Targeting Casein Kinase 1 (CK1) in Hematological Cancers



Abstract
1. Introduction
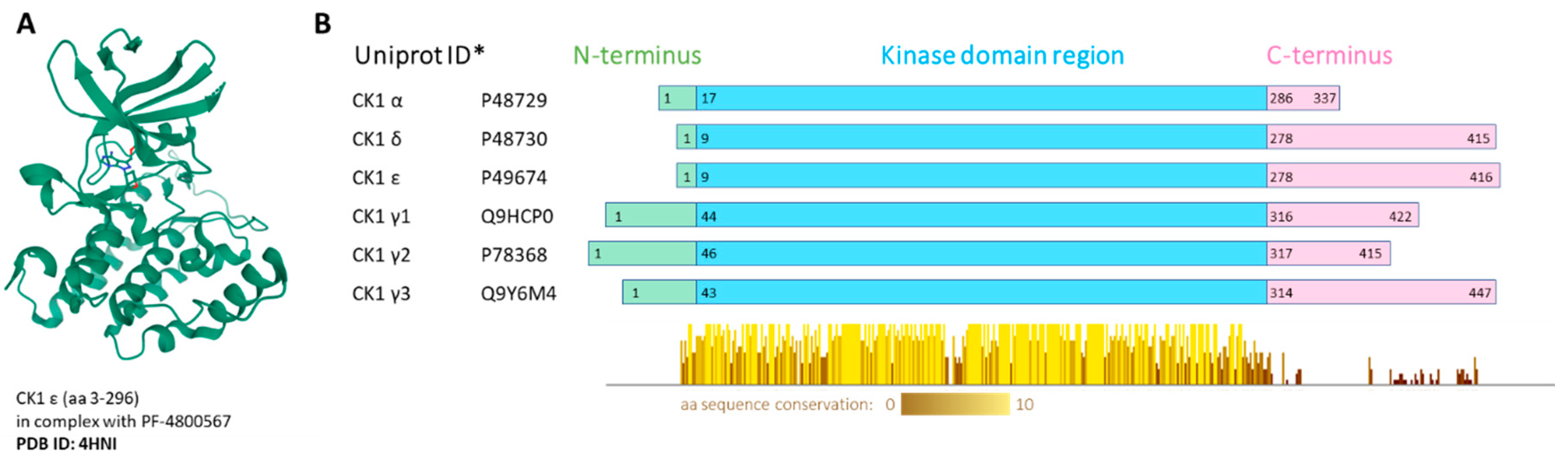
2. Biological Functions of the CK1 Family
3. CK1 Inhibitors and Their Therapeutic Potential in Hematologic Malignancies
4. CK1ε in Chronic Lymphocytic Leukemia (CLL) and Other Non-Hodgkin Lymphomas (NHLs)
4.1. CLL and the Non-Canonical Wnt Pathways
4.2. Inhibition of CK1 δ/ε in the Preclinical Models of CLL
4.3. Umbralisib, A Dual PI3Kδ-CK1ε Inhibitor, Demonstrates Safety of CK1ε Inhibition
4.4. Potential of the Combinations of the B-Cell Receptor (BCR) and CK1 Inhibition in CLL
5. CK1α in Myelodysplastic Syndrome (MDS) and Acute Myeloid Leukemia (AML)
5.1. CK1α Activity is Critical for AML Leukemic Stem Cells (LSCs) Survival and Growth In Vivo
5.2. Targeting CK1α in MDS/AML Patients—Lenalidomide and BTX-A51
6. Summary and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| aa | Amino acid |
| AML | Acute myeloid leukemia |
| ATP | Adenosine triphosphate |
| BCR | B cell receptor |
| BM | Bone marrow |
| BTK | Bruton tyrosine kinase |
| CDK | Cyclin-dependent kinase |
| CK1 | Casein kinase 1 |
| CLL | Chronic lymphocytic leukemia |
| CRC | Colorectal cancer |
| DVL | Dishevelled |
| 4E-BP1 | Eukaryotic translation initiation factor 4E-binding protein 1 |
| FDA | U.S. Food and Drug Administration |
| FL | Follicular lymphoma |
| FZD | Frizzled |
| HSC | Hematopoietic stem cell |
| IgH | Immunoglobulin heavy chain |
| LRP | Low-density lipoprotein receptor-related protein |
| LSC | Leukemic stem cell |
| MCL | Mantle cell lymphoma |
| MDS | Myelodysplastic syndrome |
| MM | Multiple myeloma |
| mRNA | Messenger RNA |
| MZL | Marginal zone lymphoma |
| NHL | Non-Hodgkin lymphoma |
| PB | Peripheral blood |
| PCP | Planar cell polarity |
| PI3K | Phosphoinositide 3-kinase |
| ROR1 | Receptor tyrosine kinase-like orphan receptor-1 |
| shRNA | Short hairpin RNA |
| Treg | Regulatory T-cell |
References
- Lemeer, S.; Heck, A.J. The phosphoproteomics data explosion. Curr. Opin. Chem. Biol. 2009, 13, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Sikes, R.A. Chemistry and pharmacology of anticancer drugs. Br. J. Cancer 2007, 97, 1713. [Google Scholar] [CrossRef]
- Roskoski, R. Properties of FDA-approved small molecule protein kinase inhibitors: A 2020 update. Pharmacol. Res. 2020, 152, 104609. [Google Scholar] [CrossRef] [PubMed]
- Knippschild, U.; Krüger, M.; Richter, J.; Xu, P.; García-Reyes, B.; Peifer, C.; Halekotte, J.; Bakulev, V.; Bischof, J. The CK1 Family: Contribution to Cellular Stress Response and Its Role in Carcinogenesis. Front. Oncol. 2014, 4, 96. [Google Scholar] [CrossRef]
- Cegielska, A.; Gietzen, K.F.; Rivers, A.; Virshup, D.M. Autoinhibition of casein kinase I epsilon (CKI epsilon) is relieved by protein phosphatases and limited proteolysis. J. Biol Chem 1998, 273, 1357–1364. [Google Scholar] [CrossRef]
- Fish, K.J.; Cegielska, A.; Getman, M.E.; Landes, G.M.; Virshup, D.M. Isolation and Characterization of Human Casein Kinase I-Epsilon (Cki), a Novel Member of the Cki Gene Family. J. Biol. Chem. 1995, 270, 14875–14883. [Google Scholar] [CrossRef]
- Petzold, G.; Fischer, E.S.; Thomä, N.H. Structural basis of lenalidomide-induced CK1α degradation by the CRL4 CRBN ubiquitin ligase. Nature 2016, 532, 127–130. [Google Scholar] [CrossRef]
- Minzel, W.; Venkatachalam, A.; Fink, A.; Hung, E.; Brachya, G.; Burstain, I.; Shaham, M.; Rivlin, A.; Omer, I.; Zinger, A.; et al. Small Molecules Co-targeting CKIα and the Transcriptional Kinases CDK7/9 Control AML in Preclinical Models. Cell 2018. [Google Scholar] [CrossRef]
- Long, A.; Zhao, H.; Huang, X. Structural Basis for the Interaction between Casein Kinase 1 Delta and a Potent and Selective Inhibitor. J. Med. Chem. 2012, 55, 956–960. [Google Scholar] [CrossRef]
- Long, A.M.; Zhao, H.; Huang, X. Structural basis for the potent and selective inhibition of casein kinase 1 epsilon. J. Med. Chem. 2012, 55, 10307–10311. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Schittek, B.; Sinnberg, T. Biological functions of casein kinase 1 isoforms and putative roles in tumorigenesis. Mol. Cancer 2014, 13, 231. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Ianes, C.; Gärtner, F.; Liu, C.; Burster, T.; Bakulev, V.; Rachidi, N.; Knippschild, U.; Bischof, J. Structure, regulation, and (patho-)physiological functions of the stress-induced protein kinase CK1 delta (CSNK1D). Gene 2019, 715, 144005. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Zhang, M.; Sun, J.; Yang, X. Casein kinase 1α: Biological mechanisms and theranostic potential. Cell Commun. Signal. 2018, 16, 1–24. [Google Scholar] [CrossRef]
- Perez, D.I.; Gil, C.; Martinez, A. Protein kinases CK1 and CK2 as new targets for neurodegenerative diseases. Med. Res. Rev. 2011, 31, 924–954. [Google Scholar] [CrossRef]
- Bernatik, O.; Ganji, R.S.; Dijksterhuis, J.P.; Konik, P.; Cervenka, I.; Polonio, T.; Krejci, P.; Schulte, G.; Bryja, V. Sequential activation and inactivation of Dishevelled in the Wnt/beta-catenin pathway by casein kinases. J. Biol. Chem. 2011, 286, 10396–10410. [Google Scholar] [CrossRef]
- Bryja, V.; Schulte, G.; Rawal, N.; Grahn, A.; Arenas, E. Wnt-5a induces Dishevelled phosphorylation and dopaminergic differentiation via a CK1-dependent mechanism. J. Cell Sci. 2007, 120, 586–595. [Google Scholar] [CrossRef]
- Greer, Y.E.; Rubin, J.S. Casein kinase 1 delta functions at the centrosome to mediate Wnt-3a-dependent neurite outgrowth. J. Cell Biol. 2011, 192, 993–1004. [Google Scholar] [CrossRef]
- Peters, J.M.; McKay, R.M.; McKay, J.P.; Graff, J.M. Casein kinase I transduces Wnt signals. Nature 1999, 401, 345–350. [Google Scholar] [CrossRef]
- Vinyoles, M.; Del Valle-Pérez, B.; Curto, J.; Padilla, M.; Villarroel, A.; Yang, J.; de Herreros, A.G.; Duñach, M. Activation of CK1ɛ by PP2A/PR61ɛ is required for the initiation of Wnt signaling. Oncogene 2017, 36, 429–438. [Google Scholar] [CrossRef]
- Zeng, C.M.; Chen, Z.; Fu, L. Frizzled Receptors as Potential Therapeutic Targets in Human Cancers. Int J. Mol. Sci 2018, 19, 1543. [Google Scholar] [CrossRef] [PubMed]
- Dijksterhuis, J.P.; Baljinnyam, B.; Stanger, K.; Sercan, H.O.; Ji, Y.; Andres, O.; Rubin, J.S.; Hannoush, R.N.; Schulte, G. Systematic mapping of WNT-FZD protein interactions reveals functional selectivity by distinct WNT-FZD pairs. J. Biol. Chem. 2015, 290, 6789–6798. [Google Scholar] [CrossRef] [PubMed]
- Suraweera, N.; Robinson, J.; Volikos, E.; Guenther, T.; Talbot, I.; Tomlinson, I.; Silver, A. Mutations within Wnt pathway genes in sporadic colorectal cancers and cell lines. Int. J. Cancer 2006, 119, 1837–1842. [Google Scholar] [CrossRef] [PubMed]
- Fodde, R. The APC gene in colorectal cancer. Eur. J. Cancer 2002, 38, 867–871. [Google Scholar] [CrossRef]
- Kim, S.; Jeong, S. Mutation hotspots in the β-catenin gene: Lessons from the human cancer genome databases. Mol. Cells 2019. [Google Scholar] [CrossRef]
- Anvarian, Z.; Nojima, H.; van Kappel, E.C.; Madl, T.; Spit, M.; Viertler, M.; Jordens, I.; Low, T.Y.; van Scherpenzeel, R.C.; Kuper, I.; et al. Axin cancer mutants form nanoaggregates to rewire the Wnt signaling network. Nat. Struct. Mol. Biol. 2016. [Google Scholar] [CrossRef]
- Foldynova-Trantirkova, S.; Sekyrova, P.; Tmejova, K.; Brumovska, E.; Bernatik, O.; Blankenfeldt, W.; Krejci, P.; Kozubik, A.; Dolezal, T.; Trantirek, L.; et al. Breast cancer-specific mutations in CK1epsilon inhibit Wnt/beta-catenin and activate the Wnt/Rac1/JNK and NFAT pathways to decrease cell adhesion and promote cell migration. Breast Cancer Res. 2010, 12, R30. [Google Scholar] [CrossRef]
- Kaucka, M.; Plevova, K.; Pavlova, S.; Janovska, P.; Mishra, A.; Verner, J.; Prochazkova, J.; Krejci, P.; Kotaskova, J.; Ovesna, P.; et al. The planar cell polarity pathway drives pathogenesis of chronic lymphocytic leukemia by the regulation of b-lymphocyte migration. Cancer Res. 2013, 73, 1491–1501. [Google Scholar] [CrossRef]
- Baskar, S.; Kwong, K.Y.; Hofer, T.; Levy, J.M.; Kennedy, M.G.; Lee, E.; Staudt, L.M.; Wilson, W.H.; Wiestner, A.; Rader, C. Unique cell surface expression of receptor tyrosine kinase ROR1 in human B-cell chronic lymphocytic leukemia. Clin. Cancer Res. 2008, 14, 396–404. [Google Scholar] [CrossRef]
- Janovska, P.; Verner, J.; Kohoutek, J.; Bryjova, L.; Gregorova, M.; Dzimkova, M.; Skabrahova, H.; Radaszkiewicz, T.; Ovesna, P.; Vondalova Blanarova, O.; et al. Casein Kinase 1 is a Therapeutic Target in Chronic Lymphocytic Leukemia. Blood 2018. [Google Scholar] [CrossRef]
- Wang, L.; Shalek, A.K.; Lawrence, M.; Ding, R.; Gaublomme, J.T.; Pochet, N.; Stojanov, P.; Sougnez, C.; Shukla, S.A.; Stevenson, K.E.; et al. Somatic mutation as a mechanism of Wnt/β-catenin pathway activation in CLL. Blood 2014, 124. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.S.; Hojjat-Farsangi, M.; Daneshmanesh, A.H.; Hansson, L.; Kokhaei, P.; Österborg, A.; Mellstedt, H.; Moshfegh, A. Dishevelled proteins are significantly upregulated in chronic lymphocytic leukaemia. Tumour Biol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Cong, F.; Schweizer, L.; Varmus, H. Casein kinase Iepsilon modulates the signaling specificities of dishevelled. Mol. Cell Biol. 2004, 24, 2000–2011. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.H.; Seeling, J.M.; Hill, V.; Yochum, A.; Virshup, D.M. Casein kinase I phosphorylates and destabilizes the beta-catenin degradation complex. Proc. Natl. Acad. Sci. USA 2002, 99, 1182–1187. [Google Scholar] [CrossRef]
- Polakis, P. The many ways of Wnt in cancer. Curr. Opin. Genet. Dev. 2007, 17, 45–51. [Google Scholar] [CrossRef]
- Okamura, H.; Garcia-Rodriguez, C.; Martinson, H.; Qin, J.; Virshup, D.M.; Rao, A. A conserved docking motif for CK1 binding controls the nuclear localization of NFAT1. Mol. Cell Biol. 2004, 24, 4184–4195. [Google Scholar] [CrossRef]
- Utz, A.C.; Hirner, H.; Blatz, A.; Hillenbrand, A.; Schmidt, B.; Deppert, W.; Henne-Bruns, D.; Fischer, D.; Thal, D.R.; Leithäuser, F.; et al. Analysis of cell type-specific expression of CK1 epsilon in various tissues of young adult BALB/c Mice and in mammary tumors of SV40 T-Ag-transgenic mice. J. Histochem. Cytochem. 2010, 58, 1–15. [Google Scholar] [CrossRef]
- Grainger, S.; Traver, D.; Willert, K. Wnt Signaling in Hematological Malignancies. Prog. Mol. Biol. Transl. Sci. 2018, 153, 321–341. [Google Scholar] [CrossRef]
- Staal, F.J.T.; Chhatta, A.; Mikkers, H. Caught in a Wnt storm: Complexities of Wnt signaling in hematopoiesis. Exp. Hematol. 2016, 44, 451–457. [Google Scholar] [CrossRef]
- Janovská, P.; Bryja, V. Wnt signalling pathways in chronic lymphocytic leukaemia and B-cell lymphomas. Br. J. Pharmacol. 2017, 174, 4701–4715. [Google Scholar] [CrossRef]
- Cozza, G.; Pinna, L.A. Casein kinases as potential therapeutic targets. Expert Opin. Ther. Targets 2015, 20, 319–340. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, P.S.; Ahern, S.A.; Smith, L.C.; da Silva Santos, C.S.; Wager, T.T.; Bechtold, D.A. Targeting of the circadian clock via CK1δ/ϵ to improve glucose homeostasis in obesity. Sci. Rep. 2016. [Google Scholar] [CrossRef] [PubMed]
- Arey, R.; McClung, C.A. An inhibitor of casein kinase 1 ε/δ partially normalizes the manic-like behaviors of the ClockΔ19 mouse. Behav. Pharmacol. 2012, 23, 392–396. [Google Scholar] [CrossRef] [PubMed]
- Perreau-Lenz, S.; Vengeliene, V.; Noori, H.R.; Merlo-Pich, E.V.; Corsi, M.A.; Corti, C.; Spanagel, R. Inhibition of the casein-kinase-1-ε/δ/prevents relapse-like alcohol drinking. Neuropsychopharmacology 2012, 37, 2121–2131. [Google Scholar] [CrossRef] [PubMed]
- Wager, T.T.; Chandrasekaran, R.Y.; Bradley, J.; Rubitski, D.; Berke, H.; Mente, S.; Butler, T.; Doran, A.; Chang, C.; Fisher, K.; et al. Casein kinase 1δ/ε inhibitor PF-5006739 attenuates opioid drug-seeking behavior. ACS Chem. Neurosci. 2014. [Google Scholar] [CrossRef]
- Rosenberg, L.H.; Lafitte, M.; Quereda, V.; Grant, W.; Chen, W.; Bibian, M.; Noguchi, Y.; Fallahi, M.; Yang, C.; Chang, J.C.; et al. Therapeutic targeting of casein kinase 1δ in breast cancer. Sci. Transl. Med. 2015, 7, 318ra202. [Google Scholar] [CrossRef]
- Liu, C.; Witt, L.; Ianes, C.; Bischof, J.; Bammert, M.T.; Baier, J.; Kirschner, S.; Henne-Bruns, D.; Xu, P.; Kornmann, M.; et al. Newly developed CK1-specific inhibitors show specifically stronger effects on ck1 mutants and colon cancer cell lines. Int. J. Mol. Sci. 2019, 20, 6184. [Google Scholar] [CrossRef]
- Rena, G.; Bain, J.; Elliott, M.; Cohen, P. D4476, a cell-permeant inhibitor of CK1, suppresses the site-specific phosphorylation and nuclear exclusion of FOXO1a. EMBO Rep. 2004, 5, 60–65. [Google Scholar] [CrossRef]
- Mashhoon, N.; DeMaggio, A.J.; Tereshko, V.; Bergmeier, S.C.; Egli, M.; Hoekstra, M.F.; Kuret, J. Crystal structure of a conformation-selective casein kinase-1 inhibitor. J. Biol. Chem. 2000. [Google Scholar] [CrossRef]
- Brockschmidt, C.; Hirner, H.; Huber, N.; Eismann, T.; Hillenbrand, A.; Giamas, G.; Radunsky, B.; Ammerpohl, O.; Bohm, B.; Henne-Bruns, D.; et al. Anti-apoptotic and growth-stimulatory functions of CK1 delta and epsilon in ductal adenocarcinoma of the pancreas are inhibited by IC261 in vitro and in vivo. Gut 2008, 57, 799–806. [Google Scholar] [CrossRef]
- Toyoshima, M.; Howie, H.L.; Imakura, M.; Walsh, R.M.; Annis, J.E.; Chang, A.N.; Frazier, J.; Chau, B.N.; Loboda, A.; Linsley, P.S.; et al. Functional genomics identifies therapeutic targets for MYC-driven cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 9545–9550. [Google Scholar] [CrossRef] [PubMed]
- Cheong, J.K.; Nguyen, T.H.; Wang, H.; Tan, P.; Voorhoeve, P.M.; Lee, S.H.; Virshup, D.M. IC261 induces cell cycle arrest and apoptosis of human cancer cells via CK1delta/varepsilon and Wnt/beta-catenin independent inhibition of mitotic spindle formation. Oncogene 2011, 30, 2558–2569. [Google Scholar] [CrossRef] [PubMed]
- Anastassiadis, T.; Deacon, S.W.; Devarajan, K.; Ma, H.; Peterson, J.R. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat. Biotechnol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Walton, K.M.; Fisher, K.; Rubitski, D.; Marconi, M.; Meng, Q.-J.; Adams, J.; Bass, M.; Chandrasekaran, R.; Butler, T.; Griffor, M.; et al. Selective Inhibition of Casein Kinase 1ε Minimally Alters Circadian Clock Period. J. Pharmacol. Exp. Ther. 2009, 330, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.-J.; Maywood, E.S.; Bechtold, D.A.; Lu, W.-Q.; Li, J.; Gibbs, J.E.; Dupré, S.M.; Chesham, J.E.; Rajamohan, F.; Knafels, J.; et al. Entrainment of disrupted circadian behavior through inhibition of casein kinase 1 (CK1) enzymes. Proc. Natl. Acad. Sci. USA 2010, 107, 15240–15245. [Google Scholar] [CrossRef]
- Badura, L.; Swanson, T.; Adamowicz, W.; Adams, J.; Cianfrogna, J.; Fisher, K.; Holland, J.; Kleiman, R.; Nelson, F.; Reynolds, L.; et al. An inhibitor of casein kinase I epsilon induces phase delays in circadian rhythms under free-running and entrained conditions. J. Pharmacol. Exp. Ther. 2007, 322, 730–738. [Google Scholar] [CrossRef]
- Deng, C.; Lipstein, M.R.; Scotto, L.; Jirau Serrano, X.O.; Mangone, M.A.; Li, S.; Vendome, J.; Hao, Y.; Xu, X.; Deng, S.X.; et al. Silencing c-Myc translation as a therapeutic strategy through targeting PI3Kdelta and CK1epsilon in hematological malignancies. Blood 2017, 129, 88–99. [Google Scholar] [CrossRef]
- Huang, H.; Acquaviva, L.; Berry, V.; Bregman, H.; Chakka, N.; O’Connor, A.; Dimauro, E.F.; Dovey, J.; Epstein, O.; Grubinska, B.; et al. Structure-based design of potent and selective CK1γ inhibitors. ACS Med. Chem. Lett. 2012. [Google Scholar] [CrossRef]
- Zhang, T.; Davidson-Moncada, J.K.; Mukherjee, P.; Furman, R.R.; Bhavsar, E.; Chen, Z.; Hakimpour, P.; Papavasiliou, N.; Tam, W. MicroRNA-155 regulates casein kinase 1 gamma 2: A potential pathogenetic role in chronic lymphocytic leukemia. Blood Cancer J. 2017. [Google Scholar] [CrossRef]
- Hallek, M. Chronic lymphocytic leukemia: 2020 update on diagnosis, risk stratification and treatment. Am. J. Hematol. 2019, 94, 1266–1287. [Google Scholar] [CrossRef]
- Sant, M.; Allemani, C.; Tereanu, C.; De Angelis, R.; Capocaccia, R.; Visser, O.; Marcos-Gragera, R.; Maynadié, M.; Simonetti, A.; Lutz, J.-M.; et al. Incidence of hematologic malignancies in Europe by morphologic subtype: Results of the HAEMACARE project. Blood 2010, 116, 3724–3734. [Google Scholar] [CrossRef] [PubMed]
- Janovska, P.; Poppova, L.; Plevova, K.; Plesingerova, H.; Behal, M.; Kaucka, M.; Ovesna, P.; Hlozkova, M.; Borsky, M.; Stehlikova, O.; et al. Autocrine signaling by Wnt-5a deregulates chemotaxis of leukemic cells and predicts clinical outcome in chronic lymphocytic leukemia. Clin. Cancer Res. 2016, 22, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Plešingerová, H.; Janovská, P.; Mishra, A.; Smyčková, L.; Poppová, L.; Libra, A.; Plevová, K.; Ovesná, P.; Radová, L.; Doubek, M.; et al. Expression of COBLL1 encoding novel ROR1 binding partner is robust predictor of survival in chronic lymphocytic leukemia. Haematologica 2018, 103, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Daneshmanesh, A.H.; Mikaelsson, E.; Jeddi-Tehrani, M.; Bayat, A.A.; Ghods, R.; Ostadkarampour, M.; Akhondi, M.; Lagercrantz, S.; Larsson, C.; Osterborg, A.; et al. Ror1, a cell surface receptor tyrosine kinase is expressed in chronic lymphocytic leukemia and may serve as a putative target for therapy. Int. J. Cancer 2008, 123, 1190–1195. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, T.; Chen, L.; Endo, T.; Tang, L.; Lu, D.; Castro, J.E.; Widhopf, G.F.; Rassenti, L.Z.; Cantwell, M.J.; Prussak, C.E.; et al. Antisera induced by infusions of autologous Ad-CD154-leukemia B cells identify ROR1 as an oncofetal antigen and receptor for Wnt5a. Proc. Natl. Acad. Sci. USA 2008, 105, 3047–3052. [Google Scholar] [CrossRef]
- Daneshmanesh, A.H.; Porwit, A.; Hojjat-Farsangi, M.; Jeddi-Tehrani, M.; Tamm, K.P.; Grandér, D.; Lehmann, S.; Norin, S.; Shokri, F.; Rabbani, H.; et al. Orphan receptor tyrosine kinases ROR1 and ROR2 in hematological malignancies. Leuk. Lymphoma 2013, 8194, 843–850. [Google Scholar] [CrossRef]
- Karvonen, H.; Chiron, D.; Niininen, W.; Ek, S.; Jerkeman, M.; Moradi, E.; Nykter, M.; Heckman, C.A.; Kallioniemi, O.; Murumägi, A.; et al. Crosstalk between ROR1 and BCR pathways defines novel treatment strategies in mantle cell lymphoma. Blood Adv. 2017, 1, 2257–2268. [Google Scholar] [CrossRef]
- Campo, E.; Raffeld, M.; Jaffe, E.S. Mantle-cell lymphoma. Semin. Hematol 1999, 36, 115–127. [Google Scholar]
- Choi, M.Y.; Widhopf, G.F.; Wu, C.C.N.; Cui, B.; Lao, F.; Sadarangani, A.; Cavagnaro, J.; Prussak, C.; Carson, D.A.; Jamieson, C.; et al. Pre-clinical Specificity and Safety of UC-961, a First-In-Class Monoclonal Antibody Targeting ROR1. Clin. Lymphoma Myeloma Leuk. 2015, 15, S167–S169. [Google Scholar] [CrossRef]
- Berger, C.; Sommermeyer, D.; Hudecek, M.; Berger, M.; Balakrishnan, A.; Paszkiewicz, P.J.; Kosasih, P.L.; Rader, C.; Riddell, S.R. Safety of Targeting ROR1 in Primates with Chimeric Antigen Receptor-Modified T Cells. Cancer Immunol. Res. 2015, 3, 206–216. [Google Scholar] [CrossRef]
- Liu, X.; Pu, W.; He, H.; Fan, X.; Zheng, Y.; Zhou, J.K.; Ma, R.; He, J.; Zheng, Y.; Wu, K.; et al. Novel ROR1 inhibitor ARI-1 suppresses the development of non-small cell lung cancer. Cancer Lett. 2019. [Google Scholar] [CrossRef] [PubMed]
- Fultang, N.; Illendula, A.; Chen, B.; Wu, C.; Jonnalagadda, S.; Baird, N.; Klase, Z.; Peethambaran, B. Strictinin, a novel ROR1-inhibitor, represses triple negative breast cancer survival and migration via modulation of PI3K/AKT/GSK3ß activity. PLoS ONE 2019. [CrossRef] [PubMed]
- Daneshmanesh, A.H.; Hojjat-Farsangi, M.; Ghaderi, A.; Moshfegh, A.; Hansson, L.; Schultz, J.; Vågberg, J.; Byström, S.; Olsson, E.; Olin, T.; et al. A receptor tyrosine kinase ROR1 inhibitor (KAN0439834) induced significant apoptosis of pancreatic cells which was enhanced by erlotinib and ibrutinib. PLoS ONE 2018. [Google Scholar] [CrossRef] [PubMed]
- Mellstedt, H.; Ghaderi, A.; Aschan, J.; Mozaffari, F.; Moshfegh, A.; Sander, B.; Schultz, J.; Norin, M.; Olin, T.; Drakos, E.; et al. ROR1 Small Molecule Inhibitor (KAN0441571C) Induced Significant Apoptosis of Mantle Cell Lymphoma (MCL) Cells. Blood 2019. [Google Scholar] [CrossRef]
- Karvonen, H.; Perttilä, R.; Niininen, W.; Barker, H.; Ungureanu, D. Targeting Wnt signaling pseudokinases in hematological cancers. Eur. J. Haematol. 2018. [Google Scholar] [CrossRef]
- Zhang, S.; Chen, L.; Cui, B.; Chuang, H.-Y.; Yu, J.; Wang-Rodriguez, J.; Tang, L.; Chen, G.; Basak, G.W.; Kipps, T.J. ROR1 is expressed in human breast cancer and associated with enhanced tumor-cell growth. PLoS ONE 2012, 7, e31127. [Google Scholar] [CrossRef]
- Modak, C.; Bryant, P. Casein Kinase I epsilon positively regulates the Akt pathway in breast cancer cell lines. Biochem. Biophys. Res. Commun. 2008, 368, 801–807. [Google Scholar] [CrossRef]
- Kani, S.; Oishi, I.; Yamamoto, H.; Yoda, A.; Suzuki, H.; Nomachi, A.; Iozumi, K.; Nishita, M.; Kikuchi, A.; Takumi, T.; et al. The receptor tyrosine kinase Ror2 associates with and is activated by casein kinase Iepsilon. J. Biol. Chem. 2004. [Google Scholar] [CrossRef]
- Kim, J.; Kim, D.W.; Chang, W.; Choe, J.; Kim, J.; Park, C.-S.; Song, K.; Lee, I. Wnt5a is secreted by follicular dendritic cells to protect germinal center B cells via Wnt/Ca2+/NFAT/NF-κB-B cell lymphoma 6 signaling. J. Immunol. 2012, 188, 182–189. [Google Scholar] [CrossRef]
- Yu, J.; Chen, L.; Cui, B.; Widhopf, G.F., II; Shen, Z.; Wu, R.; Zhang, L.; Zhang, S.; Briggs, S.P.; Kipps, T.J. Wnt5a induces ROR1/ROR2 heterooligomerization to enhance leukemia chemotaxis and proliferation. J. Clin. Investig. 2016, 126, 585–598. [Google Scholar] [CrossRef]
- Hasan, M.K.; Yu, J.; Chen, L.; Cui, B.; Widhopf Ii, G.F.; Rassenti, L.; Shen, Z.; Briggs, S.P.; Kipps, T.J. Wnt5a induces ROR1 to complex with HS1 to enhance migration of chronic lymphocytic leukemia cells. Leukemia 2017, 31, 2615–2622. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.K.; Yu, J.; Widhopf, G.F., II; Rassenti, L.Z.; Chen, L.; Shen, Z.; Briggs, S.P.; Neuberg, D.S.; Kipps, T.J. Wnt5a induces ROR1 to recruit DOCK2 to activate Rac1/2 in chronic lymphocytic leukemia. Blood 2018, 132, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Chen, L.; Chen, Y.; Hasan, M.K.; Ghia, E.M.; Zhang, L.; Wu, R.; Rassenti, L.Z.; Widhopf, G.F.; Shen, Z.; et al. Wnt5a induces ROR1 to associate with 14-3-3ζ for enhanced chemotaxis and proliferation of chronic lymphocytic leukemia cells. Leukemia 2017, 31, 2608–2614. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wang, H.Y.; Liu, X.; Nunez-Cruz, S.; Jillab, M.; Melnikov, O.; Nath, K.; Glickson, J.; Wasik, M.A. Cutting Edge: ROR1/CD19 Receptor Complex Promotes Growth of Mantle Cell Lymphoma Cells Independently of the B Cell Receptor–BTK Signaling Pathway. J. Immunol. 2019, 203, 2043–2048. [Google Scholar] [CrossRef]
- Hasan, M.K.; Rassenti, L.; Widhopf, G.F.; Yu, J.; Kipps, T.J. Wnt5a causes ROR1 to complex and activate cortactin to enhance migration of chronic lymphocytic leukemia cells. Leukemia 2019. [Google Scholar] [CrossRef]
- Rudelius, M.; Pittaluga, S.; Nishizuka, S.; Pham, T.H.; Fend, F.; Jaffe, E.S.; Quintanilla-Martinez, L.; Raffeld, M. Constitutive activation of Akt contributes to the pathogenesis and survival of mantle cell lymphoma. Blood 2006, 108, 1668–1676. [Google Scholar] [CrossRef]
- Linke, F.; Zaunig, S.; Nietert, M.M.; VonBonin, F.; Lutz, S.; Dullin, C.; Janovská, P.; Beissbarth, T.; Alves, F.; Klapper, W.; et al. WNT5A: A motility-promoting factor in Hodgkin lymphoma. Oncogene 2017, 36, 13–23. [Google Scholar] [CrossRef]
- Cui, B.; Chen, L.; Rassenti, L.Z.; Ghia, E.M.; Yu, J.; Zhang, L.; Neuberg, D.S.; Wierda, W.; Keating, M.; Rai, K.; et al. High-level expression of ROR1 associates with early disease progression in patients with chronic lymphocytic leukemia. Blood 2015, 126, 1713. [Google Scholar] [CrossRef]
- Widhopf 2nd, G.F.; Cui, B.; Ghia, E.M.; Chen, L.; Messer, K.; Shen, Z.; Briggs, S.P.; Croce, C.M.; Kipps, T.J. ROR1 can interact with TCL1 and enhance leukemogenesis in Emu-TCL1 transgenic mice. Proc. Natl. Acad. Sci. USA 2014, 111, 793–798. [Google Scholar] [CrossRef]
- Wu, Q.-L. Dysregulation of Frizzled 6 is a critical component of Bcell leukemogenesis in a mouse model of chronic lymphocytic leukemia. Blood 2009. [Google Scholar] [CrossRef]
- Kaucká, M.; Petersen, J.; Janovská, P.; Radaszkiewicz, T.; Smyčková, L.; Daulat, A.M.; Borg, J.P.; Schulte, G.; Bryja, V. Asymmetry of VANGL2 in migrating lymphocytes as a tool to monitor activity of the mammalian WNT/planar cell polarity pathway. Cell Commun. Signal. 2015, 13, 2. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, E.M.; Rudelius, M.; Burger, J.A.; Rosenwald, A. CCL3 chemokine expression by chronic lymphocytic leukemia cells orchestrates the composition of the microenvironment in lymph node infiltrates. Leuk. Lymphoma 2016, 57, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Burris 3rd, H.A.; Flinn, I.W.; Patel, M.R.; Fenske, T.S.; Deng, C.; Brander, D.M.; Gutierrez, M.; Essell, J.H.; Kuhn, J.G.; Miskin, H.P.; et al. Umbralisib, a novel PI3Kdelta and casein kinase-1epsilon inhibitor, in relapsed or refractory chronic lymphocytic leukaemia and lymphoma: An open-label, phase 1, dose-escalation, first-in-human study. Lancet Oncol. 2018, 19, 486–496. [Google Scholar] [CrossRef]
- Lunning, M.; Vose, J.; Nastoupil, L.; Fowler, N.; Burger, J.A.; Wierda, W.G.; Schreeder, M.T.; Siddiqi, T.; Flowers, C.R.; Cohen, J.B.; et al. Ublituximab and umbralisib in relapsed/refractory B-cell non-Hodgkin lymphoma and chronic lymphocytic leukemia. Blood 2019, 134, 1811–1820. [Google Scholar] [CrossRef] [PubMed]
- Davids, M.S.; Kim, H.T.; Nicotra, A.; Savell, A.; Francoeur, K.; Hellman, J.M.; Bazemore, J.; Miskin, H.P.; Sportelli, P.; Stampleman, L.; et al. Umbralisib in combination with ibrutinib in patients with relapsed or refractory chronic lymphocytic leukaemia or mantle cell lymphoma: A multicentre phase 1-1b study. Lancet Haematol. 2019, 6, e38–e47. [Google Scholar] [CrossRef]
- Maharaj, K.; Powers, J.J.; Achille, A.; Mediavilla-Varela, M.; Gamal, W.; Burger, K.L.; Fonseca, R.; Jiang, K.; Miskin, H.P.; Maryanski, D.; et al. The dual PI3Kdelta/CK1epsilon inhibitor umbralisib exhibits unique immunomodulatory effects on CLL T cells. Blood Adv. 2020, 4, 3072–3084. [Google Scholar] [CrossRef]
- Shin, S.; Wolgamott, L.; Roux, P.P.; Yoon, S.O. Casein kinase 1epsilon promotes cell proliferation by regulating mRNA translation. Cancer Res. 2014, 74, 201–211. [Google Scholar] [CrossRef]
- van Loosdregt, J.; Fleskens, V.; Tiemessen, M.M.; Mokry, M.; van Boxtel, R.; Meerding, J.; Pals, C.E.; Kurek, D.; Baert, M.R.; Delemarre, E.M.; et al. Canonical Wnt signaling negatively modulates regulatory T cell function. Immunity 2013, 39, 298–310. [Google Scholar] [CrossRef]
- Yu, J.; Chen, L.; Cui, B.; Wu, C.; Choi, M.Y.; Chen, Y.; Zhang, L.; Rassenti, L.Z.; Widhopf Ii, G.F.; Kipps, T.J. Cirmtuzumab inhibits Wnt5a-induced Rac1 activation in chronic lymphocytic leukemia treated with ibrutinib. Leukemia 2017, 31, 1333–1339. [Google Scholar] [CrossRef]
- Nastoupil, L.J.; Lunning, M.A.; Vose, J.M.; Schreeder, M.T.; Siddiqi, T.; Flowers, C.R.; Cohen, J.B.; Burger, J.A.; Wierda, W.G.; O’Brien, S.; et al. Tolerability and activity of ublituximab, umbralisib, and ibrutinib in patients with chronic lymphocytic leukaemia and non-Hodgkin lymphoma: A phase 1 dose escalation and expansion trial. Lancet Haematol. 2019, 6, e100–e109. [Google Scholar] [CrossRef]
- Hellström-Lindberg, E.; Tobiasson, M.; Greenberg, P. Myelodysplastic syndromes: Moving towards personalized management. Haematologica 2020. [Google Scholar] [CrossRef] [PubMed]
- Estey, E.H. Acute myeloid leukemia: 2019 update on risk-stratification and management. Am. J. Hematol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Heuser, M.; Ofran, Y.; Boissel, N.; Brunet Mauri, S.; Craddock, C.; Janssen, J.; Wierzbowska, A.; Buske, C. Acute myeloid leukaemia in adult patients: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S. Genetics of MDS. Blood 2019. [Google Scholar] [CrossRef] [PubMed]
- Short, N.J.; Konopleva, M.; Kadia, T.M.; Borthakur, G.; Ravandi, F.; DiNardo, C.D.; Daver, N. Advances in the treatment of acute myeloid leukemia: New drugs and new challenges. Cancer Discov. 2020, 10, 506–525. [Google Scholar] [CrossRef]
- Maynadié, M.; Girodon, F.; Manivet-Janoray, I.; Mounier, M.; Mugneret, F.; Bailly, F.; Favre, B.; Caillot, D.; Petrella, T.; Flesch, M.; et al. Twenty-five years of epidemiological recording on myeloid malignancies: Data from the specialized registry of hematologic malignancies of côte d’or (Burgundy, France). Haematologica 2011. [Google Scholar] [CrossRef]
- Schneider, R.K.; Ademà, V.; Heckl, D.; Järås, M.; Mallo, M.; Lord, A.M.; Chu, L.P.; McConkey, M.E.; Kramann, R.; Mullally, A.; et al. Role of casein kinase 1A1 in the biology and targeted therapy of del(5q) MDS. Cancer Cell 2014. [Google Scholar] [CrossRef]
- Krönke, J.; Fink, E.C.; Hollenbach, P.W.; MacBeth, K.J.; Hurst, S.N.; Udeshi, N.D.; Chamberlain, P.P.; Mani, D.R.; Man, H.W.; Gandhi, A.K.; et al. Lenalidomide induces ubiquitination and degradation of CK1α in del(5q) MDS. Nature 2015. [Google Scholar] [CrossRef]
- Ebert, B.L. Molecular Dissection of the 5q Deletion in Myelodysplastic Syndrome. Semin. Oncol. 2011. [Google Scholar] [CrossRef]
- Haase, D.; Germing, U.; Schanz, J.; Pfeilstöcker, M.; Nösslinger, T.; Hildebrandt, B.; Kundgen, A.; Lübbert, M.; Kunzmann, R.; Giagounidis, A.A.N.; et al. New insights into the prognostic impact of the karyotype in MDS and correlation with subtypes: Evidence from a core dataset of 2124 patients. Blood 2007. [Google Scholar] [CrossRef]
- Hasserjian, R.P. Myelodysplastic Syndrome Updated. Pathobiology 2019. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Sheng, Y.; Li, W.; Hu, C.; Mittal, N.; Tohyama, K.; Seba, A.; Zhao, Y.Y.; Ozer, H.; Zhu, T.; et al. β-catenin is a candidate therapeutic target for myeloid neoplasms with del(5q). Cancer Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Järås, M.; Miller, P.G.; Chu, L.P.; Puram, R.V.; Fink, E.C.; Schneider, R.K.; Al-Shahrour, F.; Peña, P.; Breyfogle, L.J.; Hartwell, K.A.; et al. Csnk1a1 inhibition has p53-dependent therapeutic efficacy in acute myeloid leukemia. J. Exp. Med. 2014, 211, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Huart, A.S.; MacLaine, N.J.; Meek, D.W.; Hupp, T.R. CK1α plays a central role in mediating MDM2 control of p53 and E2F-1 protein stability. J. Biol. Chem. 2009. [Google Scholar] [CrossRef]
- Wei, X. Secondary interaction between MDMX and p53 core domain inhibits p53 DNA binding. Proc. Natl. Acad. Sci. USA 2016. [Google Scholar] [CrossRef]
- Wu, S.; Chen, L.; Becker, A.; Schonbrunn, E.; Chen, J. Casein Kinase 1 Regulates an MDMX Intramolecular Interaction To Stimulate p53 Binding. Mol. Cell. Biol. 2012. [Google Scholar] [CrossRef]
- Elyada, E.; Pribluda, A.; Goldstein, R.E.; Morgenstern, Y.; Brachya, G.; Cojocaru, G.; Snir-Alkalay, I.; Burstain, I.; Haffner-Krausz, R.; Jung, S.; et al. CKIα ablation highlights a critical role for p53 in invasiveness control. Nature 2011, 470, 409–413. [Google Scholar] [CrossRef]
- Khalaileh, A.; Dreazen, A.; Khatib, A.; Apel, R.; Swisa, A.; Kidess-Bassir, N.; Maitra, A.; Meyuhas, O.; Dor, Y.; Zamir, G. Phosphorylation of ribosomal protein S6 attenuates DNA damage and tumor suppression during development of pancreatic cancer. Cancer Res. 2013. [Google Scholar] [CrossRef]
- Takam Kamga, P.; Dal Collo, G.; Cassaro, A.; Bazzoni, R.; Delfino, P.; Adamo, A.; Bonato, A.; Carbone, C.; Tanasi, I.; Bonifacio, M.; et al. Small Molecule Inhibitors of Microenvironmental Wnt/β-Catenin Signaling Enhance the Chemosensitivity of Acute Myeloid Leukemia. Cancers 2020, 12, 2696. [Google Scholar] [CrossRef]
- Wang, Y.; Krivtsov, A.V.; Sinha, A.U.; North, T.E.; Goessling, W.; Feng, Z.; Zon, L.I.; Armstrong, S.A. The wnt/β-catenin pathway is required for the development of leukemia stem cells in AML. Science 2010. [Google Scholar] [CrossRef]
- Yeung, J.; Esposito, M.T.; Gandillet, A.; Zeisig, B.B.; Griessinger, E.; Bonnet, D.; So, C.W.E. β-Catenin Mediates the Establishment and Drug Resistance of MLL Leukemic Stem Cells. Cancer Cell 2010. [Google Scholar] [CrossRef] [PubMed]
- Miller, P.G.; Al-Shahrour, F.; Hartwell, K.A.; Chu, L.P.; Järås, M.; Puram, R.V.; Puissant, A.; Callahan, K.P.; Ashton, J.; McConkey, M.E.; et al. InVivo RNAi Screening Identifies a Leukemia-Specific Dependence on Integrin Beta 3 Signaling. Cancer Cell 2013. [Google Scholar] [CrossRef] [PubMed]
- Gruszka, A.M.; Valli, D.; Alcalay, M. Wnt Signalling in Acute Myeloid Leukaemia. Cells 2019, 8, 1403. [Google Scholar] [CrossRef]
- Liu, C.; Li, Y.; Semenov, M.; Han, C.; Baeg, G.H.; Tan, Y.; Zhang, Z.; Lin, X.; He, X. Control of β-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 2002. [Google Scholar] [CrossRef]
- Kadia, T.M.; Jain, P.; Ravandi, F.; Garcia-Manero, G.; Andreef, M.; Takahashi, K.; Borthakur, G.; Jabbour, E.; Konopleva, M.; Daver, N.G.; et al. TP53 mutations in newly diagnosed acute myeloid leukemia: Clinicomolecular characteristics, response to therapy, and outcomes. Cancer 2016. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, S.; Bykov, V.J.; Ali, D.; Andrén, O.; Cherif, H.; Tidefelt, U.; Uggla, B.; Yachnin, J.; Juliusson, G.; Moshfegh, A.; et al. Targeting p53 in vivo: A first-in-human study with p53-targeting compound APR-246 in refractory hematologic malignancies and prostate cancer. J. Clin. Oncol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Maslah, N.; Salomao, N.; Drevon, L.; Verger, E.; Partouche, N.; Ly, P.; Aubin, P.; Naoui, N.; Schlageter, M.H.; Bally, C.; et al. Synergistic effects of PRIMA-1Met (APR-246) and 5-azacitidine in TP53-mutated myelodysplastic syndromes and acute myeloid leukemia. Haematologica 2020. [Google Scholar] [CrossRef]
- Matsuoka, A.; Tochigi, A.; Kishimoto, M.; Nakahara, T.; Kondo, T.; Tsujioka, T.; Tasaka, T.; Tohyama, Y.; Tohyama, K. Lenalidomide induces cell death in an MDS-derived cell line with deletion of chromosome 5q by inhibition of cytokinesis. Leukemia 2010. [Google Scholar] [CrossRef]
- Chen, Y.; Borthakur, G. Lenalidomide as a novel treatment of acute myeloid leukemia. Expert Opin. Investig. Drugs 2013. [Google Scholar] [CrossRef]
- List, A.; Dewald, G.; Bennett, J.; Giagounidis, A.; Raza, A.; Feldman, E.; Powell, B.; Greenberg, P.; Thomas, D.; Stone, R.; et al. Lenalidomide in the myelodysplastic syndrome with chromosome 5q deletion. N. Engl. J. Med. 2006. [Google Scholar] [CrossRef]
- Pellagatti, A.; Jädersten, M.; Forsblom, A.M.; Cattan, H.; Christensson, B.; Emanuelsson, E.K.; Merup, M.; Nilsson, L.; Samuelsson, J.; Sander, B.; et al. Lenalidomide inhibits the malignant clone and up-regulates the SPARC gene mapping to the commonly deleted region in 5q- syndrome patients. Proc. Natl. Acad. Sci. USA 2007. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.H.; Wei, M.; Yang, F.Y.; Wu, F.Z.; Chen, L.; Wang, J.K.; Liu, Q.; Huang, J.X. Efficacy and safety of lenalidomide for thtreatment of acute myeloid leukemia: A systematic review and meta-analysis. Cancer Manag. Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Chellappa, S.; Kushekhar, K.; Munthe, L.A.; Tjonnfjord, G.E.; Aandahl, E.M.; Okkenhaug, K.; Tasken, K. The PI3K p110delta Isoform Inhibitor Idelalisib Preferentially Inhibits Human Regulatory T Cell Function. J. Immunol. 2019, 202, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Bibian, M. Development of highly selective casein kinase 1δ/1ε (CK1δ/ε) inhibitors with potent antiproliferative properties. Bioorg. Med. Chem. Lett. 2013, 23, 4374–4380. [Google Scholar] [CrossRef] [PubMed]
- Morgenstern, Y.; Das Adhikari, U.; Ayyash, M.; Elyada, E.; Tóth, B.; Moor, A.; Itzkovitz, S.; Ben-Neriah, Y. Casein kinase 1-epsilon or 1-delta required for Wnt-mediated intestinal stem cell maintenance. EMBO J. 2017, 36, 3046–3061. [Google Scholar] [CrossRef] [PubMed]
- Sen, M.; Chamorro, M.; Reifert, J.; Corr, M.; Carson, D.A. Blockade of Wnt-5A/frizzled 5 signaling inhibits rheumatoid synoviocyte activation. Arthritis Rheum. 2001, 44, 772–781. [Google Scholar] [CrossRef]
- He, T.; Wu, D.; He, L.; Wang, X.; Yang, B.; Li, S.; Chen, Y.; Wang, K.; Chen, R.; Liu, B.; et al. Casein kinase 1 epsilon facilitates cartilage destruction in osteoarthritis through JNK pathway. FASEB J. 2020. [Google Scholar] [CrossRef]
- Reischl, J.; Schwenke, S.; Beekman, J.M.; Stürzebecher, J.; Mrowietz, U.; Heubach, J.F. Increased expression of Wnt5a in psoriatic plaques. J. Investig. Dermatol 2007, 127, 163–169. [Google Scholar] [CrossRef]
- Choi, E.Y.; Park, H.H.; Kim, H.; Kim, H.N.; Kim, I.; Jeon, S.; Kim, W.; Bae, J.-S.; Lee, W. Wnt5a and Wnt11 as acute respiratory distress syndrome biomarkers for SARS-CoV-2 patients. Eur. Respir. J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Zyss, D.; Ebrahimi, H.; Gergely, F. Casein kinase I delta controls centrosome positioning during T cell activation. J. Cell Biol. 2011, 195, 781–797. [Google Scholar] [CrossRef][Green Version]
- Hu, Y.; Song, W.; Cirstea, D.; Lu, D.; Munshi, N.C.; Anderson, K.C. CSNK1α1 mediates malignant plasma cell survival. Leukemia 2015, 29, 474–482. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Manni, S.; Carrino, M.; Manzoni, M.; Gianesin, K.; Nunes, S.C.; Costacurta, M.; Tubi, L.Q.; Macaccaro, P.; Taiana, E.; Cabrelle, A.; et al. Inactivation of CK1α in multiple myeloma empowers drug cytotoxicity by affecting AKT and ß-catenin survival signaling pathways. Oncotarget 2017. [Google Scholar] [CrossRef] [PubMed]
- Carrino, M.; Quotti Tubi, L.; Fregnani, A.; Canovas Nunes, S.; Barilà, G.; Trentin, L.; Zambello, R.; Semenzato, G.; Manni, S.; Piazza, F. Prosurvival autophagy is regulated by protein kinase CK1 alpha in multiple myeloma. Cell Death Discov. 2019. [Google Scholar] [CrossRef] [PubMed]
- Cheong, J.K.; Zhang, F.; Chua, P.J.; Bay, B.H.; Thorburn, A.; Virshup, D.M. Casein kinase 1α-dependent feedback loop controls autophagy in RAS-driven cancers. J. Clin. Investig. 2015. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| IC50 (nM)/ % Kinase Activity | CK1ε | CK1δ | CK1α | CK1γ1/2/3 | Other Targets | Source Publication | Stage |
|---|---|---|---|---|---|---|---|
| PF-4800567 | 32 | 711 | - | No | EGFR | [54] | Preclinical |
| PF-670462 | 7.7 | 14 | - | No | EGFR, p38α, | [54,56] | Preclinical |
| D4476 | 270 | 300 | 37% at 0.5 μM * | No | ALK5 | [48,53,56] | Preclinical |
| BTX-A51 | 4.4 | 1.8 | 5.3 | 20/0.5/6 | CDK7/9 | [8] | Clinical, Phase I |
| Umbralisib | 40% at 1 μM * | 105% at 1 μM * | 111% at 1 μM * | 96–104% at 1 μM * | PI3Kδ | [57] | Clinical, Phase III |
| Kd (nM) | PI3Kδ | PI3Kγ | PI3Kα | PI3Kβ | CK1ε |
|---|---|---|---|---|---|
| Duvelisib | 0.047 | 0.21 | 40 | 0.89 | >30000 |
| Idelalisib | 1.2 | 9.1 | 600 | 19 | >30000 |
| Umbralisib | 6.2 | 1400 | >10000 | >10000 | 180 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janovská, P.; Normant, E.; Miskin, H.; Bryja, V. Targeting Casein Kinase 1 (CK1) in Hematological Cancers. Int. J. Mol. Sci. 2020, 21, 9026. https://doi.org/10.3390/ijms21239026
Janovská P, Normant E, Miskin H, Bryja V. Targeting Casein Kinase 1 (CK1) in Hematological Cancers. International Journal of Molecular Sciences. 2020; 21(23):9026. https://doi.org/10.3390/ijms21239026
Chicago/Turabian StyleJanovská, Pavlína, Emmanuel Normant, Hari Miskin, and Vítězslav Bryja. 2020. "Targeting Casein Kinase 1 (CK1) in Hematological Cancers" International Journal of Molecular Sciences 21, no. 23: 9026. https://doi.org/10.3390/ijms21239026
APA StyleJanovská, P., Normant, E., Miskin, H., & Bryja, V. (2020). Targeting Casein Kinase 1 (CK1) in Hematological Cancers. International Journal of Molecular Sciences, 21(23), 9026. https://doi.org/10.3390/ijms21239026