need for a deeper understanding of cancer-specific splicing programs in order to provide new insights in oncology. Particularly, we highlight the relevance of identifying cancer-specific AS events for the development of novel biomarkers and discuss part of the current therapeutic landscape regarding splicing-based therapies for cancer treatment.
2. Alternative RNA Splicing: An Overview
Pre-mRNA splicing consists of a multistep process orchestrated by the spliceosome, a huge RNA/protein complex comprising five small nuclear ribonucleoproteins (snRNPs; U1, U2, U4, U5, and U6) and numerous associated proteins [12,13]. Briefly, the reaction initiates with the assembly of an initial spliceosome complex through recognition of critical consensus splice sites at the pre-mRNA transcript, as schematically represented in Figure 1A. It comprises a stepwise process that begins with the recruitment of U1 snRNP to the 5′ splice site. Then, the splicing factor 1 (SF1), U2 snRNP auxiliary factor 2 (U2AF2), and U2 snRNP auxiliary factor (U2AF) 2, and U2AF 1recognize the branch point site (BPS), the polypyrimidine (poly-Y) tract, and the AG dinucleotide of the 3′ splice site region, respectively. The occupancy of these three consensus sequences induces the association of U2 snRNP with the BPS, which is further stabilized by the U2 snRNP component SF3B1. Consequently, intronic recognition prompts the engagement of U4/U6/U5 tri-snRNP with the complex, and subsequent formation of a catalytically inactive complex. This leads to several conformational and compositional rearrangements of spliceosomal components, including the dissociation of U1 and U4 snRNPs, which in turn promotes the formation of the activated spliceosome that catalyzes the splicing reaction [14]. Transcripts from nearly all protein-coding genes undergo one or more types of AS, giving rise to different mRNAs that differ in transcript degradation or are translated into alternative protein isoforms in a cell type-, organ-, or tissue-specific manner [2,4,15]. In higher eukaryotes, among the currently known AS events represented in Figure 1B, the most common is exon skipping [16], accounting for approximately 40% of all AS events, in which a cassette exon is removed from the pre-mRNA together with its flaking introns. Besides this, switching between alternative 5′ and 3′ splice site positions, mutually exclusive splicing of adjacent exons and differential retention of introns are also important variations of AS (Figure 1B). Other types of AS events include the use of alternative transcription start sites and alternative polyadenylation.

Figure 1.
Regulation of pre-mRNA splicing. (A) Stepwise assembly of spliceosome on the pre-mRNA and catalysis of the splicing reaction to generate mature spliced mRNA. (B) Schematic representation of the most common alternative splicing AS events. The grey, yellow, red, and blue boxes represent different exons. The solid black and dotted grey lines indicate distinct splicing events. (C) Complex interplay between cis- and trans-acting factors in the regulation of AS. RNA-binding motif (RBM) proteins, serine/arginine-rich (SR) proteins, and heterogeneous (hn) ribonucleoproteins (hnRNPs) bind to exonic or intronic regulatory elements to promote or prevent the recognition of either 3′ or 5′ splice sites (ss) by the small nuclear (sn) RNPs (snRNPs) and splicing factors. The solid and dotted black arrows represent binding stimulation and inhibition, respectively; (ss—splice sites; BPS—branch point site; poly-Y—polypyrimidine tract; pre-mRNA—precursor messenger RNA; snRNPs—small nuclear ribonucleoprotein particle; SF1—splicing factor 1; U2AF—U2 snRNP auxiliary factor).
In AS, the regulated process consists of the recognition of an exon by the spliceosome. For this, splice site utilization is further regulated by cis-acting splicing-regulatory elements, which either promote or inhibit the use of adjacent splice sites by recruiting trans-acting splicing factors [17]. Thus, they are classified into exonic or intronic splicing enhancers (ESE/ISE) or silencers (ESS/ISS), depending on their positions and functions (Figure 1C). In general, enhancers are recognized by trans-acting factors belonging to the serine/arginine-rich (SR) protein family to facilitate splice site recognition and exon inclusion [18]. On the other hand, silencers usually interact with other types of trans-acting factors such as heterogeneous ribonucleoproteins (hnRNPs) to inhibit splice site recognition and promote exon skipping [2]. However, several AS events exist in which SR or hnRNP proteins act as inhibitors or enhancers of splicing, respectively.
2.1. Dysregulation of Alternative Splicing in Cancer
Cancer mainly evolves through successive genetic alterations and genomic dysregulation, but is also affected by the tumor microenvironment. These render oncogenes constitutively active and inactivate tumor-suppressor genes. As a result, cancer cells acquire specific abilities during tumor development, including self-sufficiency in growth signals, insensitivity to growth inhibitory signals, evasion of apoptosis, limitless replicative potential, sustained angiogenesis, and tissue invasion and metastasis [19]. These processes can also be dysregulated by AS, which in turn can generate variant proteins with altered physiological function [3]. Particularly, a recent systematic study performed by Kahles et al. reported that AS events are more frequent in cancer tissues compared to normal ones, and many of them are cancer-type specific [20]. Among the factors that can trigger aberrant AS, somatic mutations that disrupt splicing regulatory motifs, as well as mutations or expression changes in components of the core splicing machinery or splicing auxiliary factors, are frequently described [6,7,21,22,23,24].
Aberrant splicing in cancer has been widely linked to mutations creating cis-regulatory motifs that generate novel splice sites, as demonstrated by the discovery of almost 2000 splice site-creating mutations through a robust whole-exome analysis encompassing more than 8000 tumor samples across 33 cancer types [25]. One of the AS events frequently associated with these somatic mutations is intron retention, and mainly affects tumor suppressor genes such as TP53, ARID1A, and PTEN [7]. Importantly, most of the intron retention events are able to induce frameshifts in pre-mRNA sequence, resulting in the generation of premature termination codons (PTCs), which in turn leads to the degradation of the transcript through nonsense-mediated mRNA decay (NMD) or to the production of truncated proteins (e.g., dominant negative isoforms or neo-antigens). Interestingly, somatic exonic mutations have also been reported in oncogenes, particularly in ESE and ESS sequences [6], and associated with the generation of pro-tumorigenic variants.
Recurrent somatic mutations affecting the components of the early spliceosome complex formation have frequently been described in cancer, particularly in hematological malignancies, including myelodysplastic syndromes (MDS), other myeloid neoplasms, and chronic lymphocytic leukemia (CLL) [26,27,28]. Among the genes most affected by these mutations that almost always occur in a mutually exclusive manner are SF3B1 (splicing factor 3b subunit 1), SRSF2 (serine/arginine-rich splicing factor 2), U2AF1 (U2 small nuclear RNA auxiliary factor 1), and ZRSR2 (zinc finger RNA binding motif and serine/arginine rich 2) [26]. SF3B1, a subunit of the U2 snRNP that recognizes the BPS, is the most commonly mutated splicing regulator in numerous cancers, with a prevalence ranging from 5% in breast cancer to 81% in an MDS subtype [29]. Cancer-associated SF3B1 mutations are located within HEAT (Huntingtin, Elongation factor 3, protein phosphatase 2A, Targets of rapamycin 1) domains, which are involved in protein–protein interactions and clustered in hotspots, namely K700, E622, R625, H662, and K666. Specifically, they are mainly related with the binding of SF3B1 to cryptic 3′ splice sites, located in regions with shorter and weaker poli-Y tracts, and consequently linked to aberrant BPS usage [22,30,31]. This abnormal assembly of spliceosome originates many mRNAs with a PTC, which are subsequently degraded by NMD.
Although the mechanism that induces the change of 3′ splice site usage by SF3B1 is not fully elucidated, it is hypothesized that these mutations alter the interaction of SF3B1 with other spliceosomal components required for BPS recognition. SRSF2 is a member of the SR protein family that binds to specific ESE sequences, namely CCNG or GGNG, through its RNA recognition motif (RRM) domain, and recruits U1 snRNP and U2AF to the 5′ and 3′ flanking splice sites, respectively [32]. This splicing regulator has also been found recurrently mutated, particularly in patients with MDS and chronic myelomonocytic leukemia (CMML) [26]. SRSF2 mutations predominantly occur at the P95 residue, which is located near the RRM domain [26]. According to several reports, these mutations change the RNA-binding affinity of SRSF2, favoring the recognition of C-rich CCNG over G-rich GGNG motifs in ESE consensus sites, which in turn leads to misregulation of exon inclusion [33,34]. The gene encoding UA2F1 is also mutated in myeloid malignancies, as well as in lung adenocarcinomas [26,35,36]. U2AF1 hotspot mutations occur almost exclusively at S34 and Q157 residues within the two conserved zinc-finger domains, thus affecting the recognition of the 3′ splice site AG motif [37,38]. In contrast to mutually exclusive hotspot mutations described for SF3B1, SRSF2, and U2AF1, ZRSR2 mutations are distributed throughout the gene and most are consistent with a loss-of-function phenotype [23]. In 2015, in addition to the major (or U2) spliceosome, ZRSR2 was also characterized as an essential component of the minor (or U12) spliceosome that catalyzes the processing of a distinct class of introns (U12-type introns). Particularly, it is involved in 3′ splice site recognition in U12 snRNA-dependent splicing, so that mutations in this gene are associated with an increase in the retention of U12-type introns [23].
Apart from genomic mutations, the pre-mRNA splicing of many genes related to cancer pathogenesis can also be disturbed by changes of the copy number or expression levels of splicing factors [39]. Actually, abnormal expression of several splicing factors have frequently been reported in solid tumors and closely associated with cancer development and progression, even in the absence of mutations [40,41,42,43]. One of the best characterized is the serine-arginine splicing factor 1 (SRSF1; formerly known as ASF or SF2), an SR protein involved in both constitutive and AS, as well as in other cellular processes. It is upregulated in several human tumors, including colon, breast, thyroid, small intestine, kidney, and lung, and its experimentally induced overexpression leads to the transformation of human and mouse mammary epithelial cells, suggesting that it acts as a proto-oncogene [44,45,46]. Until now, SRSF1 upregulation has been shown to affect many AS events in cancer-associated genes. In particular, SRSF1 overexpression induces an increase in the levels of oncogenic protein isoforms of RON [47], MNK2, and S6K1 [44] and of the anti-apoptotic isoforms Bcl-xL and MCL-1L [48], and a loss of the tumor suppressor isoform of BIN1 [44]. Curiously, the overexpression of hnRNP A1 and hnRNP A2/B1, two factors previously suggested to antagonize SR proteins, was also reported in lung, breast, and brain tumors [49,50,51,52]. Interestingly, in glioblastoma (GBM) cells, hnRNP A2/B1 showed splicing effects similar to the proto-oncogenic SR protein SRSF1 [52]. More recently, hnRNP A2 (as well as B1 and K) has been associated with enhanced expression of anti-apoptotic variants of BIN1 and CASP9, and decreased expression of the pro-apoptotic variant Bcl-xS [48], promoting the same phenotypic response as SRSF1 overexpression.
The major drivers of aberrant splicing profiles appear to be changes in the expression levels of splicing factors; however, the mechanisms behind the altered expression of the splicing factors in tumors are not yet fully understood. Although sporadic somatic mutations in genes encoding splicing factors have already been recurrently detected in solid tumors [43], it is widely recognized that oncogenic signaling has a central role [53]. Actually, abnormal activation of signaling pathways has been extensively reported in cancer. For instance, in colon cancer, oncogenic Kirsten rat sarcoma viral (KRAS) activates the RAS–MAPK pathway, leading to an increase in the expression levels of the AS factor polypyrimidine tract-binding protein 1 (PTBP1), activated via transcription factor ELK1. In turn, increased PTBP1 levels induce a shift in the AS of tumor-associated transcripts, namely, the small GTPase Ras-related C3 botulinum toxin substrate 1 (RAC1), adaptor protein NUMB, and PKM [54]. In addition to transcriptional stimulation of PTBP1 downstream of RAS, ERK was reported to phosphorylate the splicing factor SAM68, thereby inducing the binding of phospho-SAM68 to the 3′UTR of the SRSF1 transcript [55]. This binding promotes the retention of an intron required for production of full-length SRSF1 and prevents the downregulation of SRSF1 transcripts through the NMD pathway. Consequently, the increased SRSF1 levels, comparable in effect to the above described SRSF1 gene amplifications [44], induce a switch in AS of the RON gene transcripts, favoring the production of the oncogenic isoform RONΔex11. Phosphorylated SAM68 further stimulates inclusion of the variable exon 5 sequence into the CD44 mRNA, generating a pro-invasive cell adhesion protein variant [56].
Another MAPK pathway responds when cells experience physiologic stress. Osmotic stress triggers the MKK(3/6)-signaling cascade, leading to p38-activation, which upon nuclear translocation induces hnRNP A1 phosphorylation, followed by its export into the cytoplasm [57,58]. The corresponding decrease in nuclear splicing factor abundance is sufficient to change AS patterns. The PI3K/AKT signaling is another key pathway involved in cell survival and escape from apoptosis in numerous solid tumors. In non-small cell lung cancers (NSCLC), it was demonstrated that the activation of the PI3K/AKT pathway by oncogenic factors mediates the exclusion of the exon 3,4,5,6 cassette of CASP9 transcripts’ via the phosphorylation state of SRSF1, thus generating the anti-apoptotic Casp-9b isoform [59]. At the same time, AKT-mediated phosphorylation of hnRNPL induces its binding to a splice silencer element in Casp-9 pre-mRNA, further enhancing the exclusion of the exon cassette [60,61]. AKT activation also leads to phosphorylation and nuclear translocation of SR proteins, causing alternative exon inclusion in the fibronectin pre-mRNA [62]. Interestingly, in colorectal cells, inhibition of PI3K/AKT signaling led to increased expression of endogenous SRSF1, leading to the inclusion of an alternative exon, termed 3b, in the mRNA of the small GTPase RAC1, which generates the pro-tumorigenic splice variant RAC1B [63]. Later, it was described that SRPK1 and GSK3β act upstream of SRSF1, and are required to sustain RAC1B splicing in colorectal cancer (CRC) cells [64]. Particularly, it was shown that GSK3β indirectly regulates the levels of SRSF1 and RAC1B via SRPK1, since its depletion leads to a reduction of SRPK1 activity towards SRSF1, and a concomitant decrease in nuclear SRSF1 levels, resulting in less RAC1B generated. Another central hub of oncogenic signaling is the Wnt pathway, which is activated in many colorectal tumors. Remarkably, this pathway also modulates RAC1B splicing in CRC cells: It was described that the SRSF3 gene encoding splicing factor SRSF3/SRp20 is a transcriptional target for activated β-catenin/TCF4 complexes, leading to increased SRSF3 protein levels [65]. In a subsequent work, it was demonstrated that increased SRSF3 transcription following activation of the β-catenin/TCF4 pathway suppresses RAC1B splicing through SRSF3-mediated exclusion of exon 3b from the RAC1 mRNA [63]. Together, these examples show how signaling mechanisms affect alternative pre-mRNA splicing and change tumor-related gene expression.
2.2. Examples of Cancer-Associated Alternatively Spliced Variants
Several splice variants have been associated with different hallmarks of cancer, including initiation, progression, and metastasis. In Table 1, we highlight some of the most relevant AS events in cancer-associated genes involved in different steps of oncogenic transformation, as well as the types of cancer they are most often associated with. Other examples were listed in a recent review [66].

Table 1.
Tumor-associated AS variants and the respective cancer-promoting process.
3. RNA Splice Variants as Potential Biomarkers in Cancer
Early detection and diagnosis of cancer as well as the identification of the most effective personalized therapy for each patient remain the main challenges in oncology. Over the past few years, cancer biomarkers have emerged as valuable screening, diagnostic, and prognostic tools, enabling us to classify the extent of disease, define the prognosis, select the most appropriate treatment regimens, or follow up on the clinical response after treatment or surgical intervention [113]. Despite the advances, the development of more efficient biomarkers is still needed. Indeed, the amount of candidate cancer biomarkers that have been approved for clinical practice is too low, indicating that the majority of them are poor predictors of disease and treatment outcome, and are thus not reliable clinical tools [114]. In order to fill this gap, the biomarker potential of AS in cancer is currently being explored. Notably, the technological developments in sequencing and bioinformatics have provided extensive information to identify AS targets on a genome-wide scale, and in turn pathways and cellular programs that are differentially regulated in cancer cells [115,116,117,118,119,120,121]. However, from this large-scale approach, hundreds of splicing alterations are obtained that result either from mutations or abnormal expression of splicing factors, but do not readily allow for the identification of the critical cancer-driving splicing events. On the other hand, although individual pathogenic splicing events have already been described, systematic studies of the functional impact of widespread splicing alterations in cancer have yet to be performed. Actually, it is crucial to determine the outcome induced by the observed splicing changes in a tumor-specific manner with corresponding resolution at the proteome level. Therefore, in order to overcome this issue of data science and explore the splicing opportunities in precision medicine, it is extremely important to use robust analysis methods able to predict and validate reliable cancer-associated splicing changes.
To date, several cancer-specific alternative transcripts with potential prognostic and predictive value in clinical settings were identified. For instance, the presence of the alternatively spliced androgen receptor variant 7 (AR-V7) in castration-resistant prostate cancer patients has been linked to a decrease in the effectiveness of hormone-directed therapy [122]. In pancreatic ductal adenocarcinoma patients receiving radical surgery and adjuvant chemotherapy, it was suggested that the tumors with higher basal expression of PKM2 exhibit more aggressive behavior and worse response to chemotherapy [123]. Additionally, EGFR variants have been widely reported in various tumor types and related to tumor progression [103,104,105,124,125]. In some cases, however, the evidence is less clear. For example, the prognostic value of the CD44 variant 6 (CD44v6) in CRC was debated for years due to contradictory results [126,127,128]. Nevertheless, further studies reinforced the relevance of CD44v6 as an independent negative prognostic factor and a promising therapeutic target in CRC [94,95,129]. Another example of a splicing biomarker with predictive potential in CRC is the upregulation of RAC1B. The overexpression of this RAC1 splice variant is frequent in CRCs carrying BRAFV600E mutation, which in advanced-stage tumors is a recognized poor prognostic biomarker [130]. Moreover, it was also reported that RAC1B expression impacts the clinical outcome of metastatic CRC patients treated with first-line 5-fluorouracil/leucovorin plus oxaliplatin or capecitabine plus oxaliplatin (FOLFOX/XELOX) chemotherapy. Indeed, the results obtained indicate that RAC1B overexpression represents an independent predictive marker of poor outcome in KRAS/BRAF wild-type metastatic CRC patients treated with this adjuvant therapy [131]. In 2013, the overexpression of RAC1B in papillary thyroid carcinomas (PTCs) was documented for the first time, and a possible interplay between BRAFV600E mutation and RAC1B postulated that may contribute to an unfavorable prognosis [132], which was also proposed for follicular thyroid carcinomas [101]. Later, the pro-tumorigenic advantage of RAC1B overexpression in thyroid carcinomas was linked to the induction of apoptosis resistance through NFκB activation [102]. Curiously, in CRC, RAC1B expression was also described as conferring chemoresistance to oxaliplatin through activation of NFκB signaling [133]. Despite being preliminary, these results indicate that RAC1B may be a clinically useful prognostic molecular biomarker for disease progression as well as a marker of resistance to therapy.
Genome-Wide Identification of Cancer-Associated Splicing Signatures
Recent advances in high-throughput screening (HTS) technologies covering whole-genome and -exome sequencing, such as RNA sequencing (RNA-seq), have greatly contributed to improving the diagnosis and treatment of human diseases. Particularly, RNA-seq currently represents one of the most powerful tools to investigate AS at the genome-wide level [134,135,136,137,138,139]. Compared with gene expression microarrays, also designed to sample AS events on a genome scale, RNA-seq exhibits various potential advantages, including the ability of estimating the abundance of known and novel alternative transcripts, and providing better resolution, deeper coverage, and higher accuracy [139]. However, this technology still presents some drawbacks, namely the high cost of sequencing at deep coverage and the need to continually optimize the bioinformatics protocols for processing and analyzing RNA-seq data. On the other hand, most standard RNA-seq-based analyses have mainly focused on changes in gene expression level, thus lacking information about slight differences in alternative isoform usage and exon inclusion or exclusion. The importance of investigating AS profiles in RNA-seq data was recently highlighted by a study in a preclinical model of progressive diabetic nephropathy [135]. Using the isoform- and exon-level analysis of RNA-seq data, the authors identified AS patterns in genes implicated in disease pathogenesis, such as SHC1, SERPINC1, EPB4.1L5, and IL-33, which would have been overlooked by standard gene-level analysis.
Similarly, the profiling of AS signatures can be expected to provide insight into the disease process or identify potential prognostic or therapeutic biomarkers for cancer. Indeed, with the advent of HTS technologies, several studies have focused on detecting cancer-specific AS events by comparing cancer tissues with normal controls. In 2013, Eswaran et al. described for the first time splicing signatures specific to one of the three breast cancer sub-types. They further revealed that exon skipping and intron retention were the predominantly occurring splicing changes and also uncovered previously unknown isoforms of CDK4, LARP1, ADD3, and PHLPP2 [140]. This example highlights how the accumulation of RNA-seq data derived from clinical samples holds great potential to yield not only cancer-specific isoforms but also biomarkers of patient prognosis or response to therapy. In fact, certain other AS events have recently been reported to show prognostic value for ovarian, lung, pancreatic, prostate, and colorectal cancer patients [141,142,143,144,145]. For instance, in lung cancer, a genome-wide profiling identified various AS events significantly associated with patient survival [142], including EGFR, CD44, AR, RRAS2, MAPKAP1, and FGFR2. In CRC, two differently expressed AS events, namely, CSTF3-RI (intron retention) and CXCL12-AT (alternate terminator), were validated as independent prognostic indicators for both overall survival (OS) and disease-free survival [145]. Recently, the combination of high expression levels of COL6A3 E5-E6 junction and HKDC1 E1-E2 junction was for the first time associated with a better CRC patient OS [146]. Interestingly, it was previously reported that high gene expression of COL6A3 in stroma is linked to poor OS in CRC [147], while high expression of the HKDC1 gene is related to poor OS in hepatocarcinoma [148], indicating that the biomarker value of some AS events is tumor-type or -stage specific. Additionally, some reports have focused on the identification of predictive biomarkers for drug response. For instance, Safikhani et al., based on a combined approach between pharmacological data and genome-wide transcriptomics, validated AS isoforms of IGF2BP2, NECTIN4, ITGB6, and KLHDC9 as predictive biomarkers for drug response to AZD6244, lapatinib, erlotinib, and paclitaxel, respectively [149]. As a whole, despite the potential biomarkers identified to date, they still require validation in independent patient cohorts and translation into clinical practice.
4. Therapeutic Strategies Targeting Alternative Splicing in Cancer
The identification of cancer-specific AS variations has guided the development of a multitude of promising therapeutic strategies. Actually, due to the different origins of AS dysregulation, previously discussed in Section 2.1, aberrant splicing programs in cancer can be targeted in diverse ways, as exemplified in Figure 2, including strategies such as blocking of protein kinases that post-translationally regulate splicing factors, disruption of signaling pathways regulating AS programs, use of oligonucleotides that modulate splicing factor recruitment to the pre-mRNA, targeting of protein isoforms derived from aberrant AS events, or targeting of the components of RNA spliceosome machinery. The latter has been discussed in detail elsewhere [29,150,151], and thus will not be addressed here.
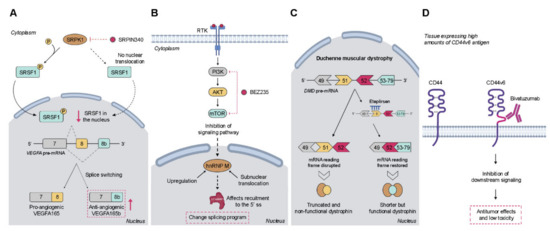
Figure 2.
Examples of therapeutic strategies targeting alternative splicing (AS). (A) Targeting of protein kinases by small molecules to inhibit the post-translational phosphorylation of splicing factors. (B) Inhibition of signaling pathways by small molecules. (C) Splice-switching antisense oligonucleotides. (D) Targeting of cancer-specific isoforms by therapeutic monoclonal antibodies; (pre-mRNA—precursor messenger RNA; ss—splice site; RTK—receptor tyrosine kinase; hnRNP—heterogeneous nuclear ribonucleoprotein; DMD—dystrophin gene).
4.1. Inhibiting Post-Translational Modifications of Splicing Factors or RNA Binding Proteins
Although several types of post-translational modifications of splicing factors were described, their phosphorylation has a key role, and the development of small-molecule inhibitors targeting protein kinases has emerged as a promising therapeutic strategy to reverse aberrant RNA splicing [152,153]. The two main targets of these molecules are the SR-rich protein-specific kinases (SRPKs) and the dual-specificity Cdc2-like kinases (CLKs), which primarily regulate pre-mRNA splicing by phosphorylating SR proteins, controlling both their nucleo-cytoplasmic shuttling and their interactions with the spliceosome [154]. Increased levels of these splicing regulatory protein kinases have been found in several types of cancers, which highlights the therapeutic potential of their pharmacological targeting. Particularly, upregulated expression of SRPK1 is frequently associated with an oncogenic activity in a variety of cancer types [155]. Accordingly, pharmacological inhibition of SRPK1 with the first-generation drug SRPIN340 induced splice switching of pro-angiogenic VEGFA165 to anti-angiogenic VEGFA165b (Figure 2A) in prostate cancer and leukemic cells [156,157]. Another study also showed that SRPIN340 significantly reduces tumor growth in metastatic melanoma in vivo via reduced expression of pro-angiogenic VEGF isoforms [158]. More recently, a covalent inhibitor of SPRK1 and SPRK2—SRPKIN-1—was developed, which efficiently reduced SR protein phosphorylation, promoted splice switching of VEGFA165 to VEGFA165b, and blocked neovascularization. This was achieved by local application in mice [159], but a corresponding benefit in tumor therapy remains to be demonstrated. The first CLK inhibitor to be discovered was the benzothiazole compound TG-003, which demonstrated selective potency toward CLK1, CLK2, and CLK4 and regulated splicing by reducing the phosphorylation of several SR proteins, including SRSF1 [160]. Two other compounds, leucettine L41 and T-025, were also identified as CLK inhibitors that modulate AS by the same mechanism of action [161,162]. Lastly, in a large-scale screening, a set of related compounds was identified, namely Cpd-1, Cpd-2, and Cpd-3, capable of targeting CLK1 and CLK2 and, to a lesser extent, SPRK1 and SRPK2 [163]. Despite the apparent success of these compounds in vitro, further studies are needed in order to improve their efficacy and narrow the window of off-target effects on splicing before moving to the clinical trial setting.
Another strategy to target splicing is exemplified by the use of sulfonamides, including E7820, indisulam, tasisulam, and chloroquinoxaline sulphonamide. These agents are known to show antitumor activity, and some of them have already been tested in clinical trials [164,165,166]. Later, it was confirmed that several sulphonamides interfere with splicing by promoting ubiquitin-mediated degradation of U2AF-related splicing factor RBM39 (also called CAPERα) via CRL4 E3 ubiquitin ligase complex [167,168]. For example, it was found that after indisulam treatment of cultured cancer cells, RBM39 degradation led to altered pre-mRNA splicing, including intron retention and exon skipping, in hundreds of genes [167]. However, it was also shown that RBM39 degradation is limited to certain cancer cells. Actually, mutations in RBM39, specifically in RRM2 domain, prevent its proteasomal degradation, thus conferring sulphonamide resistance. Sensitivity to these compounds also correlates with the expression levels of DCAF15 in hematopoietic and lymphoid lineages because the CUL4-DCAF15 complex regulates the ubiquitination and degradation of RBM39 [167].
4.2. Modulation of Signaling Pathways Regulating Alternative Splicing Events
The involvement of signaling pathways in the regulation of AS is well recognized, as referred to above. The mechanisms through which signal transduction pathways interfere directly or indirectly with splicing, typically involve regulation of either the cellular localization or the activation status of splicing-regulatory proteins. Therefore, modulation of signaling pathways represents a promising approach to target dysregulated AS. Importantly, despite the existence of a wide range of compounds able to target these pathways (reviewed in [169]), some of which have already been tested in clinical trials, they were not specifically developed to modulate AS. However, they proved to be valuable tools to further elucidate the mechanisms involved in the regulation of AS by oncogenic signaling pathways. For instance, the AKT inhibitor MK2206 was used to validate the results obtained in a study that aimed to unravel the differential regulation of the phosphoproteome by AKT isoforms [170]. Briefly, it was demonstrated that the specific RNA processing protein IWS1 is phosphorylated by AKT1 and AKT3 in lung cancer. IWS1 phosphorylation allows the recruitment of SETD2 to the RNA polymerase II complex. SETD2 trimethylates histone H3 during transcription, creating a docking site for PTBP1 splicing factor. In turn, PTBP1 promotes the skipping of exon IIIb in the fibroblast growth factor receptor 2 (FGFR-2) gene, shifting the balance of FGFR-2 splicing from the IIIb to the IIIc isoform, which promotes cell proliferation, migration, and invasiveness in response to FGF-2. Moreover, in a work carried out in Ewing sarcoma cells, it was found that hnRNP M was strongly upregulated both at the mRNA and protein level upon inhibition of the PI3K/AKT/mTOR pathway with BEZ235, and located in the soluble nucleoplasmic fraction, where it modulated U1 snRNP recruitment to a 5′ splice site and triggered a splicing program contributing to drug resistance (Figure 2B) [171]. Overall, these types of inhibitory drugs have the ability to change splicing outcomes; however, it is crucial to invest in the development of compounds targeting specific abnormal AS events in order to limit the occurrence of undesired side effects.
4.3. Antisense Oligonucleotides
RNA-targeted therapies emerged in 1978 when Zamecnik and Stephenson described for the first time a chemically modified oligonucleotide that inhibited gene expression and viral replication of Rous sarcoma virus [172,173]. From there, antisense oligonucleotides (ASOs) have been extensively explored in the process of drug development and proven to be a useful alternative approach for the target-specific treatment of splicing-related human diseases, including cancer. Briefly, ASOs are synthetic molecules consisting of short single-stranded nucleic-acid sequences, generally 15–25 nucleotides in length, that specifically bind through Watson–Crick base-pairing to complementary pre-mRNA sequences [174]. RNA-targeted therapies are already used in the clinic and numerous clinical trials with therapeutic ASOs are currently underway [175,176,177].
The antisense therapies can be subdivided into two groups according to their downstream mechanisms of action and functional outcomes. The majority of ASOs are designed to promote the cleavage of targeted mRNA by endogenous cellular nucleases, such as RNase H, which recognizes double-stranded RNA:DNA hybrids and subsequently degrades the disease-causing gene product. A different strategy aims to interfere with the access of the splicing machinery to the regulatory sequences in the pre-mRNA instead of causing the transcript degradation. So-called splice-switching antisense oligonucleotides (SSOs) are designed to compete with and sterically block the binding of certain splicing factors to their specific sites in the pre-mRNA, which in turn changes exon recognition by the spliceosome [178]. As such, this strategy intends to specifically shift the splicing pattern of a targeted pre-mRNA transcript, favoring the production of one of the splicing variants with potential therapeutic benefits. To date, two SSOs were approved by the US Food and Drug Administration (FDA), Eteplirsen and Nusinersen, for the clinical treatment of the genetic diseases Duchenne muscular dystrophy and spinal muscular atrophy, respectively [179,180]. Eteplirsen hybridizes at exon 51 of the DMD pre-mRNA (which encodes the dystrophin protein), sterically blocking the recognition of this exon by the spliceosome and thereby promoting the skipping of exon 51 to correct the disease-causing frameshift mutation and generating a shorter but functional variant of the protein (Figure 2C) [181]. In a distinct way, Nusinersen binds to an intronic region upstream of exon 7 in the SMN2 pre-mRNA that encodes the survival motor neuron protein [180]. The binding blocks recruitment of an inhibitory splicing factor that would normally impede the recognition of exon 7 by the spliceosome, thus enhancing the inclusion of the formerly missing exon 7 of SMN2, and the subsequent production of a fully functional protein that is absent in patients with spinal muscular atrophy. Although the application of SSOs in anticancer therapy is still under evaluation, the modulation of RNA splicing of cancer-related genes has been successfully achieved in various pre-clinical cancer models. One of the most used strategies targets the BCL2L1 gene that is alternatively spliced, originating either anti-apoptotic Bcl-xL or pro-apoptotic Bcl-xS proteins. Thus, in order to abolish the high expression levels of Bcl-xL reported in many cancers, Bcl-x SSOs were designed to induce a splicing switch, favoring the production of the pro-apoptotic isoform Bcl-xS. It was shown in vitro that treatment with these SSOs shifted splicing from Bcl-xL to Bcl-xS in various cancer cell lines [182]. Moreover, it was found that Bcl-xS proteins induced by the SSOs sensitized the cancer cells to treatment with chemotherapeutic agents or ultraviolet (UV) radiation [70]. Additionally, the antitumor activity of SSOs was also demonstrated in vivo in a mouse model of melanoma lung metastases where the systemic administration of Bcl-x SSO using a lipid nanoparticle redirected Bcl-x splicing and led to a significant reduction in tumor burden in treated mice [183]. Another important antitumor target is the hnRNP-regulated splicing of the PKM gene [184], which is a critical player in the regulation of glucose metabolism by producing either the PKM1 isoform (that stimulates oxidative phosphorylation) or the PKM2 isoform (that promotes aerobic glycolysis, a metabolic shift also recognized as the Warburg effect). PKM2 is frequently upregulated in cancer cell lines and various tumor types, including CRC, and SSOs used to switch the expression back to PKM1 induced apoptosis [185]. Further examples of SSO-mediated splicing modulation of other genes, including BCL2L11, BRCA1, ERBB2, MDM4, MKNK2, and STAT3, were recently reviewed in [150]. A related SSO approach is the design of decoy oligonucleotides composed of the RNA motif recognized by a given splicing factor, which can downmodulate its splicing activity. This could be a promising therapeutic approach whenever a splicing factor is either overexpressed or hyperactived in cancer cells [186].
4.4. Targeting the Alternative Protein Isoform
The presence of specific AS variant proteins in tumor cells suggest them as potential therapeutic targets. Some variants may result in the translation of immunogenic neoantigens, either as a result of frameshifts or re-expression of developmental variants. As such, some strategies have been developed to target cancer-specific isoforms by immunotherapies. One of the most explored therapeutic targets are EGFR variants de4 and vIII. Although in GBM and other cancers EGFRvIII results from a genomic deletion of exons 2–7 [187], an AS variant with skipping exon 4 leads to a comparable phenotype in other tumors: a lack of amino acids in the extracellular ligand-binding domain, resulting in a constitutively active variant able to stimulate downstream signaling in a ligand-independent manner. Several studies have supported the oncogenic role of these EGFR variants and their association with a poor prognosis [103,104,105,124,125,188,189,190,191,192]. Being tumor-specific cell surface molecules, these receptor variants were successfully targeted by therapeutic antibodies [104,189]. Notably, in 2015, the vaccine rindopepimut (also known as CDX-110), consisting of an EGFRvIII-specific peptide conjugated to keyhole limpet haemocyanin, was approved by FDA for the treatment of GBM. Actually, the results obtained in phase I and II clinical trials showed that the treatment with rindopepimut increases both OS and progression-free survival of GBM patients expressing EGFRvIII [189]. Additionally, the role of cell adhesion molecule CD44 and its isoforms containing the exon v6 have been broadly implicated in the metastatic tumor process, and as such they have also been explored as targets for anticancer therapy [193]. One of the most recognized anti-CD44v6 therapy consists of using bivatuzumab, a humanized IgG1 monoclonal antibody labelled with rhenium-186 (Figure 2D). Particularly in phase I clinical trials for patients with the head and neck squamous cell carcinoma (HNSCC), a tissue that expresses high amounts of CD44v6 antigen, bivatuzumab showed promising antitumor effects with consistent stable disease at higher radioactive dose levels and with low toxicity [194,195]. Based on these results, a novel strategy comprising the coupling of bivatuzumab with a non-radioactive cytotoxic drug, mertansine, was developed [196]. Interestingly, in phase I clinical trials, the intravenous injection of bivatuzumab mertansine in adult patients with recurrent or metastatic HNSCC induced a partial response in three of the 30 patients tested, which presented a stabilization of the disease and regression of tumors [197]. Despite the promising results, the toxic side effects observed in the skin led to the discontinuation of the clinical trials with bivatuzumab mertansine. Another example is the tight junction molecule claudin-18 isoform 2 (CLDN18.2). In 2008, CLDN18.2 was identified as a highly selective cell lineage marker, whose expression in normal tissues is restricted to differentiated epithelial cells of the gastric mucosa, being absent from the gastric stem cell zone [198]. Additionally, it was also reported that CLDN18.2 is expressed in a significant proportion of primary gastric cancers and their metastases. Since CLDN18.2 exposes extracellular loops available for antibody binding, a targeted therapy based on the monoclonal antibody IMAB362 (claudiximab) was developed [199]. According to the promising results obtained in previous clinical trials, a phase III global study of IMAB362 plus FOLFOX versus FOLFOX plus placebo as first-line treatment was initiated in 2018 in gastric cancer patients (NCT03504397).
Besides these immunotherapeutic approaches, protein–protein interaction inhibitors could become a promising precision-medicine approach for targeting AS-derived protein isoforms. Many AS variants generate proteins following exon inclusion or intron retention and can contain extra protein domains that participate in protein–protein interactions involved in their downstream function. Small-molecule drugs that compete with these interactions are being developed [200,201]. For example, the BCL-2-selective inhibitor ABT-199 competes for anti-apoptotic interaction with BAK/BAD proteins [202], and inhibitors of the MDM2–p53 complex can restore p53 function in cancerous cells, leading to their growth arrest and apoptosis [203].
5. Concluding Remarks and Future Perspectives
From a large body of experimental data, it has become increasingly clear that AS is tightly associated with human health and disease [3]. However, despite AS being the major driver of biological diversity and playing a role in every hallmark of cancer, it was neglected for a long time in the profiling of tumor characteristics and overlooked as a source of new biomarkers and therapeutic targets for drug development. Nevertheless, with the emergence of advanced sequencing technologies that provide a landscape of AS at a genome-wide level, additional insights into the splicing programs were achieved [6,7,8,9]. Remarkably, these studies have contributed in a decisive way to the identification of aberrant AS events in cancer development and progression, a prerequisite for the identification of potential biomarkers and development of new therapeutic strategies towards cancer precision medicine. Despite the described progress, the tumor-specific splicing alterations are far from being characterized and further efforts are needed to provide a comprehensive view of splicing regulation and of its dysregulation in cancer.
Another important aspect that has emerged from advanced sequencing technology is the need to move our understanding from individual AS variants to the overall pattern of splicing changes in tumors. Any change in activity or localization of a splicing factor will potentially trigger a plethora of AS decisions in many different genes. Thus, AS signature profiles or patterns may represent more meaningful biomarkers. Regarding AS-targeting drug development, existing small-molecule compounds do mostly interfere with early spliceosome assembly or post-translational modification of SR proteins, but lack efficient antitumor activity. As such, the recent efforts focused on the targeting of pathological RNA isoforms or tumor-specific protein variants represent the most promising attempts to develop more effective drug candidates. Unfortunately, these targeted anticancer therapies based on AS are still far from reaching the clinic. To address this issue, it is a priority to reveal in more detail how altered AS actually drives tumorigenesis, and how it is connected to altered genotypes or signaling pathways that characterize tumor phenotypes.
Author Contributions
Conceptualization, P.J. and V.G.; writing—original draft preparation, C.B.; writing—review and editing, P.M., V.G., and P.J. All authors have read and agreed to the published version of the manuscript.
Funding
Work in the authors’ laboratory was supported by Fundação para a Ciência e a Tecnologia (FCT), Portugal (through grant UID/MULTI/04046/2019 to Research Unit BioISI since January 2019 and grant PTDC/BIA-MOL/28386/2017 to V.G. since October 2018) and by the Portuguese association Maratona da Saúde—Cancro 2014 to P.J. since January 2016.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the collection of data, the writing of the manuscript, or in the decision to publish the manuscript.
References
- Blencowe, B.J. The Relationship between Alternative Splicing and Proteomic Complexity. Trends Biochem. Sci. 2017, 42, 407–408. [Google Scholar] [CrossRef]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef]
- Kim, H.K.; Pham, M.H.C.; Ko, K.S.; Rhee, B.D.; Han, J. Alternative splicing isoforms in health and disease. Pflug. Arch. 2018, 470, 995–1016. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef] [PubMed]
- Venables, J.P. Aberrant and alternative splicing in cancer. Cancer Res. 2004, 64, 7647–7654. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Minana, B.; Valcarcel, J.; Gabaldon, T.; Lehner, B. Synonymous mutations frequently act as driver mutations in human cancers. Cell 2014, 156, 1324–1335. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Lee, D.; Lee, J.; Park, D.; Kim, Y.J.; Park, W.Y.; Hong, D.; Park, P.J.; Lee, E. Intron retention is a widespread mechanism of tumor-suppressor inactivation. Nat. Genet. 2015, 47, 1242–1248. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Tovar-Corona, J.M.; Urrutia, A.O. Increased levels of noisy splicing in cancers, but not for oncogene-derived transcripts. Hum. Mol. Genet. 2011, 20, 4422–4429. [Google Scholar] [CrossRef]
- Dvinge, H.; Bradley, R.K. Widespread intron retention diversifies most cancer transcriptomes. Genome Med. 2015, 7, 45. [Google Scholar] [CrossRef]
- Dvinge, H.; Kim, E.; Abdel-Wahab, O.; Bradley, R.K. RNA splicing factors as oncoproteins and tumour suppressors. Nat. Rev. Cancer 2016, 16, 413–430. [Google Scholar] [CrossRef]
- Paronetto, M.P.; Passacantilli, I.; Sette, C. Alternative splicing and cell survival: From tissue homeostasis to disease. Cell Death Differ. 2016, 23, 1919–1929. [Google Scholar] [CrossRef] [PubMed]
- Rappsilber, J.; Ryder, U.; Lamond, A.I.; Mann, M. Large-scale proteomic analysis of the human spliceosome. Genome Res. 2002, 12, 1231–1245. [Google Scholar] [CrossRef] [PubMed]
- Jurica, M.S.; Moore, M.J. Pre-mRNA splicing: Awash in a sea of proteins. Mol. Cell 2003, 12, 5–14. [Google Scholar] [CrossRef]
- Coltri, P.P.; Dos Santos, M.G.P.; da Silva, G.H.G. Splicing and cancer: Challenges and opportunities. Wiley Interdiscip. Rev. RNA 2019, 10, e1527. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gonzalez-Porta, M.; Santos, S.; Brazma, A.; Marioni, J.C.; Aebersold, R.; Venkitaraman, A.R.; Wickramasinghe, V.O. Impact of Alternative Splicing on the Human Proteome. Cell Rep. 2017, 20, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- Keren, H.; Lev-Maor, G.; Ast, G. Alternative splicing and evolution: Diversification, exon definition and function. Nat. Rev. Genet. 2010, 11, 345–355. [Google Scholar] [CrossRef]
- Wang, Z.; Burge, C.B. Splicing regulation: From a parts list of regulatory elements to an integrated splicing code. RNA 2008, 14, 802–813. [Google Scholar] [CrossRef]
- Lee, Y.; Rio, D.C. Mechanisms and Regulation of Alternative Pre-mRNA Splicing. Annu Rev. Biochem. 2015, 84, 291–323. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Kahles, A.; Lehmann, K.V.; Toussaint, N.C.; Huser, M.; Stark, S.G.; Sachsenberg, T.; Stegle, O.; Kohlbacher, O.; Sander, C.; Cancer Genome Atlas Research Network; et al. Comprehensive Analysis of Alternative Splicing Across Tumors from 8,705 Patients. Cancer Cell 2018, 34, 211–224. [Google Scholar] [CrossRef]
- Bechara, E.G.; Sebestyen, E.; Bernardis, I.; Eyras, E.; Valcarcel, J. RBM5, 6, and 10 differentially regulate NUMB alternative splicing to control cancer cell proliferation. Mol. Cell 2013, 52, 720–733. [Google Scholar] [CrossRef] [PubMed]
- Darman, R.B.; Seiler, M.; Agrawal, A.A.; Lim, K.H.; Peng, S.; Aird, D.; Bailey, S.L.; Bhavsar, E.B.; Chan, B.; Colla, S.; et al. Cancer-Associated SF3B1 Hotspot Mutations Induce Cryptic 3′ Splice Site Selection through Use of a Different Branch Point. Cell Rep. 2015, 13, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Madan, V.; Kanojia, D.; Li, J.; Okamoto, R.; Sato-Otsubo, A.; Kohlmann, A.; Sanada, M.; Grossmann, V.; Sundaresan, J.; Shiraishi, Y.; et al. Aberrant splicing of U12-type introns is the hallmark of ZRSR2 mutant myelodysplastic syndrome. Nat. Commun. 2015, 6, 6042. [Google Scholar] [CrossRef] [PubMed]
- Zong, F.Y.; Fu, X.; Wei, W.J.; Luo, Y.G.; Heiner, M.; Cao, L.J.; Fang, Z.; Fang, R.; Lu, D.; Ji, H.; et al. The RNA-binding protein QKI suppresses cancer-associated aberrant splicing. PLoS Genet. 2014, 10, e1004289. [Google Scholar] [CrossRef] [PubMed]
- Jayasinghe, R.G.; Cao, S.; Gao, Q.; Wendl, M.C.; Vo, N.S.; Reynolds, S.M.; Zhao, Y.; Climente-Gonzalez, H.; Chai, S.; Wang, F.; et al. Systematic Analysis of Splice-Site-Creating Mutations in Cancer. Cell Rep. 2018, 23, 270–281. [Google Scholar] [CrossRef]
- Yoshida, K.; Sanada, M.; Shiraishi, Y.; Nowak, D.; Nagata, Y.; Yamamoto, R.; Sato, Y.; Sato-Otsubo, A.; Kon, A.; Nagasaki, M.; et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011, 478, 64–69. [Google Scholar] [CrossRef]
- Bejar, R.; Stevenson, K.E.; Caughey, B.A.; Abdel-Wahab, O.; Steensma, D.P.; Galili, N.; Raza, A.; Kantarjian, H.; Levine, R.L.; Neuberg, D.; et al. Validation of a prognostic model and the impact of mutations in patients with lower-risk myelodysplastic syndromes. J. Clin. Oncol. 2012, 30, 3376–3382. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Chronic Myeloid Disorders Working Group of the International Cancer Genome, C., Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3627. [Google Scholar] [CrossRef]
- Bonnal, S.C.; Lopez-Oreja, I.; Valcarcel, J. Roles and mechanisms of alternative splicing in cancer—implications for care. Nat. Rev. Clin. Oncol. 2020, 17, 457–474. [Google Scholar] [CrossRef]
- DeBoever, C.; Ghia, E.M.; Shepard, P.J.; Rassenti, L.; Barrett, C.L.; Jepsen, K.; Jamieson, C.H.; Carson, D.; Kipps, T.J.; Frazer, K.A. Transcriptome sequencing reveals potential mechanism of cryptic 3′ splice site selection in SF3B1-mutated cancers. PLoS Comput. Biol. 2015, 11, e1004105. [Google Scholar] [CrossRef]
- Alsafadi, S.; Houy, A.; Battistella, A.; Popova, T.; Wassef, M.; Henry, E.; Tirode, F.; Constantinou, A.; Piperno-Neumann, S.; Roman-Roman, S.; et al. Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat. Commun. 2016, 7, 10615. [Google Scholar] [CrossRef] [PubMed]
- Daubner, G.M.; Clery, A.; Jayne, S.; Stevenin, J.; Allain, F.H. A syn-anti conformational difference allows SRSF2 to recognize guanines and cytosines equally well. EMBO J. 2012, 31, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Ilagan, J.O.; Liang, Y.; Daubner, G.M.; Lee, S.C.; Ramakrishnan, A.; Li, Y.; Chung, Y.R.; Micol, J.B.; Murphy, M.E.; et al. SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell 2015, 27, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lieu, Y.K.; Ali, A.M.; Penson, A.; Reggio, K.S.; Rabadan, R.; Raza, A.; Mukherjee, S.; Manley, J.L. Disease-associated mutation in SRSF2 misregulates splicing by altering RNA-binding affinities. Proc. Natl. Acad. Sci. USA 2015, 112, 4726–4734. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef]
- Seiler, M.; Peng, S.; Agrawal, A.A.; Palacino, J.; Teng, T.; Zhu, P.; Smith, P.G.; Cancer Genome Atlas Research, N.; Buonamici, S.; Yu, L. Somatic Mutational Landscape of Splicing Factor Genes and Their Functional Consequences across 33 Cancer Types. Cell Rep. 2018, 23, 282–296. [Google Scholar] [CrossRef]
- Ilagan, J.O.; Ramakrishnan, A.; Hayes, B.; Murphy, M.E.; Zebari, A.S.; Bradley, P.; Bradley, R.K. U2AF1 mutations alter splice site recognition in hematological malignancies. Genome Res. 2015, 25, 14–26. [Google Scholar] [CrossRef]
- Fei, D.L.; Motowski, H.; Chatrikhi, R.; Prasad, S.; Yu, J.; Gao, S.; Kielkopf, C.L.; Bradley, R.K.; Varmus, H. Wild-Type U2AF1 Antagonizes the Splicing Program Characteristic of U2AF1-Mutant Tumors and Is Required for Cell Survival. PLoS Genet. 2016, 12, e1006384. [Google Scholar] [CrossRef]
- Zhang, J.; Manley, J.L. Misregulation of pre-mRNA alternative splicing in cancer. Cancer Discov. 2013, 3, 1228–1237. [Google Scholar] [CrossRef]
- Ghigna, C.; Valacca, C.; Biamonti, G. Alternative splicing and tumor progression. Curr. Genom. 2008, 9, 556–570. [Google Scholar] [CrossRef]
- Ghigna, C.; Moroni, M.; Porta, C.; Riva, S.; Biamonti, G. Altered expression of heterogenous nuclear ribonucleoproteins and SR factors in human colon adenocarcinomas. Cancer Res. 1998, 58, 5818–5824. [Google Scholar] [PubMed]
- Grosso, A.R.; Martins, S.; Carmo-Fonseca, M. The emerging role of splicing factors in cancer. EMBO Rep. 2008, 9, 1087–1093. [Google Scholar] [CrossRef] [PubMed]
- Sveen, A.; Kilpinen, S.; Ruusulehto, A.; Lothe, R.A.; Skotheim, R.I. Aberrant RNA splicing in cancer; expression changes and driver mutations of splicing factor genes. Oncogene 2016, 35, 2413–2427. [Google Scholar] [CrossRef] [PubMed]
- Karni, R.; de Stanchina, E.; Lowe, S.W.; Sinha, R.; Mu, D.; Krainer, A.R. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat. Struct. Mol. Biol. 2007, 14, 185–193. [Google Scholar] [CrossRef]
- Anczukow, O.; Rosenberg, A.Z.; Akerman, M.; Das, S.; Zhan, L.; Karni, R.; Muthuswamy, S.K.; Krainer, A.R. The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat. Struct. Mol. Biol. 2012, 19, 220–228. [Google Scholar] [CrossRef]
- Anczukow, O.; Akerman, M.; Clery, A.; Wu, J.; Shen, C.; Shirole, N.H.; Raimer, A.; Sun, S.; Jensen, M.A.; Hua, Y.; et al. SRSF1-Regulated Alternative Splicing in Breast Cancer. Mol. Cell 2015, 60, 105–117. [Google Scholar] [CrossRef]
- Ghigna, C.; Giordano, S.; Shen, H.; Benvenuto, F.; Castiglioni, F.; Comoglio, P.M.; Green, M.R.; Riva, S.; Biamonti, G. Cell motility is controlled by SF2/ASF through alternative splicing of the Ron protooncogene. Mol. Cell 2005, 20, 881–890. [Google Scholar] [CrossRef]
- Kedzierska, H.; Piekielko-Witkowska, A. Splicing factors of SR and hnRNP families as regulators of apoptosis in cancer. Cancer Lett. 2017, 396, 53–65. [Google Scholar] [CrossRef]
- Fielding, P.; Turnbull, L.; Prime, W.; Walshaw, M.; Field, J.K. Heterogeneous nuclear ribonucleoprotein A2/B1 up-regulation in bronchial lavage specimens: A clinical marker of early lung cancer detection. Clin. Cancer Res. 1999, 5, 4048–4052. [Google Scholar]
- Zhou, J.; Nong, L.; Wloch, M.; Cantor, A.; Mulshine, J.L.; Tockman, M.S. Expression of early lung cancer detection marker: hnRNP-A2/B1 and its relation to microsatellite alteration in non-small cell lung cancer. Lung Cancer 2001, 34, 341–350. [Google Scholar] [CrossRef]
- Zhou, J.; Allred, D.C.; Avis, I.; Martinez, A.; Vos, M.D.; Smith, L.; Treston, A.M.; Mulshine, J.L. Differential expression of the early lung cancer detection marker, heterogeneous nuclear ribonucleoprotein-A2/B1 (hnRNP-A2/B1) in normal breast and neoplastic breast cancer. Breast Cancer Res. Treat. 2001, 66, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Golan-Gerstl, R.; Cohen, M.; Shilo, A.; Suh, S.S.; Bakacs, A.; Coppola, L.; Karni, R. Splicing factor hnRNP A2/B1 regulates tumor suppressor gene splicing and is an oncogenic driver in glioblastoma. Cancer Res. 2011, 71, 4464–4472. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, V.; Pereira, J.F.S.; Jordan, P. Signaling Pathways Driving Aberrant Splicing in Cancer Cells. Genes 2017, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Hollander, D.; Donyo, M.; Atias, N.; Mekahel, K.; Melamed, Z.; Yannai, S.; Lev-Maor, G.; Shilo, A.; Schwartz, S.; Barshack, I.; et al. A network-based analysis of colon cancer splicing changes reveals a tumorigenesis-favoring regulatory pathway emanating from ELK1. Genome Res. 2016, 26, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Valacca, C.; Bonomi, S.; Buratti, E.; Pedrotti, S.; Baralle, F.E.; Sette, C.; Ghigna, C.; Biamonti, G. Sam68 regulates EMT through alternative splicing-activated nonsense-mediated mRNA decay of the SF2/ASF proto-oncogene. J. Cell Biol. 2010, 191, 87–99. [Google Scholar] [CrossRef]
- Matter, N.; Herrlich, P.; Konig, H. Signal-dependent regulation of splicing via phosphorylation of Sam68. Nature 2002, 420, 691–695. [Google Scholar] [CrossRef]
- van der Houven van Oordt, W.; Diaz-Meco, M.T.; Lozano, J.; Krainer, A.R.; Moscat, J.; Caceres, J.F. The MKK(3/6)-p38-signaling cascade alters the subcellular distribution of hnRNP A1 and modulates alternative splicing regulation. J. Cell Biol. 2000, 149, 307–316. [Google Scholar] [CrossRef]
- Allemand, E.; Guil, S.; Myers, M.; Moscat, J.; Caceres, J.F.; Krainer, A.R. Regulation of heterogenous nuclear ribonucleoprotein A1 transport by phosphorylation in cells stressed by osmotic shock. Proc. Natl. Acad. Sci. USA 2005, 102, 3605–3610. [Google Scholar] [CrossRef]
- Shultz, J.C.; Goehe, R.W.; Wijesinghe, D.S.; Murudkar, C.; Hawkins, A.J.; Shay, J.W.; Minna, J.D.; Chalfant, C.E. Alternative splicing of caspase 9 is modulated by the phosphoinositide 3-kinase/Akt pathway via phosphorylation of SRp30a. Cancer Res. 2010, 70, 9185–9196. [Google Scholar] [CrossRef]
- Goehe, R.W.; Shultz, J.C.; Murudkar, C.; Usanovic, S.; Lamour, N.F.; Massey, D.H.; Zhang, L.; Camidge, D.R.; Shay, J.W.; Minna, J.D.; et al. hnRNP L regulates the tumorigenic capacity of lung cancer xenografts in mice via caspase-9 pre-mRNA processing. J. Clin. Investig. 2010, 120, 3923–3939. [Google Scholar] [CrossRef]
- Vu, N.T.; Park, M.A.; Shultz, J.C.; Goehe, R.W.; Hoeferlin, L.A.; Shultz, M.D.; Smith, S.A.; Lynch, K.W.; Chalfant, C.E. hnRNP U enhances caspase-9 splicing and is modulated by AKT-dependent phosphorylation of hnRNP L. J. Biol. Chem. 2013, 288, 8575–8584. [Google Scholar] [CrossRef] [PubMed]
- Blaustein, M.; Pelisch, F.; Tanos, T.; Munoz, M.J.; Wengier, D.; Quadrana, L.; Sanford, J.R.; Muschietti, J.P.; Kornblihtt, A.R.; Caceres, J.F.; et al. Concerted regulation of nuclear and cytoplasmic activities of SR proteins by AKT. Nat. Struct. Mol. Biol. 2005, 12, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, V.; Matos, P.; Jordan, P. Antagonistic SR proteins regulate alternative splicing of tumor-related Rac1b downstream of the PI3-kinase and Wnt pathways. Hum. Mol. Genet. 2009, 18, 3696–3707. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, V.; Henriques, A.F.; Pereira, J.F.; Neves Costa, A.; Moyer, M.P.; Moita, L.F.; Gama-Carvalho, M.; Matos, P.; Jordan, P. Phosphorylation of SRSF1 by SRPK1 regulates alternative splicing of tumor-related Rac1b in colorectal cells. RNA 2014, 20, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, V.; Matos, P.; Jordan, P. The beta-catenin/TCF4 pathway modifies alternative splicing through modulation of SRp20 expression. RNA 2008, 14, 2538–2549. [Google Scholar] [CrossRef]
- El Marabti, E.; Younis, I. The Cancer Spliceome: Reprograming of Alternative Splicing in Cancer. Front. Mol. Biosci. 2018, 5, 80. [Google Scholar] [CrossRef]
- Xerri, L.; Hassoun, J.; Devilard, E.; Birnbaum, D.; Birg, F. BCL-X and the apoptotic machinery of lymphoma cells. Leuk. Lymphoma 1998, 28, 451–458. [Google Scholar] [CrossRef]
- Li, Z.; Li, Q.; Han, L.; Tian, N.; Liang, Q.; Li, Y.; Zhao, X.; Du, C.; Tian, Y. Pro-apoptotic effects of splice-switching oligonucleotides targeting Bcl-x pre-mRNA in human glioma cell lines. Oncol. Rep. 2016, 35, 1013–1019. [Google Scholar] [CrossRef]
- Olopade, O.I.; Adeyanju, M.O.; Safa, A.R.; Hagos, F.; Mick, R.; Thompson, C.B.; Recant, W.M. Overexpression of BCL-x protein in primary breast cancer is associated with high tumor grade and nodal metastases. Cancer J. Sci. Am. 1997, 3, 230–237. [Google Scholar]
- Mercatante, D.R.; Mohler, J.L.; Kole, R. Cellular response to an antisense-mediated shift of Bcl-x pre-mRNA splicing and antineoplastic agents. J. Biol. Chem. 2002, 277, 49374–49382. [Google Scholar] [CrossRef]
- Takehara, T.; Liu, X.; Fujimoto, J.; Friedman, S.L.; Takahashi, H. Expression and role of Bcl-xL in human hepatocellular carcinomas. Hepatology 2001, 34, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Eliav, M.; Golan-Gerstl, R.; Siegfried, Z.; Andersen, C.L.; Thorsen, K.; Orntoft, T.F.; Mu, D.; Karni, R. The splicing factor SRSF6 is amplified and is an oncoprotein in lung and colon cancers. J. Pathol. 2013, 229, 630–639. [Google Scholar] [CrossRef] [PubMed]
- Maimon, A.; Mogilevsky, M.; Shilo, A.; Golan-Gerstl, R.; Obiedat, A.; Ben-Hur, V.; Lebenthal-Loinger, I.; Stein, I.; Reich, R.; Beenstock, J.; et al. Mnk2 alternative splicing modulates the p38-MAPK pathway and impacts Ras-induced transformation. Cell Rep. 2014, 7, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Chao, T.K.; Huang, T.S.; Liao, Y.P.; Huang, R.L.; Su, P.H.; Shen, H.Y.; Lai, H.C.; Wang, Y.C. Pyruvate kinase M2 is a poor prognostic marker of and a therapeutic target in ovarian cancer. PLoS ONE 2017, 12, e0182166. [Google Scholar] [CrossRef] [PubMed]
- Shiroki, T.; Yokoyama, M.; Tanuma, N.; Maejima, R.; Tamai, K.; Yamaguchi, K.; Oikawa, T.; Noguchi, T.; Miura, K.; Fujiya, T.; et al. Enhanced expression of the M2 isoform of pyruvate kinase is involved in gastric cancer development by regulating cancer-specific metabolism. Cancer Sci. 2017, 108, 931–940. [Google Scholar] [CrossRef]
- Liu, W.R.; Tian, M.X.; Yang, L.X.; Lin, Y.L.; Jin, L.; Ding, Z.B.; Shen, Y.H.; Peng, Y.F.; Gao, D.M.; Zhou, J.; et al. PKM2 promotes metastasis by recruiting myeloid-derived suppressor cells and indicates poor prognosis for hepatocellular carcinoma. Oncotarget 2015, 6, 846–861. [Google Scholar] [CrossRef]
- Takahashi, H.; Nishimura, J.; Kagawa, Y.; Kano, Y.; Takahashi, Y.; Wu, X.; Hiraki, M.; Hamabe, A.; Konno, M.; Haraguchi, N.; et al. Significance of Polypyrimidine Tract-Binding Protein 1 Expression in Colorectal Cancer. Mol. Cancer Ther. 2015, 14, 1705–1716. [Google Scholar] [CrossRef]
- Zhou, Y.Q.; He, C.; Chen, Y.Q.; Wang, D.; Wang, M.H. Altered expression of the RON receptor tyrosine kinase in primary human colorectal adenocarcinomas: Generation of different splicing RON variants and their oncogenic potential. Oncogene 2003, 22, 186–197. [Google Scholar] [CrossRef]
- Mayer, S.; Hirschfeld, M.; Jaeger, M.; Pies, S.; Iborra, S.; Erbes, T.; Stickeler, E. RON alternative splicing regulation in primary ovarian cancer. Oncol. Rep. 2015, 34, 423–430. [Google Scholar] [CrossRef][Green Version]
- Eckerich, C.; Schulte, A.; Martens, T.; Zapf, S.; Westphal, M.; Lamszus, K. RON receptor tyrosine kinase in human gliomas: Expression, function, and identification of a novel soluble splice variant. J. Neurochem. 2009, 109, 969–980. [Google Scholar] [CrossRef]
- Krishnaswamy, S.; Mohammed, A.K.; Tripathi, G.; Alokail, M.S.; Al-Daghri, N.M. Splice variants of the extracellular region of RON receptor tyrosine kinase in lung cancer cell lines identified by PCR and sequencing. BMC Cancer 2017, 17, 738. [Google Scholar] [CrossRef] [PubMed]
- Collesi, C.; Santoro, M.M.; Gaudino, G.; Comoglio, P.M. A splicing variant of the RON transcript induces constitutive tyrosine kinase activity and an invasive phenotype. Mol. Cell. Biol. 1996, 16, 5518–5526. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ben-Hur, V.; Denichenko, P.; Siegfried, Z.; Maimon, A.; Krainer, A.; Davidson, B.; Karni, R. S6K1 alternative splicing modulates its oncogenic activity and regulates mTORC1. Cell Rep. 2013, 3, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Mei, H.; Wang, Y.; Fan, J.; Lin, Z. Alternative splicing of S6K1 promotes non-small cell lung cancer survival. Tumour Biol. 2016, 37, 13369–13376. [Google Scholar] [CrossRef]
- Burd, C.J.; Petre, C.E.; Morey, L.M.; Wang, Y.; Revelo, M.P.; Haiman, C.A.; Lu, S.; Fenoglio-Preiser, C.M.; Li, J.; Knudsen, E.S.; et al. Cyclin D1b variant influences prostate cancer growth through aberrant androgen receptor regulation. Proc. Natl. Acad. Sci. USA 2006, 103, 2190–2195. [Google Scholar] [CrossRef]
- Li, R.; An, S.J.; Chen, Z.H.; Zhang, G.C.; Zhu, J.Q.; Nie, Q.; Xie, Z.; Guo, A.L.; Mok, T.S.; Wu, Y.L. Expression of cyclin D1 splice variants is differentially associated with outcome in non-small cell lung cancer patients. Hum. Pathol. 2008, 39, 1792–1801. [Google Scholar] [CrossRef]
- Wang, Y.; Dean, J.L.; Millar, E.K.; Tran, T.H.; McNeil, C.M.; Burd, C.J.; Henshall, S.M.; Utama, F.E.; Witkiewicz, A.; Rui, H.; et al. Cyclin D1b is aberrantly regulated in response to therapeutic challenge and promotes resistance to estrogen antagonists. Cancer Res. 2008, 68, 5628–5638. [Google Scholar] [CrossRef]
- Varey, A.H.; Rennel, E.S.; Qiu, Y.; Bevan, H.S.; Perrin, R.M.; Raffy, S.; Dixon, A.R.; Paraskeva, C.; Zaccheo, O.; Hassan, A.B.; et al. VEGF 165 b, an antiangiogenic VEGF-A isoform, binds and inhibits bevacizumab treatment in experimental colorectal carcinoma: Balance of pro- and antiangiogenic VEGF-A isoforms has implications for therapy. Br. J. Cancer 2008, 98, 1366–1379. [Google Scholar] [CrossRef]
- Rennel, E.; Waine, E.; Guan, H.; Schuler, Y.; Leenders, W.; Woolard, J.; Sugiono, M.; Gillatt, D.; Kleinerman, E.; Bates, D.; et al. The endogenous anti-angiogenic VEGF isoform, VEGF165b inhibits human tumour growth in mice. Br. J. Cancer 2008, 98, 1250–1257. [Google Scholar] [CrossRef]
- Bates, D.O.; Cui, T.G.; Doughty, J.M.; Winkler, M.; Sugiono, M.; Shields, J.D.; Peat, D.; Gillatt, D.; Harper, S.J. VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, is down-regulated in renal cell carcinoma. Cancer Res. 2002, 62, 4123–4131. [Google Scholar]
- Pritchard-Jones, R.O.; Dunn, D.B.; Qiu, Y.; Varey, A.H.; Orlando, A.; Rigby, H.; Harper, S.J.; Bates, D.O. Expression of VEGF(xxx)b, the inhibitory isoforms of VEGF, in malignant melanoma. Br. J. Cancer 2007, 97, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Ieda, J.; Yokoyama, S.; Tamura, K.; Takifuji, K.; Hotta, T.; Matsuda, K.; Oku, Y.; Nasu, T.; Kiriyama, S.; Yamamoto, N.; et al. Re-expression of CEACAM1 long cytoplasmic domain isoform is associated with invasion and migration of colorectal cancer. Int. J. Cancer 2011, 129, 1351–1361. [Google Scholar] [CrossRef] [PubMed]
- Ortenberg, R.; Galore-Haskel, G.; Greenberg, I.; Zamlin, B.; Sapoznik, S.; Greenberg, E.; Barshack, I.; Avivi, C.; Feiler, Y.; Zan-Bar, I.; et al. CEACAM1 promotes melanoma cell growth through Sox-2. Neoplasia 2014, 16, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Todaro, M.; Gaggianesi, M.; Catalano, V.; Benfante, A.; Iovino, F.; Biffoni, M.; Apuzzo, T.; Sperduti, I.; Volpe, S.; Cocorullo, G.; et al. CD44v6 is a marker of constitutive and reprogrammed cancer stem cells driving colon cancer metastasis. Cell Stem Cell 2014, 14, 342–356. [Google Scholar] [CrossRef]
- Saito, S.; Okabe, H.; Watanabe, M.; Ishimoto, T.; Iwatsuki, M.; Baba, Y.; Tanaka, Y.; Kurashige, J.; Miyamoto, Y.; Baba, H. CD44v6 expression is related to mesenchymal phenotype and poor prognosis in patients with colorectal cancer. Oncol. Rep. 2013, 29, 1570–1578. [Google Scholar] [CrossRef]
- Yan, Y.; Zuo, X.; Wei, D. Concise Review: Emerging Role of CD44 in Cancer Stem Cells: A Promising Biomarker and Therapeutic Target. Stem Cells Transl. Med. 2015, 4, 1033–1043. [Google Scholar] [CrossRef]
- Singh, A.; Karnoub, A.E.; Palmby, T.R.; Lengyel, E.; Sondek, J.; Der, C.J. Rac1b, a tumor associated, constitutively active Rac1 splice variant, promotes cellular transformation. Oncogene 2004, 23, 9369–9380. [Google Scholar] [CrossRef]
- Pelisch, F.; Khauv, D.; Risso, G.; Stallings-Mann, M.; Blaustein, M.; Quadrana, L.; Radisky, D.C.; Srebrow, A. Involvement of hnRNP A1 in the matrix metalloprotease-3-dependent regulation of Rac1 pre-mRNA splicing. J. Cell Biochem. 2012, 113, 2319–2329. [Google Scholar] [CrossRef]
- Melzer, C.; Hass, R.; Lehnert, H.; Ungefroren, H. RAC1B: A Rho GTPase with Versatile Functions in Malignant Transformation and Tumor Progression. Cells 2019, 8, 21. [Google Scholar] [CrossRef]
- Matos, P.; Jordan, P. Increased Rac1b expression sustains colorectal tumor cell survival. Mol. Cancer Res. 2008, 6, 1178–1184. [Google Scholar] [CrossRef]
- Faria, M.; Capinha, L.; Simoes-Pereira, J.; Bugalho, M.J.; Silva, A.L. Extending the Impact of RAC1b Overexpression to Follicular Thyroid Carcinomas. Int. J. Endocrinol. 2016, 2016, 1972367. [Google Scholar] [CrossRef] [PubMed]
- Faria, M.; Matos, P.; Pereira, T.; Cabrera, R.; Cardoso, B.A.; Bugalho, M.J.; Silva, A.L. RAC1b overexpression stimulates proliferation and NF-kB-mediated anti-apoptotic signaling in thyroid cancer cells. PLoS ONE 2017, 12, e0172689. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhou, M.; Shi, B.; Zhang, Q.; Jiang, H.; Sun, Y.; Liu, J.; Zhou, K.; Yao, M.; Gu, J.; et al. Identification of an exon 4-deletion variant of epidermal growth factor receptor with increased metastasis-promoting capacity. Neoplasia 2011, 13, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Shi, B.; Zhang, Q.; Jiang, H.; Hu, S.; Kong, J.; Yao, M.; Yang, S.; Li, Z. Growth and metastasis suppression of glioma xenografts expressing exon 4-deletion variant of epidermal growth factor receptor by monoclonal antibody CH12-mediated receptor degradation. FASEB J. 2012, 26, 73–80. [Google Scholar] [CrossRef]
- Zhang, P.; Zhang, P.; Zhou, M.; Jiang, H.; Zhang, H.; Shi, B.; Pan, X.; Gao, H.; Sun, H.; Li, Z. Exon 4 deletion variant of epidermal growth factor receptor enhances invasiveness and cisplatin resistance in epithelial ovarian cancer. Carcinogenesis 2013, 34, 2639–2646. [Google Scholar] [CrossRef]
- Hatami, R.; Sieuwerts, A.M.; Izadmehr, S.; Yao, Z.; Qiao, R.F.; Papa, L.; Look, M.P.; Smid, M.; Ohlssen, J.; Levine, A.C.; et al. KLF6-SV1 drives breast cancer metastasis and is associated with poor survival. Sci. Transl. Med. 2013, 5, 169. [Google Scholar] [CrossRef]
- DiFeo, A.; Feld, L.; Rodriguez, E.; Wang, C.; Beer, D.G.; Martignetti, J.A.; Narla, G. A functional role for KLF6-SV1 in lung adenocarcinoma prognosis and chemotherapy response. Cancer Res. 2008, 68, 965–970. [Google Scholar] [CrossRef]
- Hartel, M.; Narla, G.; Wente, M.N.; Giese, N.A.; Martignoni, M.E.; Martignetti, J.A.; Friess, H.; Friedman, S.L. Increased alternative splicing of the KLF6 tumour suppressor gene correlates with prognosis and tumour grade in patients with pancreatic cancer. Eur. J. Cancer 2008, 44, 1895–1903. [Google Scholar] [CrossRef]
- Narla, G.; DiFeo, A.; Yao, S.; Banno, A.; Hod, E.; Reeves, H.L.; Qiao, R.F.; Camacho-Vanegas, O.; Levine, A.; Kirschenbaum, A.; et al. Targeted inhibition of the KLF6 splice variant, KLF6 SV1, suppresses prostate cancer cell growth and spread. Cancer Res. 2005, 65, 5761–5768. [Google Scholar] [CrossRef]
- Yea, S.; Narla, G.; Zhao, X.; Garg, R.; Tal-Kremer, S.; Hod, E.; Villanueva, A.; Loke, J.; Tarocchi, M.; Akita, K.; et al. Ras promotes growth by alternative splicing-mediated inactivation of the KLF6 tumor suppressor in hepatocellular carcinoma. Gastroenterology 2008, 134, 1521–1531. [Google Scholar] [CrossRef]
- Wang, Z.N.; Liu, D.; Yin, B.; Ju, W.Y.; Qiu, H.Z.; Xiao, Y.; Chen, Y.J.; Peng, X.Z.; Lu, C.M. High expression of PTBP1 promote invasion of colorectal cancer by alternative splicing of cortactin. Oncotarget 2017, 8, 36185–36202. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Li, K.; Peng, W.; Lin, Q.; Li, S.; Hu, X.; Zheng, X.; Shao, Z. An aberrant spliced transcript of focal adhesion kinase is exclusively expressed in human breast cancer. J. Transl. Med. 2014, 12, 136. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, J.A.; Weinstein, J.N. Biomarkers in cancer staging, prognosis and treatment selection. Nat. Rev. Cancer 2005, 5, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Drucker, E.; Krapfenbauer, K. Pitfalls and limitations in translation from biomarker discovery to clinical utility in predictive and personalised medicine. EPMA J. 2013, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Manley, J.L. Alternative pre-mRNA splicing regulation in cancer: Pathways and programs unhinged. Genes Dev. 2010, 24, 2343–2364. [Google Scholar] [CrossRef] [PubMed]
- Venables, J.P.; Klinck, R.; Koh, C.; Gervais-Bird, J.; Bramard, A.; Inkel, L.; Durand, M.; Couture, S.; Froehlich, U.; Lapointe, E.; et al. Cancer-associated regulation of alternative splicing. Nat. Struct. Mol. Biol. 2009, 16, 670–676. [Google Scholar] [CrossRef]
- Gardina, P.J.; Clark, T.A.; Shimada, B.; Staples, M.K.; Yang, Q.; Veitch, J.; Schweitzer, A.; Awad, T.; Sugnet, C.; Dee, S.; et al. Alternative splicing and differential gene expression in colon cancer detected by a whole genome exon array. BMC Genomics 2006, 7, 325. [Google Scholar] [CrossRef]
- Shapiro, I.M.; Cheng, A.W.; Flytzanis, N.C.; Balsamo, M.; Condeelis, J.S.; Oktay, M.H.; Burge, C.B.; Gertler, F.B. An EMT-driven alternative splicing program occurs in human breast cancer and modulates cellular phenotype. PLoS Genet. 2011, 7, e1002218. [Google Scholar] [CrossRef]
- Misquitta-Ali, C.M.; Cheng, E.; O’Hanlon, D.; Liu, N.; McGlade, C.J.; Tsao, M.S.; Blencowe, B.J. Global profiling and molecular characterization of alternative splicing events misregulated in lung cancer. Mol. Cell. Biol. 2011, 31, 138–150. [Google Scholar] [CrossRef]
- Germann, S.; Gratadou, L.; Dutertre, M.; Auboeuf, D. Splicing programs and cancer. J. Nucleic Acids 2012, 2012, 269570. [Google Scholar] [CrossRef]
- Lapuk, A.; Marr, H.; Jakkula, L.; Pedro, H.; Bhattacharya, S.; Purdom, E.; Hu, Z.; Simpson, K.; Pachter, L.; Durinck, S.; et al. Exon-level microarray analyses identify alternative splicing programs in breast cancer. Mol. Cancer Res. 2010, 8, 961–974. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Dai, B.; Ye, D.; Kong, Y.; Chang, K.; Jia, Z.; Yang, X.; Zhang, H.; Zhu, Y.; Shi, G. Constitutively active AR-V7 plays an essential role in the development and progression of castration-resistant prostate cancer. Sci. Rep. 2015, 5, 7654. [Google Scholar] [CrossRef] [PubMed]
- Calabretta, S.; Bielli, P.; Passacantilli, I.; Pilozzi, E.; Fendrich, V.; Capurso, G.; Fave, G.D.; Sette, C. Modulation of PKM alternative splicing by PTBP1 promotes gemcitabine resistance in pancreatic cancer cells. Oncogene 2016, 35, 2031–2039. [Google Scholar] [CrossRef] [PubMed]
- Nagane, M.; Coufal, F.; Lin, H.; Bogler, O.; Cavenee, W.K.; Huang, H.J. A common mutant epidermal growth factor receptor confers enhanced tumorigenicity on human glioblastoma cells by increasing proliferation and reducing apoptosis. Cancer Res. 1996, 56, 5079–5086. [Google Scholar] [PubMed]
- Zhu, H.; Acquaviva, J.; Ramachandran, P.; Boskovitz, A.; Woolfenden, S.; Pfannl, R.; Bronson, R.T.; Chen, J.W.; Weissleder, R.; Housman, D.E.; et al. Oncogenic EGFR signaling cooperates with loss of tumor suppressor gene functions in gliomagenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 2712–2716. [Google Scholar] [CrossRef]
- Gotley, D.C.; Fawcett, J.; Walsh, M.D.; Reeder, J.A.; Simmons, D.L.; Antalis, T.M. Alternatively spliced variants of the cell adhesion molecule CD44 and tumour progression in colorectal cancer. Br. J. Cancer 1996, 74, 342–351. [Google Scholar] [CrossRef]
- Coppola, D.; Hyacinthe, M.; Fu, L.; Cantor, A.B.; Karl, R.; Marcet, J.; Cooper, D.L.; Nicosia, S.V.; Cooper, H.S. CD44V6 expression in human colorectal carcinoma. Hum. Pathol. 1998, 29, 627–635. [Google Scholar] [CrossRef]
- Mikami, T.; Mitomi, H.; Hara, A.; Yanagisawa, N.; Yoshida, T.; Tsuruta, O.; Okayasu, I. Decreased expression of CD44, alpha-catenin, and deleted colon carcinoma and altered expression of beta-catenin in ulcerative colitis-associated dysplasia and carcinoma, as compared with sporadic colon neoplasms. Cancer 2000, 89, 733–740. [Google Scholar] [CrossRef]
- Fan, C.W.; Wen, L.; Qiang, Z.D.; Chen, T.; Zhou, Z.G.; Mo, X.M.; Hu, J.K. Prognostic significance of relevant markers of cancer stem cells in colorectal cancer—A meta analysis. Hepatogastroenterology 2012, 59, 1421–1427. [Google Scholar]
- Matos, P.; Oliveira, C.; Velho, S.; Goncalves, V.; da Costa, L.T.; Moyer, M.P.; Seruca, R.; Jordan, P. B-Raf(V600E) cooperates with alternative spliced Rac1b to sustain colorectal cancer cell survival. Gastroenterology 2008, 135, 899–906. [Google Scholar] [CrossRef]
- Alonso-Espinaco, V.; Cuatrecasas, M.; Alonso, V.; Escudero, P.; Marmol, M.; Horndler, C.; Ortego, J.; Gallego, R.; Codony-Servat, J.; Garcia-Albeniz, X.; et al. RAC1b overexpression correlates with poor prognosis in KRAS/BRAF WT metastatic colorectal cancer patients treated with first-line FOLFOX/XELOX chemotherapy. Eur. J. Cancer 2014, 50, 1973–1981. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.L.; Carmo, F.; Bugalho, M.J. RAC1b overexpression in papillary thyroid carcinoma: A role to unravel. Eur. J. Endocrinol. 2013, 168, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Goka, E.T.; Chaturvedi, P.; Lopez, D.T.M.; Garza, A.; Lippman, M.E. RAC1b Overexpression Confers Resistance to Chemotherapy Treatment in Colorectal Cancer. Mol. Cancer Ther. 2019, 18, 957–968. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Fung-Leung, W.P.; Bittner, A.; Ngo, K.; Liu, X. Comparison of RNA-Seq and microarray in transcriptome profiling of activated T cells. PLoS ONE 2014, 9, e78644. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Dower, K.; Zhang, B.; Martinez, R.V.; Lin, L.L.; Zhao, S. Computational identification and validation of alternative splicing in ZSF1 rat RNA-seq data, a preclinical model for type 2 diabetic nephropathy. Sci. Rep. 2018, 8, 7624. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhang, B.; Lin, L.L.; Zhao, S. Evaluation and comparison of computational tools for RNA-seq isoform quantification. BMC Genom. 2017, 18, 583. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Byron, S.A.; Van Keuren-Jensen, K.R.; Engelthaler, D.M.; Carpten, J.D.; Craig, D.W. Translating RNA sequencing into clinical diagnostics: Opportunities and challenges. Nat. Rev. Genet. 2016, 17, 257–271. [Google Scholar] [CrossRef]
- Eswaran, J.; Horvath, A.; Godbole, S.; Reddy, S.D.; Mudvari, P.; Ohshiro, K.; Cyanam, D.; Nair, S.; Fuqua, S.A.; Polyak, K.; et al. RNA sequencing of cancer reveals novel splicing alterations. Sci. Rep. 2013, 3, 1689. [Google Scholar] [CrossRef]
- Zhu, J.; Chen, Z.; Yong, L. Systematic profiling of alternative splicing signature reveals prognostic predictor for ovarian cancer. Gynecol. Oncol. 2018, 148, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Sun, N.; Lu, Z.; Sun, S.; Huang, J.; Chen, Z.; He, J. Prognostic alternative mRNA splicing signature in non-small cell lung cancer. Cancer Lett. 2017, 393, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Wu, Q.; Huang, K.; Wang, X.; Yu, T.; Liao, X.; Huang, J.; Zhu, G.; Gong, Y.; Han, C.; et al. Genome-Wide Profiling Reveals the Landscape of Prognostic Alternative Splicing Signatures in Pancreatic Ductal Adenocarcinoma. Front. Oncol. 2019, 9, 511. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Hu, Q.; Liu, X.; Ji, Y.; Chao, H.P.; Liu, Y.; Tracz, A.; Kirk, J.; Buonamici, S.; Zhu, P.; et al. Intron retention is a hallmark and spliceosome represents a therapeutic vulnerability in aggressive prostate cancer. Nat. Commun. 2020, 11, 2089. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Deng, Y.; Wang, K.; Zhou, H.; Zheng, X.; Si, L.; Fu, Z. Profiles of alternative splicing in colorectal cancer and their clinical significance: A study based on large-scale sequencing data. EBioMedicine 2018, 36, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Lian, H.; Wang, A.; Shen, Y.; Wang, Q.; Zhou, Z.; Zhang, R.; Li, K.; Liu, C.; Jia, H. Identification of novel alternative splicing isoform biomarkers and their association with overall survival in colorectal cancer. BMC Gastroenterol. 2020, 20, 171. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Fang, C.Y.; Chen, S.X.; Wang, X.Q.; Cui, S.J.; Liu, X.H.; Jiang, Y.H.; Wang, J.; Zhang, Y.; Yang, P.Y.; et al. Stroma derived COL6A3 is a potential prognosis marker of colorectal carcinoma revealed by quantitative proteomics. Oncotarget 2015, 6, 29929–29946. [Google Scholar] [CrossRef]
- Li, G.H.; Huang, J.F. Inferring therapeutic targets from heterogeneous data: HKDC1 is a novel potential therapeutic target for cancer. Bioinformatics 2014, 30, 748–752. [Google Scholar] [CrossRef]
- Safikhani, Z.; Smirnov, P.; Thu, K.L.; Silvester, J.; El-Hachem, N.; Quevedo, R.; Lupien, M.; Mak, T.W.; Cescon, D.; Haibe-Kains, B. Gene isoforms as expression-based biomarkers predictive of drug response in vitro. Nat. Commun. 2017, 8, 1126. [Google Scholar] [CrossRef]
- Desterro, J.; Bak-Gordon, P.; Carmo-Fonseca, M. Targeting mRNA processing as an anticancer strategy. Nat. Rev. Drug Discov. 2020, 19, 112–129. [Google Scholar] [CrossRef]
- Rahman, M.A.; Nasrin, F.; Bhattacharjee, S.; Nandi, S. Hallmarks of Splicing Defects in Cancer: Clinical Applications in the Era of Personalized Medicine. Cancers 2020, 12, 1381. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; Abdel-Wahab, O. Therapeutic targeting of splicing in cancer. Nat. Med. 2016, 22, 976–986. [Google Scholar] [CrossRef] [PubMed]
- Antonopoulou, E.; Ladomery, M. Targeting Splicing in Prostate Cancer. Int. J. Mol. Sci. 2018, 19, 1287. [Google Scholar] [CrossRef] [PubMed]
- Aubol, B.E.; Wu, G.; Keshwani, M.M.; Movassat, M.; Fattet, L.; Hertel, K.J.; Fu, X.D.; Adams, J.A. Release of SR Proteins from CLK1 by SRPK1: A Symbiotic Kinase System for Phosphorylation Control of Pre-mRNA Splicing. Mol. Cell 2016, 63, 218–228. [Google Scholar] [CrossRef]
- Toker, A.; Chin, Y.R. Akt-ing up on SRPK1: Oncogene or tumor suppressor? Mol. Cell 2014, 54, 329–330. [Google Scholar] [CrossRef]
- Amin, E.M.; Oltean, S.; Hua, J.; Gammons, M.V.; Hamdollah-Zadeh, M.; Welsh, G.I.; Cheung, M.K.; Ni, L.; Kase, S.; Rennel, E.S.; et al. WT1 mutants reveal SRPK1 to be a downstream angiogenesis target by altering VEGF splicing. Cancer Cell 2011, 20, 768–780. [Google Scholar] [CrossRef]
- Siqueira, R.P.; Barbosa Ede, A.; Poleto, M.D.; Righetto, G.L.; Seraphim, T.V.; Salgado, R.L.; Ferreira, J.G.; Barros, M.V.; de Oliveira, L.L.; Laranjeira, A.B.; et al. Potential Antileukemia Effect and Structural Analyses of SRPK Inhibition by N-(2-(Piperidin-1-yl)-5-(Trifluoromethyl)Phenyl)Isonicotinamide (SRPIN340). PLoS ONE 2015, 10, e0134882. [Google Scholar] [CrossRef]
- Gammons, M.V.; Lucas, R.; Dean, R.; Coupland, S.E.; Oltean, S.; Bates, D.O. Targeting SRPK1 to control VEGF-mediated tumour angiogenesis in metastatic melanoma. Br. J. Cancer 2014, 111, 477–485. [Google Scholar] [CrossRef]
- Hatcher, J.M.; Wu, G.; Zeng, C.; Zhu, J.; Meng, F.; Patel, S.; Wang, W.; Ficarro, S.B.; Leggett, A.L.; Powell, C.E.; et al. SRPKIN-1: A Covalent SRPK1/2 Inhibitor that Potently Converts VEGF from Pro-angiogenic to Anti-angiogenic Isoform. Cell Chem. Biol. 2018, 25, 460–470. [Google Scholar] [CrossRef]
- Muraki, M.; Ohkawara, B.; Hosoya, T.; Onogi, H.; Koizumi, J.; Koizumi, T.; Sumi, K.; Yomoda, J.; Murray, M.V.; Kimura, H.; et al. Manipulation of alternative splicing by a newly developed inhibitor of Clks. J. Biol. Chem. 2004, 279, 24246–24254. [Google Scholar] [CrossRef]
- Debdab, M.; Carreaux, F.; Renault, S.; Soundararajan, M.; Fedorov, O.; Filippakopoulos, P.; Lozach, O.; Babault, L.; Tahtouh, T.; Baratte, B.; et al. Leucettines, a class of potent inhibitors of cdc2-like kinases and dual specificity, tyrosine phosphorylation regulated kinases derived from the marine sponge leucettamine B: Modulation of alternative pre-RNA splicing. J. Med. Chem. 2011, 54, 4172–4186. [Google Scholar] [CrossRef] [PubMed]
- Iwai, K.; Yaguchi, M.; Nishimura, K.; Yamamoto, Y.; Tamura, T.; Nakata, D.; Dairiki, R.; Kawakita, Y.; Mizojiri, R.; Ito, Y.; et al. Anti-tumor efficacy of a novel CLK inhibitor via targeting RNA splicing and MYC-dependent vulnerability. EMBO Mol. Med. 2018, 10, e8289. [Google Scholar] [CrossRef] [PubMed]
- Araki, S.; Dairiki, R.; Nakayama, Y.; Murai, A.; Miyashita, R.; Iwatani, M.; Nomura, T.; Nakanishi, O. Inhibitors of CLK protein kinases suppress cell growth and induce apoptosis by modulating pre-mRNA splicing. PLoS ONE 2015, 10, e0116929. [Google Scholar] [CrossRef] [PubMed]
- Assi, R.; Kantarjian, H.M.; Kadia, T.M.; Pemmaraju, N.; Jabbour, E.; Jain, N.; Daver, N.; Estrov, Z.; Uehara, T.; Owa, T.; et al. Final results of a phase 2, open-label study of indisulam, idarubicin, and cytarabine in patients with relapsed or refractory acute myeloid leukemia and high-risk myelodysplastic syndrome. Cancer 2018, 124, 2758–2765. [Google Scholar] [CrossRef] [PubMed]
- Talbot, D.C.; von Pawel, J.; Cattell, E.; Yule, S.M.; Johnston, C.; Zandvliet, A.S.; Huitema, A.D.; Norbury, C.J.; Ellis, P.; Bosquee, L.; et al. A randomized phase II pharmacokinetic and pharmacodynamic study of indisulam as second-line therapy in patients with advanced non-small cell lung cancer. Clin. Cancer Res. 2007, 13, 1816–1822. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Indisulam: An anticancer sulfonamide in clinical development. Expert Opin. Investig. Drugs 2003, 12, 283–287. [Google Scholar] [CrossRef]
- Han, T.; Goralski, M.; Gaskill, N.; Capota, E.; Kim, J.; Ting, T.C.; Xie, Y.; Williams, N.S.; Nijhawan, D. Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science 2017, 356, eaal3755. [Google Scholar] [CrossRef]
- Uehara, T.; Minoshima, Y.; Sagane, K.; Sugi, N.H.; Mitsuhashi, K.O.; Yamamoto, N.; Kamiyama, H.; Takahashi, K.; Kotake, Y.; Uesugi, M.; et al. Selective degradation of splicing factor CAPERalpha by anticancer sulfonamides. Nat. Chem. Biol. 2017, 13, 675–680. [Google Scholar] [CrossRef]
- Black, A.J.; Gamarra, J.R.; Giudice, J. More than a messenger: Alternative splicing as a therapeutic target. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 194395. [Google Scholar] [CrossRef]
- Sanidas, I.; Polytarchou, C.; Hatziapostolou, M.; Ezell, S.A.; Kottakis, F.; Hu, L.; Guo, A.; Xie, J.; Comb, M.J.; Iliopoulos, D.; et al. Phosphoproteomics screen reveals akt isoform-specific signals linking RNA processing to lung cancer. Mol. Cell 2014, 53, 577–590. [Google Scholar] [CrossRef]
- Passacantilli, I.; Frisone, P.; De Paola, E.; Fidaleo, M.; Paronetto, M.P. hnRNPM guides an alternative splicing program in response to inhibition of the PI3K/AKT/mTOR pathway in Ewing sarcoma cells. Nucleic Acids Res. 2017, 45, 12270–12284. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, M.L.; Zamecnik, P.C. Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Zamecnik, P.C.; Stephenson, M.L. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Dean, N.M.; Bennett, C.F. Antisense oligonucleotide-based therapeutics for cancer. Oncogene 2003, 22, 9087–9096. [Google Scholar] [CrossRef]
- Levin, A.A. Treating Disease at the RNA Level with Oligonucleotides. N. Engl. J. Med. 2019, 380, 57–70. [Google Scholar] [CrossRef]
- Bennett, C.F. Therapeutic Antisense Oligonucleotides Are Coming of Age. Annu Rev. Med. 2019, 70, 307–321. [Google Scholar] [CrossRef]
- Liu, J.; Guo, B. RNA-based therapeutics for colorectal cancer: Updates and future directions. Pharmacol. Res. 2020, 152, 104550. [Google Scholar] [CrossRef]
- Havens, M.A.; Hastings, M.L. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. 2016, 44, 6549–6563. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Krieg, A.M. FDA Approves Eteplirsen for Duchenne Muscular Dystrophy: The Next Chapter in the Eteplirsen Saga. Nucleic Acid Ther. 2017, 27, 1–3. [Google Scholar] [CrossRef]
- Michelson, D.; Ciafaloni, E.; Ashwal, S.; Lewis, E.; Narayanaswami, P.; Oskoui, M.; Armstrong, M.J. Evidence in focus: Nusinersen use in spinal muscular atrophy: Report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology. Neurology 2018, 91, 923–933. [Google Scholar] [CrossRef]
- Charleston, J.S.; Schnell, F.J.; Dworzak, J.; Donoghue, C.; Lewis, S.; Chen, L.; Young, G.D.; Milici, A.J.; Voss, J.; DeAlwis, U.; et al. Eteplirsen treatment for Duchenne muscular dystrophy: Exon skipping and dystrophin production. Neurology 2018, 90, e2146–e2154. [Google Scholar] [CrossRef] [PubMed]
- Mercatante, D.R.; Bortner, C.D.; Cidlowski, J.A.; Kole, R. Modification of alternative splicing of Bcl-x pre-mRNA in prostate and breast cancer cells. analysis of apoptosis and cell death. J. Biol. Chem. 2001, 276, 16411–16417. [Google Scholar] [CrossRef] [PubMed]
- Bauman, J.A.; Kole, R. Modulation of RNA splicing as a potential treatment for cancer. Bioeng. Bugs 2011, 2, 125–128. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhang, J.; Manley, J.L. Turning on a fuel switch of cancer: hnRNP proteins regulate alternative splicing of pyruvate kinase mRNA. Cancer Res. 2010, 70, 8977–8980. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Jeon, H.Y.; Rigo, F.; Bennett, C.F.; Krainer, A.R. Manipulation of PK-M mutually exclusive alternative splicing by antisense oligonucleotides. Open Biol. 2012, 2, 120133. [Google Scholar] [CrossRef] [PubMed]
- Denichenko, P.; Mogilevsky, M.; Clery, A.; Welte, T.; Biran, J.; Shimshon, O.; Barnabas, G.D.; Danan-Gotthold, M.; Kumar, S.; Yavin, E.; et al. Specific inhibition of splicing factor activity by decoy RNA oligonucleotides. Nat. Commun. 2019, 10, 1590. [Google Scholar] [CrossRef]
- Moscatello, D.K.; Holgado-Madruga, M.; Godwin, A.K.; Ramirez, G.; Gunn, G.; Zoltick, P.W.; Biegel, J.A.; Hayes, R.L.; Wong, A.J. Frequent expression of a mutant epidermal growth factor receptor in multiple human tumors. Cancer Res. 1995, 55, 5536–5539. [Google Scholar]
- Aldape, K.D.; Ballman, K.; Furth, A.; Buckner, J.C.; Giannini, C.; Burger, P.C.; Scheithauer, B.W.; Jenkins, R.B.; James, C.D. Immunohistochemical detection of EGFRvIII in high malignancy grade astrocytomas and evaluation of prognostic significance. J. Neuropathol. Exp. Neurol. 2004, 63, 700–707. [Google Scholar] [CrossRef]
- Swartz, A.M.; Li, Q.J.; Sampson, J.H. Rindopepimut: A promising immunotherapeutic for the treatment of glioblastoma multiforme. Immunotherapy 2014, 6, 679–690. [Google Scholar] [CrossRef]
- Johns, T.G.; Perera, R.M.; Vernes, S.C.; Vitali, A.A.; Cao, D.X.; Cavenee, W.K.; Scott, A.M.; Furnari, F.B. The efficacy of epidermal growth factor receptor-specific antibodies against glioma xenografts is influenced by receptor levels, activation status, and heterodimerization. Clin. Cancer Res. 2007, 13, 1911–1925. [Google Scholar] [CrossRef]
- Fukai, J.; Nishio, K.; Itakura, T.; Koizumi, F. Antitumor activity of cetuximab against malignant glioma cells overexpressing EGFR deletion mutant variant III. Cancer Sci. 2008, 99, 2062–2069. [Google Scholar] [CrossRef] [PubMed]
- Heimberger, A.B.; Learn, C.A.; Archer, G.E.; McLendon, R.E.; Chewning, T.A.; Tuck, F.L.; Pracyk, J.B.; Friedman, A.H.; Friedman, H.S.; Bigner, D.D.; et al. Brain tumors in mice are susceptible to blockade of epidermal growth factor receptor (EGFR) with the oral, specific, EGFR-tyrosine kinase inhibitor ZD1839 (iressa). Clin. Cancer Res. 2002, 8, 3496–3502. [Google Scholar] [PubMed]
- Orian-Rousseau, V. CD44, a therapeutic target for metastasising tumours. Eur. J. Cancer 2010, 46, 1271–1277. [Google Scholar] [CrossRef] [PubMed]
- Borjesson, P.K.; Postema, E.J.; Roos, J.C.; Colnot, D.R.; Marres, H.A.; van Schie, M.H.; Stehle, G.; de Bree, R.; Snow, G.B.; Oyen, W.J.; et al. Phase I therapy study with (186)Re-labeled humanized monoclonal antibody BIWA 4 (bivatuzumab) in patients with head and neck squamous cell carcinoma. Clin. Cancer Res. 2003, 9, 3961S–3972S. [Google Scholar] [PubMed]
- Colnot, D.R.; Roos, J.C.; de Bree, R.; Wilhelm, A.J.; Kummer, J.A.; Hanft, G.; Heider, K.H.; Stehle, G.; Snow, G.B.; van Dongen, G.A. Safety, biodistribution, pharmacokinetics, and immunogenicity of 99mTc-labeled humanized monoclonal antibody BIWA 4 (bivatuzumab) in patients with squamous cell carcinoma of the head and neck. Cancer Immunol. Immunother. 2003, 52, 576–582. [Google Scholar] [CrossRef]
- Tijink, B.M.; Buter, J.; de Bree, R.; Giaccone, G.; Lang, M.S.; Staab, A.; Leemans, C.R.; van Dongen, G.A. A phase I dose escalation study with anti-CD44v6 bivatuzumab mertansine in patients with incurable squamous cell carcinoma of the head and neck or esophagus. Clin. Cancer Res. 2006, 12, 6064–6072. [Google Scholar] [CrossRef]
- Riechelmann, H.; Sauter, A.; Golze, W.; Hanft, G.; Schroen, C.; Hoermann, K.; Erhardt, T.; Gronau, S. Phase I trial with the CD44v6-targeting immunoconjugate bivatuzumab mertansine in head and neck squamous cell carcinoma. Oral Oncol. 2008, 44, 823–829. [Google Scholar] [CrossRef]
- Sahin, U.; Koslowski, M.; Dhaene, K.; Usener, D.; Brandenburg, G.; Seitz, G.; Huber, C.; Tureci, O. Claudin-18 splice variant 2 is a pan-cancer target suitable for therapeutic antibody development. Clin. Cancer Res. 2008, 14, 7624–7634. [Google Scholar] [CrossRef]
- Singh, P.; Toom, S.; Huang, Y. Anti-claudin 18.2 antibody as new targeted therapy for advanced gastric cancer. J. Hematol. Oncol. 2017, 10, 105. [Google Scholar] [CrossRef]
- Arkin, M.R.; Tang, Y.; Wells, J.A. Small-molecule inhibitors of protein-protein interactions: Progressing toward the reality. Chem. Biol. 2014, 21, 1102–1114. [Google Scholar] [CrossRef]
- Jin, L.; Wang, W.; Fang, G. Targeting protein-protein interaction by small molecules. Annu Rev. Pharmacol. Toxicol. 2014, 54, 435–456. [Google Scholar] [CrossRef] [PubMed]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Aguilar, A.; Bernard, D.; Wang, S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction (MDM2 Inhibitors) in clinical trials for cancer treatment. J. Med. Chem. 2015, 58, 1038–1052. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).