NAA10 p.(D10G) and NAA10 p.(L11R) Variants Hamper Formation of the NatA N-Terminal Acetyltransferase Complex


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
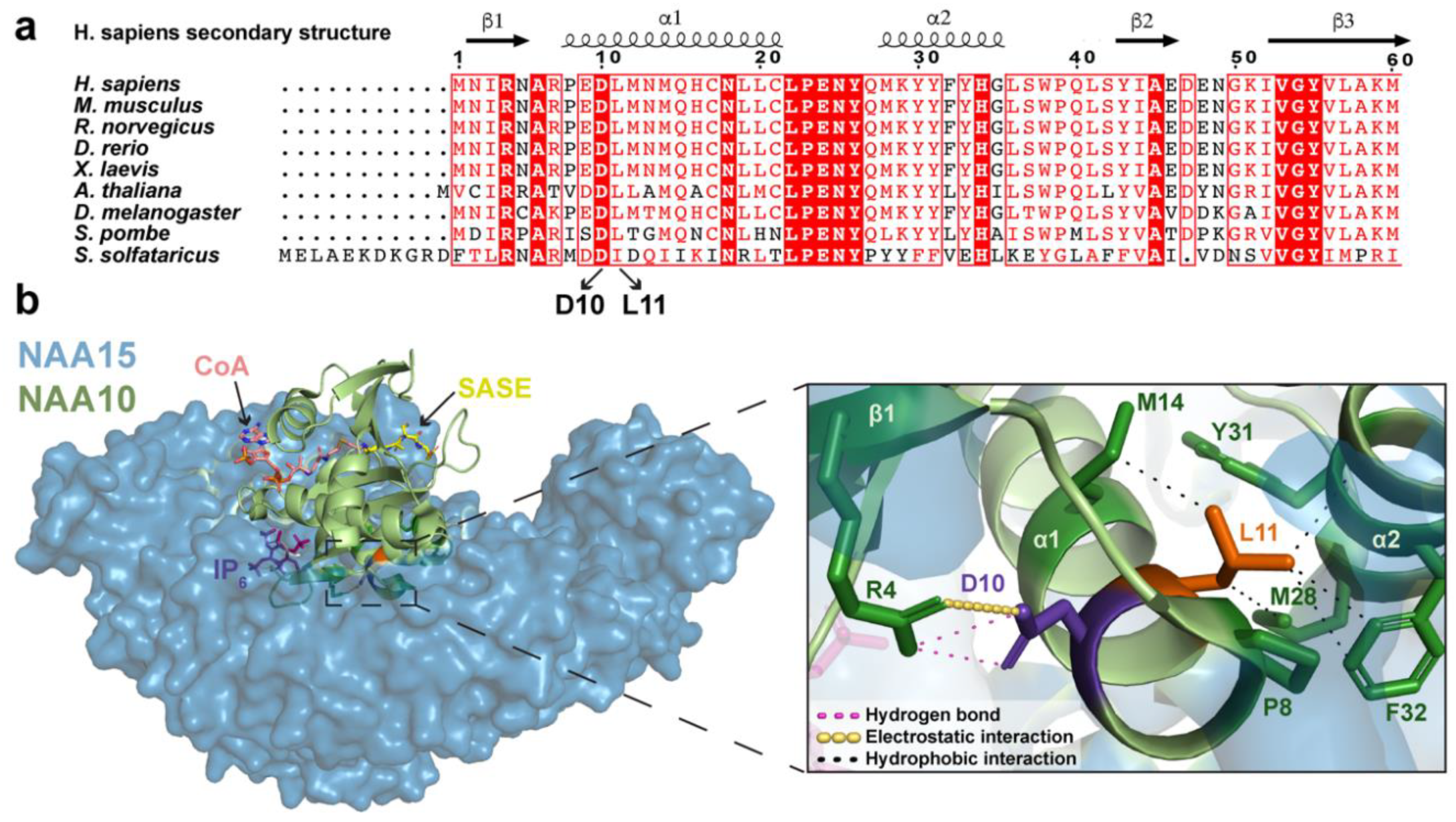
2.1. Analysis of NAA10 Sequence and NatA Structure
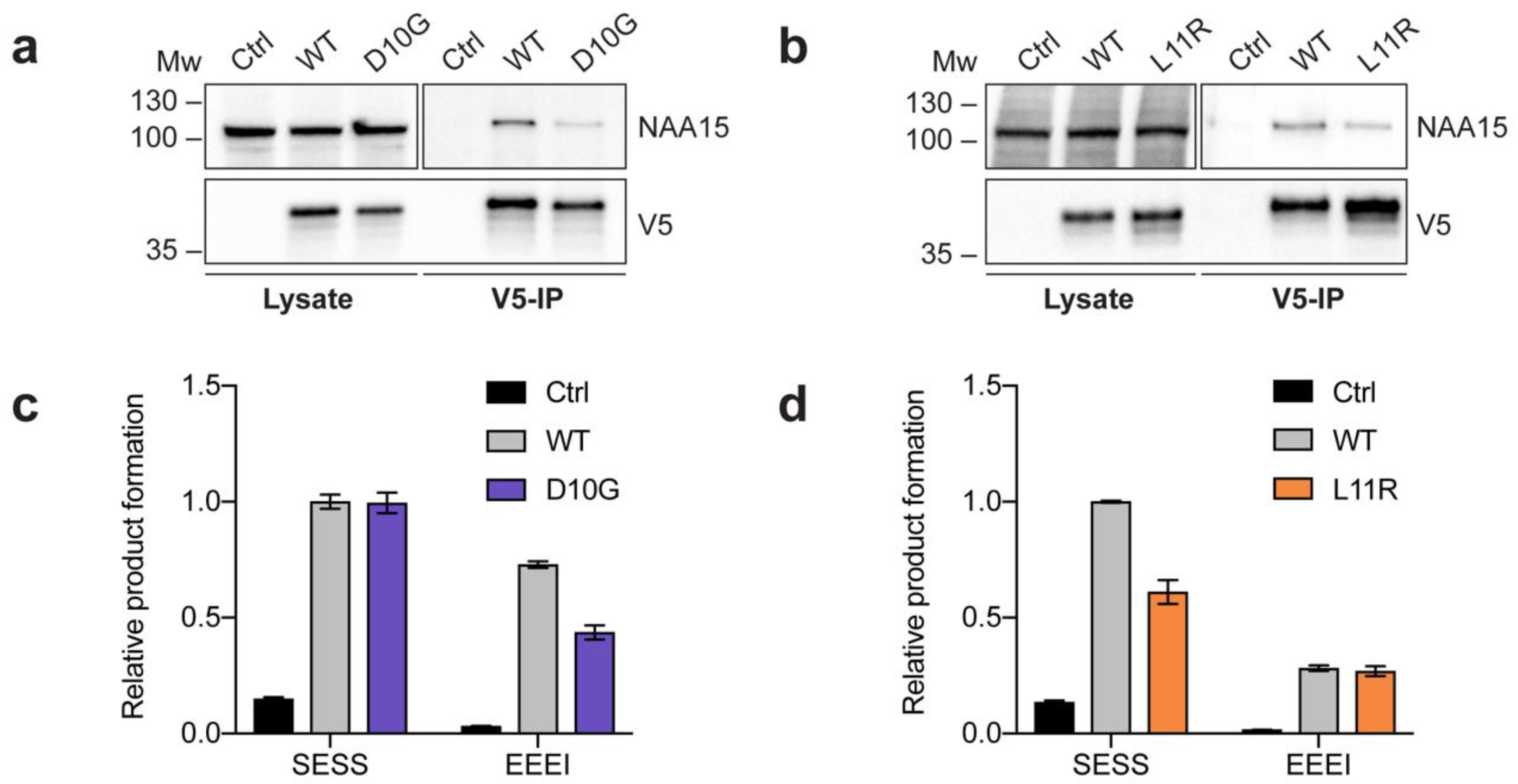
2.2. Complex Formation and Catalytic Activity
3. Discussion
4. Materials and Methods
4.1. Construction of Vectors
4.2. Bioinformatic Analyses
4.3. Transfection, Immunoprecipitation and Western Blot Analysis
4.4. In Vitro N-terminal (Nt)-Acetylation Assays
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Nt | N-terminal |
| NAT | N-terminal acetyltransferase |
| KAT | lysine acetyltransferase |
| DD | developmental delay |
| ID | intellectual disability |
| IP | immunoprecipitation |
References
- Arnesen, T.; Van Damme, P.; Polevoda, B.; Helsens, K.; Evjenth, R.; Colaert, N.; Varhaug, J.E.; Vandekerckhove, J.; Lillehaug, J.R.; Sherman, F.; et al. Proteomics analyses reveal the evolutionary conservation and divergence of N-terminal acetyltransferases from yeast and humans. Proc. Natl. Acad. Sci. USA 2009, 106, 8157–8162. [Google Scholar] [CrossRef] [PubMed]
- Bienvenut, W.V.; Sumpton, D.; Martinez, A.; Lilla, S.; Espagne, C.; Meinnel, T.; Giglione, C. Comparative large scale characterization of plant versus mammal proteins reveals similar and idiosyncratic N-α-acetylation features. Mol. Cell. Proteom. 2012, 11, M111.015131. [Google Scholar] [CrossRef] [PubMed]
- Aksnes, H.; Ree, R.; Arnesen, T. Co-translational, Post-translational, and Non-catalytic Roles of N-Terminal Acetyltransferases. Mol. Cell 2019, 73, 1097–1114. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.C.; Monda, J.K.; Bennett, E.J.; Harper, J.W.; Schulman, B.A. N-terminal acetylation acts as an avidity enhancer within an interconnected multiprotein complex. Science 2011, 334, 674–678. [Google Scholar] [CrossRef]
- Behnia, R.; Panic, B.; Whyte, J.R.; Munro, S. Targeting of the Arf-like GTPase Arl3p to the Golgi requires N-terminal acetylation and the membrane protein Sys1p. Nat. Cell Biol. 2004, 6, 405–413. [Google Scholar] [CrossRef]
- Setty, S.R.; Strochlic, T.I.; Tong, A.H.; Boone, C.; Burd, C.G. Golgi targeting of ARF-like GTPase Arl3p requires its Nalpha-acetylation and the integral membrane protein Sys1p. Nat. Cell Biol. 2004, 6, 414–419. [Google Scholar] [CrossRef]
- Dikiy, I.; Eliezer, D. N-terminal acetylation stabilizes N-terminal helicity in lipid- and micelle-bound alpha-synuclein and increases its affinity for physiological membranes. J. Biol. Chem. 2014, 289, 3652–3665. [Google Scholar] [CrossRef]
- Forte, G.M.; Pool, M.R.; Stirling, C.J. N-terminal acetylation inhibits protein targeting to the endoplasmic reticulum. PLoS Biol. 2011, 9, e1001073. [Google Scholar] [CrossRef]
- Holmes, W.M.; Mannakee, B.K.; Gutenkunst, R.N.; Serio, T.R. Loss of amino-terminal acetylation suppresses a prion phenotype by modulating global protein folding. Nat. Commun. 2014, 5, 4383. [Google Scholar] [CrossRef]
- Shemorry, A.; Hwang, C.S.; Varshavsky, A. Control of protein quality and stoichiometries by N-terminal acetylation and the N-end rule pathway. Mol. Cell 2013, 50, 540–551. [Google Scholar] [CrossRef]
- Kats, I.; Khmelinskii, A.; Kschonsak, M.; Huber, F.; Knieß, R.A.; Bartosik, A.; Knop, M. Mapping Degradation Signals and Pathways in a Eukaryotic N-terminome. Mol. Cell 2018, 70, 488–501. [Google Scholar] [CrossRef] [PubMed]
- Arnesen, T.; Anderson, D.; Baldersheim, C.; Lanotte, M.; Varhaug, J.E.; Lillehaug, J.R. Identification and characterization of the human ARD1-NATH protein acetyltransferase complex. Biochem. J. 2005, 386, 433–443. [Google Scholar] [CrossRef]
- Mullen, J.R.; Kayne, P.S.; Moerschell, R.P.; Tsunasawa, S.; Gribskov, M.; Colavito-Shepanski, M.; Grunstein, M.; Sherman, F.; Sternglanz, R. Identification and characterization of genes and mutants for an N-terminal acetyltransferase from yeast. EMBO J. 1989, 8, 2067–2075. [Google Scholar] [CrossRef] [PubMed]
- Park, E.C.; Szostak, J.W. ARD1 and NAT1 proteins form a complex that has N-terminal acetyltransferase activity. EMBO J. 1992, 11, 2087–2093. [Google Scholar] [CrossRef] [PubMed]
- Liszczak, G.; Goldberg, J.M.; Foyn, H.; Petersson, E.J.; Arnesen, T.; Marmorstein, R. Molecular basis for N-terminal acetylation by the heterodimeric NatA complex. Nat. Struct. Mol. Biol. 2013, 20, 1098–1105. [Google Scholar] [CrossRef] [PubMed]
- Gautschi, M.; Just, S.; Mun, A.; Ross, S.; Rucknagel, P.; Dubaquie, Y.; Ehrenhofer-Murray, A.; Rospert, S. The Yeast Nα-Acetyltransferase NatA Is Quantitatively Anchored to the Ribosome and Interacts with Nascent Polypeptides. Mol. Cell Biol. 2003, 23, 7403–7414. [Google Scholar] [CrossRef]
- Magin, R.S.; Deng, S.; Zhang, H.; Cooperman, B.; Marmorstein, R. Probing the interaction between NatA and the ribosome for co-translational protein acetylation. PLoS ONE 2017, 12, e0186278. [Google Scholar] [CrossRef]
- Van Damme, P.; Evjenth, R.; Foyn, H.; Demeyer, K.; De Bock, P.-J.; Lillehaug, J.R.; Vandekerckhove, J.; Arnesen, T.; Gevaert, K. Proteome-derived peptide libraries allow detailed analysis of the substrate specificities of Nα-acetyltransferases and point to hNaa10p as the post-translational actin Nα-acetyltransferase. Mol. Cell. Proteom. 2011, 10, M110.004580. [Google Scholar] [CrossRef]
- Arnesen, T.; Starheim, K.K.; Van Damme, P.; Evjenth, R.; Dinh, H.; Betts, M.J.; Ryningen, A.; Vandekerckhove, J.; Gevaert, K.; Anderson, D. The Chaperone-Like Protein HYPK Acts Together with NatA in Cotranslational N-Terminal Acetylation and Prevention of Huntingtin Aggregation. Mol. Cell. Biol. 2010, 30, 1898–1909. [Google Scholar] [CrossRef]
- Weyer, F.A.; Gumiero, A.; Lapouge, K.; Bange, G.; Kopp, J.; Sinning, I. Structural basis of HypK regulating N-terminal acetylation by the NatA complex. Nat. Commun. 2017, 8, 15726. [Google Scholar] [CrossRef]
- Gottlieb, L.; Marmorstein, R. Structure of Human NatA and Its Regulation by the Huntingtin Interacting Protein HYPK. Structure 2018, 26, 925–935. [Google Scholar] [CrossRef]
- Lim, J.-H.; Park, J.-W.; Chun, Y.-S. Human arrest defective 1 acetylates and activates β-catenin, promoting lung cancer cell proliferation. Cancer Res. 2006, 66, 10677–10682. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.-H.; Chun, Y.-S.; Park, J.-W. Hypoxia-Inducible Factor-1α Obstructs a Wnt Signaling Pathway by Inhibiting the hARD1-Mediated Activation of β-Catenin. Cancer Res. 2008, 68, 5177–5184. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.H.; Park, J.-H.; Lee, E.J.; Vo, T.T.L.; Choi, H.; Kim, J.Y.; Jang, J.K.; Wee, H.-J.; Lee, H.S.; Jang, S.H.; et al. ARD1-mediated Hsp70 acetylation balances stress-induced protein refolding and degradation. Nat. Commun. 2016, 7, 12882. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Seo, J.H.; Park, J.-H.; Vo, T.T.L.; An, S.; Bae, S.-J.; Le, H.; Lee, H.S.; Wee, H.-J.; Lee, D.; et al. SAMHD1 acetylation enhances its deoxynucleotide triphosphohydrolase activity and promotes cancer cell proliferation. Oncotarget 2017, 8, 68517–68529. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Min, L.; Han, Y.; Meng, L.; Liu, C.; Xie, Y.; Dong, B.; Wang, L.; Jiang, B.; Xu, H.; et al. Inhibition of STAT5a by Naa10p contributes to decreased breast cancer metastasis. Carcinogenesis 2014, 35, 2244–2253. [Google Scholar] [CrossRef]
- Lee, C.-F.; Ou, D.S.C.; Lee, S.-B.; Chang, L.-H.; Lin, R.-K.; Li, Y.-S.; Upadhyay, A.K.; Cheng, X.; Wang, Y.-C.; Hsu, H.-S.; et al. HNaa10p contributes to tumorigenesis by facilitating DNMT1-mediated tumor suppressor gene silencing. J. Clin. Investig. 2010, 120, 2920–2930. [Google Scholar] [CrossRef]
- Lee, C.-C.; Peng, S.-H.; Shen, L.; Lee, C.-F.; Du, T.-H.; Kang, M.-L.; Xu, G.-L.; Upadhyay, A.K.; Cheng, X.; Yan, Y.-T.; et al. The Role of N-α-acetyltransferase 10 Protein in DNA Methylation and Genomic Imprinting. Mol. Cell 2017, 68, 89–103. [Google Scholar] [CrossRef]
- Lee, M.-N.; Kweon, H.Y.; Oh, G.T. N-α-acetyltransferase 10 (NAA10) in development: The role of NAA10. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef]
- Kalvik, T.V.; Arnesen, T. Protein N-terminal acetyltransferases in cancer. Oncogene 2013, 32, 269–276. [Google Scholar] [CrossRef]
- Wu, Y.; Lyon, G.J. NAA10-related syndrome. Exp. Mol. Med. 2018, 50, 85. [Google Scholar] [CrossRef] [PubMed]
- Rope, A.F.; Wang, K.; Evjenth, R.; Xing, J.; Johnston, J.J.; Swensen, J.J.; Johnson, W.E.; Moore, B.; Huff, C.D.; Bird, L.M.; et al. Using VAAST to Identify an X-Linked Disorder Resulting in Lethality in Male Infants Due to N-Terminal Acetyltransferase Deficiency. Am. J. Hum. Genet. 2011, 89, 28–43. [Google Scholar] [CrossRef] [PubMed]
- Myklebust, L.M.; Van Damme, P.; Støve, S.I.; Dörfel, M.J.; Abboud, A.; Kalvik, T.V.; Grauffel, C.; Jonckheere, V.; Wu, Y.; Swensen, J.; et al. Biochemical and cellular analysis of Ogden syndrome reveals downstream Nt-acetylation defects. Hum. Mol. Genet. 2015, 24, 1956–1976. [Google Scholar] [CrossRef] [PubMed]
- Esmailpour, T.; Riazifar, H.; Liu, L.; Donkervoort, S.; Huang, V.H.; Madaan, S.; Shoucri, B.M.; Busch, A.; Wu, J.; Towbin, A.; et al. A splice donor mutation in results in the dysregulation of the retinoic acid signalling pathway and causes Lenz microphthalmia syndrome. J. Med. Genet. 2014, 51, 185–196. [Google Scholar] [CrossRef]
- Saunier, C.; Støve, S.I.; Popp, B.; Gérard, B.; Blenski, M.; Ahmew, N.; Bie, C.; Goldenberg, P.; Isidor, B.; Keren, B.; et al. Expanding the Phenotype Associated with NAA10-Related N-Terminal Acetylation Deficiency. Hum. Mutat. 2016, 37, 755–764. [Google Scholar] [CrossRef]
- Casey, J.P.; Støve, S.I.; McGorrian, C.; Galvin, J.; Blenski, M.; Dunne, A.; Ennis, S.; Brett, F.; King, M.D.; Arnesen, T.; et al. NAA10 mutation causing a novel intellectual disability syndrome with Long QT due to N-terminal acetyltransferase impairment. Sci. Rep. 2015, 5, 16022. [Google Scholar] [CrossRef]
- McTiernan, N.; Støve, S.I.; Aukrust, I.; Mårli, M.T.; Myklebust, L.M.; Houge, G.; Arnesen, T. NAA10 dysfunction with normal NatA-complex activity in a girl with non-syndromic ID and a de novo NAA10 p.(V111G) variant—A case report. BMC Med. Genet. 2018, 19, 47. [Google Scholar] [CrossRef]
- Støve, S.I.; Blenski, M.; Stray-Pedersen, A.; Wierenga, K.J.; Jhangiani, S.N.; Akdemir, Z.C.; Crawford, D.; McTiernan, N.; Myklebust, L.M.; Purcarin, G.; et al. A novel NAA10 variant with impaired acetyltransferase activity causes developmental delay, intellectual disability, and hypertrophic cardiomyopathy. Eur. J. Hum. Genet. 2018, 26, 1294–1305. [Google Scholar] [CrossRef]
- Bader, I.; McTiernan, N.; Darbakk, C.; Boltshauser, E.; Ree, R.; Ebner, S.; Mayr, J.A.; Arnesen, T. Severe syndromic ID and skewed X-inactivation in a girl with NAA10 dysfunction and a novel heterozygous de novo NAA10 p.(His16Pro) variant—A case report. BMC Med. Genet. 2020, 21, 153. [Google Scholar] [CrossRef]
- Cheng, H.; Gottlieb, L.; Marchi, E.; Kleyner, R.; Bhardwaj, P.; Rope, A.F.; Rosenheck, S.; Moutton, S.; Philippe, C.; Eyaid, W.; et al. Phenotypic and biochemical analysis of an international cohort of individuals with variants in NAA10 and NAA15. Hum. Mol. Genet. 2019, 28, 2900–2919. [Google Scholar] [CrossRef]
- Popp, B.; Støve, S.I.; Endele, S.; Myklebust, L.M.; Hoyer, J.; Sticht, H.; Azzarello-Burri, S.; Rauch, A.; Arnesen, T.; Reis, A. De novo missense mutations in the NAA10 gene cause severe non-syndromic developmental delay in males and females. Eur. J. Hum. Genet. 2015, 23, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Ree, R.; Geithus, A.S.; Tørring, P.M.; Sørensen, K.P.; Damkjær, M.; Lynch, S.A.; Arnesen, T. A novel NAA10 p.(R83H) variant with impaired acetyltransferase activity identified in two boys with ID and microcephaly. BMC Med. Genet. 2019, 20, 101. [Google Scholar] [CrossRef] [PubMed]
- McTiernan, N.; Gill, H.; Prada, C.E.; Pachajoa, H.; Lores, J.; Arnesen, T. NAA10 p.(N101K) disrupts N-terminal acetyltransferase complex NatA and is associated with developmental delay and hemihypertrophy. Eur. J. Hum. Genet. 2020, 1–9. [Google Scholar] [CrossRef]
- Rodrigues, C.H.; Pires, D.E.; Ascher, D.B. DynaMut: Predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res. 2018, 46, W350–W355. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef]
- Drazic, A.; Arnesen, T. [14C]-Acetyl-Coenzyme A-Based In Vitro N-Terminal Acetylation Assay. In Protein Terminal Profiling: Methods and Protocols; Schilling, O., Ed.; Springer: New York, NY, USA, 2017; pp. 1–8. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McTiernan, N.; Darbakk, C.; Ree, R.; Arnesen, T. NAA10 p.(D10G) and NAA10 p.(L11R) Variants Hamper Formation of the NatA N-Terminal Acetyltransferase Complex. Int. J. Mol. Sci. 2020, 21, 8973. https://doi.org/10.3390/ijms21238973
McTiernan N, Darbakk C, Ree R, Arnesen T. NAA10 p.(D10G) and NAA10 p.(L11R) Variants Hamper Formation of the NatA N-Terminal Acetyltransferase Complex. International Journal of Molecular Sciences. 2020; 21(23):8973. https://doi.org/10.3390/ijms21238973
Chicago/Turabian StyleMcTiernan, Nina, Christine Darbakk, Rasmus Ree, and Thomas Arnesen. 2020. "NAA10 p.(D10G) and NAA10 p.(L11R) Variants Hamper Formation of the NatA N-Terminal Acetyltransferase Complex" International Journal of Molecular Sciences 21, no. 23: 8973. https://doi.org/10.3390/ijms21238973
APA StyleMcTiernan, N., Darbakk, C., Ree, R., & Arnesen, T. (2020). NAA10 p.(D10G) and NAA10 p.(L11R) Variants Hamper Formation of the NatA N-Terminal Acetyltransferase Complex. International Journal of Molecular Sciences, 21(23), 8973. https://doi.org/10.3390/ijms21238973