Endothelial Damage in Acute Respiratory Distress Syndrome



Abstract
1. Introduction
2. Pathogenesis
3. Pulmonary Endothelial Functions
3.1. Endothelium Barrier and Transport Functions
3.2. Vascular Tone
3.2.1. Endothelin-1
3.2.2. Renin–Angiotensin–Aldosterone System (RAAS)
3.2.3. Pulmonary Endothelial ACE Activity as a Measure of Endothelial Function
3.2.4. Endothelial Nitric Oxide Synthase (eNOS)
3.2.5. Prostacyclin
3.3. Host Defence
3.3.1. Interleukins
IL-6
IL-8
IL-10
3.3.2. Leukocytes and Pulmonary Endothelium
The Selectin Family
Soluble Intercellular Adhesion Molecule-1 (sICAM-1)
3.4. Haemostasis and Coagulation
3.4.1. von Willebrand Factor (vWf)
3.4.2. Coagulation
3.4.3. Fibrinolysis
3.4.4. Platelet–EC Interaction
3.5. Angiogenesis
3.5.1. VEGF
3.5.2. Angiopoietin-2 (Ang-2)
3.6. COVID-19 and ARDS
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ARDS | acute respiratory distress syndrome |
| COVID-19 | coronavirus disease 19 |
| ALI | acute lung injury |
| VILI | ventilator-induced lung injury |
| LPS | lipopolysaccharide |
| BLM | bleomycin |
| EC | endothelial cell |
| IEJ | interendothelial junction |
| TJ | tight junction |
| AJ | adherens junction |
| GJ | gap junction |
| JAM | junctional adhesion molecule |
| VE-cadherin | vascular endothelial cadherin |
| LMVEC | lung microvascular endothelial cell |
| EG | endothelial glycocalyx |
| ET-1 | endothelin-1 |
| NO | nitric oxide |
| Ang-1 | angiopoietin-1 |
| RAAS | renin–angiotensin–aldosterone system |
| ACE | angiotensin converting enzyme |
| BALF | bronchoalveolar lavage fluid |
| EMP | endothelial microparticle |
| SARS | severe acute respiratory syndrome |
| SARS-CoV-2 | severe acute respiratory syndrome coronavirus 2 |
| rACE2 | recombinant ACE2 |
| PCEB | pulmonary capillary endothelium-bound |
| eNOS | endothelial nitric oxide synthase |
| iNOS | inducible nitric oxide synthase |
| iEPO | inhaled prostacyclin |
| IL | interleukin |
| ICU | intensive care unit |
| PMN | polymorphonuclear cell |
| ROS | reactive oxygen species |
| NET | neutrophil extracellular trap |
| sE-selectin | soluble E-selectin |
| sP-selectin | soluble P-selectin |
| sICAM-1 | soluble intercellular adhesion molecule-1 |
| PECAM-1 | platelet-endothelial cell adhesion molecule-1 |
| vWF | von Willebrand factor |
| TF | tissue factor |
| PC | protein C |
| APC | activated protein C |
| TM | thrombomodulin |
| EPCR | endothelial protein C receptor |
| VAP | ventilator-associated pneumonia |
| PAI-1 | plasminogen activator inhibitor 1 |
| PMEC | pulmonary microvascular endothelial cell |
| rhTM | recombinant human TM |
| rTM | recombinant TM |
| VEGF | vascular endothelial growth factor |
| VEGFR1 | VEGF receptor 1 |
| VEGFR2 | VEGF receptor 2 |
| FE | fat embolism |
| AKI | acute kidney injury |
| COVID-19 | coronavirus disease-19 |
| MicroCLOTS | microvascular COVID-19 lung vessels obstructive thromboinflammatory syndrome |
References
- Force, A.D.T.; Ranieri, V.M.; Rubenfeld, G.D.; Thompson, B.T.; Ferguson, N.D.; Caldwell, E.; Fan, E.; Camporota, L.; Slutsky, A.S. Acute Respiratory Distress Syndrome. JAMA 2012, 307, 2526–2533. [Google Scholar] [CrossRef]
- Martin, T.R. Lung Cytokines and ARDS. Chest 1999, 116, 2S–8S. [Google Scholar] [CrossRef] [PubMed]
- Bernard, G.R.; Artigas, A.; Brigham, K.L.; Carlet, J.; Falke, K.; Hudson, L.; Lamy, M.; Legall, J.R.; Morris, A.; Spragg, R. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am. J. Respir. Crit. Care Med. 1994, 149 Pt 1, 818–824. [Google Scholar] [CrossRef] [PubMed]
- Sweatt, A.J.; Levitt, J.E. Evolving Epidemiology and Definitions of the Acute Respiratory Distress Syndrome and Early Acute Lung Injury. Clin. Chest Med. 2014, 35, 609–624. [Google Scholar] [CrossRef] [PubMed]
- Maniatis, N.A.; Kotanidou, A.; Catravas, J.D.; Orfanos, S.E. Endothelial pathomechanisms in acute lung injury. Vasc. Pharmacol. 2008, 49, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Maniatis, N.A.; Orfanos, S.E. The endothelium in acute lung injury/acute respiratory distress syndrome. Curr. Opin. Crit. Care 2008, 14, 22–30. [Google Scholar] [CrossRef]
- Orfanos, S.E.; Mavrommati, I.; Korovesi, I.; Roussos, C. Pulmonary endothelium in acute lung injury: From basic science to the critically ill. Intensive Care Med. 2004, 30, 1702–1714. [Google Scholar] [CrossRef]
- Mehta, D.; Malik, A.B. Signaling Mechanisms Regulating Endothelial Permeability. Physiol. Rev. 2006, 86, 279–367. [Google Scholar] [CrossRef]
- Galley, H.F.; Webster, N.R. Physiology of the endothelium. Br. J. Anaesth. 2004, 93, 105–113. [Google Scholar] [CrossRef]
- Matute-Bello, G.; Frevert, C.W.; Martin, T.R. Animal models of acute lung injury. Am. J. Physiol. Cell. Mol. Physiol. 2008, 295, L379–L399. [Google Scholar] [CrossRef]
- Vandenbroucke, E.; Mehta, D.; Minshall, R.; Malik, A.B. Regulation of endothelial junctional permeability. Ann. N. Y. Acad. Sci. 2008, 1123, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Komarova, Y.; Malik, A.B. Regulation of Endothelial Permeability via Paracellular and Transcellular Transport Pathways. Annu. Rev. Physiol. 2010, 72, 463–493. [Google Scholar] [CrossRef] [PubMed]
- Sukriti, S.; Tauseef, M.; Yazbeck, P.; Mehta, D. Mechanisms Regulating Endothelial Permeability. Pulm. Circ. 2014, 4, 535–551. [Google Scholar] [CrossRef] [PubMed]
- Matthay, M.A.; Zemans, R.L.; Zimmerman, G.A.; Arabi, Y.M.; Beitler, J.R.; Mercat, A.; Herridge, M.; Randolph, A.G.; Calfee, C.S. Acute respiratory distress syndrome. Nat. Rev. Dis. Primers 2019, 5, 18. [Google Scholar] [CrossRef]
- Rippe, B.; Rosengren, B.-I.; Carlsson, O.; Venturoli, D. Transendothelial Transport: The Vesicle Controversy. J. Vasc. Res. 2002, 39, 375–390. [Google Scholar] [CrossRef]
- Tsushima, K.; King, L.S.; Aggarwal, N.R.; De Gorordo, A.; D’Alessio, F.R.; Kubo, K. Acute Lung Injury Review. Intern. Med. 2009, 48, 621–630. [Google Scholar] [CrossRef]
- Liu, Y.; Mu, S.; Li, X.; Liang, Y.; Wang, L.; Ma, X. Unfractionated Heparin Alleviates Sepsis-Induced Acute Lung Injury by Protecting Tight Junctions. J. Surg. Res. 2019, 238, 175–185. [Google Scholar] [CrossRef]
- Benatti, M.N.; Fabro, A.T.; Miranda, C.H. Endothelial glycocalyx shedding in the acute respiratory distress syndrome after flu syndrome. J. Intensive Care 2020, 8, 1–10. [Google Scholar] [CrossRef]
- Zhang, D.; Qi, B.-Y.; Zhu, W.W.; Huang, X.; Wang, X. Crocin alleviates lipopolysaccharide-induced acute respiratory distress syndrome by protecting against glycocalyx damage and suppressing inflammatory signaling pathways. Inflamm. Res. 2020, 69, 267–278. [Google Scholar] [CrossRef]
- Epstein, F.H.; Vane, J.R.; Änggård, E.E.; Botting, R.M. Regulatory Functions of the Vascular Endothelium. N. Engl. J. Med. 1990, 323, 27–36. [Google Scholar] [CrossRef]
- Moloney, E.D.; Evans, T.W. Pathophysiology and pharmacological treatment of pulmonary hypertension in acute respiratory distress syndrome. Eur. Respir. J. 2003, 21, 720–727. [Google Scholar] [CrossRef] [PubMed]
- Druml, W.; Steltzer, H.; Waldhäusl, W.; Lenz, K.; Hammerle, A.; Vierhapper, H.; Gasic, S.; Wagner, O.F. Endothelin-1 in Adult Respiratory Distress Syndrome. Am. Rev. Respir. Dis. 1993, 148, 1169–1173. [Google Scholar] [CrossRef] [PubMed]
- Sanai, L.; Haynes, W.G.; MacKenzie, A.; Grant, I.S.; Webb, D.J. Endothelin production in sepsis and the adult respiratory distress syndrome. Intensive Care Med. 1996, 22, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Langleben, D.; DeMarchie, M.; Laporta, D.; Spanier, A.H.; Schlesinger, R.D.; Stewart, D.J. Endothelin-1 in acute lung injury and the adult respiratory distress syndrome. Am. Rev. Respir. Dis. 1993, 148 Pt 1, 1646–1650. [Google Scholar] [CrossRef]
- Mitaka, C.; Hirata, Y.; Nagura, T.; Tsunoda, Y.; Amaha, K. Circulating Endothelin-1 Concentrations in Acute Respiratory Failure. Chest 1993, 104, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Albertine, K.H.; Wang, Z.M.; Michael, J.R. Expression of endothelial nitric oxide synthase, inducible nitric oxide synthase, and endothelin-1 in lungs of subjects who died with ARDS. Chest 1999, 116 (Suppl. 1), 101S–102S. [Google Scholar] [CrossRef] [PubMed]
- Nakano, Y.; Tasaka, S.; Saito, F.; Yamada, W.; Shiraishi, Y.; Ogawa, Y.; Koh, H.; Hasegawa, N.; Fujishima, S.; Hashimoto, S.; et al. Endothelin-1 level in epithelial lining fluid of patients with acute respiratory distress syndrome. Respirology 2007, 12, 740–743. [Google Scholar] [CrossRef]
- Pritze, S.; Peskar, B.A.; Simmet, T. Release of eicosanoids and endothelin in an experimental model of adult respiratory distress syndrome. Agents Actions Suppl. 1992, 37, 41–46. [Google Scholar]
- Lai, T.-S.; Cai, S.-X.; Guo, Z.-H. Serum and lung endothelin-1 increased in a canine model of ventilator-induced lung injury. Chin. Med. J. 2010, 123, 1021–1027. [Google Scholar]
- Simmet, T.; Pritze, S.; Thelen, K.I.; Peskar, B.A. Release of endothelin in the oleic acid-induced respiratory distress syndrome in rats. Eur. J. Pharmacol. 1992, 211, 319–322. [Google Scholar] [CrossRef]
- McCarter, S.D.; Lai, P.F.H.; Suen, R.S.; Stewart, D.J. Regulation of endothelin-1 by angiopoietin-1: Implications for inflammation. Exp. Biol. Med. 2006, 231, 985–991. [Google Scholar]
- Bhavsar, T.M.; Cerreta, J.M.; Liu, M.; Reznik, S.E.; Cantor, J.O. Phosphoramidon, an endothelin-converting enzyme inhibitor, attenuates lipopolysaccharide-induced acute lung injury. Exp. Lung Res. 2008, 34, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Kuklin, V.; Kirov, M.; Sovershaev, M.; Andreasen, T.; Ingebretsen, O.C.; Ytrehus, K.; Bjertnaes, L.J. Tezosentan-induced attenuation of lung injury in endotoxemic sheep is associated with reduced activation of protein kinase C. Crit. Care 2005, 9, R211–R217. [Google Scholar] [CrossRef] [PubMed]
- Atalay, F.; Yurdakan, G.; Yilmaz-Sipahi, E. Effect of the endothelin receptor antagonist tezosentan on alpha-naphthylthiourea-induced lung injury in rats. Kaohsiung J. Med. Sci. 2012, 28, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Fujii, Y.; Magder, S.; Cernacek, P.; Goldberg, P.; Guo, Y.; Hussain, S.N.A. Endothelin Receptor Blockade Attenuates Lipopolysaccharide-induced Pulmonary Nitric Oxide Production. Am. J. Respir. Crit. Care Med. 2000, 161 Pt 1, 982–989. [Google Scholar] [CrossRef]
- Manitsopoulos, N.; Nikitopoulou, I.; Maniatis, N.A.; Magkou, C.; Kotanidou, A.; Orfanos, S.E. Highly Selective Endothelin-1 Receptor A Inhibition Prevents Bleomycin-Induced Pulmonary Inflammation and Fibrosis in Mice. Respiration 2018, 95, 122–136. [Google Scholar] [CrossRef] [PubMed]
- Aird, W.C. Phenotypic heterogeneity of the endothelium: II. Representative vascular beds. Circ. Res. 2007, 100, 174–190. [Google Scholar] [CrossRef] [PubMed]
- Pagliaro, P.; Penna, C. ACE/ACE2 Ratio: A Key Also in 2019 Coronavirus Disease (Covid-19)? Front. Med. 2020, 7, 335. [Google Scholar] [CrossRef]
- Chambers, S.; Bhatia, M. ACE and ACE2 in Inflammation: A Tale of Two Enzymes. Inflamm. Allergy Drug Targets 2014, 13, 224–234. [Google Scholar] [CrossRef]
- Casey, L.; Krieger, B.; Kohler, J.; Rice, C.; Oparil, S.; Szidon, P. Decreased serum angiotensin converting enzyme in adult respiratory distress syndrome associated with sepsis. Crit. Care Med. 1981, 9, 651–654. [Google Scholar] [CrossRef]
- Fourrier, F.; Chopi, C.; Wallaert, B.; Mazurier, C.; Mangalahoyi, J.; Durocher, A. Compared Evolution of Plasma Fibronectin and Angiotensin-converting Enzyme Levels in Septic ARDS. Chest 1985, 87, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Reddy, R.; Asante, I.; Liu, S.; Parikh, P.; Liebler, J.; Borok, Z.; Rodgers, K.; Baydur, A.; Louie, S.G. Circulating angiotensin peptides levels in Acute Respiratory Distress Syndrome correlate with clinical outcomes: A pilot study. PLoS ONE 2019, 14, e0213096. [Google Scholar] [CrossRef] [PubMed]
- Idell, S.; Kueppers, F.; Lippmann, M.; Rosen, H.; Niederman, M.; Fein, A. Angiotensin Converting Enzyme in Bronchoalveolar Lavage in ARDS. Chest 1987, 91, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Kaparianos, A. Local Renin-Angiotensin II Systems, Angiotensin-Converting Enzyme and its Homologue ACE2: Their Potential Role in the Pathogenesis of Chronic Obstructive Pulmonary Diseases, Pulmonary Hypertension and Acute Respiratory Distress Syndrome. Curr. Med. Chem. 2011, 18, 3506–3515. [Google Scholar] [CrossRef] [PubMed]
- Takei, Y.; Yamada, M.; Saito, K.; Kameyama, Y.; Sugiura, H.; Makiguchi, T.; Fujino, N.; Koarai, A.; Toyama, H.; Saito, K.; et al. Increase in circulating ACE-positive endothelial microparticles during acute lung injury. Eur. Respir. J. 2019, 54, 1801188. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Wang, L.-M.; Shimakura, K.; Sanaka, M.; Koike, Y.; Mineshita, S. Angiotensin II-Induced Pulmonary Edema in a Rabbit Model. Jpn. J. Pharmacol. 1997, 73, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Yilin, Z.; Yandong, N.; Faguang, J. Role of angiotensin-converting enzyme (ACE) and ACE2 in a rat model of smoke inhalation induced acute respiratory distress syndrome. Burns 2015, 41, 1468–1477. [Google Scholar] [CrossRef] [PubMed]
- Hermanns, M.I.; Müller, A.M.; Tsokos, M.; Kirkpatrick, C.J. LPS-induced effects on angiotensin I-converting enzyme expression and shedding in human pulmonary microvascular endothelial cells. In Vitro Cell. Dev. Biol. Anim. 2013, 50, 287–295. [Google Scholar] [CrossRef]
- Rey-Parra, G.J.; Vadivel, A.; Coltan, L.; Hall, A.; Eaton, F.; Schuster, M.; Loibner, H.; Penninger, J.M.; Kassiri, Z.; Oudit, G.Y.; et al. Angiotensin converting enzyme 2 abrogates bleomycin-induced lung injury. J. Mol. Med. 2012, 90, 637–647. [Google Scholar] [CrossRef]
- Asperen, R.M.W.-V.; Lutter, R.; Specht, P.A.; Moll, G.N.; Van Woensel, J.B.; Van Der Loos, C.M.; Van Goor, H.; Kamilic, J.; Florquin, S.; Bos, A.P. Acute respiratory distress syndrome leads to reduced ratio of ACE/ACE2 activities and is prevented by angiotensin-(1-7) or an angiotensin II receptor antagonist. J. Pathol. 2011, 225, 618–627. [Google Scholar] [CrossRef]
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H.; et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nat. Cell Biol. 2005, 436, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zeng, Z.; Cao, Y.; Liu, Y.; Ping, F.; Liang, M.; Xue, Y.; Xi, C.; Zhou, M.; Jiang, W. Angiotensin-converting enzyme 2 prevents lipopolysaccharide-induced rat acute lung injury via suppressing the ERK1/2 and NF-kappaB signaling pathways. Sci. Rep. 2016, 6, 27911. [Google Scholar] [CrossRef] [PubMed]
- Harmer, D.; Gilbert, M.; Borman, R.; Clark, K.L. Quantitative mRNA expression profiling of ACE 2, a novel homologue of angiotensin converting enzyme. FEBS Lett. 2002, 532, 107–110. [Google Scholar] [CrossRef]
- Li, M.-Y.; Li, L.; Zhang, Y.; Wang, X. Expression of the SARS-CoV-2 cell receptor gene ACE2 in a wide variety of human tissues. Infect. Dis. Poverty 2020, 9, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Rigat, B.; Hubert, C.; Alhenc-Gelas, F.; Cambien, F.; Corvol, P.; Soubrier, F. An insertion/deletion polymorphism in the angiotensin I-converting enzyme gene accounting for half the variance of serum enzyme levels. J. Clin. Investig. 1990, 86, 1343–1346. [Google Scholar] [CrossRef]
- Marshall, R.P.; Webb, S.; Bellingan, G.J.; Montgomery, H.E.; Chaudhari, B.; McAnulty, R.J.; Humphries, S.E.; Hill, M.R.; Laurent, G.J. Angiotensin Converting Enzyme Insertion/Deletion Polymorphism Is Associated with Susceptibility and Outcome in Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2002, 166, 646–650. [Google Scholar] [CrossRef]
- Adamzik, M.; Frey, M.U.H.; Sixt, S.; Knemeyer, L.; Beiderlinden, M.; Peters, J.; Siffert, W. ACE I/D but not AGT (-6)A/G polymorphism is a risk factor for mortality in ARDS. Eur. Respir. J. 2007, 29, 482–488. [Google Scholar] [CrossRef]
- Matsuda, A.; Kishi, T.; Jacob, A.; Aziz, M.; Wang, P. Association between insertion/deletion polymorphism in angiotensin-converting enzyme gene and acute lung injury/acute respiratory distress syndrome: A meta-analysis. BMC Med. Genet. 2012, 13, 76. [Google Scholar] [CrossRef]
- Jerng, J.-S.; Yu, C.-J.; Wang, H.-C.; Chen, K.-Y.; Cheng, S.-L.; Yang, P.-C. Polymorphism of the angiotensin-converting enzyme gene affects the outcome of acute respiratory distress syndrome. Crit. Care Med. 2006, 34, 1001–1006. [Google Scholar] [CrossRef]
- Cruces, P.; Diaz, F.; Puga, A.; Erranz, B.; Donoso, A.; Carvajal, C.; Wilhelm, J.; Repetto, G.M. Angiotensin-converting enzyme insertion/deletion polymorphism is associated with severe hypoxemia in pediatric ARDS. Intensive Care Med. 2012, 38, 113–119. [Google Scholar] [CrossRef]
- Villar, J.; Flores, C.; Pérez-Méndez, L.; Maca-Meyer, N.; Espinosa, E.; Blanco, J.; Sangüesa, R.; Muriel, A.; Tejera, P.; Muros, M.; et al. Angiotensin-converting enzyme insertion/deletion polymorphism is not associated with susceptibility and outcome in sepsis and acute respiratory distress syndrome. Intensive Care Med. 2008, 34, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Chai, X.-Q.; Magnussen, C.G.; Zosky, G.R.; Shu, S.-H.; Wei, X.; Hu, S.-S. Renin-angiotensin-system, a potential pharmacological candidate, in acute respiratory distress syndrome during mechanical ventilation. Pulm. Pharmacol. Ther. 2019, 58, 101833. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Baker, A. Recombinant human ACE2: Acing out angiotensin II in ARDS therapy. Crit. Care 2017, 21, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus–induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Yan, Y.; Shu, Y.; Gao, R.; Sun, Y.; Li, X.; Ju, X.; Liang, Z.; Liu, Q.; Zhao, Y.; et al. Angiotensin-converting enzyme 2 protects from lethal avian influenza A H5N1 infections. Nat. Commun. 2014, 5, 3594. [Google Scholar] [CrossRef]
- Orfanos, S.; Ehrhart, I.; Barman, S.; Hofman, W.; Catravas, J. Endothelial Ectoenzyme Assays Estimate Perfused Capillary Surface Area in the Dog Lung. Microvasc. Res. 1997, 54, 145–155. [Google Scholar] [CrossRef]
- Orfanos, S.E.; Langleben, D.; Khoury, J.; Schlesinger, R.D.; Dragatakis, L.; Roussos, C.; Ryan, J.W.; Catravas, J.D. Pulmonary capillary endothelium-bound angiotensin-converting enzyme activity in humans. Circulation 1999, 99, 1593–1599. [Google Scholar] [CrossRef]
- Kaziani, K.; Vassiliou, A.; Kotanidou, A.; Athanasiou, C.; Korovesi, I.; Glynos, K.; Orfanos, S.E. Activated Protein C has No Effect on Pulmonary Capillary Endothelial Function in Septic Patients with Acute Respiratory Distress Syndrome: Association of Endothelial Dysfunction with Mortality. Infect. Dis. Ther. 2018, 7 (Suppl. 1), 15–25. [Google Scholar] [CrossRef]
- Langleben, D.; Orfanos, S.E.; Giovinazzo, M.; Schlesinger, R.D.; Hirsch, A.M.; Blenkhorn, F.; Lesenko, L.; Armaganidis, A.; Catravas, J.D. Acute Vasodilator Responsiveness and Microvascular Recruitment in Idiopathic Pulmonary Arterial Hypertension. Ann. Intern. Med. 2015, 162, 154. [Google Scholar] [CrossRef]
- Glynos, C.; Athanasiou, C.; Kotanidou, A.; Korovesi, I.; Kaziani, K.; Livaditi, O.; Dimopoulou, I.; Maniatis, N.A.; Tsangaris, I.; Roussos, C.; et al. Preclinical Pulmonary Capillary Endothelial Dysfunction is Present in Brain Dead Subjects. Pulm. Circ. 2013, 3, 419–425. [Google Scholar] [CrossRef]
- Orfanos, S.E.; Armaganidis, A.; Glynos, C.; Psevdi, E.; Kaltsas, P.; Sarafidou, P.; Catravas, J.D.; Dafni, U.G.; Langleben, D.; Roussos, C. Pulmonary Capillary Endothelium-Bound Angiotensin-Converting Enzyme Activity in Acute Lung Injury. Circulation 2000, 102, 2011–2018. [Google Scholar] [CrossRef] [PubMed]
- Orfanos, S.E.; Hirsch, A.M.; Giovinazzo, M.; Armaganidis, A.; Catravas, J.D.; Langleben, D. Pulmonary capillary endothelial metabolic function in chronic thromboembolic pulmonary hypertension. J. Thromb. Haemost. 2008, 6, 1275–1280. [Google Scholar] [CrossRef] [PubMed]
- Orfanos, S.E.; Psevdi, E.; Stratigis, N.; Langleben, D.; Catravas, J.D.; Kyriakidis, M.; Moutsopoulos, H.M.; Roussos, C.; Vlachoyiannopoulos, P.G. Pulmonary capillary endothelial dysfunction in early systemic sclerosis. Arthritis Rheum. 2001, 44, 902–911. [Google Scholar] [CrossRef]
- Langleben, D.; Orfanos, S.E.; Giovinazzo, M.; Hirsch, A.; Baron, M.; Armaganidis, A.; Catravas, J.D.; Senécal, J.-L. Pulmonary capillary endothelial metabolic dysfunction: Severity in pulmonary arterial hypertension related to connective tissue disease versus idiopathic pulmonary arterial hypertension. Arthritis Rheum. 2008, 58, 1156–1164. [Google Scholar] [CrossRef] [PubMed]
- Dudzinski, D.M.; Igarashi, J.; Greif, D.; Michel, T. The regulation and pharmacology of endothelial nitric oxide synthase. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 235–276. [Google Scholar] [CrossRef]
- Abdih, H.; Kelly, C.J.; Bouchier-Hayes, D.J.; Watson, R.W.; Redmond, H.; Burke, P.; Bouchier-Hayes, D.J.; William, R. Nitric Oxide (Endothelium-Derived Relaxing Factor) Attenuates Revascularization-Induced Lung Injury. J. Surg. Res. 1994, 57, 39–43. [Google Scholar] [CrossRef]
- Garrean, S.; Gao, X.; Brovkovych, V.; Shimizu, J.; Zhao, Y.-Y.; Vogel, S.M.; Malik, A.B. Caveolin-1 regulates NF-kappaB activation and lung inflammatory response to sepsis induced by lipopolysaccharide. J. Immunol. 2006, 177, 4853–4860. [Google Scholar] [CrossRef]
- Kaminski, A.; Pohl, C.B.; Sponholz, C.; Ma, N.; Stamm, C.; Vollmar, B.; Steinhoff, G. Up-Regulation of Endothelial Nitric Oxide Synthase Inhibits Pulmonary Leukocyte Migration Following Lung Ischemia-Reperfusion in Mice. Am. J. Pathol. 2004, 164, 2241–2249. [Google Scholar] [CrossRef]
- Kaminski, A.; Kasch, C.; Zhang, L.; Kumar, S.; Sponholz, C.; Choi, Y.-H.; Ma, N.; Liebold, A.; Ladilov, Y.; Steinhoff, G.; et al. Endothelial nitric oxide synthase mediates protective effects of hypoxic preconditioning in lungs. Respir. Physiol. Neurobiol. 2007, 155, 280–285. [Google Scholar] [CrossRef]
- Takenaka, K.; Nishimura, Y.; Nishiuma, T.; Sakashita, A.; Yamashita, T.; Kobayashi, K.; Satouchi, M.; Ishida, T.; Kawashima, S.; Yokoyama, M. Ventilator-induced lung injury is reduced in transgenic mice that overexpress endothelial nitric oxide synthase. Am. J. Physiol. Cell. Mol. Physiol. 2006, 290, L1078–L1086. [Google Scholar] [CrossRef]
- Yamashita, T.; Kawashima, S.; Ohashi, Y.; Ozaki, M.; Ueyama, T.; Ishida, T.; Inoue, N.; Hirata, K.-I.; Akita, H.; Yokoyama, M. Resistance to endotoxin shock in transgenic mice overexpressing endothelial nitric oxide synthase. Circulation 2000, 101, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Kumar, S.; Kaminski, A.; Kasch, C.; Sponholz, C.; Stamm, C.; Ladilov, Y.; Steinhoff, G. Importance of endothelial nitric oxide synthase for the hypothermic protection of lungs against ischemia-reperfusion injury. J. Thorac. Cardiovasc. Surg. 2006, 131, 969–974. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gielis, J.F.; Quirynen, L.; Briedé, J.J.; Roelant, E.; Cos, P.; Van Schil, P.E. Pathogenetic role of endothelial nitric oxide synthase uncoupling during lung ischaemia–reperfusion injury. Eur. J. Cardio-Thoracic Surg. 2017, 52, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Drucker, N.A.; Jensen, A.R.; Winkel, J.P.T.; Ferkowicz, M.J.; Markel, T.A. Loss of endothelial nitric oxide synthase exacerbates intestinal and lung injury in experimental necrotizing enterocolitis. J. Pediatr. Surg. 2018, 53, 1208–1214. [Google Scholar] [CrossRef] [PubMed]
- Forstermann, U.; Munzel, T. Endothelial Nitric Oxide Synthase in Vascular Disease. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef] [PubMed]
- Gow, A.J.; Thom, S.R.; Ischiropoulos, H. Nitric oxide and peroxynitrite-mediated pulmonary cell death. Am. J. Physiol. Content 1998, 274, L112–L118. [Google Scholar] [CrossRef]
- Weinberger, B.; Djerad, A.; Monier, C.; Houzé, P.; Borron, S.W.; Lefauconnier, J.-M.; Baud, F.J. The Toxicology of Inhaled Nitric Oxide. Toxicol. Sci. 2001, 59, 5–16. [Google Scholar] [CrossRef]
- Kristof, A.S.; Goldberg, P.; Laubach, V.; Hussain, S.N.A. Role of Inducible Nitric Oxide Synthase in Endotoxin-induced Acute Lung Injury. Am. J. Respir. Crit. Care Med. 1998, 158, 1883–1889. [Google Scholar] [CrossRef]
- Gross, C.M.; Rafikov, R.; Kumar, S.; Aggarwal, S.; Iii, P.B.H.; Meadows, M.L.; Cherian-Shaw, M.; Kangath, A.; Sridhar, S.; Lucas, R.; et al. Endothelial Nitric Oxide Synthase Deficient Mice Are Protected from Lipopolysaccharide Induced Acute Lung Injury. PLoS ONE 2015, 10, e0119918. [Google Scholar] [CrossRef]
- Song, J.; Palmer, K.; Sun, B. Effects of inhaled nitric oxide and surfactant with extracorporeal life support in recovery phase of septic acute lung injury in piglets. Pulm. Pharmacol. Ther. 2010, 23, 78–87. [Google Scholar] [CrossRef]
- Hart, C.M. Nitric Oxide in Adult Lung Disease. Chest 1999, 115, 1407–1417. [Google Scholar] [CrossRef] [PubMed]
- Zayek, M.; Wild, L.; Roberts, J.D.; Morin, F.C. Effect of nitric oxide on the survival rate and incidence of lung injury in newborn lambs with persistent pulmonary hypertension. J. Pediatr. 1993, 123, 947–952. [Google Scholar] [CrossRef]
- Rossaint, R.; Falke, K.; Lopez, F.; Slama, K.; Pison, U.; Zapol, W.M. Inhaled Nitric Oxide for the Adult Respiratory Distress Syndrome. N. Engl. J. Med. 1993, 328, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Benzing, A.; Geiger, K. Inhaled nitric oxide lowers pulmonary capillary pressure and changes longitudinal distribution of pulmonary vascular resistance in patients with acute lung injury. Acta Anaesthesiol. Scand. 1994, 38, 640–645. [Google Scholar] [CrossRef] [PubMed]
- Michael, J.R.; Barton, R.G.; Saffle, J.R.; Mone, M.; Markewitz, B.A.; Hillier, K.; Elstad, M.R.; Campbell, E.J.; Troyer, B.E.; Whatley, R.E.; et al. Inhaled Nitric Oxide Versus Conventional Therapy. Am. J. Respir. Crit. Care Med. 1998, 157 Pt 1, 1372–1380. [Google Scholar] [CrossRef]
- Troncy, E.; Collet, J.-P.; Shapiro, S.; Guimond, J.-G.; Blair, L.; Ducruet, T.; Francoeur, M.; Charbonneau, M.; Blaise, G. Inhaled Nitric Oxide in Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 1998, 157 Pt 1, 1483–1488. [Google Scholar] [CrossRef]
- Dellinger, R.P.; Zimmerman, J.L.; Taylor, R.W.; Straube, R.C.; Hauser, D.L.; Criner, G.J.; Davis, K.; Hyers, T.M.; Papadakos, P. Effects of inhaled nitric oxide in patients with acute respiratory distress syndrome. Crit. Care Med. 1998, 26, 15–23. [Google Scholar] [CrossRef]
- Lundin, S.; Mang, H.; Smithies, M.; Stenqvist, O.; Frostell, C. Inhalation of nitric oxide in acute lung injury: Results of a European multicentre study. Intensive Care Med. 1999, 25, 911–919. [Google Scholar] [CrossRef]
- Taylor, R.W.; Zimmerman, J.L.; Dellinger, R.P.; Straube, R.C.; Criner, G.J.; Davis, J.K.; Kelly, K.M.; Smith, T.C.; Small, R.J. Low-Dose Inhaled Nitric Oxide in Patients with Acute Lung InjuryA Randomized Controlled Trial. JAMA 2004, 291, 1603–1609. [Google Scholar] [CrossRef]
- Adhikari, N.K.J.; Burns, K.E.; Friedrich, J.O.; Granton, J.T.; Cook, D.J.; Meade, M.O. Effect of nitric oxide on oxygenation and mortality in acute lung injury: Systematic review and meta-analysis. BMJ 2007, 334, 779. [Google Scholar] [CrossRef]
- Afshari, A.; Brok, J.; Moller, A.M.; Wetterslev, J. Inhaled nitric oxide for acute respiratory distress syndrome (ARDS) and acute lung injury in children and adults. Cochrane Database Syst. Rev. 2010, 7, CD002787. [Google Scholar] [CrossRef]
- Adhikari, N.K.J.; Dellinger, R.P.; Lundin, S.; Payen, D.; Vallet, B.; Gerlach, H.; Park, K.J.; Mehta, S.; Slutsky, A.S.; Friedrich, J.O. Inhaled Nitric Oxide Does Not Reduce Mortality in Patients with Acute Respiratory Distress Syndrome Regardless of Severity. Crit. Care Med. 2014, 42, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Gebistorf, F.; Karam, O.; Wetterslev, J.; Afshari, A. Inhaled nitric oxide for acute respiratory distress syndrome (ARDS) in children and adults. Cochrane Database Syst. Rev. 2016, 2016, CD002787. [Google Scholar] [CrossRef] [PubMed]
- Ruan, S.-Y.; Huang, T.-M.; Wu, H.-Y.; Wu, H.-D.; Jann-Yuan, W.; Lai, M.-S. Inhaled nitric oxide therapy and risk of renal dysfunction: A systematic review and meta-analysis of randomized trials. Crit. Care 2015, 19, 137. [Google Scholar] [CrossRef]
- Ruan, S.-Y.; Wu, H.-Y.; Lin, H.-H.; Wu, H.-D.; Yu, C.-J.; Lai, M.-S. Inhaled nitric oxide and the risk of renal dysfunction in patients with acute respiratory distress syndrome: A propensity-matched cohort study. Crit. Care 2016, 20, 389. [Google Scholar] [CrossRef]
- Mitchell, J.A.; Ali, F.; Bailey, L.; Moreno, L.; Harrington, L.S. Role of nitric oxide and prostacyclin as vasoactive hormones released by the endothelium. Exp. Physiol. 2008, 93, 141–147. [Google Scholar] [CrossRef]
- Walmrath, D.; Schneider, T.; Pilch, J.; Grimminger, F.; Seeger, W. Aerosolised prostacyclin in adult respiratory distress syndrome. Lancet 1993, 342, 961–962. [Google Scholar] [CrossRef]
- Zwissler, B.; Kemming, G.; Habler, O.; Kleen, M.; Merkel, M.; Haller, M.; Briegel, J.; Welte, M.; Peter, K. Inhaled prostacyclin (PGI2) versus inhaled nitric oxide in adult respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 1996, 154 Pt 1, 1671–1677. [Google Scholar] [CrossRef]
- Kallet, R.H.; Burns, G.; Zhuo, H.; Ho, K.; Phillips, J.S.; Pangilinan, L.P.; Yip, V.; Gomez, A.; Lipnick, M.S. Severity of Hypoxemia and Other Factors That Influence the Response to Aerosolized Prostacyclin in ARDS. Respir. Care 2017, 62, 1014–1022. [Google Scholar] [CrossRef]
- Searcy, R.J.; Morales, J.R.; Ferreira, J.; Johnson, D.W. The role of inhaled prostacyclin in treating acute respiratory distress syndrome. Ther. Adv. Respir. Dis. 2015, 9, 302–312. [Google Scholar] [CrossRef]
- Afshari, A.; Bille, A.B.; Allingstrup, M. Aerosolized prostacyclins for acute respiratory distress syndrome (ARDS). Cochrane Database Syst. Rev. 2017, 7, CD007733. [Google Scholar] [CrossRef] [PubMed]
- Levi, M.; Cate, H.T.; Van Der Poll, T. Endothelium: Interface between coagulation and inflammation. Crit. Care Med. 2002, 30 (Suppl. 5), S220–S224. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Bussolino, F.; Introna, M. Cytokine regulation of endothelial cell function: From molecular level to the bedside. Immunol. Today 1997, 18, 231–240. [Google Scholar] [CrossRef]
- Muller, W.A. How Endothelial Cells Regulate Transmigration of Leukocytes in the Inflammatory Response. Am. J. Pathol. 2014, 184, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Levi, M.; Schultz, M. The inflammation-coagulation axis as an important intermediate pathway in acute lung injury. Crit. Care 2008, 12, 144. [Google Scholar] [CrossRef]
- Meduri, G.U.; Kohler, G.; Headley, S.; Tolley, E.; Stentz, F.; Postlethwaite, A. Inflammatory Cytokines in the BAL of Patients With ARDS. Chest 1995, 108, 1303–1314. [Google Scholar] [CrossRef]
- Park, W.Y.; Goodman, R.B.; Steinberg, K.P.; Ruzinski, J.T.; Radella, F.; Park, D.R.; Pugin, J.; Skerrett, S.J.; Hudson, L.D.; Martin, T.R. Cytokine Balance in the Lungs of Patients with Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2001, 164 Pt 1, 1896–1903. [Google Scholar] [CrossRef]
- Meduri, G.U.; Headley, S.; Kohler, G.; Stentz, F.; Tolley, E.; Umberger, R.; Leeper, K. Persistent elevation of inflammatory cytokines predicts a poor outcome in ARDS. Plasma IL-1 beta and IL-6 levels are consistent and efficient predictors of outcome over time. Chest 1995, 107, 1062–1073. [Google Scholar] [CrossRef]
- Cepkova, M.; Brady, S.; Sapru, A.; Matthay, M.A.; Church, G.D. Biological markers of lung injury before and after the institution of positive pressure ventilation in patients with acute lung injury. Crit. Care 2006, 10, R126. [Google Scholar] [CrossRef]
- Nakamura, T.; Sato, E.; Fujiwara, N.; Kawagoe, Y.; Maeda, S.; Yamagishi, S.-I. Increased levels of soluble receptor for advanced glycation end products (sRAGE) and high mobility group box 1 (HMGB1) are associated with death in patients with acute respiratory distress syndrome. Clin. Biochem. 2011, 44, 601–604. [Google Scholar] [CrossRef]
- Swaroopa, D.; Bhaskar, K.; Mahathi, T.; Katkam, S.; Raju, Y.S.; Chandra, N.; Kutala, V.K. Association of serum interleukin-6, interleukin-8, and Acute Physiology and Chronic Health Evaluation II score with clinical outcome in patients with acute respiratory distress syndrome. Indian J. Crit. Care Med. 2016, 20, 518–525. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Donnelly, T.J.; Meade, P.; Jagels, M.; Cryer, H.G.; Law, M.M.; Hugli, T.E.; Shoemaker, W.C.; Abraham, E. Cytokine, complement, and endotoxin profiles associated with the development of the adult respiratory distress syndrome after severe injury. Crit. Care Med. 1994, 22, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Meade, P.; Shoemaker, W.C.; Donnelly, T.J.; Abraham, E.; Jagels, M.A.; Cryer, H.G.; Hugli, T.E.; Bishop, M.H.; Wo, C.C.J. Temporal patterns of hemodynamics, oxygen transport, cytokine activity, and complement activity in the development of adult respiratory distress syndrome after severe injury. J. Trauma Inj. Infect. Crit. Care 1994, 36, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Stüber, F.; Wrigge, H.; Schroeder, S.; Wetegrove, S.; Zinserling, J.; Hoeft, A.; Putensen, C. Kinetic and reversibility of mechanical ventilation-associated pulmonary and systemic inflammatory response in patients with acute lung injury. Intensive Care Med. 2002, 28, 834–841. [Google Scholar] [CrossRef] [PubMed]
- Ranieri, V.M.; Suter, P.M.; Tortorella, C.; De Tullio, R.; Dayer, J.M.; Brienza, A.; Bruno, F.; Slutsky, A.S. Effect of Mechanical Ventilation on Inflammatory Mediators in Patients with Acute Respiratory Distress Syndrome. JAMA 1999, 282, 54–61. [Google Scholar] [CrossRef]
- Voiriot, G.; Razazi, K.; Amsellem, V.; Van Nhieu, J.T.; Abid, S.; Adnot, S.; Dessap, A.M.; Maitre, B. Interleukin-6 displays lung anti-inflammatory properties and exerts protective hemodynamic effects in a double-hit murine acute lung injury. Respir. Res. 2017, 18, 1–14. [Google Scholar] [CrossRef]
- Qin, M.; Qiu, Z. Changes in TNF-α, IL-6, IL-10 and VEGF in rats with ARDS and the effects of dexamethasone. Exp. Ther. Med. 2019, 17, 383–387. [Google Scholar] [CrossRef]
- Donnelly, S.C.; Haslett, C.; Strieter, R.M.; Kunkel, S.L.; Walz, A.; Robertson, C.R.; Carter, D.C.; Pollok, A.J.; Grant, I.S. Interleukin-8 and development of adult respiratory distress syndrome in at-risk patient groups. Lancet 1993, 341, 643–647. [Google Scholar] [CrossRef]
- Calfee, C.S.; Ware, L.B.; Glidden, D.V.; Eisner, M.D.; Parsons, P.E.; Thompson, B.T.; Matthay, M.A. Use of risk reclassification with multiple biomarkers improves mortality prediction in acute lung injury. Crit. Care Med. 2011, 39, 711–717. [Google Scholar] [CrossRef]
- McClintock, D.; Zhuo, H.; Wickersham, N.; Matthay, M.A.; Ware, L.B. Biomarkers of inflammation, coagulation and fibrinolysis predict mortality in acute lung injury. Crit. Care 2008, 12, R41. [Google Scholar] [CrossRef]
- Ware, L.B.; Koyama, T.; Billheimer, D.D.; Wu, W.; Bernard, G.R.; Thompson, B.T.; Brower, R.G.; Standiford, T.J.; Martin, T.R.; Matthay, M.A. Prognostic and Pathogenetic Value of Combining Clinical and Biochemical Indices in Patients with Acute Lung Injury. Chest 2010, 137, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Flori, H.R.; Sapru, A.; Quasney, M.W.; Gildengorin, G.; Curley, M.A.; Matthay, M.A.; Dahmer, M.K. A prospective investigation of interleukin-8 levels in pediatric acute respiratory failure and acute respiratory distress syndrome. Crit. Care 2019, 23, 128. [Google Scholar] [CrossRef] [PubMed]
- Laffon, M.; Pittet, J.-F.; Modelska, K.; Matthay, M.A.; Young, D.M. Interleukin-8 Mediates Injury from Smoke Inhalation to both the Lung Endothelial and the Alveolar Epithelial Barriers in Rabbits. Am. J. Respir. Crit. Care Med. 1999, 160 Pt 1, 1443–1449. [Google Scholar] [CrossRef]
- Osman, M.O.; Kristensen, J.U.; Jacobsen, N.O.; Lausten, S.B.; Deleuran, B.; Gesser, B.; Matsushima, K.; Larsen, C.G.; Jensen, S.L. A monoclonal anti-interleukin 8 antibody (WS-4) inhibits cytokine response and acute lung injury in experimental severe acute necrotising pancreatitis in rabbits. Gut 1998, 43, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Sekido, N.; Mukaida, N.; Harada, A.; Nakanishi, I.; Watanabe, Y.; Matsushima, K. Prevention of lung reperfusion injury in rabbits by a monoclonal antibody against interleukin-8. Nat. Cell Biol. 1993, 365, 654–657. [Google Scholar] [CrossRef] [PubMed]
- Bao, Z.; Ye, Q.; Gong, W.; Xiang, Y.; Wan, H. Humanized monoclonal antibody against the chemokine CXCL-8 (IL-8) effectively prevents acute lung injury. Int. Immunopharmacol. 2010, 10, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Hoegl, S.; Boost, K.A.; Czerwonka, H.; Dolfen, A.; Scheiermann, P.; Mühl, H.; Zwissler, B.; Hofstetter, C. Inhaled IL-10 reduces biotrauma and mortality in a model of ventilator-induced lung injury. Respir. Med. 2009, 103, 463–470. [Google Scholar] [CrossRef]
- Wu, J.; Xiong, Z.; Xiong, G.; Ding, F.; Lei, J.; Lu, S.; Li, Y.; He, G.; Zhao, L.; Liu, Z. Protective effect of interleukin-10 and recombinant human keratinocyte growth factor-2 on ventilation-induced lung injury in rats. Genet. Mol. Res. 2015, 14, 15642–15651. [Google Scholar] [CrossRef]
- Chen, J.; Lin, J.; Luo, H.; Li, M. Effects of Human Interleukin-10 on Ventilator-Associated Lung Injury in Rats. Inflammation 2019, 42, 538–547. [Google Scholar] [CrossRef]
- Aisiku, I.P.; Yamal, J.-M.; Doshi, P.; Benoit, J.S.; Gopinath, S.; Goodman, J.C.; Robertson, C.S. Plasma cytokines IL-6, IL-8, and IL-10 are associated with the development of acute respiratory distress syndrome in patients with severe traumatic brain injury. Crit. Care 2016, 20, 1–10. [Google Scholar] [CrossRef]
- Belopolskaya, O.B.; Smelaya, T.V.; Moroz, V.V.; Golubev, A.M.; Salnikova, L.E. Clinical associations of host genetic variations in the genes of cytokines in critically ill patients. Clin. Exp. Immunol. 2015, 180, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Abraham, E. Neutrophils and acute lung injury. Crit. Care Med. 2003, 31, S195–S199. [Google Scholar] [CrossRef] [PubMed]
- Rebetz, J.; Semple, J.W.; Kapur, R. The Pathogenic Involvement of Neutrophils in Acute Respiratory Distress Syndrome and Transfusion-Related Acute Lung Injury. Transfus. Med. Hemother. 2018, 45, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Grunwell, J.R.; Stephenson, S.T.; Mohammad, A.F.; Jones, K.; Mason, C.; Opolka, C.; Fitzpatrick, A.M. Differential type I interferon response and primary airway neutrophil extracellular trap release in children with acute respiratory distress syndrome. Sci. Rep. 2020, 10, 19049. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhang, G.; Cui, W.; Tian, B. Progress of neutrophil extracellular traps in airway inflammation of acute lung injury/acute respiratory distress syndrome: Review. Chin. J. Cell. Mol. Immunol. 2020, 36, 664–670. [Google Scholar]
- Middleton, E.A.; He, X.-Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood 2020, 136, 1169–1179. [Google Scholar] [CrossRef]
- Yildiz, C.; Palaniyar, N.; Otulakowski, G.; Khan, M.A.; Post, M.; Kuebler, W.M.; Tanswell, K.; Belcastro, R.; Masood, A.; Engelberts, D.; et al. Mechanical Ventilation Induces Neutrophil Extracellular Trap Formation. Anesthesiology 2015, 122, 864–875. [Google Scholar] [CrossRef]
- Tedder, T.F.; Steeber, D.A.; Chen, A.; Engel, P. The selecting: Vascular adhesion molecules. FASEB J. 1995, 9, 866–873. [Google Scholar] [CrossRef]
- Patel, K.D.; Cuvelier, S.L.; Wiehlera, S. Selectins: Critical mediators of leukocyte recruitment. Semin. Immunol. 2002, 14, 73–81. [Google Scholar] [CrossRef]
- Donnelly, S.C.; Haslett, C.; Dransfield, I.; Robertson, C.E.; Carter, D.C.; Ross, J.A.; Grant, I.S.; Tedder, T.F. Role of selectins in development of adult respiratory distress syndrome. Lancet 1994, 344, 215–219. [Google Scholar] [CrossRef]
- Sakamaki, F.; Ishizaka, A.; Handa, M.; Fujishima, S.; Urano, T.; Sayama, K.; Nakamura, H.; Kanazawa, M.; Kawashiro, T.; Katayama, M. Soluble form of P-selectin in plasma is elevated in acute lung injury. Am. J. Respir. Crit. Care Med. 1995, 151, 1821–1826. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Wu, G.-M.; Li, Q.; Ji, F.-Y.; Shi, Z.; Guo, H.; Yin, J.-B.; Zhou, J.; Gong, L.; Mei, C.-X.; et al. Predictive Value of Combined LIPS and ANG-2 Level in Critically Ill Patients with ARDS Risk Factors. Mediat. Inflamm. 2018, 2018, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Okajima, K.; Harada, N.; Sakurai, G.; Soga, Y.; Suga, H.; Terada, T.; Nakagawa, T. Rapid assay for plasma soluble E-selectin predicts the development of acute respiratory distress syndrome in patients with systemic inflammatory response syndrome. Transl. Res. 2006, 148, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Burnham, E.L.; Moss, M.; Harris, F.; Brown, L.A.S. Elevated plasma and lung endothelial selectin levels in patients with acute respiratory distress syndrome and a history of chronic alcohol abuse. Crit. Care Med. 2004, 32, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Ruchaud-Sparagano, M.-H.; Drost, E.M.; Donnelly, S.C.; Bird, M.I.; Haslett, C.; Dransfield, I. Potential pro-inflammatory effects of soluble E-selectin upon neutrophil function. Eur. J. Immunol. 1998, 28, 80–89. [Google Scholar] [CrossRef]
- Al-Biltagi, M.; Abo-Elezz, A.A.E.; Elshafiey, R.M.G.; Suliman, G.A.; Mabrouk, M.M.; Mourad, H.A. The predictive value of soluble endothelial selectin plasma levels in children with acute lung injury. J. Crit. Care 2016, 32, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Carraway, M.S.; Welty-Wolf, K.E.; Kantrow, S.P.; Huang, Y.-C.T.; Simonson, S.G.; Que, L.G.; Kishimoto, T.K.; Piantadosi, C.A. Antibody to E- and L-Selectin Does Not Prevent Lung Injury or Mortality in Septic Baboons. Am. J. Respir. Crit. Care Med. 1998, 157 Pt 1, 938–949. [Google Scholar] [CrossRef]
- Ridings, P.C.; Windsor, A.C.J.; Jutila, M.A.; Blocher, C.R.; Fisher, B.J.; Sholley, M.M.; Sugerman, H.J.; Fowler, A.A. A dual-binding antibody to E- and L-selectin attenuates sepsis-induced lung injury. Am. J. Respir. Crit. Care Med. 1995, 152, 247–253. [Google Scholar] [CrossRef]
- Chandra, A.; Katahira, J.; Schmalstieg, F.C.; Murakami, K.; Enkhbaatar, P.; Cox, R.A.; Hawkins, H.K.; Traber, L.D.; Herndon, D.N.; Traber, D.L. P-selectin blockade fails to improve acute lung injury in sheep. Clin. Sci. 2003, 104, 313–321. [Google Scholar] [CrossRef]
- Doerschuk, C.M.; Quinlan, W.M.; Doyle, N.A.; Bullard, D.C.; Vestweber, D.; Jones, M.L.; Takei, F.; Ward, P.A.; Beaudet, A.L. The role of P-selectin and ICAM-1 in acute lung injury as determined using blocking antibodies and mutant mice. J. Immunol. 1996, 157, 4609–4614. [Google Scholar]
- Hayashi, H.; Koike, H.; Kurata, Y.; Imanishi, N.; Tojo, S.J. Protective effects of sialyl Lewis X and anti-P-selectin antibody against lipopolysaccharide-induced acute lung injury in rabbits. Eur. J. Pharmacol. 1999, 370, 47–56. [Google Scholar] [CrossRef]
- Zarbock, A.; Singbartl, K.; Ley, K. Complete reversal of acid-induced acute lung injury by blocking of platelet-neutrophil aggregation. J. Clin. Investig. 2006, 116, 3211–3219. [Google Scholar] [CrossRef] [PubMed]
- Bime, C.; Pouladi, N.; Sammani, S.; Batai, K.; Casanova, N.; Zhou, T.; Kempf, C.L.; Sun, X.; Camp, S.M.; Wang, T.; et al. Genome-Wide Association Study in African Americans with Acute Respiratory Distress Syndrome Identifies the Selectin P Ligand Gene as a Risk Factor. Am. J. Respir. Crit. Care Med. 2018, 197, 1421–1432. [Google Scholar] [CrossRef] [PubMed]
- Reutershan, J.; Ley, K. Bench-to-bedside review: Acute respiratory distress syndrome—How neutrophils migrate into the lung. Crit. Care 2004, 8, 453–461. [Google Scholar] [CrossRef][Green Version]
- Moss, M.; Gillespie, M.K.; Ackerson, L.; Moore, F.A.; Moore, E.E.; Parsons, P.E. Endothelial cell activity varies in patients at risk for the adult respiratory distress syndrome. Crit. Care Med. 1996, 24, 1782–1786. [Google Scholar] [CrossRef]
- Skiba-Choińska, I.; Rogowski, F. Adhesion molecules and their role in pathogenesis of ARDS. Prz. Lek. 1996, 53, 627–630. [Google Scholar]
- Kimura, D.; Saravia, J.; Rovnaghi, C.R.; Meduri, G.U.; Schwingshackl, A.; Cormier, S.A.; Anand, K.J. Plasma Biomarker Analysis in Pediatric ARDS: Generating Future Framework from a Pilot Randomized Control Trial of Methylprednisolone: A Framework for Identifying Plasma Biomarkers Related to Clinical Outcomes in Pediatric ARDS. Front. Pediatr. 2016, 4. [Google Scholar] [CrossRef]
- Müller, A.M.; Cronen, C.; Müller, K.-M.; Kirkpatrick, C.J. Heterogeneous expression of cell adhesion molecules by endothelial cells in ARDS. J. Pathol. 2002, 198, 270–275. [Google Scholar] [CrossRef]
- Reiss, L.K.; Uhlig, U.; Uhlig, S. Models and mechanisms of acute lung injury caused by direct insults. Eur. J. Cell Biol. 2012, 91, 590–601. [Google Scholar] [CrossRef]
- Bedirli, A.; Kerem, M.; Pasaoglu, H.; Akyurek, N.; Tezcaner, T.; Elbeg, S.; Memis, L.; Sakrak, O. Beta-glucan attenuates inflammatory cytokine release and prevents acute lung injury in an experimental model of sepsis. Shock 2007, 27, 397–401. [Google Scholar] [CrossRef]
- Chen, L.; Li, W.; Qi, D.; Lu, L.; Zhang, Z.; Wang, D. Honokiol protects pulmonary microvascular endothelial barrier against lipopolysaccharide-induced ARDS partially via the Sirt3/AMPK signaling axis. Life Sci. 2018, 210, 86–95. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Geng, S.; Zhou, W.; Rui, Y.; Mu, X.; Zhang, C.; You, Q.; Su, X. MMI-0100 ameliorates lung inflammation in a mouse model of acute respiratory distress syndrome by reducing endothelial expression of ICAM-1. Drug Des. Dev. Ther. 2018, 12, 4253–4260. [Google Scholar] [CrossRef] [PubMed]
- Lang, S.; Li, L.; Wang, X.; Sun, J.; Xue, X.; Xiao, Y.; Zhang, M.; Ao, T.; Wang, J. CXCL10/IP-10 Neutralization Can Ameliorate Lipopolysaccharide-Induced Acute Respiratory Distress Syndrome in Rats. PLoS ONE 2017, 12, e0169100. [Google Scholar] [CrossRef] [PubMed]
- Félétou, M.; Vanhoutte, P.M. Endothelial dysfunction: A multifaceted disorder (The Wiggers Award Lecture). Am. J. Physiol. Circ. Physiol. 2006, 291, H985–H1002. [Google Scholar] [CrossRef] [PubMed]
- Edelberg, J.M.; Christie, P.D.; Rosenberg, R.D. Regulation of Vascular Bed–Specific Prothrombotic Potential. Circ. Res. 2001, 89, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Sabharwal, A.K.; Bajaj, S.P.; Ameri, A.; Tricomi, S.M.; Hyers, T.M.; Dahms, T.E.; Taylor, F.B., Jr.; Bajaj, M.S. Tissue factor pathway inhibitor and von Willebrand factor antigen levels in adult respiratory distress syndrome and in a primate model of sepsis. Am. J. Respir. Crit. Care Med. 1995, 151 Pt 1, 758–767. [Google Scholar] [CrossRef]
- Scarpati, E.M.; Sadler, J.E. Regulation of endothelial cell coagulant properties. Modulation of tissue factor, plasminogen activator inhibitors, and thrombomodulin by phorbol 12-myristate 13-acetate and tumor necrosis factor. J. Biol. Chem. 1989, 264, 20705–20713. [Google Scholar]
- Siemiatkowski, A.; Kłoczko, J.; Galar, M.; Czaban, S.L. Von Willebrand Factor Antigen as a Prognostic Marker in Posttraumatic Acute Lung Injury. Pathophysiol. Haemost. Thromb. 2000, 30, 189–195. [Google Scholar] [CrossRef]
- Ware, L.B.; Eisner, M.D.; Thompson, B.T.; Parsons, P.E.; Matthay, M.A. Significance of Von Willebrand Factor in Septic and Nonseptic Patients with Acute Lung Injury. Am. J. Respir. Crit. Care Med. 2004, 170, 766–772. [Google Scholar] [CrossRef]
- Hou, P.C.; Filbin, M.R.; Wang, H.; Ngo, L.; Huang, D.T.; Aird, W.C.; Yealy, D.M.; Angus, D.C.; Kellum, J.A.; Shapiro, N.I. Endothelial Permeability and Hemostasis in Septic Shock. Chest 2017, 152, 22–31. [Google Scholar] [CrossRef]
- Afshar, M.; Burnham, E.L.; Joyce, C.; Gagnon, R.; Dunn, R.; Albright, J.M.; Ramirez, L.; Repine, J.E.; Netzer, G.; Kovacs, E.J. Injury Characteristics and von Willebrand Factor for the Prediction of Acute Respiratory Distress Syndrome in Patients With Burn Injury: Development and Internal Validation. Ann. Surg. 2019, 270, 1186–1193. [Google Scholar] [CrossRef] [PubMed]
- Rubin, D.B.; Wiener-Kronish, J.P.; Murray, J.F.; Green, D.R.; Turner, J.; Luce, J.M.; Montgomery, A.B.; Marks, J.D.; Matthay, M.A. Elevated von Willebrand factor antigen is an early plasma predictor of acute lung injury in nonpulmonary sepsis syndrome. J. Clin. Investig. 1990, 86, 474–480. [Google Scholar] [CrossRef] [PubMed]
- El Basset Abo El Ezz, A.A.; Abd El Hafez, M.A.; El Amrousy, D.M.; El Momen Suliman, G.A. The predictive value of Von Willebrand factor antigen plasma levels in children with acute lung injury. Pediatr. Pulmonol. 2017, 52, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Moss, M.; Ackerson, L.; Gillespie, M.K.; Moore, F.A.; Moore, E.E.; Parsons, P.E. Von Willebrand factor antigen levels are not predictive for the adult respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 1995, 151, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, M.S.; Tricomi, S.M. Plasma levels of the three endothelial-specific proteins von Willebrand factor, tissue factor pathway inhibitor, and thrombomodulin do not predict the development of acute respiratory distress syndrome. Intensive Care Med. 1999, 25, 1259–1266. [Google Scholar] [CrossRef]
- Agrawal, A.; Matthay, M.A.; Kangelaris, K.N.; Stein, J.; Chu, J.C.; Imp, B.M.; Cortez, A.; Abbott, J.; Liu, K.D.; Calfee, C.S. Plasma Angiopoietin-2 Predicts the Onset of Acute Lung Injury in Critically Ill Patients. Am. J. Respir. Crit. Care Med. 2013, 187, 736–742. [Google Scholar] [CrossRef]
- Hendrickson, C.M.; Matthay, M.A. Endothelial biomarkers in human sepsis: Pathogenesis and prognosis for ARDS. Pulm. Circ. 2018, 8, 2045894018769876. [Google Scholar] [CrossRef]
- Frantzeskaki, F.; Armaganidis, A.; Orfanos, S.E. Immunothrombosis in Acute Respiratory Distress Syndrome: Cross Talks between Inflammation and Coagulation. Respiration 2017, 93, 212–225. [Google Scholar] [CrossRef]
- Yau, J.W.; Teoh, H.; Verma, S. Endothelial cell control of thrombosis. BMC Cardiovasc. Disord. 2015, 15, 1–11. [Google Scholar] [CrossRef]
- Monroe, D.M.; Key, N.S. The tissue factor-factor VIIa complex: Procoagulant activity, regulation, and multitasking. J. Thromb. Haemost. 2007, 5, 1097–1105. [Google Scholar] [CrossRef]
- Esmon, C.T. The Protein C Pathway. Chest 2003, 124 (Suppl. 3), 26S–32S. [Google Scholar] [CrossRef] [PubMed]
- Vassiliou, A.G.; Kotanidou, A.; Mastora, Z.; Maniatis, N.A.; Albani, P.; Jahaj, E.; Koutsoukou, A.; Armaganidis, A.; Orfanos, S.E. Elevated soluble endothelial protein C receptor levels at ICU admission are associated with sepsis development. Minerva Anestesiol. 2015, 81, 125–134. [Google Scholar] [PubMed]
- Block, E.R. Pulmonary Endothelial Cell Pathobiology: Implications for Acute Lung Injury. Am. J. Med Sci. 1992, 304, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Millar, F.R.; Summers, C.; Griffiths, M.J.; Toshner, M.R.; Proudfoot, A.G. The pulmonary endothelium in acute respiratory distress syndrome: Insights and therapeutic opportunities. Thorax 2016, 71, 462–473. [Google Scholar] [CrossRef] [PubMed]
- Horn, S.; Lang, S.; Fukudome, K.; Nahrup, A.S.; Hoffmann, U.; Kaden, J.J.; Borggrefe, M.; Haase, K.K.; Brueckmann, M.; Huhle, G. Recombinant human activated protein C upregulates cyclooxygenase-2 expression in endothelial cells via binding to endothelial cell protein C receptor and activation of protease-activated receptor-1. Thromb. Haemost. 2005, 93, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Mosnier, L.O.; Yang, X.V.; Griffin, J.H. Activated Protein C Mutant with Minimal Anticoagulant Activity, Normal Cytoprotective Activity, and Preservation of Thrombin Activable Fibrinolysis Inhibitor-dependent Cytoprotective Functions. J. Biol. Chem. 2007, 282, 33022–33033. [Google Scholar] [CrossRef] [PubMed]
- Gando, S.; Nanzaki, S.; Morimoto, Y.; Kobayashi, S.; Kemmotsu, O. Systemic Activation of Tissue-Factor Dependent Coagulation Pathway in Evolving Acute Respiratory Distress Syndrome in Patients with Trauma and Sepsis. J. Trauma Inj. Infect. Crit. Care 1999, 47, 719–723. [Google Scholar] [CrossRef]
- Carraway, M.; Ortel, T.; Piantadosi, C.; Welty-Wolf, K.E. Coagulation and Inflammation in Acute Lung Injury. Thromb. Haemost. 2002, 88, 17–25. [Google Scholar] [CrossRef]
- Van Der Poll, T. Tissue factor as an initiator of coagulation and inflammation in the lung. Crit. Care 2008, 12, S3. [Google Scholar] [CrossRef]
- Idell, S.; Gonzalez, K.; Bradford, H.; MacArthur, C.K.; Fein, A.M.; Maunder, R.J.; Garcia, J.G.; Griffith, D.E.; Weiland, J.; Martin, T.R.; et al. Procoagulant activity in bronchoalveolar lavage in the adult respiratory distress syndrome. Contribution of tissue factor associated with factor VII. Am. Rev. Respir. Dis. 1987, 136, 1466–1474. [Google Scholar] [CrossRef]
- Ware, L.B.; Fang, X.; Matthay, M.A. Protein C and thrombomodulin in human acute lung injury. Am. J. Physiol. Cell. Mol. Physiol. 2003, 285, L514–L521. [Google Scholar] [CrossRef] [PubMed]
- MacGregor, I.R.; Perrie, A.M.; Donnelly, S.C.; Haslett, C. Modulation of human endothelial thrombomodulin by neutrophils and their release products. Am. J. Respir. Crit. Care Med. 1997, 155, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Gando, S.; Kameue, T.; Matsuda, N.; Sawamura, A.; Hayakawa, M.; Kato, H. Systemic Inflammation and Disseminated Intravascular Coagulation in Early Stage of ALI and ARDS: Role of Neutrophil and Endothelial Activation. Inflammation 2004, 28, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Distefano, G.; Romeo, M.G.; Betta, P.; Rodono’, A.; Amato, M. Thrombomodulin serum levels in ventilated preterm babies with respiratory distress syndrome. Eur. J. Nucl. Med. Mol. Imaging 1998, 157, 327–330. [Google Scholar] [CrossRef] [PubMed]
- Orwoll, B.; Spicer, A.C.; Zinter, M.S.; Alkhouli, M.F.; Khemani, R.G.; Flori, H.R.; Neuhaus, J.; Calfee, C.S.; Matthay, M.A.; Sapru, M.A. Elevated soluble thrombomodulin is associated with organ failure and mortality in children with acute respiratory distress syndrome (ARDS): A prospective observational cohort study. Crit. Care 2015, 19, 435. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Taniguchi, H.; Kondoh, Y.; Ando, M.; Watanabe, N.; Kimura, T.; Kataoka, K.; Yokoyama, T.; Sakamoto, K.; Hasegawa, Y. Soluble thrombomodulin in bronchoalveolar lavage fluid is an independent predictor of severe drug-induced lung injury. Respirology 2017, 22, 744–749. [Google Scholar] [CrossRef]
- Sapru, A.; Network, T.N.A.; Calfee, C.S.; Liu, K.D.; Kangelaris, K.; Hansen, H.; Pawlikowska, L.; Ware, L.B.; Alkhouli, M.F.; Abbott, J.; et al. Plasma soluble thrombomodulin levels are associated with mortality in the acute respiratory distress syndrome. Intensive Care Med. 2015, 41, 470–478. [Google Scholar] [CrossRef]
- Ware, L.B.; Matthay, M.A.; Parsons, P.E.; Thompson, B.T.; Januzzi, J.L.; Eisner, M.D. Pathogenetic and prognostic significance of altered coagulation and fibrinolysis in acute lung injury/acute respiratory distress syndrome. Crit. Care Med. 2007, 35, 1821–1828. [Google Scholar] [CrossRef]
- Ortolan, L.S.; Sercundes, M.K.; Moura, G.C.; Quirino, T.D.C.; Debone, D.; Costa, D.D.S.; Murillo, O.; Marinho, C.R.F.; Epiphanio, S. Endothelial Protein C Receptor Could Contribute to Experimental Malaria-Associated Acute Respiratory Distress Syndrome. J. Immunol. Res. 2019, 2019, 3105817. [Google Scholar] [CrossRef]
- Maknitikul, S.; Luplertlop, N.; Grau, G.E.R.; Ampawong, S. Dysregulation of pulmonary endothelial protein C receptor and thrombomodulin in severe falciparum malaria-associated ARDS relevant to hemozoin. PLoS ONE 2017, 12, e0181674. [Google Scholar] [CrossRef]
- Sapru, A.; Network, T.N.A.; Liu, K.D.; Wiemels, J.; Hansen, H.; Pawlikowska, L.; Poon, A.; Jorgenson, E.; Witte, J.S.; Calfee, C.S.; et al. Association of common genetic variation in the protein C pathway genes with clinical outcomes in acute respiratory distress syndrome. Crit. Care 2016, 20, 151. [Google Scholar] [CrossRef] [PubMed]
- Ware, L.B.; Bastarache, J.A.; Wang, L. Coagulation and fibrinolysis in human acute lung injury-New therapeutic targets? Keio J. Med. 2005, 54, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Whyte, C.S.; Morrow, G.B.; Mitchell, J.L.; Chowdary, P.; Mutch, N.J. Fibrinolytic abnormalities in acute respiratory distress syndrome (ARDS) and versatility of thrombolytic drugs to treat COVID-19. J. Thromb. Haemost. 2020, 18, 1548–1555. [Google Scholar] [CrossRef] [PubMed]
- Grau, G.E.; De Moerloose, P.; Bulla, O.; Lou, J.; Lei, Z.; Reber, G.; Mili, N.; Ricou, B.; Morel, D.R.; Suter, P.M. Haemostatic Properties of Human Pulmonary and Cerebral Microvascular Endothelial Cells. Thromb. Haemost. 1997, 77, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Ozolina, A.; Sarkele, M.; Sabelnikovs, O.; Skesters, A.; Jaunalksne, I.; Serova, J.; Ievins, T.; Bjertnaes, L.J.; Vanags, I. Activation of Coagulation and Fibrinolysis in Acute Respiratory Distress Syndrome: A Prospective Pilot Study. Front. Med. 2016, 3, 64. [Google Scholar] [CrossRef]
- El Solh, A.A.; Bhora, M.; Pineda, L.; Aquilina, A.; Abbetessa, L.; Berbary, E. Alveolar plasminogen activator inhibitor-1 predicts ARDS in aspiration pneumonitis. Intensive Care Med. 2006, 32, 110–115. [Google Scholar] [CrossRef]
- Prabhakaran, P.; Ware, L.B.; White, K.E.; Cross, M.T.; Matthay, M.A.; Olman, M.A. Elevated levels of plasminogen activator inhibitor-1 in pulmonary edema fluid are associated with mortality in acute lung injury. Am. J. Physiol. Cell. Mol. Physiol. 2003, 285, L20–L28. [Google Scholar] [CrossRef]
- Günther, A.; Mosavi, P.; Heinemann, S.; Ruppert, C.; Muth, H.; Markart, P.; Grimminger, F.; Walmrath, D.; Temmesfeld-Wollbrück, B.; Seeger, W. Alveolar Fibrin Formation Caused by Enhanced Procoagulant and Depressed Fibrinolytic Capacities in Severe Pneumonia. Am. J. Respir. Crit. Care Med. 2000, 161, 454–462. [Google Scholar] [CrossRef]
- Schultz, M.J.; Millo, J.; Levi, M.; Hack, C.E.; Weverling, G.J.; Garrard, C.S.; van der Poll, T. Local activation of coagulation and inhibition of fibrinolysis in the lung during ventilator associated pneumonia. Thorax 2004, 59, 130–135. [Google Scholar] [CrossRef]
- Koyama, K.; Katayama, S.; Tonai, K.; Shima, J.; Koinuma, T.; Nunomiya, S. Biomarker profiles of coagulopathy and alveolar epithelial injury in acute respiratory distress syndrome with idiopathic/immune-related disease or common direct risk factors. Crit. Care 2019, 23, 283. [Google Scholar] [CrossRef]
- Miyoshi, S.; Ito, R.; Katayama, H.; Dote, K.; Aibiki, M.; Hamada, H.; Okura, T.; Higaki, J. Combination therapy with sivelestat and recombinant human soluble thrombomodulin for ARDS and DIC patients. Drug Des. Dev. Ther. 2014, 8, 1211–1219. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Takahashi, Y.; Matsutani, N.; Dejima, H.; Nakayama, T.; Okamura, R.; Uehara, H.; Kawamura, M. Therapeutic potential of recombinant thrombomodulin for lung injury after pneumonectomy via inhibition of high-mobility group box 1 in mice. J. Trauma Acute Care Surg. 2016, 81, 868–875. [Google Scholar] [CrossRef] [PubMed]
- Kudo, D.; Toyama, M.; Aoyagi, T.; Akahori, Y.; Yamamoto, H.; Ishii, K.; Kanno, E.; Maruyama, R.; Kaku, M.; Kushimoto, S.; et al. Involvement of high mobility group box 1 and the therapeutic effect of recombinant thrombomodulin in a mouse model of severe acute respiratory distress syndrome. Clin. Exp. Immunol. 2013, 173, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Juschten, J.; Ingelse, S.A.; Maas, M.A.W.; Girbes, A.R.J.; Juffermans, N.P.; Schultz, M.J.; Tuinman, P.R. Antithrombin plus alpha-1 protease inhibitor does not affect coagulation and inflammation in two murine models of acute lung injury. Intensive Care Med. Exp. 2019, 7 (Suppl. 1), 36. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Acosta, Y.; Gonzalez, M.; Fernandez, M.; Valance, W.A. Emerging Roles for Platelets in Inflammation and Disease. J. Infect. Dis. Ther. 2014, 2, 1–7. [Google Scholar] [CrossRef]
- Gale, N.W.; Yancopoulos, G.D. Growth factors acting via endothelial cell-specific receptor tyrosine kinases: VEGFs, Angiopoietins, and ephrins in vascular development. Genes Dev. 1999, 13, 1055–1066. [Google Scholar] [CrossRef]
- Plouët, J.; Schilling, J.; Gospodarowicz, D. Isolation and characterization of a newly identified endothelial cell mitogen produced by AtT-20 cells. EMBO J. 1989, 8, 3801–3806. [Google Scholar] [CrossRef]
- Senger, D.R.; Galli, S.J.; Dvorak, A.M.; Perruzzi, C.A.; Harvey, V.S.; Dvorak, H.F. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 1983, 219, 983–985. [Google Scholar] [CrossRef]
- Medford, A.R.L. Vascular endothelial growth factor (VEGF) in acute lung injury (ALI) and acute respiratory distress syndrome (ARDS): Paradox or paradigm? Thorax 2006, 61, 621–626. [Google Scholar] [CrossRef]
- Maitre, B.; Boussat, S.; Jean, D.; Gouge, M.; Brochard, L.; Housset, B.; Adnot, S.; Delclaux, C. Vascular endothelial growth factor synthesis in the acute phase of experimental and clinical lung injury. Eur. Respir. J. 2001, 18, 100–106. [Google Scholar] [CrossRef]
- Abadie, Y.; Bregeon, F.; Papazian, L.; Lange, F.; Chailley-Heu, B.; Thomas, P.A.; Duvaldestin, P.; Adnot, S.; Maitre, B.; Delclaux, C. Decreased VEGF concentration in lung tissue and vascular injury during ARDS. Eur. Respir. J. 2005, 25, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Azamfirei, L.; Gurzu, S.; Solomon, R.; Copotoiu, R.; Copotoiu, S.; Jung, I.; Tilinca, M.; Branzaniuc, K.; Corneci, D.; Szederjesi, J.; et al. Vascular endothelial growth factor: A possible mediator of endothelial activation in acute respiratory distress syndrome. Minerva Anestesiol. 2010, 76, 609–616. [Google Scholar] [PubMed]
- Medford, A.R.; Ibrahim, N.B.; Millar, A.B. Vascular endothelial growth factor receptor and coreceptor expression in human acute respiratory distress syndrome. J. Crit. Care 2009, 24, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Perkins, G.; Roberts, J.; McAuley, D.; Armstrong, L.; Millar, A.; Gao, F.; Thickett, D.R. Regulation of vascular endothelial growth factor bioactivity in patients with acute lung injury. Thorax 2005, 60, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Thickett, D.R.; Armstrong, L.; Christie, S.J.; Millar, A.B. Vascular Endothelial Growth Factor May Contribute to Increased Vascular Permeability in Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2001, 164, 1601–1605. [Google Scholar] [CrossRef] [PubMed]
- Gaudry, M.; Bregerie, O.; Andrieu, V.; El Benna, J.; Pocidalo, M.A.; Hakim, J. Intracellular Pool of Vascular Endothelial Growth Factor in Human Neutrophils. Blood 1997, 90, 4153–4161. [Google Scholar] [CrossRef] [PubMed]
- Thickett, D.R.; Armstrong, L.; Millar, A.B. A Role for Vascular Endothelial Growth Factor in Acute and Resolving Lung Injury. Am. J. Respir. Crit. Care Med. 2002, 166, 1332–1337. [Google Scholar] [CrossRef] [PubMed]
- Varet, J.; Douglas, S.K.; Gilmartin, L.; Medford, A.R.L.; Bates, D.O.; Harper, S.J.; Millar, A.B. VEGF in the lung: A role for novel isoforms. Am. J. Physiol. Cell. Mol. Physiol. 2010, 298, L768–L774. [Google Scholar] [CrossRef] [PubMed]
- Medford, A.R.L. Vascular endothelial growth factor gene polymorphism and acute respiratory distress syndrome. Thorax 2005, 60, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Zhai, R.; Gong, M.N.; Zhou, W.; Thompson, T.B.; Kraft, P.; Su, L.; Christiani, D.C. Genotypes and haplotypes of the VEGF gene are associated with higher mortality and lower VEGF plasma levels in patients with ARDS. Thorax 2007, 62, 718–722. [Google Scholar] [CrossRef]
- Yang, S.; Cao, S.; Li, J.; Chang, J. Association between Vascular Endothelial Growth Factor + 936 Genotype and Acute Respiratory Distress Syndrome in a Chinese Population. Genet. Test. Mol. Biomark. 2011, 15, 737–740. [Google Scholar] [CrossRef] [PubMed]
- Medford, A.R.L.; Douglas, S.K.; Godinho, S.I.; Uppington, K.M.; Armstrong, L.; Gillespie, K.M.; Van Zyl, B.; Tetley, T.D.; Ibrahim, N.; Millar, A.B. Vascular Endothelial Growth Factor (VEGF) isoform expression and activity in human and murine lung injury. Respir. Res. 2009, 10, 27. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-K.; Lin, Y.-H.; Huang, T.-C.; Shi, C.-S.; Yang, C.-T.; Yang, Y.-L. VEGF mediates fat embolism-induced acute lung injury via VEGF receptor 2 and the MAPK cascade. Sci. Rep. 2019, 9, 11713. [Google Scholar] [CrossRef] [PubMed]
- Kaner, R.J.; Ladetto, J.V.; Singh, R.; Fukuda, N.; Matthay, M.A.; Crystal, R.G. Lung Overexpression of the Vascular Endothelial Growth Factor Gene Induces Pulmonary Edema. Am. J. Respir. Cell Mol. Biol. 2000, 22, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Godzich, M.; Hodnett, M.; Frank, J.A.; Su, G.; Pespeni, M.; Angel, A.; Howard, M.B.; Matthay, M.A.; Pittet, J.-F. Activation of the stress protein response prevents the development of pulmonary edema by inhibiting VEGF cell signaling in a model of lung ischemia-reperfusion injury in rats. FASEB J. 2006, 20, 1519–1521. [Google Scholar] [CrossRef]
- Watanabe, M.; Boyer, J.L.; Crystal, R.G. Genetic Delivery of Bevacizumab to Suppress Vascular Endothelial Growth Factor-Induced High-Permeability Pulmonary Edema. Hum. Gene Ther. 2009, 20, 598–610. [Google Scholar] [CrossRef]
- Compernolle, V.; Brusselmans, K.; Acker, T.; Hoet, P.; Tjwa, M.; Beck, H.; Plaisance, S.; Dor, Y.; Keshet, E.; Lupu, F.; et al. Loss of HIF-2α and inhibition of VEGF impair fetal lung maturation, whereas treatment with VEGF prevents fatal respiratory distress in premature mice. Nat. Med. 2002, 8, 702–710. [Google Scholar] [CrossRef]
- Maisonpierre, P.C.; Suri, C.; Jones, P.F.; Bartunkova, S.; Wiegand, S.J.; Radziejewski, C.; Compton, D.; McClain, J.; Aldrich, T.H.; Papadopoulos, N.; et al. Angiopoietin-2, a Natural Antagonist for Tie2 That Disrupts in vivo Angiogenesis. Science 1997, 277, 55–60. [Google Scholar] [CrossRef]
- Thurston, G.; Rudge, J.S.; Ioffe, E.; Zhou, H.; Ross, L.; Croll, S.D.; Glazer, N.; Holash, J.; McDonald, D.M.; Yancopoulos, G.D. Angiopoietin-1 protects the adult vasculature against plasma leakage. Nat. Med. 2000, 6, 460–463. [Google Scholar] [CrossRef]
- Fiedler, U.; Reiss, Y.; Scharpfenecker, M.; Grunow, V.; Koidl, S.; Thurston, G.; Gale, N.W.; Witzenrath, M.; Rosseau, S.; Suttorp, N.; et al. Angiopoietin-2 sensitizes endothelial cells to TNF-α and has a crucial role in the induction of inflammation. Nat. Med. 2006, 12, 235–239. [Google Scholar] [CrossRef]
- Roviezzo, F.; Tsigkos, S.; Kotanidou, A.; Bucci, M.; Brancaleone, V.; Cirino, G.; Papapetropoulos, A. Angiopoietin-2 Causes Inflammation in Vivo by Promoting Vascular Leakage. J. Pharmacol. Exp. Ther. 2005, 314, 738–744. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.M.; Mammoto, T.; Schultz, A.; Yuan, H.-T.; Christiani, D.C.; Karumanchi, S.A.; Sukhatme, V.P. Excess Circulating Angiopoietin-2 May Contribute to Pulmonary Vascular Leak in Sepsis in Humans. PLoS Med. 2006, 3, e46. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, D.C.; Parikh, S.M.; Balonov, K.; Miller, A.; Gautam, S.; Talmor, D.; Sukhatme, V.P. Circulating angiopoietin 2 correlates with mortality in a surgical population with acute lung injury/adult respiratory distress syndrome. Shock 2008, 29, 656–661. [Google Scholar] [CrossRef] [PubMed]
- Van Der Heijden, M.; Amerongen, G.P.V.N.; Koolwijk, P.; Van Hinsbergh, V.W.M.; Groeneveld, A.B.J. Angiopoietin-2, permeability oedema, occurrence and severity of ALI/ARDS in septic and non-septic critically ill patients. Thorax 2008, 63, 903–909. [Google Scholar] [CrossRef] [PubMed]
- Ando, M.; Miyazaki, E.; Abe, T.; Ehara, C.; Goto, A.; Masuda, T.; Nishio, S.; Fujisaki, H.; Yamasue, M.; Ishii, T.; et al. Angiopoietin-2 expression in patients with an acute exacerbation of idiopathic interstitial pneumonias. Respir. Med. 2016, 117, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Zhong, M.; Zhang, L.; Wang, F.; Peng, S.; Zhang, J.; Xuan, G. The levels of angiopoietin-2 in patients with acute respiratory distress syndrome and its value on prognosis. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue 2014, 26, 804–809. [Google Scholar]
- Zinter, M.S.; Spicer, A.C.; Orwoll, B.; Alkhouli, M.; Dvorak, C.C.; Calfee, C.S.; Matthay, M.A.; Sapru, A. Plasma angiopoietin-2 outperforms other markers of endothelial injury in prognosticating pediatric ARDS mortality. Am. J. Physiol. Cell. Mol. Physiol. 2016, 310, L224–L231. [Google Scholar] [CrossRef]
- Tsangaris, I.; Tsantes, A.; Vrigkou, E.; Kopterides, P.; Pelekanou, A.; Zerva, K.; Antonakos, G.; Konstantonis, D.; Mavrou, I.; Tsaknis, G.; et al. Angiopoietin-2 Levels as Predictors of Outcome in Mechanically Ventilated Patients with Acute Respiratory Distress Syndrome. Dis. Markers 2017, 2017, 1–6. [Google Scholar] [CrossRef]
- Calfee, C.S.; Gallagher, D.; Abbott, J.; Thompson, B.T.; Matthay, M.A. Plasma angiopoietin-2 in clinical acute lung injury. Crit. Care Med. 2012, 40, 1731–1737. [Google Scholar] [CrossRef]
- Ma, S.; Zhao, M.-L.; Wang, K.; Yue, Y.-F.; Sun, R.-Q.; Zhang, R.-M.; Wang, S.-F.; Sun, G.; Xie, H.-Q.; Yu, Y.; et al. Association of Ang-2, vWF, and EVLWI with risk of mortality in sepsis patients with concomitant ARDS: A retrospective study. J. Formos. Med. Assoc. 2020, 119, 950–956. [Google Scholar] [CrossRef]
- Reilly, J.P.; Wang, F.; Jones, T.K.; Palakshappa, J.A.; Anderson, B.J.; Shashaty, M.G.S.; Dunn, T.G.; Johansson, E.D.; Riley, T.R.; Lim, B.; et al. Plasma angiopoietin-2 as a potential causal marker in sepsis-associated ARDS development: Evidence from Mendelian randomization and mediation analysis. Intensive Care Med. 2018, 44, 1849–1858. [Google Scholar] [CrossRef] [PubMed]
- Araujo, C.B.; de Oliveira Neves, F.M.; de Freitas, D.F.; Arruda, B.F.T.; de Macedo Filho, L.J.M.; Salles, V.B.; Meneses, G.C.; Martins, A.M.C.; Liborio, A.B. Angiopoietin-2 as a predictor of acute kidney injury in critically ill patients and association with ARDS. Respirology 2019, 24, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Yin, R.; Guo, Q. Circulating angiopoietin-2 and the risk of mortality in patients with acute respiratory distress syndrome: A systematic review and meta-analysis of 10 prospective cohort studies. Ther. Adv. Respir. Dis. 2020, 14, 1753466620905274. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Zhai, R.; Sheu, C.-C.; Gallagher, D.C.; Gong, M.N.; Tejera, P.; Thompson, B.T.; Christiani, D.C. Genetic variants in the angiopoietin-2 gene are associated with increased risk of ARDS. Intensive Care Med. 2009, 35, 1024–1030. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wada, T.; Jesmin, S.; Gando, S.; Yanagida, Y.; Mizugaki, A.; Sultana, S.N.; Zaedi, S.; Yokota, H. The role of angiogenic factors and their soluble receptors in acute lung injury (ALI)/acute respiratory distress syndrome (ARDS) associated with critical illness. J. Inflamm. 2013, 10, 526. [Google Scholar] [CrossRef] [PubMed]
- Ware, L.B.; Zhao, Z.; Koyama, T.; Brown, R.M.; Semler, M.W.; Janz, D.R.; May, A.K.; Fremont, R.D.; Matthay, M.A.; Cohen, M.J.; et al. Derivation and validation of a two-biomarker panel for diagnosis of ARDS in patients with severe traumatic injuries. Trauma Surg. Acute Care Open 2017, 2, e000121. [Google Scholar] [CrossRef]
- Xu, W.; Song, Y. Biomarkers for patients with trauma associated acute respiratory distress syndrome. Mil. Med. Res. 2017, 4, 25. [Google Scholar] [CrossRef]
- Kümpers, P.; Gueler, F.; David, S.; Van Slyke, P.; Dumont, D.J.; Park, J.-K.; Bockmeyer, C.L.; Parikh, S.M.; Pavenstädt, H.; Haller, H.; et al. The synthetic Tie2 agonist peptide vasculotide protects against vascular leakage and reduces mortality in murine abdominal sepsis. Crit. Care 2011, 15, R261. [Google Scholar] [CrossRef]
- David, S.; Ghosh, C.C.; Kümpers, P.; Shushakova, N.; Van Slyke, P.; Khankin, E.V.; Karumanchi, S.A.; Dumont, D.; Parikh, S.M. Effects of a synthetic PEG-ylated Tie-2 agonist peptide on endotoxemic lung injury and mortality. Am. J. Physiol. Cell. Mol. Physiol. 2011, 300, L851–L862. [Google Scholar] [CrossRef]
- Lomas-Neira, J.L.; Heffernan, D.S.; Ayala, A.; Monaghan, S.F. Blockade of Endothelial Growth Factor, Angiopoietin-2, Reduces Indices of Ards and Mortality in Mice Resulting from the Dual-Insults of Hemorrhagic Shock and Sepsis. Shock 2016, 45, 157–165. [Google Scholar] [CrossRef]
- Stiehl, T.; Thamm, K.; Kaufmann, J.; Schaeper, U.; Kirsch, T.; Haller, H.; Santel, A.; Ghosh, C.C.; Parikh, S.M.; David, S. Lung-Targeted RNA Interference Against Angiopoietin-2 Ameliorates Multiple Organ Dysfunction and Death in Sepsis. Crit. Care Med. 2014, 42, e654–e662. [Google Scholar] [CrossRef] [PubMed]
- van der Heijden, M.; van Nieuw Amerongen, G.P.; Chedamni, S.; van Hinsbergh, V.W.; Johan Groeneveld, A.B. The angiopoietin-Tie2 system as a therapeutic target in sepsis and acute lung injury. Expert Opin. Ther. Targets 2009, 13, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Amraei, R.; Rahimi, N. COVID19, Renin-Angiotensin System and Endothelial Dysfunction. Cells 2020, 9, 1652. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Narayan, R.K.; Kumari, C.; Faiq, M.A.; Kulandhasamy, M.; Kant, K.; Pareek, V. SARS-CoV-2 cell entry receptor ACE2 mediated endothelial dysfunction leads to vascular thrombosis in COVID-19 patients. Med. Hypotheses 2020, 145, 110320. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Smadja, D.M.; Guerin, C.L.; Chocron, R.; Yatim, N.; Boussier, J.; Gendron, N.; Khider, L.; Hadjadj, J.; Goudot, G.; DeBuc, B.; et al. Angiopoietin-2 as a marker of endothelial activation is a good predictor factor for intensive care unit admission of COVID-19 patients. Angiogenesis 2020, 23, 611–620. [Google Scholar] [CrossRef]
- Gustafson, D.; Raju, S.; Wu, R.; Ching, C.; Veitch, S.; Rathnakumar, K.; Boudreau, E.; Howe, K.L.; Fish, J.E. Overcoming Barriers: The Endothelium as a Linchpin of Coronavirus Disease 2019 Pathogenesis? Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1818–1829. [Google Scholar] [CrossRef]
- Giamarellos-Bourboulis, E.J.; Netea, M.G.; Rovina, N.; Akinosoglou, K.; Antoniadou, A.; Antonakos, N.; Damoraki, G.; Gkavogianni, T.; Adami, M.-E.; Katsaounou, P.; et al. Complex Immune Dysregulation in COVID-19 Patients with Severe Respiratory Failure. Cell Host Microbe 2020, 27, 992–1000.e3. [Google Scholar] [CrossRef]
- Ciceri, F.; Beretta, L.; Scandroglio, A.M.; Colombo, S.; Landoni, G.; Ruggeri, A.; Peccatori, J.; D’Angelo, A.; De Cobelli, F.; Rovere-Querini, P.; et al. Microvascular COVID-19 lung vessels obstructive thromboinflammatory syndrome (MicroCLOTS): An atypical acute respiratory distress syndrome working hypothesis. Crit. Care Resusc. 2020, 22, 95–97. [Google Scholar]
- Goshua, G.; Pine, A.B.; Meizlish, M.L.; Chang, C.-H.; Zhang, H.; Bahel, P.; Baluha, A.; Bar, N.; Bona, R.D.; Burns, A.J.; et al. Endotheliopathy in COVID-19-associated coagulopathy: Evidence from a single-centre, cross-sectional study. Lancet Haematol. 2020, 7, e575–e582. [Google Scholar] [CrossRef]
- Frank, J.A.; McAuley, D.F.; Gutierrez, J.A.; Daniel, B.M.; Dobbs, L.; Matthay, M.A. Differential effects of sustained inflation recruitment maneuvers on alveolar epithelial and lung endothelial injury. Crit. Care Med. 2005, 33, 181–188. [Google Scholar] [CrossRef] [PubMed]


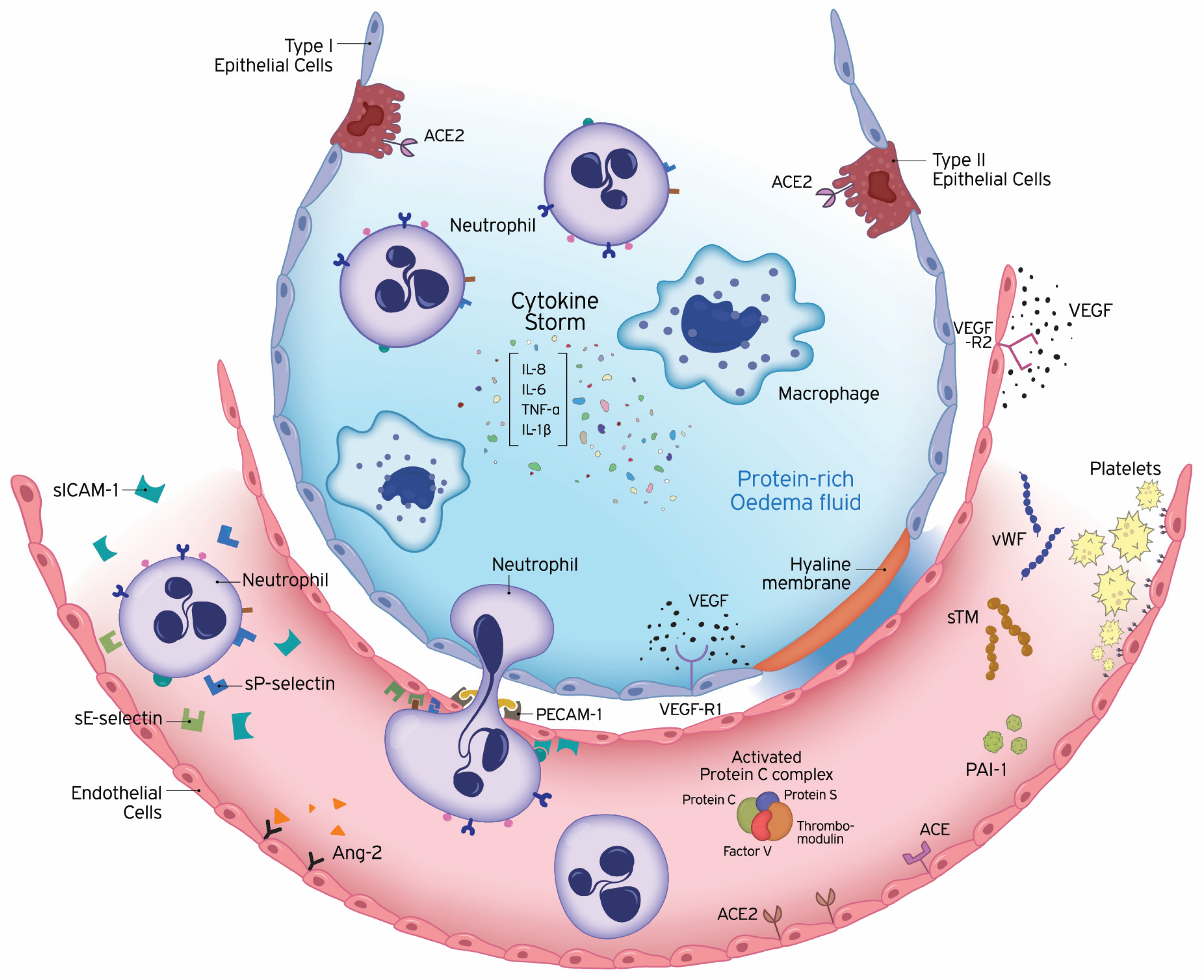{kind=link}
| Barrier and transport functions |
| Synthesis of vasoactive compounds–maintenance of vascular tone |
| Host defence—production of cytokines and chemokines |
| Haemostasis and coagulation |
| Angiogenesis—production of growth factors |
| Expression of receptors and signal transduction molecules |
| Expression of adhesion molecules |
| Production of reactive oxygen species |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vassiliou, A.G.; Kotanidou, A.; Dimopoulou, I.; Orfanos, S.E. Endothelial Damage in Acute Respiratory Distress Syndrome. Int. J. Mol. Sci. 2020, 21, 8793. https://doi.org/10.3390/ijms21228793
Vassiliou AG, Kotanidou A, Dimopoulou I, Orfanos SE. Endothelial Damage in Acute Respiratory Distress Syndrome. International Journal of Molecular Sciences. 2020; 21(22):8793. https://doi.org/10.3390/ijms21228793
Chicago/Turabian StyleVassiliou, Alice G., Anastasia Kotanidou, Ioanna Dimopoulou, and Stylianos E. Orfanos. 2020. "Endothelial Damage in Acute Respiratory Distress Syndrome" International Journal of Molecular Sciences 21, no. 22: 8793. https://doi.org/10.3390/ijms21228793
APA StyleVassiliou, A. G., Kotanidou, A., Dimopoulou, I., & Orfanos, S. E. (2020). Endothelial Damage in Acute Respiratory Distress Syndrome. International Journal of Molecular Sciences, 21(22), 8793. https://doi.org/10.3390/ijms21228793