Abstract
The aberrant aggregation of proteins is implicated in the onset and pathogenesis of a wide range of neurodegenerative disorders, including Alzheimer’s and Parkinson’s diseases. Mounting evidence indicates that misfolded protein oligomers produced as intermediates in the aggregation process are potent neurotoxic agents in these diseases. Because of the transient and heterogeneous nature of these elusive aggregates, however, it has proven challenging to develop therapeutics that can effectively target them. Here, we review approaches aimed at reducing oligomer toxicity, including (1) modulating the oligomer populations (e.g., by altering the kinetics of aggregation by inhibiting, enhancing, or redirecting the process), (2) modulating the oligomer properties (e.g., through the size–hydrophobicity–toxicity relationship), (3) modulating the oligomer interactions (e.g., by protecting cell membranes by displacing oligomers), and (4) reducing oligomer toxicity by potentiating the protein homeostasis system. We analyze examples of these complementary approaches, which may lead to the development of compounds capable of preventing or treating neurodegenerative disorders associated with protein aggregation.
1. Protein Aggregation and Its Links with Neurodegenerative Diseases
A wide variety of human disorders, including Alzheimer’s disease (AD) and other forms of dementia, are associated with ageing and unhealthy lifestyles [1]. These diseases are still largely incurable and represent a terrible burden for our societies and healthcare systems [2,3]. Over 50 million people currently suffer from dementia worldwide, a number estimated to triple within the next three decades unless effective treatments become available [4]. These conditions are characterized by the presence of aberrant proteinaceous deposits in affected tissues [5,6,7,8,9,10].
To understand the molecular origins of these widespread disorders, we highlight two observations. The first observation is about the generic nature of the phenomenon of protein misfolding and aggregation [11]. The amide and carbonyl groups of the backbone of the polypeptide chains of protein molecules have a strong tendency to form hydrogen bonds. Because the polypeptide backbone is common to all proteins independent of their amino acid sequences, under suitable conditions, almost any protein can access the amyloid state [1,12]. While at low protein concentrations, hydrogen bonds form intra-molecularly, leading to the formation of protein secondary structures, such as α-helices and β-sheets that are characteristic of the native states of proteins, at high concentrations, the formation of inter-molecular hydrogen bonds is favored, so that proteins tend to self-assemble into a highly ordered structures known as amyloid fibrils, which are stabilized by a network of hydrogen bonds [12]. The second observation is that the cellular concentrations of proteins are typically close to the critical values that define whether the most stable state is the native or the amyloid state [13,14,15]. This observation, which has been referred to as “life at the edge of solubility” [13], has an evolutionary origin. Proteins that are highly soluble are subject to an evolutionary drift in which random mutations that are neutral in functional terms decrease the overall solubility of the proteins. This type of mutation goes unchallenged by natural selection until the solubility decreases to the critical level, beyond which negative selection prevents further reductions in solubility [13]. As a result, proteins tend to be supersaturated in the cellular environment and they are, therefore, on the edge of aggregation [16,17]. Although it is perhaps surprising, we now know that proteins commonly aggregate under physiological conditions [15], and it is only the presence of a strong protein homeostasis system that prevents the progressive accumulation of large protein deposits [12]. Overall, the natural tendency of proteins to aggregate when they are supersaturated predisposes certain cell and tissues to protein aggregation upon stress or ageing [18,19,20,21].
These observations lead to the conclusion that in order to prevent or treat protein misfolding diseases, one should find ways to pharmacologically augment or protect the protein homeostasis system that regulates protein aggregation. The pharmacological interventions that we review in the following sections are meant to address precisely this point.
2. Misfolded Protein Oligomers and Their Cytotoxic Effects
The abundance of amyloid deposits in the brains of patients afflicted by neurodegenerative diseases initially implicated mature amyloid fibrils as the central toxic agents. Similar deposits are observed for numerous other neurodegenerative diseases, including chronic traumatic encephalopathy, dementia with Lewy bodies, and Parkinson’s disease (PD). Although in this review we generally analyze the links between protein aggregation and human disease, we focus predominantly on the proteins associated with AD and PD, because they have been investigated more extensively. More recently, it has been realized that amyloid fibrils may not be directly responsible for toxic gain-of-function. Instead, transient, metastable oligomeric intermediates may be more neurotoxic [22]. An important role for these aggregates started to emerge over two decades ago, when pre-fibrillar species comprised of the 42-residue form of amyloid-β peptide (Aβ42), termed Aβ-derived diffusible ligands (ADDLs), were reported as potent neurotoxic species and shown to induce cellular death at nM concentrations [23]. Antibodies against ADDLs, but not monomeric or fibrillar Aβ, were found to interact with AD brain samples [24,25,26]. Soon afterwards, other soluble assemblies were similarly described as toxic forms of Aβ [27,28]. Many studies have since shown the presence of oligomers in the brain of patients affected by AD [29] and PD [30] and have reported that these aggregates are seminal in the induction of cellular dysfunction [31,32,33,34,35]. In a similar way, the onset of PD is characterized by the presence of Lewy bodies and Lewy neurites comprised of the α-synuclein protein (αS) in the brains of patients with the disease, and evidence indicates the source of neuronal death is the oligomeric phase of α-synuclein [36]. It is now well accepted that oligomers are key pathological species in protein misfolding diseases associated with amyloid deposition [7,31,37,38,39].
Misfolded protein oligomers can be formed as intermediates in the aggregation reaction of various amyloidogenic peptides or proteins from primary and secondary nucleation processes or from fragmentation of mature fibrils [37]. The heterogeneous nature of the oligomers that form during the aggregation process is reflected in the report of many different types of these assemblies, including Aβ*56 [32], ADDLs [23], amylospheroids [40], annular protofibrils of αS [41], dimers [29], fibrillar oligomers [42], covalently stabilized oligomers [43], globulomers [44], kinetically trapped αS oligomers [45], mice 3×Tg-AD oligomers [46], prefibrillar oligomers [47], secreted oligomers [48], spherical amyloid intermediates [49], and zinc-stabilized Aβ40 oligomers [50] (Figure 1a).
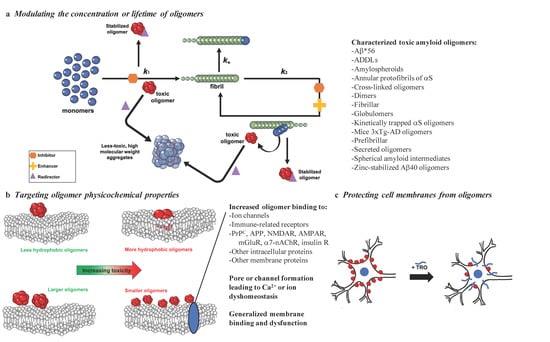
Figure 1.
Possible strategies to target the toxicity of misfolded protein oligomers. With the goal of reducing the toxicity of misfolded protein oligomers, promising methods include: (a) targeting oligomer populations by inhibiting (orange), enhancing (yellow), or redirecting (purple) the protein aggregation reaction (k1, primary nucleation; k2, secondary nucleation; k+, elongation), (b) targeting the properties responsible for the ability of oligomers to induce cell membrane dysfunction by binding to specific receptors or generally to cell membranes, or (c) modifying cell membranes to prevent disruptive oligomer interactions, as exemplified by trodusquemine (TRO). A subset of the toxic oligomers that have been characterized thus far [23,29,32,40,41,42,43,44,45,46,47,48,49,50] are listed in (a) to illustrate the striking heterogeneity in their structures, physiochemical properties, and biological activities.
These species have been found to cause cellular dysfunction in a wide range of manners, including by interacting with the lipid and protein components of the cell [31], resulting in the disruption of homeostatic mechanisms and the induction of cell death [51,52]. Oligomeric species of disease-related aggregated proteins are highly toxic owing to their structural properties, such as small size and high hydrophobicity [53,54,55,56,57], which makes them highly diffusible and prone to interact with cell proteins and membranes. Notably, a direct relationship has been established between the cytotoxicity of amyloid aggregates and their molecular weight [57]. In particular, it has been illustrated that small A11 antibody-positive aggregates reduced the viability of cells by up to 80% in comparison to fibrils, which only reduced cellular health by approximately 20% [31].
A great deal of effort has therefore been devoted to the discovery of inhibitors of protein aggregation, which suppress oligomer generation through a variety of mechanisms [58,59,60,61,62,63,64], leading to reports of hundreds of compounds that can potentially reduce the rate of protein self-assembly. It has been much more difficult, however, to demonstrate the efficacy of these compounds in the clinical setting to become FDA-approved, disease-modifying therapeutics [3,65]. This situation can be attributed, in part, to the limited understanding of the mechanisms by which aggregation occurs, of its effects on the different cell types that comprise specific vulnerable tissues, of the means by which these compounds modify the aggregation process, and, critically, to their administration at too late a stage in a clinical situation where the amyloid load has already reached high levels [37].
In this review, we analyze recent strategies to suppress the toxicity linked to neurodegenerative diseases with a critical focus on demonstrated molecular agents that have shown the ability to combat the toxicity caused by oligomers. In particular, we assess species that (1) modulate populations of oligomers by perturbing their respective protein aggregation reactions via inhibiting, enhancing, or redirecting its reactive flux network (Figure 1a and Section 3), (2) target directly the properties of oligomers linked to their ability to induce cell damage (Figure 1b and Section 4), (3) protect cell membranes by targeting oligomer interactions (Figure 1c and Section 5), or (4) preserve the health of cells through all of the above by regulation of the protein homeostasis network (see Section 6). These approaches demonstrate examples of potential therapeutics designed to suppress the negative effects of toxic oligomeric assemblies, as well as highlight the diversity of mechanisms by which amyloid aggregation can be targeted. The hope is that future work using these promising lead strategies will culminate in the development of therapeutically relevant treatments that can arrest the devasting effects of neurodegeneration by targeting specifically misfolded protein oligomers.
3. Targeting the Kinetics of Formation of Misfolded Protein Oligomers
Due to the transient nature of the oligomers produced during the aggregation processes, many therapeutics rely on modulating the kinetics of oligomer assembly to reduce the populations of oligomers that are responsible for inducing cell death. We describe here three modes by which the kinetics can be altered using therapeutics, including aggregation inhibition, acceleration, or redirection, where inhibition reduces oligomer concentrations, acceleration shifts the reactive flux towards the final fibrillar form to reduce the lifetime of oligomers, and redirection leads to the formation of off-pathway species that are less toxic than on-pathway oligomeric aggregates (Figure 1a).
3.1. Inhibiting Protein Aggregation to Reduce the Number of Oligomeric Species
A series of recent studies on the modulation of the kinetics of Aβ and αS oligomer assembly has revealed that this type of intervention can suppress the cytotoxicity associated with protein aggregation [56,58,61,62,63,64,66,67,68]. While the exact mechanism of inhibition varies significantly for the diverse array of small molecules, antibodies, molecular chaperones, and other agents that have demonstrated efficacy in delaying the aggregation process, the biological importance of inhibition is that the inhibitor prevents oligomer formation to reduce their populations by preventing some step in the aggregation process of their respective oligomer assembly reaction. Additionally, some of these molecules can not only interrupt the formation of oligomer aggregates, but also induce the disaggregation of toxic oligomers into inert monomers, therein preventing the accumulation of oligomeric species.
Following the development of a highly reproducible aggregation assay and the quantification of the microscopic steps implicit to the Aβ42 aggregation reaction [69,70], numerous studies have been reported that dissected the specific molecular processes inhibited by candidate therapeutic compounds. Aβ42 aggregation is characterized by the formation of oligomers by primary nucleation, where a relatively low population grow and become fibrillar aggregates. This process is stochastically limited until a small but critical concentration of fibrillar aggregates have formed, after which time monomer-dependent secondary nucleation on fibril surfaces dominates the proliferation of further Aβ42 aggregates through a positive feedback loop [58,69,71]. During secondary nucleation, monomeric proteins are catalytically converted to oligomeric aggregates on the fibril surface, and these oligomers can then grow into fibrillar aggregates. The nature of this multiplicative process dictates that the majority of oligomers are formed from secondary pathways during the aggregation reaction once the critical concentration threshold of fibrillar aggregates is exceeded [72,73]. We note that these kinetic assays require highly pure monomeric peptide as the starting material, as the presence of higher-order aggregates at the start of an aggregation reaction modulates the mechanism of protein assembly [56,58,74]. Moreover, all assays based on amyloid-specific fluorescent dyes have the potential to generate false positive results, in particular when a prospective therapeutic shares structural similarity to the probe being used. For example, some conformations of candidate inhibitors, including quinones of catecholamines, polyphenols, and flavonoids, have been found to quench the fluorescence of thioflavin T (ThT), an amyloid-specific dye commonly used in aggregation assays [75]. It is therefore important to run independent validation experiments to confirm the mechanism of action of a candidate therapeutic, which can be accomplished through the use of label-free methods, including atomic force microscopy (AFM) [66,67,76,77], transmission electron microscopy (TEM) [76], or dot-blot assays [66] on aggregation mixtures over time using conformation or sequence-specific antibodies in the absence and presence of centrifugation. In doing so, it is possible to gain mechanistic insight into the microscopic steps governing the protein aggregation reaction using ThT kinetics while safeguarding against the possibility of false positives that can result from molecules that bind to amyloid aggregates and induce the displacement or quenching of ThT or other amylophilic dyes rather than directly impacting amyloid fibril formation [74].
By adopting this type of approach, the anticancer drug bexarotene was characterized as a potent inhibitor of primary nucleation in the Aβ42 aggregation process [56], and the subsequent derivatization of this small molecule found that structurally similar agents could inhibit different and specific microscopic steps, to include primary and secondary nucleation, to varying degrees [58]. A related strategy leveraged the high specificity of rationally designed antibodies to generate a library scanning the sequence of monomeric Aβ42, and a subsequent kinetic analysis revealed that the antibodies selectively targeted specific microscopic steps in a similar fashion [62]. Additionally, phage display was used to optimize the antibody inhibitory power by targeting specifically fibril-dependent secondary nucleation [78].
A recent study has compared the mechanism of action of four clinical stage antibodies (aducanumab, bapinezumab, gantenerumab, and solanezumab), revealing that while they all modulate Aβ aggregation, they do so with different effects on Aβ oligomers [68]. Aducanumab was found to be particularly effective in binding Aβ fibrils, thereby blocking the production of Aβ oligomers by secondary nucleation [68], in addition to directing the fibrils themselves towards degradation. This antibody is currently under consideration by the FDA and, if approved, will be the first disease-modifying treatment for AD. By contrast, the other three antibodies did not show major effects on Aβ oligomer production. Gantenerumab and bapinezumab were found to bind both Aβ monomers and fibrils, and to inhibit mainly fibril elongation, and solanezumab was found to bind Aβ monomers, and to inhibit primary nucleation. Similarly to aducanumab, BAN2401, which is a humanized version of an antibody raised in mice immunized with Aβ42 protofibrils [79] and is in Phase 3 clinical trials, has the ability to potently inhibit Aβ protofibril accumulation in AD mice [80]. Of note, crenezumab has been reported to target different Aβ conformations, including oligomers, and to reduce oligomer levels in cerebrospinal fluid (CSF) [81,82].
Molecular chaperones have also been characterized to attenuate Aβ42 aggregation, including DNAJB6, which targets primary nucleation [83], the transport protein transthyretin, which inhibits both primary and secondary nucleation processes in Aβ aggregation [77]; a Brichos domain, which inhibits specifically the monomer-dependent secondary nucleation microscopic step [63]; and clusterin, which attenuates fibril elongation at low concentrations [71]. The latter process may not be therapeutically relevant, as inhibiting elongation can redirect monomeric proteins from fibril ends to their surface, where autocatalysis leads to the generation of additional oligomeric and fibrillar aggregates. By quantifying a structure–kinetic–activity relationship for drug discovery against Aβ42, it was found that hit compounds, including those with minimal inhibitory power in their original form, could be rationally optimized from a kinetic perspective to target the production of oligomers [84]. Collectively, these results facilitate hope that hit compounds, including small molecules and antibodies, can be further optimized using rationally designed processes to target specifically the oligomeric toxins that are implicit and central to protein aggregation [58,62,84].
It has also been reported that naturally occurring polyphenols, including epigallocatechin-3-gallate (EGCG), curcumin, resveratrol, gallic acid, and oleuropein, as well as naturally occurring polyphenolic biflavonoids, and polyphenol molecules containing two flavone groups, such as amentoflavone, bilobetin, sequoiaflavone, sotetsuflavone, podocarpuflavone, ginkgetin, isoginkgetin, and sciadopitysin, can reduce the cytotoxicity of oligomers by inhibiting protein aggregation [85,86]. Many studies have shown that both polyphenols and polyphenolic bioflavonoids interact with the aggregation profiles of Aβ and αS either by destabilizing existing β-sheet rich fibrils to create less toxic monomers or through directly inhibiting the formation of soluble oligomers [85]. Studies have revealed that polyphenolic biflavonoids interact with the Aβ aggregation reaction through inhibiting Aβ fibrillization and disaggregating pre-formed fibrils, significantly reducing their cytotoxicity. Notably, bioflavonoids possess hydrophobic and hydrophilic groups that are proposed to facilitate their binding to toxic aggregates through hydrophobic and aromatic interactions, as well as hydrogen bonds. Additionally, biflavonoids have an increased number of aromatic rings compared to monoflavonoids, which may potentially afford them increased effectiveness in inhibiting fibrillization [85,86]. The interaction of curcumin, a natural polyphenol, with αS can induce both the disaggregation of large pre-formed aggregates, effectively creating more non-toxic monomers, as well as the prevention of oligomerization by increasing the solubility of αS [86,87].
Aminosterols, a family of molecules including trodusquemine and squalamine, can inhibit and accelerate protein aggregation depending on the protein under investigation [61,64,76]. Trodusquemine and squalamine inhibit αS oligomer formation [61,64], while trodusquemine has been found to accelerate Aβ aggregation [76]. Both aminosterols inhibit αS aggregation by preventing the lipid-induced initiation process [61,64], therein attenuating the rate of the oligomerization reaction. Further studies of trodusquemine revealed its ability to also inhibit fibril amplification in αS aggregation, demonstrating its ability to exert a multi-step inhibition mechanism [64].
Another class of molecules exhibiting anti-amyloidogenic behavior are tetracyclines and anthracyclines [86,88,89]. Tetracyclines, notably tetracycline and doxycycline, and the anthracycline iododoxorubicin, can disrupt pre-formed oligomers and reduce their toxicity [88,89]. Due to structural similarities between tetracyclines and anthracyclines, it has been proposed that the inhibition tendencies of these molecules originate from their binding to fibril structures. Moreover, a variety of probe and diagnostic dyes, including Congo red, methylene blue, crystal violet, acid fuchsin, and fast green FCF, have revealed the inhibitory capacity of these molecules [86,90,91,92,93].
Finally, recent studies have shown that it may be possible to target Aβ42 and αS in their monomeric forms to prevent their aggregation through the use of small molecules [66] or nanobodies [67]. From a kinetic perspective, monomer sequestration would be expected to reduce primary nucleation, secondary nucleation, and elongation [83], and despite the challenges associated with binding an intrinsically disorder protein, these approaches hold promise towards combatting the formation and proliferation of oligomeric species. Collectively, all of the inhibitors described in this section can function to suppress amyloid toxicity by reducing the number of toxins that are formed during the aberrant protein aggregation reactions central to numerous proteins and peptides.
3.2. Enhancing Protein Aggregation to Reduce the Populations of Oligomeric Species
Studies focused on preventing oligomer cytotoxicity through controlling the kinetics of their formation have revealed a limited set of molecules that act in a net beneficial way by accelerating the protein aggregation reaction. Molecules acting through the acceleration mechanism are postulated to function by shortening the lifetime of the oligomeric phase of the aggregation reaction by stimulating their conversion to the less toxic fibrillar form. Despite the possibility of the enhancement strategy as a potential mode to suppress oligomer toxicity, a relatively small number of studies have demonstrated the enhancement of fibril formation with the result that toxicity is reduced in comparison to the body of work that is established for inhibitory compounds.
Congo red has been reported to promote β-sheet formation and Aβ aggregation, which may act to reduce the lifetime and corresponding populations of oligomeric species, but it has also been suggested to attenuate aggregation by stabilizing monomeric or partially folded intermediates of the peptide [86,93]. Another dye, methylene blue, has demonstrated differential effects dependent upon the protein under investigation, where it was found to mitigate Aβ oligomerization through the promotion of Aβ fibrillization [92] and to inhibit prion protein aggregation [94]. The nonsteroidal anti-inflammatory drug sulindac sulfide similarly was characterized to deplete toxic Aβ oligomers by enhancing the rate of fibrillization in vitro [95], and the luminescent-conjugated oligothiophene p-FTAA was determined to suppress the number of toxic Aβ aggregates by generating amyloid fibrils that in this case were less hydrophobic and more resistant to proteinase K digestion [96].
Through a creative strategy, the racemic combination of mirror-image enantiomers of Aβ42 was shown to cause an acceleration in its rate of aggregation with a reduced propensity to form soluble oligomers, therein demonstrating that the enantiomeric mixing stimulated the formation of non-toxic fibrils [97]. The orcein-related small molecule O4 has also been characterized for its ability to bind hydrophobic amino acids in Aβ and catalyze the Aβ polymerization reaction. Therein, it was demonstrated that the O4-mediated acceleration of fibril formation decreases the concentration of oligomers in vitro and that O4 prevented oligomer-induced dysfunction in hippocampal brain slices, therein revealing that shifting the reactive flux of the aggregation reaction can suppress toxicity [98].
Recently, it was shown that the aminosterol trodusquemine, a natural product originally isolated from the dogfish shark, stimulates predominantly secondary nucleation in Aβ42 aggregation by solving analytically for the perturbation induced by the molecule using the master equation formalism [76]. This kinetic model was validated by measuring the morphology of the final fibrillar products formed in the absence and presence of trodusquemine using high-resolution and phase-controlled atomic force microscopy [99,100,101], which demonstrated that the aminosterol generated a multitude of shorter and wider fibrils that were consistent with the predicted molecule-induced shift in reactive flux [76]. As discussed in more detail in Section 5 of this review, aminosterols also possess the ability to displace toxic oligomers of multiple proteins from cell membranes [61,64,76,102]. In a C. elegans model of AD, we found that trodusquemine decreased the toxicity induced by Aβ42 aggregation alongside stimulating the rate of its aggregation [76], in stark contrast to its multistep mechanism of inhibition that was observed in vitro and in vivo with respect to αS aggregation [64]. These results for trodusquemine suggest that both the conversion of Aβ42 oligomeric aggregates to less toxic, higher-order fibrillar forms, and also their displacement from cell membranes can work in tandem with a combinatorial effect to suppress oligomer cytotoxicity [76,102].
3.3. Redirecting the Protein Aggregation Process to Sequester Oligomeric Species
In this section, we focus on potential therapeutics that can modulate the aggregation reaction in a way that redirects oligomeric aggregates towards less toxic, higher-order forms that are often off-pathway, e.g., high molecular weight aggregates, rather than mature fibrils. Despite the differing end species in these two processes, both off-pathway high molecular weight aggregates and fibrils can achieve the same result of reducing the lifetime or populations of oligomeric aggregates. Specifically, the formation of higher-order species by the redirection mechanism can consume toxic oligomeric intermediates, thereby depleting the populations of oligomers that are present and able to induce cellular dysfunction.
Resveratrol has the ability to redirect three separate conformers in the aggregation reaction to form unstructured off-pathway aggregates that are both non-toxic and of a high molecular weight [103]. These non-toxic aggregates can be formed by resveratrol from the soluble oligomers, fibrillar intermediates, and amyloid fibril conformers of the aggregation reaction, therein reducing toxicity by limiting the number of oligomers and other conformers that can participate in the aggregation reaction.
In addition to the polyphenol resveratrol, EGCG also reduces toxicity by remodeling mature amyloid fibrils and aggregates of Aβ and αS [104,105,106]. Studies show that EGCG remodels the large, mature Aβ and αS fibrils into unstructured and nontoxic aggregates, decreasing the amount of amyloidogenic species without creating a toxic product or reforming toxic oligomers in its mechanism of action [105]. A similar mechanism has also been displayed for other polyphenols in addition to resveratrol and EGCG, including trihydroxybenzophenone in tau protein aggregation and myricetin in Aβ aggregation [107,108].
The biological consequences of forming large deposits of aggregates in vivo as a result of redirecting the aggregation reaction away from fibrillar species, as well as the ability of the protein homeostasis machinery of the cell to store or degrade such high molecular weight aggregates, such as through the clearance of Aβ peptides through a proteasome-dependent mechanism, are processes likely to play an important role in determining the physiological relevance of this approach in patients afflicted by neurodegenerative disease. For example, fibrils have been shown to act as reservoirs of oligomers, thereby storing them for their ultimate release [37].
Collectively, there is a wide range of heterogeneity with respect to the function and specificity of molecular agents designed to disrupt the protein aggregation process with the result that toxicity is reduced through inhibiting, accelerating, or redirecting the aggregation of specific proteins. As can be observed by the appearance of certain molecules in the various classes of kinetic modulators of aggregation described herein, the Aβ, αS, and tau protein aggregation reactions have many steps by which molecules can interact with and perturb the direction and speed of the reaction in ways that can reduce oligomer toxicity. As described for small molecule inhibitors [58,84], rational strategies to optimize molecular potency could lead to species that can more efficiently accelerate or redirect the aggregation reaction with the potential to further reduce toxicity associated with the oligomeric state.
4. Targeting the Physicochemical Properties of Misfolded Protein Oligomers
Owing to the transient and metastable nature of oligomeric species formed during an aggregation reaction in vitro or in vivo, these aggregates represent a very small relative proportion of the total protein concentration. For example, it has been shown that oligomeric aggregates can reach a maximum of 1% of the total monomer concentration during an in vitro aggregation process for Aβ42 at relevant concentrations of the peptide [69,73]. In addition to their short lifetimes, oligomers of various proteins are highly heterogeneous in terms of their structures, morphologies, mechanisms of formation, and biological activates [109]. In light of these facts, a multitude of studies have used chemical or physical means to isolate or stabilize oligomeric aggregates in order to facilitate their investigation at pragmatic timescales and concentrations [45,50,110,111,112].
The exposure of hydrophobic patches and size are structural determinants of oligomer toxicity, and a high level of hydrophobicity and small size are associated with the ability to cause cellular dysfunction (Figure 1b) [31,37,57,113,114,115]. Multiple studies have established the contribution of increased solvent-exposed hydrophobicity to the toxicity of Aβ oligomers [57,116,117]. Additionally, the size of the oligomers themselves is a key factor, with larger oligomers being less toxic than their smaller counterparts [57,117,118]. Consequently, numerous studies have examined methods for reducing the solvent-exposed hydrophobicity and increasing the size of oligomers for therapeutic purposes. We therefore focus herein on the size–hydrophobicity–toxicity relationship.
By generating 12 oligomer variants of the N-terminal domain of the E. coli HypF protein (HypF-N), it was demonstrated that the induced variations in oligomer size and hydrophobicity rationalized the differential empirically observed cellular dysfunction caused by the aggregates. Moreover, an equation was proposed based on size and solvent-exposed hydrophobicity that could predict the toxicity of oligomers using relatively high-throughput methods [57]. This equation was recently leveraged to explain the effects of rationally designed antibodies that were used as molecular tools to probe the size–hydrophobicity–toxicity relationship for zinc-stabilized Aβ40 oligomers [50], where concomitant increases in size and hydrophobicity induced by the antibodies were predicted and observed not to change the toxicity of the stabilized oligomers due to their offsetting effects [117]. Moreover, two forms of αS oligomers have been resolved to embed themselves in cell membranes to different degrees based upon their levels of solvent-exposed hydrophobicity, therein disrupting membrane integrity only in the case of the embedded variant that was more significantly more hydrophobic [55].
The concept of reducing the cellular toxicity of oligomeric species by their sequestration into larger, innocuous species with reduced diffusional mobility has been accomplished previously through the use of molecular chaperones [113,114,119,120] and small molecules [103,106]. In particular, a variety of chaperone and non-chaperone proteins have been shown to suppress the toxicity of oligomers by promoting their assembly into bigger and less mobile species [114], including Hsp27 with Aβ42 oligomers [120] and other molecular chaperones with Aβ42, IAPP, and HypF-N oligomers [113]. In some cases, the mechanism of action of these molecular chaperones involves the inhibition of the interaction of oligomers with cellular membranes [121,122]. Given their propensity to target oligomeric intermediates, many of these chaperones also modulate the kinetics of protein polymerization, as discussed in the preceding sections. Indeed, multiple chaperones can play an important role in regulating the aggregation reactions innate to AD [63,123,124,125]. Molecular chaperones, such as clusterin and crystallin, also protect cells from the aggregation of misfolded proteins by modulating the folding of their respective substrate proteins [126].
Heat shock proteins are a class of molecular chaperones that have received attention for their ability to reduce Aβ42 cytotoxicity; HspB1 and Hsp70 have both been shown to modulate the cytotoxicity of Aβ42 in vitro [120,127], although they operate through different mechanisms. Of the two, HspB1 appears to function by modulating the size–hydrophobicity–cytotoxicity relationship, sequestering oligomers into relatively inert aggregates [120], whereas Hsp70 appears to disrupt the primary nucleation process of oligomerization [127] by interacting with the hydrophobic residues of Aβ42 [128]. Since heat shock proteins are found to be co-located with amyloid plaques in the brains of AD patients, it is possible that increasing their endogenous concentration in vivo could improve outcomes for patients. In support of this notion, transgenic mouse models for AD that are deficient in heat shock proteins express significantly worse symptoms than their counterparts who do have the ability to produce heat shock proteins [129], suggesting that these chaperones play an important role in regulating Aβ42 aggregation.
In addition to heat shock proteins, a number of other species have shown promise in modulating the size or hydrophobicity of Aβ42 aggregates. For example, monomeric human transthyretin has been shown to produce large co-aggregates with reduced cytotoxicity and inhibit nucleation processes [130]. Clusterin can also bind to hydrophobic patches in Aβ42 oligomers and inhibit its aggregation [131]. The aminosterol trodusquemine accelerates the aggregation of Aβ42 in vitro, reducing the concentration of toxic oligomers in solution and encouraging the formation of fibrillar aggregates [76]. Analogous to the case of the rationally designed antibodies against oligomers of Aβ40 stabilized by zinc ions, trodusquemine at super-stoichiometric concentrations that are not physiologically relevant was observed to linearly increase the size and hydrophobicity of the stabilized aggregates. These physiochemical chemical changes would therefore be predicted not to change the toxicity of the oligomers [117], and it was shown that these biophysical parameters were not overtly changed at physiological concentrations [102]. Collectively, these studies support the existence of a size–hydrophobicity–toxicity relationship and suggest that targeting these biophysical parameters can result in alleviating the cytotoxicity innate to protein misfolded oligomers by reducing oligomer binding to cell membranes or specific receptors.
5. Targeting the Aberrant Interactions of Misfolded Protein Oligomers
A relatively distinct mechanism of counteracting the cytotoxicity of misfolded protein oligomers is through the use of molecules to prevent their deleterious interactions with cell membranes. Oligomeric species of Aβ42 are thought to damage cells by binding to their membranes, therein forming ion channels and disrupting the integrity of the cell membrane [132,133]. Recent work suggests that cellular membranes can act as nucleation sites for Aβ42 oligomerization at physiological concentrations of the protein [134]. Cell membranes themselves are highly heterogenous in their compositions of various biomolecules, including sterols, fatty acids, lipids, and scaffolding proteins, amongst many other molecules, and it has been suggested that age-related changes in membrane lipid rafts correlate to the loss of neuronal function observed in neurodegenerative disease [135]. Furthermore, Aβ42 oligomers bind to a wide variety of receptors on membranes, which has contributed significantly towards the challenge of blocking the effects of oligomer binding to membranes by targeting a specific receptor or set of receptors [136].
The environment of the cellular membrane, with its heterogeneous regions of differing lipids and receptors, provides evidence for the proposed general mechanism of oligomeric membrane interactions. For one, extensive research has demonstrated that the ganglioside GM1 (and ganglioside GT1b to a lesser degree) amplifies Aβ42 cytotoxicity by promoting its aggregation cascade [137]. Additionally, evidence suggests lipid rafts as regions of increased oligomer interaction with the membrane, as well as the site of the cleavage of amyloid precursor protein (APP) [138]. Furthermore, studies have identified relatively expanded regions of the membrane characterized by increased cholesterol content as more favorable for the insertion of APP [139], which may be related to the increased distribution of cholesterol on the exterior of lipid rafts as cells age [138]. Moreover, the sphingolipid sphingomyelin is thought to be a driver of lipid raft formation due to its interaction with cholesterol, pointing to its potential influence on Aβ42–membrane interactions [140]. The aggregation of Aβ42 activates protein homeostasis systems in the cell, which downregulates sphingomyelin production, further implicating sphingomyelin and by extension lipid rafts in the progression of AD [141]. Finally, an increase in sphingomyelin synthesis is associated with oxidative stress and increased concentrations of long-chain ceramides, which can induce cellular stress responses [142]. These interactions, when taken together, indicate that while oligomer binding to specific membrane receptors does indeed occur, the mechanism of membrane interaction appears to be more generic in nature.
While the structures of Aβ42 oligomers are currently not well resolved, with many different shapes and structures having been characterized simply in the case of Aβ oligomers, such as dimers [143,144,145], timers [146,147], Aβ*56 oligomers [32,148], ADDLs [25,26,149], and amylospheroids as spherical Aβ oligomers [150], where each type of oligomer can exhibit differing biological effects [151], a common principle exists: toxic oligomeric species bind to cellular membranes and disrupt membrane integrity [37]. Due to their high degree of solvent-exposed hydrophobicity, oligomers embed into the interior of the membrane, remaining relatively immobile with respect to the rest of the membrane, with a majority of oligomers remaining in place rather than diffusing throughout the membrane [152,153]. The oligomers, once embedded, disrupt the function of the cell by increasing the molar volume of acyl chains in the interior of the membrane, thereby increasing the conductivity of the membrane, most notably to calcium ions [53,154]. Additionally, the method of binding of oligomers to cell membranes has been observed to be heterogeneous in nature, with the same peptide binding to different regions of the membrane based on its specific quaternary structure, which further points toward a general mechanism by which oligomers interact with membranes, rather than one beholden to a specific lipid moiety [152,153]. Therefore, molecules that displace misfolded protein oligomers from the cell membrane stand to potentially disrupt both aggregation pathways and directly protect cells from the effects of already assembled oligomers [61,155].
One promising molecule that operates according to this mechanism is again the aminosterol trodusquemine, which can cross the blood–brain barrier [156]. Trodusquemine displaces a variety of toxic oligomers from cell membranes in vitro, including those of Aβ40, Aβ42, HypF-N, and αS, suggesting that the aminosterol-induced protection of cell membranes from oligomers occurs through a generic mechanism (Figure 1c) [64,76,102]. This oligomer displacement hypothesis for the mechanism by which trodusquemine attenuates oligomeric cytotoxicity is also supported by research observing a marked difference in the impact of trodusquemine on the aggregation of Aβ42 and αS, where the former was enhanced and the latter inhibited, coupled with similar reductions in the cytotoxicity of isolated or stabilized oligomers [64,76,102]. These studies together suggest that trodusquemine is able to outcompete oligomers for binding sites on cell membranes. Importantly, this mechanism does not seem to effect the normal binding of monomeric αS to cell membranes [64].
Similar to trodusquemine, other aminosterols have been shown to function through a comparable mechanism. Specifically, squalamine reduces oligomer toxicity in a similar fashion [61]. In addition to the aminosterol family, cholesterol has also been shown to impact the binding of Aβ40 oligomers to cellular membranes at super-stoichiometric concentrations [157], and a clear correlation was found between the concentration of GM1 and binding and subsequent toxicity of Aβ42 and HypF-N oligomers [158]. Additionally, the small molecule anle138b appears to affect Aβ42 oligomers in the cell membrane, binding to the pores Aβ42 creates in membranes and thereby preventing the loss of membrane integrity normally associated with Aβ42 membrane interactions [159].
Collectively, these studies illustrate the importance of membrane composition with a wide range of biomolecules and show that molecules that interact directly with cell membranes, rather than binding oligomeric aggregate themselves, represent an approach distinct from the previous strategies. By bypassing the heterogeneity innate to protein oligomerization and the host of specific receptors that may play a role in the oligomer-mediated dysfunction of cell membranes via targeting membranes in a ubiquitous manner, molecules with a common therapeutic effect on a variety of toxic oligomers have been identified [102]. Further exploration of molecules that compose and interact with membranes, therefore, could be a promising avenue by which to arrive at cellular targets for oligomeric species and potential countermeasures against their deleterious effects.
6. Targeting Misfolded Protein Oligomers by Potentiating the Protein Homeostasis System
In order to be functional, the proteins that constitute the cellular proteome need to fold properly and remain soluble. The maintenance of a functional proteome is achieved through the action of the protein homeostasis system, which regulates the synthesis, folding, transport, and degradation of proteins, guaranteeing their functionality and mitigating their aggregation [160]. This system assists in protein folding, remodeling misfolded proteins back to folding intermediates, disaggregating intermediate aggregates, and assisting in protein turnover or aggregate removal via the ubiquitin–proteasome or autophagosomal–lysosomal systems (Figure 2) [51,161]. This quality control system declines over time as a function of ageing, often resulting in age-related comorbidities [161,162]. Over the course of a life, the gradual accumulation of certain mutations can result in a larger pool of aggregation-prone species. Evidence from model organisms suggests that as genetic and epigenetic longevity controls affect a downregulation in the expression of molecular chaperones, it becomes increasingly difficult for the protein homeostasis network to adequately keep pace with the growing demand of proteotoxic species [51].
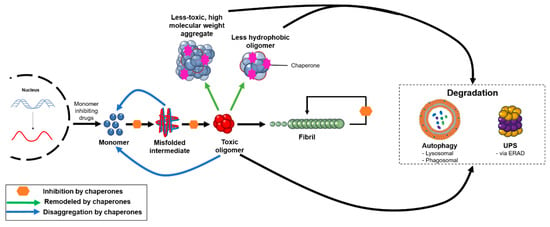
Figure 2.
The protein homeostasis system targets oligomeric aggregates through a variety of mechanisms. Molecular chaperones can redirect misfolded or aggregated proteins back to the monomeric state, remodel pre-formed oligomers into less hydrophobic or higher molecular weight aggregates that are less toxic, and inhibit various microscopic steps in the protein aggregation process. These aggregates can be targeted for degradation by autophagic and proteasomal processes, and the degree to which the body can eliminate protein aggregates plays a critical role in the onset and progression of neurodegenerative disease. In addition, therapeutics are under development to reduce the production of monomeric proteins, such as through the use of BACE1 inhibitors in AD.
The decline of the protein homeostasis network represents both a trigger and a target in a wide range of protein misfolding diseases. A study in C. elegans showed that naturally the vast majority of the proteins are expressed at concentrations close to their intrinsic solubility limits [15], and these supersaturated proteins form a metastable subproteome that is highly susceptible to aggregation related to specific neurodegenerative diseases [13,16].
Primary regulators of the protein homeostasis system are molecular chaperones, which constitute a family of structurally and functionally diverse proteins with varying mechanisms of action based on cell-specific expression [163]. According to a recent mapping, the human chaperome consists of 332 genes, of which 88 are molecular chaperones and 244 are co-chaperones [164]. As humans age, the quality of these control measures often degrades leading to the formation and propagation of toxic protein aggregates, specifically intermediate oligomers that are commonly found in neurodegenerative diseases. While the role that molecular chaperones play in maintaining protein homeostasis is well documented [165,166], recent studies have shown that they also exhibit cytoprotective characteristics through their ability to neutralize toxicity associated with aberrant protein oligomers present in neurodegenerative diseases [114,164]. Numerous studies have shown the ability of molecular chaperones to interact with oligomers, and fibrils, to neutralize their associated toxicity by preventing cell membrane interaction and inducing the formation of larger, non-toxic species [114,167,168], and others have demonstrated the ability to inhibit protein aggregation and therefore oligomer formation [63,127]. While many mechanisms of action of molecular chaperones are not fully resolved, they may be utilized therapeutically to reduce the deleterious effects of oligomers in protein misfolding diseases. Recently, the S100 family of proteins have been investigated for their role in AD, based on their ability to influence the Aβ aggregation pathway and its cellular receptors. While evidence shows that their function depends on the stage of pathology and related levels of Aβ and S100 proteins, it is possible that the S100 family could function as molecular chaperones to suppress oligomer toxicity through several mechanisms [169,170]. Development of chaperone-targeted therapeutics is limited by their stability, oral bioavailability, and blood–brain barrier permeability. Current therapeutic strategies targeting molecular chaperones include development of small molecule inhibitors or activators, viral and non-viral mediated delivery of molecular chaperones or their activators, and pharmacological chaperones [171,172,173,174,175,176].
This type of approach may be particularly helpful in the case of neurodegenerative diseases such as PD, which have been linked to the dysfunction of lipid metabolism resulting in neuroinflammation [177]. The involvement of the stress-induced unfolded protein response (UPR) of the endoplasmic reticulum (ER) is an active area of investigation, and molecular chaperones are being considered to potentiate the folding capacity of proteins in the endoplasmic reticulum or mitochondria [161]. Targeting such stress responses by modulating signaling pathways could be therapeutically efficacious against the widespread toxicity caused by protein aggregation, as stress responses can vary significantly amongst different cell types.
A major therapeutic approach towards reducing oligomer numbers is to stimulate their removal from the cellular milieu. A promising target is autophagy, a lysosomal degradation pathway already investigated in relation to diverse pathologies including cancer and infections [178]. The pathway itself also deteriorates with age, which contributes to the myriad of comorbidities in elderly populations [179,180], and stimulation of the pathways that preserve autophagy may demonstrate a protective effect against protein aggregation [181]. For example, rapamycin has been demonstrated to inhibit mTOR signaling and to reduce the amount of amyloid plaques and tau tangles in an AD mouse model [182,183]. Contrasting studies, however, have suggested that inducing autophagy could cause deleterious changes in Aβ production, and it is currently unclear if alterations in autophagy are causative, protective, or simply a result of AD [184]. In addition to the autophagosomal–lysosomal pathway, aggregate removal can also be potentiated by the ubiquitin–proteasome system (UPS) [185]. This pathway requires ATP usage to unfold proteins, and in both systems, molecular chaperones recognize misfolded proteins and hold them in conformations that can be degraded [161,186].
In addition to enhancing the clearance of oligomer aggregates, reducing the concentration of Aβ through the use of secretase inhibitors or regulating its metabolism by ApoE-modifying therapeutics are active areas of clinical research [187,188]. Apolipoprotein E4 (ApoE4) is one of the three major isoforms of apolipoprotein E (ApoE) implicated in neurodegenerative disorders, in particular AD. These lipoproteins play an essential role in lipid metabolism, as well as maintaining normal brain function [189]. Studies have shown a strong association between ApoE, especially ApoE4, and Aβ accumulation in the brain leading to AD through reducing microglial function, lipid transport, synapse integrity and stability, energy metabolism, protein aggregation, and inhibition of protein clearance [190]. In these cases, reducing the populations or lifetimes of oligomers by acting on protein expression or degradation could prove therapeutically beneficial by indirectly targeting oligomeric species.
7. Discussion
The development of therapeutics for the prevention or treatment of protein misfolding diseases is fraught with challenges. For one, as misfolded protein oligomers are intermediates in the aggregation process, their structures are transient and heterogeneous. Furthermore, there are many interconnected pathways in the aggregation process in each disease, making the exact nature of the cytotoxic aggregates difficult to discern. For similar reasons, the oligomers themselves are also difficult to control in vitro, making estimates of oligomer concentrations and cytotoxic properties difficult to compare across different experimental protocols. By increasing our understanding of the mechanisms of action of candidate compounds targeting oligomers, we anticipate that it will become possible to develop more clinically relevant therapeutics in comparison to the numerous ones described thus far that target non-specifically the protein aggregation reaction and have failed in the clinical setting [3].
The molecules with the different mechanisms of actions outlined in this review (Figure 1 and Figure 2) have advantages and disadvantages. We have highlighted numerous candidate compounds for their ability to target amyloid oligomers in order to illustrate the current state of the field. To broaden the scope of this review and to show more examples of amyloid oligomer targeting species that fit within these categorizations, Table 1 summarizes compounds discussed in this review, as well as many more not mentioned explicitly in the text, for their ability to impact oligomers and reduce their toxicity in vitro, in model organisms, and, in some cases, in clinical trials. For the compounds in Table 1, the National Institutes of Health (NIH) U.S. National Library of Medicine’s clinical trial database (ClinicalTrials.gov) was used to determine the statuses of the various listed molecules that have been, or are currently in, clinical trials.

Table 1.
Examples of compounds, including those that are currently in clinical trials, reported to target misfolded protein oligomers.
Modulating the kinetics of oligomer assembly by either acceleration, inhibition, or redirection has the advantage of working directly with the protein in question, and an extensive list of promising molecules have already been identified for their ability to suppress the toxicity innate to amyloid aggregation. Molecules that accelerate the aggregation cascade or redirect it toward the formation of larger, less cytotoxic aggregates, however, could lead to the long-term stabilization of sizable aggregates in the brain, which could cause complications over time.
Molecular chaperones, including those that modulate the size–hydrophobicity–toxicity relationship, have been intensively studied, and many promising candidates have been identified, including those that are dysfunctional in AD patients. However, these molecules are relatively large and typically unable to cross the blood–brain barrier, thus hindering their usefulness as therapeutics. Additionally, there is the question of whether these molecules, and other therapeutics that target the protein directly, will remain effective at relatively high concentrations of the protein, as most patients with neurodegenerative diseases are first diagnosed at an advanced stage of the disease.
Molecules that interact with oligomers on cell membranes have the advantage of functioning outside the aggregation cascade altogether, therein protecting cells in a general way, independent of the stage of oligomerization. Additionally, some of these molecules function through a generic mechanism and are therefore able to displace multiple oligomeric species, indicating their potential for use in treating multiple diseases [102]. However, these molecules also face challenges due to their mechanism of interaction, as they have varied effects on the aggregation cascades of different proteins related to neurodegenerative disease [61,64,76]. Additionally, since many of these molecules, such as trodusquemine, function by competing with oligomers at the site of membrane binding, their effectiveness would rely on their continual presence of the molecule for duration of the life of a patient.
Finally, modulation of the protein homeostasis network has proven therapeutically challenging, and strategies to reduce monomeric concentrations of disordered proteins have in some cases resulted in worsened cognition in AD patients, such as those for secretase inhibitors [187], highlighting the challenges of regulating the functional forms of the target proteins. For all described strategies, specificity for oligomeric aggregates remains a critical challenge in the design of effective therapeutics [86].
Looking forward, these classifications of potential therapeutic molecules illustrate that it may be possible to leverage combination therapies to combat neurodegeneration, as seen in many cancer treatments. Combining two molecules with different mechanisms of action has the potential to operate along the advantages of both methods, without engaging in competition.
Although not discussed in depth in this review, the treatment of neurodegenerative disease has proven highly challenging in significant part due to limited diagnostic assays that can detect the various pathologies early in a patient’s life. Recent efforts to rationally design antibodies against oligomeric Aβ42 have illustrated the possibility of using these highly specific molecular tools to advance diagnostics [191]. Moreover, advances in cryo-electron microscopy have recently led to the characterization of patient-derived fibrils of the tau protein in AD [192], Pick’s disease [193], chronic traumatic encephalopathy [194], corticobasal degeneration [195,196], and αS filaments from multiple system atrophy [197]. For the various tau fibrils in the above pathologies, discrete core structures were observed in a disease-specific manner. With recent advances in the resolution of this approach [198], and the possibility of isolating a homogenous population of oligomers from brain tissue, cryo-electron microscopy may in the future lead to a better understanding of the precise molecular nature of oligomers implicit in specific neurodegenerative diseases.
In conclusion, the efficacy of the different approaches described here will become clearer as the field advances to understand and combat the toxic effects of oligomers in neurodegenerative diseases.
Author Contributions
Writing, reviewing, and editing, all authors (R.P.K., A.K.W., N.R.B., J.E.H., L.T.N., K.L., B.M., M.V., and R.L.); conceptualization and supervision, M.V. and R.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by a Faculty Research Fund grant from the United States Military Academy, West Point (to R.L.), a DTRA Service Academy Research Initiative grant (HDTRA1033862 to R.L.), the Cambridge Centre for Misfolding Diseases (B.M. and M.V.), and the Wellcome Trust (203249/Z/16/Z to M.V.).
Conflicts of Interest
M.V. is a co-founder and B.M. is an employee of Wren Therapeutics Ltd., which is pursuing inhibitors of protein misfolding and aggregation. The remaining authors declare no conflicts of interest. The funders had no role in the design or writing of this review or in the decision to publish it. The views expressed herein are those of the authors and do not reflect the position of the United States Military Academy, the Department of the Army, or the Department of Defense.
Abbreviations
| αS | α-synuclein |
| Aβ | amyloid-β peptide |
| Aβ40 | 40-residue form of the amyloid-β peptide |
| Aβ42 | 42-residue form of the amyloid-β peptide |
| ADDLs | Aβ-derived diffusible ligands |
| AFM | atomic force microscopy |
| ApoE | Apolipoprotein E |
| APP CSF | amyloid precursor protein cerebrospinal fluid |
| EGCG | epigallocatechin-3-gallate |
| ER | endoplasmic reticulum |
| GM1 | monosialotetrahexosylganglioside |
| HypF-N | N-terminal fragment of the HypF protein |
| IDP | intrinsically disordered protein |
| k1 | primary nucleation (microscopic step) |
| k2 | secondary nucleation (microscopic step) |
| k+ | elongation (microscopic step) |
| PD | Parkinson’s disease |
| UPR | unfolded protein response |
| UPS | ubiquitin proteasome system |
| TEM | transmission electron microscopy |
| ThT | thioflavin T |
| TRO | trodusquemine |
References
- Dobson, C.M. Protein folding and misfolding. Nature 2003, 426, 884–890. [Google Scholar] [CrossRef]
- McFarthing, K.; Buff, S.; Rafaloff, G.; Dominey, T.; Wyse, R.K.; Stott, S.R.W. Parkinson’s disease drug therapies in the clinical trial pipeline: 2020. J. Park. Dis. 2020, 10, 757–774. [Google Scholar] [CrossRef]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimers Res. Ther. 2014, 6, 37. [Google Scholar] [CrossRef]
- International, A.D. World Alzheimer Report 2019: Attitudes to Dementia. Available online: https://www.alz.co.uk/research/world-report-2019 (accessed on 30 September 2020).
- Knowles, T.P.J.; Vendruscolo, M.; Dobson, C.M. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 2014, 15, 384–396. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.-Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-Synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. Berl. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Dobson, C.M. Protein misfolding, evolution and disease. Trends Biochem. Sci. 1999, 24, 329–332. [Google Scholar] [CrossRef]
- Knowles, T.P.; Fitzpatrick, A.W.; Meehan, S.; Mott, H.R.; Vendruscolo, M.; Dobson, C.M.; Welland, M.E. Role of intermolecular forces in defining material properties of protein nanofibrils. Science 2007, 318, 1900–1903. [Google Scholar] [CrossRef]
- Tartaglia, G.G.; Pechmann, S.; Dobson, C.M.; Vendruscolo, M. Life on the edge: A link between gene expression levels and aggregation rates of human proteins. Trends Biochem. Sci. 2007, 32, 204–206. [Google Scholar] [CrossRef]
- Baldwin, A.J.; Knowles, T.P.J.; Tartaglia, G.G.; Fitzpatrick, A.W.; Devlin, G.L.; Shammas, S.L.; Waudby, C.A.; Mossuto, M.F.; Meehan, S.; Gras, S.L.; et al. Metastability of native proteins and the phenomenon of amyloid formation. J. Am. Chem. Soc. 2011, 133, 14160–14163. [Google Scholar] [CrossRef]
- Vecchi, G.; Sormanni, P.; Mannini, B.; Vandelli, A.; Tartaglia, G.G.; Dobson, C.M.; Hartl, F.U.; Vendruscolo, M. Proteome-wide observation of the phenomenon of life on the edge of solubility. Proc. Natl. Acad. Sci. USA 2020, 117, 1015–1020. [Google Scholar] [CrossRef]
- Ciryam, P.; Kundra, R.; Morimoto, R.I.; Dobson, C.M.; Vendruscolo, M. Supersaturation is a major driving force for protein aggregation in neurodegenerative diseases. Trends Pharmacol. Sci. 2015, 36, 72–77. [Google Scholar] [CrossRef]
- Ciryam, P.; Tartaglia, G.G.; Morimoto, R.I.; Dobson, C.M.; Vendruscolo, M. Widespread aggregation and neurodegenerative diseases are associated with supersaturated proteins. Cell Rep. 2013, 5, 781–790. [Google Scholar] [CrossRef]
- Freer, R.; Sormanni, P.; Vecchi, G.; Ciryam, P.; Dobson, C.M.; Vendruscolo, M. A protein homeostasis signature in healthy brains recapitulates tissue vulnerability to Alzheimer’s disease. Sci. Adv. 2016, 2, e1600947. [Google Scholar] [CrossRef]
- Freer, R.; Sormanni, P.; Ciryam, P.; Rammner, B.; Rizzoli, S.O.; Dobson, C.M.; Vendruscolo, M. Supersaturated proteins are enriched at synapses and underlie cell and tissue vulnerability in Alzheimer’s disease. Heliyon 2019, 5, e02589. [Google Scholar] [CrossRef]
- Fu, H.; Possenti, A.; Freer, R.; Nakano, Y.; Hernandez Villegas, N.C.; Tang, M.; Cauhy, P.V.M.; Lassus, B.A.; Chen, S.; Fowler, S.L.; et al. A tau homeostasis signature is linked with the cellular and regional vulnerability of excitatory neurons to tau pathology. Nat. Neurosci. 2019, 22, 47–56. [Google Scholar] [CrossRef]
- Kundra, R.; Dobson, C.M.; Vendruscolo, M. A cell-and tissue-specific weakness of the protein homeostasis system underlies brain vulnerability to protein aggregation. iScience 2020, 23, 100934. [Google Scholar] [CrossRef]
- Um, J.W.; Nygaard, H.B.; Heiss, J.K.; Kostylev, M.A.; Stagi, M.; Vortmeyer, A.; Wisniewski, T.; Gunther, E.C.; Strittmatter, S.M. Alzheimer amyloid-β oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci. 2012, 15, 1227–1235. [Google Scholar] [CrossRef]
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA 1998, 95, 6448–6453. [Google Scholar] [CrossRef]
- Lambert, M.P.; Viola, K.L.; Chromy, B.A.; Chang, L.; Morgan, T.E.; Yu, J.; Venton, D.L.; Krafft, G.A.; Finch, C.E.; Klein, W.L. Vaccination with soluble Abeta oligomers generates toxicity-neutralizing antibodies. J. Neurochem. 2001, 79, 595–605. [Google Scholar] [CrossRef]
- Gong, Y.; Chang, L.; Viola, K.L.; Lacor, P.N.; Lambert, M.P.; Finch, C.E.; Krafft, G.A.; Klein, W.L. Alzheimer’s disease-affected brain: Presence of oligomeric Aβ ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc. Natl. Acad. Sci. USA 2003, 100, 10417–10422. [Google Scholar] [CrossRef]
- Klein, W.L. Aβ toxicity in Alzheimer’s disease: Globular oligomers (ADDLs) as new vaccine and drug targets. Neurochem. Int. 2002, 41, 345–352. [Google Scholar] [CrossRef]
- McLean, C.A.; Cherny, R.A.; Fraser, F.W.; Fuller, S.J.; Smith, M.J.; Beyreuther, K.; Bush, A.I.; Masters, C.L. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann. Neurol. 1999, 46, 860–866. [Google Scholar] [CrossRef]
- Walsh, D.M.; Hartley, D.M.; Kusumoto, Y.; Fezoui, Y.; Condron, M.M.; Lomakin, A.; Benedek, G.B.; Selkoe, D.J.; Teplow, D.B. Amyloid beta-protein fibrillogenesis. Structure and biological activity of protofibrillar intermediates. J. Biol. Chem. 1999, 274, 25945–25952. [Google Scholar] [CrossRef]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef]
- Tsigelny, I.F.; Crews, L.; Desplats, P.; Shaked, G.M.; Sharikov, Y.; Mizuno, H.; Spencer, B.; Rockenstein, E.; Trejo, M.; Platoshyn, O.; et al. Mechanisms of hybrid oligomer formation in the pathogenesis of combined Alzheimer’s and Parkinson’s diseases. PLoS ONE 2008, 3, e3135. [Google Scholar] [CrossRef]
- Bemporad, F.; Chiti, F. Protein misfolded oligomers: Experimental approaches, mechanism of formation, and structure-toxicity relationships. Chem. Biol. 2012, 19, 315–327. [Google Scholar] [CrossRef]
- Lesné, S.; Koh, M.T.; Kotilinek, L.; Kayed, R.; Glabe, C.G.; Yang, A.; Gallagher, M.; Ashe, K.H. A specific amyloid-beta protein assembly in the brain impairs memory. Nature 2006, 440, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Bucciantini, M.; Giannoni, E.; Chiti, F.; Baroni, F.; Formigli, L.; Zurdo, J.; Taddei, N.; Ramponi, G.; Dobson, C.M.; Stefani, M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 2002, 416, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Cleary, J.P.; Walsh, D.M.; Hofmeister, J.J.; Shankar, G.M.; Kuskowski, M.A.; Selkoe, D.J.; Ashe, K.H. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat. Neurosci. 2005, 8, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Winner, B.; Jappelli, R.; Maji, S.K.; Desplats, P.A.; Boyer, L.; Aigner, S.; Hetzer, C.; Loher, T.; Vilar, M.; Campioni, S.; et al. In vivo demonstration that alpha-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. USA 2011, 108, 4194–4199. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.-J.; Yu, S.-Y.; Wang, F.; Hu, Y.; Piao, Y.-S.; Du, Y.; Lian, T.-H.; Wang, R.-D.; Yu, Q.-J.; Wang, Y.-J.; et al. Parkinson’s disease with fatigue: Clinical characteristics and potential mechanisms relevant to α-synuclein oligomer. J. Clin. Neurol. 2016, 12, 172–180. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Protein misfolding, amyloid formation, and human disease: A summary of progress over the last decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Benilova, I.; Karran, E.; De Strooper, B. The toxic Aβ oligomer and Alzheimer’s disease: An emperor in need of clothes. Nat. Neurosci. 2012, 15, 349–357. [Google Scholar] [CrossRef]
- Hoshi, M.; Sato, M.; Matsumoto, S.; Noguchi, A.; Yasutake, K.; Yoshida, N.; Sato, K. Spherical aggregates of β-amyloid (amylospheroid) show high neurotoxicity and activate tau protein kinase I/glycogen synthase kinase-3β. Proc. Natl. Acad. Sci. USA 2003, 100, 6370–6375. [Google Scholar] [CrossRef]
- Lashuel, H.A.; Hartley, D.; Petre, B.M.; Walz, T.; Lansbury, P.T. Amyloid pores from pathogenic mutations. Nature 2002, 418, 291. [Google Scholar] [CrossRef]
- Kayed, R.; Head, E.; Sarsoza, F.; Saing, T.; Cotman, C.W.; Necula, M.; Margol, L.; Wu, J.; Breydo, L.; Thompson, J.L.; et al. Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol. Neurodegener. 2007, 2, 18. [Google Scholar] [CrossRef] [PubMed]
- Hayden, E.Y.; Conovaloff, J.L.; Mason, A.; Bitan, G.; Teplow, D.B. Preparation of pure populations of covalently stabilized amyloid β-protein oligomers of specific sizes. Anal. Biochem. 2017, 518, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Barghorn, S.; Nimmrich, V.; Striebinger, A.; Krantz, C.; Keller, P.; Janson, B.; Bahr, M.; Schmidt, M.; Bitner, R.S.; Harlan, J.; et al. Globular amyloid beta-peptide oligomer—A homogenous and stable neuropathological protein in Alzheimer’s disease. J. Neurochem. 2005, 95, 834–847. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.W.; Drakulic, S.; Deas, E.; Ouberai, M.; Aprile, F.A.; Arranz, R.; Ness, S.; Roodveldt, C.; Guilliams, T.; De-Genst, E.J.; et al. Structural characterization of toxic oligomers that are kinetically trapped during α-synuclein fibril formation. Proc. Natl. Acad. Sci. USA 2015, 112, E1994–E2003. [Google Scholar] [CrossRef]
- Billings, L.M.; Oddo, S.; Green, K.N.; McGaugh, J.L.; LaFerla, F.M. Intraneuronal Abeta causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron 2005, 45, 675–688. [Google Scholar] [CrossRef]
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003, 300, 486–489. [Google Scholar] [CrossRef]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef]
- Chimon, S.; Ishii, Y. Capturing intermediate structures of Alzheimer’s β-amyloid, Aβ(1−40), by solid-state NMR spectroscopy. J. Am. Chem. Soc. 2005, 127, 13472–13473. [Google Scholar] [CrossRef]
- Mannini, B.; Habchi, J.; Chia, S.; Ruggeri, F.S.; Perni, M.; Knowles, T.P.J.; Dobson, C.M.; Vendruscolo, M. Stabilization and characterization of cytotoxic Aβ40 oligomers isolated from an aggregation reaction in the presence of zinc ions. ACS Chem. Neurosci. 2018, 9, 2959–2971. [Google Scholar] [CrossRef]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef]
- Cecchi, C.; Stefani, M. The amyloid-cell membrane system. The interplay between the biophysical features of oligomers/fibrils and cell membrane defines amyloid toxicity. Biophys. Chem. 2013, 182, 30–43. [Google Scholar] [CrossRef] [PubMed]
- Kayed, R.; Sokolov, Y.; Edmonds, B.; McIntire, T.M.; Milton, S.C.; Hall, J.E.; Glabe, C.G. Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J. Biol. Chem. 2004, 279, 46363–46366. [Google Scholar] [CrossRef] [PubMed]
- Behrends, C.; Langer, C.A.; Boteva, R.; Böttcher, U.M.; Stemp, M.J.; Schaffar, G.; Rao, B.V.; Giese, A.; Kretzschmar, H.; Siegers, K.; et al. Chaperonin TRiC promotes the assembly of polyQ expansion proteins into nontoxic oligomers. Mol. Cell 2006, 23, 887–897. [Google Scholar] [CrossRef]
- Fusco, G.; Chen, S.W.; Williamson, P.T.F.; Cascella, R.; Perni, M.; Jarvis, J.A.; Cecchi, C.; Vendruscolo, M.; Chiti, F.; Cremades, N.; et al. Structural basis of membrane disruption and cellular toxicity by α-synuclein oligomers. Science 2017, 358, 1440–1443. [Google Scholar] [CrossRef] [PubMed]
- Habchi, J.; Arosio, P.; Perni, M.; Costa, A.R.; Yagi-Utsumi, M.; Joshi, P.; Chia, S.; Cohen, S.I.A.; Müller, M.B.D.; Linse, S.; et al. An anticancer drug suppresses the primary nucleation reaction that initiates the production of the toxic Aβ42 aggregates linked with Alzheimer’s disease. Sci. Adv. 2016, 2, e1501244. [Google Scholar] [CrossRef]
- Mannini, B.; Mulvihill, E.; Sgromo, C.; Cascella, R.; Khodarahmi, R.; Ramazzotti, M.; Dobson, C.M.; Cecchi, C.; Chiti, F. Toxicity of protein oligomers is rationalized by a function combining size and surface hydrophobicity. ACS Chem. Biol. 2014, 9, 2309–2317. [Google Scholar] [CrossRef] [PubMed]
- Habchi, J.; Chia, S.; Limbocker, R.; Mannini, B.; Ahn, M.; Perni, M.; Hansson, O.; Arosio, P.; Kumita, J.R.; Challa, P.K.; et al. Systematic development of small molecules to inhibit specific microscopic steps of Aβ42 aggregation in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, E200–E208. [Google Scholar] [CrossRef]
- Bartolini, M.; Bertucci, C.; Bolognesi, M.L.; Cavalli, A.; Melchiorre, C.; Andrisano, V. Insight into the kinetic of amyloid beta (1-42) peptide self-aggregation: Elucidation of inhibitors’ mechanism of action. ChemBioChem 2007, 8, 2152–2161. [Google Scholar] [CrossRef] [PubMed]
- Kroth, H.; Ansaloni, A.; Varisco, Y.; Jan, A.; Sreenivasachary, N.; Rezaei-Ghaleh, N.; Giriens, V.; Lohmann, S.; López-Deber, M.P.; Adolfsson, O.; et al. Discovery and structure activity relationship of small molecule inhibitors of toxic β-amyloid-42 fibril formation. J. Biol. Chem. 2012, 287, 34786–34800. [Google Scholar] [CrossRef]
- Perni, M.; Galvagnion, C.; Maltsev, A.; Meisl, G.; Müller, M.B.D.; Challa, P.K.; Kirkegaard, J.B.; Flagmeier, P.; Cohen, S.I.A.; Cascella, R.; et al. A natural product inhibits the initiation of α-synuclein aggregation and suppresses its toxicity. Proc. Natl. Acad. Sci. USA 2017, 114, E1009–E1017. [Google Scholar] [CrossRef]
- Aprile, F.A.; Sormanni, P.; Perni, M.; Arosio, P.; Linse, S.; Knowles, T.P.J.; Dobson, C.M.; Vendruscolo, M. Selective targeting of primary and secondary nucleation pathways in Aβ42 aggregation using a rational antibody scanning method. Sci. Adv. 2017, 3, e1700488. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.I.A.; Arosio, P.; Presto, J.; Kurudenkandy, F.R.; Biverstål, H.; Dolfe, L.; Dunning, C.; Yang, X.; Frohm, B.; Vendruscolo, M.; et al. A molecular chaperone breaks the catalytic cycle that generates toxic Aβ oligomers. Nat. Struct. Mol. Biol. 2015, 22, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Perni, M.; Flagmeier, P.; Limbocker, R.; Cascella, R.; Aprile, F.A.; Galvagnion, C.; Heller, G.T.; Meisl, G.; Chen, S.W.; Kumita, J.R.; et al. Multistep inhibition of α-synuclein aggregation and toxicity in vitro and in vivo by trodusquemine. ACS Chem. Biol. 2018, 13, 2308–2319. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s disease drug development pipeline: 2019. Alzheimers Dement. Transl. Res. Clin. Interv. 2019, 5, 272–293. [Google Scholar] [CrossRef]
- Heller, G.T.; Aprile, F.A.; Michaels, T.C.T.; Limbocker, R.; Perni, M.; Ruggeri, F.S.; Mannini, B.; Löhr, T.; Bonomi, M.; Camilloni, C.; et al. Small molecule sequestration of amyloid-β as a drug discovery strategy for Alzheimer’s disease. Sci. Adv. 2020, 6, eabb5924. [Google Scholar] [CrossRef]
- Iljina, M.; Hong, L.; Horrocks, M.H.; Ludtmann, M.H.; Choi, M.L.; Hughes, C.D.; Ruggeri, F.S.; Guilliams, T.; Buell, A.K.; Lee, J.-E.; et al. Nanobodies raised against monomeric α-synuclein inhibit fibril formation and destabilize toxic oligomeric species. BMC Biol. 2017, 15, 57. [Google Scholar] [CrossRef]
- Linse, S.; Scheidt, T.; Bernfur, K.; Vendruscolo, M.; Dobson, C.M.; Cohen, S.I.A.; Sileikis, E.; Lundqvist, M.; Qian, F.; O’Malley, T.; et al. Kinetic fingerprints differentiate the mechanisms of action of anti-Aβ antibodies. Nat. Struct. Mol. Biol. 2020, 1–9. [Google Scholar] [CrossRef]
- Cohen, S.I.A.; Linse, S.; Luheshi, L.M.; Hellstrand, E.; White, D.A.; Rajah, L.; Otzen, D.E.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P.J. Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl. Acad. Sci. USA 2013, 110, 9758–9763. [Google Scholar] [CrossRef]
- Hellstrand, E.; Boland, B.; Walsh, D.M.; Linse, S. Amyloid β-protein aggregation produces highly reproducible kinetic data and occurs by a two-phase process. ACS Chem. Neurosci. 2009, 1, 13–18. [Google Scholar] [CrossRef]
- Scheidt, T.; Łapińska, U.; Kumita, J.R.; Whiten, D.R.; Klenerman, D.; Wilson, M.R.; Cohen, S.I.A.; Linse, S.; Vendruscolo, M.; Dobson, C.M.; et al. Secondary nucleation and elongation occur at different sites on Alzheimer’s amyloid-β aggregates. Sci. Adv. 2019, 5, eaau3112. [Google Scholar] [CrossRef]
- Cohen, S.I.A.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P.J. From macroscopic measurements to microscopic mechanisms of protein aggregation. J. Mol. Biol. 2012, 421, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Michaels, T.C.T.; Šarić, A.; Curk, S.; Bernfur, K.; Arosio, P.; Meisl, G.; Dear, A.J.; Cohen, S.I.A.; Dobson, C.M.; Vendruscolo, M.; et al. Dynamics of oligomer populations formed during the aggregation of Alzheimer’s Aβ42 peptide. Nat. Chem. 2020, 12, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Malmos, K.G.; Blancas-Mejia, L.M.; Weber, B.; Buchner, J.; Ramirez-Alvarado, M.; Naiki, H.; Otzen, D. ThT 101: A primer on the use of thioflavin T to investigate amyloid formation. Amyloid 2017, 24, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Coelho-Cerqueira, E.; Pinheiro, A.S.; Follmer, C. Pitfalls associated with the use of Thioflavin-T to monitor anti-fibrillogenic activity. Bioorg. Med. Chem. Lett. 2014, 24, 3194–3198. [Google Scholar] [CrossRef] [PubMed]
- Limbocker, R.; Chia, S.; Ruggeri, F.S.; Perni, M.; Cascella, R.; Heller, G.T.; Meisl, G.; Mannini, B.; Habchi, J.; Michaels, T.C.T.; et al. Trodusquemine enhances Aβ42 aggregation but suppresses its toxicity by displacing oligomers from cell membranes. Nat. Commun. 2019, 10, 225. [Google Scholar] [CrossRef]
- Ghadami, S.A.; Chia, S.; Ruggeri, F.S.; Meisl, G.; Bemporad, F.; Habchi, J.; Cascella, R.; Dobson, C.M.; Vendruscolo, M.; Knowles, T.P.J.; et al. Transthyretin inhibits primary and secondary nucleations of amyloid-β peptide aggregation and reduces the toxicity of its oligomers. Biomacromolecules 2020, 21, 1112–1125. [Google Scholar] [CrossRef]
- Munke, A.; Persson, J.; Weiffert, T.; De Genst, E.; Meisl, G.; Arosio, P.; Carnerup, A.; Dobson, C.M.; Vendruscolo, M.; Knowles, T.P.J.; et al. Phage display and kinetic selection of antibodies that specifically inhibit amyloid self-replication. Proc. Natl. Acad. Sci. USA 2017, 114, 6444–6449. [Google Scholar] [CrossRef]
- Englund, H.; Sehlin, D.; Johansson, A.-S.; Nilsson, L.N.G.; Gellerfors, P.; Paulie, S.; Lannfelt, L.; Pettersson, F.E. Sensitive ELISA detection of amyloid-β protofibrils in biological samples. J. Neurochem. 2007, 103, 334–345. [Google Scholar] [CrossRef]
- Tucker, S.; Möller, C.; Tegerstedt, K.; Lord, A.; Laudon, H.; Sjödahl, J.; Söderberg, L.; Spens, E.; Sahlin, C.; Waara, E.R.; et al. The murine version of BAN2401 (mAb158) selectively reduces amyloid-β protofibrils in brain and cerebrospinal fluid of tg-ArcSwe mice. J. Alzheimers Dis. 2015, 43, 575–588. [Google Scholar] [CrossRef]
- Cummings, J.L.; Cohen, S.; van Dyck, C.H.; Brody, M.; Curtis, C.; Cho, W.; Ward, M.; Friesenhahn, M.; Rabe, C.; Brunstein, F.; et al. ABBY: A phase 2 randomized trial of crenezumab in mild to moderate Alzheimer disease. Neurology 2018, 90, e1889–e1897. [Google Scholar] [CrossRef]
- Yang, T.; Dang, Y.; Ostaszewski, B.; Mengel, D.; Steffen, V.; Rabe, C.; Bittner, T.; Walsh, D.M.; Selkoe, D.J. Target engagement in an alzheimer trial: Crenezumab lowers amyloid β oligomers in cerebrospinal fluid. Ann. Neurol. 2019, 86, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Arosio, P.; Michaels, T.C.T.; Linse, S.; Månsson, C.; Emanuelsson, C.; Presto, J.; Johansson, J.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P.J. Kinetic analysis reveals the diversity of microscopic mechanisms through which molecular chaperones suppress amyloid formation. Nat. Commun. 2016, 7, 10948. [Google Scholar] [CrossRef] [PubMed]
- Chia, S.; Habchi, J.; Michaels, T.C.T.; Cohen, S.I.A.; Linse, S.; Dobson, C.M.; Knowles, T.P.J.; Vendruscolo, M. SAR by kinetics for drug discovery in protein misfolding diseases. Proc. Natl. Acad. Sci. USA 2018, 115, 10245–10250. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.Y.; Kang, S.S.; Lee, S.K.; Han, B.H. Polyphenolic biflavonoids inhibit amyloid-beta fibrillation and disaggregate preformed amyloid-beta fibrils. Biomol. Ther. 2020, 28, 145–151. [Google Scholar] [CrossRef]
- Giorgetti, S.; Greco, C.; Tortora, P.; Aprile, F.A. Targeting amyloid aggregation: An overview of strategies and mechanisms. Int. J. Mol. Sci. 2018, 19, 2677. [Google Scholar] [CrossRef] [PubMed]
- Pandey, N.; Strider, J.; Nolan, W.C.; Yan, S.X.; Galvin, J.E. Curcumin inhibits aggregation of alpha-synuclein. Acta Neuropathol. Berl. 2008, 115, 479–489. [Google Scholar] [CrossRef]
- Forloni, G.; Colombo, L.; Girola, L.; Tagliavini, F.; Salmona, M. Anti-amyloidogenic activity of tetracyclines: Studies in vitro. FEBS Lett. 2001, 487, 404–407. [Google Scholar] [CrossRef]
- Howlett, D.R.; George, A.R.; Owen, D.E.; Ward, R.V.; Markwell, R.E. Common structural features determine the effectiveness of carvedilol, daunomycin and rolitetracycline as inhibitors of Alzheimer β-amyloid fibril formation. Biochem. J. 1999, 343, 419–423. [Google Scholar] [CrossRef]
- Honson, N.S.; Johnson, R.L.; Huang, W.; Inglese, J.; Austin, C.P.; Kuret, J. Differentiating Alzheimer disease-associated aggregates with small molecules. Neurobiol. Dis. 2007, 28, 251–260. [Google Scholar] [CrossRef]
- Cisek, K.; Cooper, G.L.; Huseby, C.J.; Kuret, J. Structure and mechanism of action of tau aggregation inhibitors. Curr. Alzheimer Res. 2014, 11, 918–927. [Google Scholar] [CrossRef]
- Necula, M.; Breydo, L.; Milton, S.; Kayed, R.; van der Veer, W.E.; Tone, P.; Glabe, C.G. Methylene blue inhibits amyloid Aβ oligomerization by promoting fibrillization. Biochemistry 2007, 46, 8850–8860. [Google Scholar] [CrossRef] [PubMed]
- Lendel, C.; Bolognesi, B.; Wahlström, A.; Dobson, C.M.; Gräslund, A. Detergent-like interaction of Congo Red with the amyloid β peptide. Biochemistry 2010, 49, 1358–1360. [Google Scholar] [CrossRef] [PubMed]
- Cavaliere, P.; Torrent, J.; Prigent, S.; Granata, V.; Pauwels, K.; Pastore, A.; Rezaei, H.; Zagari, A. Binding of methylene blue to a surface cleft inhibits the oligomerization and fibrillization of prion protein. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2013, 1832, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Prade, E.; Barucker, C.; Sarkar, R.; Althoff-Ospelt, G.; Lopez del Amo, J.M.; Hossain, S.; Zhong, Y.; Multhaup, G.; Reif, B. Sulindac sulfide induces the formation of large oligomeric aggregates of the Alzheimer’s disease amyloid-β peptide which exhibit reduced neurotoxicity. Biochemistry 2016, 55, 1839–1849. [Google Scholar] [CrossRef]
- Civitelli, L.; Sandin, L.; Nelson, E.; Khattak, S.I.; Brorsson, A.-C.; Kågedal, K. The luminescent oligothiophene p-FTAA converts toxic Aβ1-42 species into nontoxic amyloid fibers with altered properties. J. Biol. Chem. 2016, 291, 9233–9243. [Google Scholar] [CrossRef]
- Dutta, S.; Foley, A.R.; Warner, C.J.A.; Zhang, X.; Rolandi, M.; Abrams, B.; Raskatov, J.A. Suppression of oligomer formation and formation of non-toxic fibrils upon addition of mirror-image Aβ42 to the natural L-enantiomer. Angew. Chem. Int. Ed. Engl. 2017, 56, 11506–11510. [Google Scholar] [CrossRef] [PubMed]
- Bieschke, J.; Herbst, M.; Wiglenda, T.; Friedrich, R.P.; Boeddrich, A.; Schiele, F.; Kleckers, D.; Lopez del Amo, J.M.; Grüning, B.A.; Wang, Q.; et al. Small-molecule conversion of toxic oligomers to nontoxic β-sheet–rich amyloid fibrils. Nat. Chem. Biol. 2012, 8, 93–101. [Google Scholar] [CrossRef]
- Ruggeri, F.S.; Habchi, J.; Cerreta, A.; Dietler, G. AFM-based single molecule techniques: Unraveling the amyloid pathogenic species. Curr. Pharm. Des. 2016, 22, 3950–3970. [Google Scholar] [CrossRef]
- Ruggeri, F.S.; Adamcik, J.; Jeong, J.S.; Lashuel, H.A.; Mezzenga, R.; Dietler, G. Influence of the β-sheet content on the mechanical properties of aggregates during amyloid fibrillization. Angew. Chem. Int. Ed. 2015, 54, 2462–2466. [Google Scholar] [CrossRef]
- Ruggeri, F.S.; Vieweg, S.; Cendrowska, U.; Longo, G.; Chiki, A.; Lashuel, H.A.; Dietler, G. Nanoscale studies link amyloid maturity with polyglutamine diseases onset. Sci. Rep. 2016, 6, 31155. [Google Scholar] [CrossRef]
- Limbocker, R.; Mannini, B.; Ruggeri, F.S.; Cascella, R.; Xu, C.K.; Perni, M.; Chia, S.; Chen, S.W.; Habchi, J.; Bigi, A.; et al. Trodusquemine displaces protein misfolded oligomers from cell membranes and abrogates their cytotoxicity through a generic mechanism. Commun. Biol. 2020, 3, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ladiwala, A.R.A.; Lin, J.C.; Bale, S.S.; Marcelino-Cruz, A.M.; Bhattacharya, M.; Dordick, J.S.; Tessier, P.M. Resveratrol selectively remodels soluble oligomers and fibrils of amyloid Aβ into off-pathway conformers. J. Biol. Chem. 2010, 285, 24228–24237. [Google Scholar] [CrossRef] [PubMed]
- Palhano, F.L.; Lee, J.; Grimster, N.P.; Kelly, J.W. Toward the molecular mechanism(s) by which EGCG treatment remodels mature amyloid fibrils. J. Am. Chem. Soc. 2013, 135, 7503–7510. [Google Scholar] [CrossRef] [PubMed]
- Bieschke, J.; Russ, J.; Friedrich, R.P.; Ehrnhoefer, D.E.; Wobst, H.; Neugebauer, K.; Wanker, E.E. EGCG remodels mature alpha-synuclein and amyloid-beta fibrils and reduces cellular toxicity. Proc. Natl. Acad. Sci. USA 2010, 107, 7710–7715. [Google Scholar] [CrossRef]
- Ehrnhoefer, D.E.; Bieschke, J.; Boeddrich, A.; Herbst, M.; Masino, L.; Lurz, R.; Engemann, S.; Pastore, A.; Wanker, E.E. EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat. Struct. Mol. Biol. 2008, 15, 558–566. [Google Scholar] [CrossRef]
- Ladiwala, A.R.A.; Dordick, J.S.; Tessier, P.M. Aromatic small molecules remodel toxic soluble oligomers of amyloid beta through three independent pathways. J. Biol. Chem. 2011, 286, 3209–3218. [Google Scholar] [CrossRef]
- Taniguchi, S.; Suzuki, N.; Masuda, M.; Hisanaga, S.; Iwatsubo, T.; Goedert, M.; Hasegawa, M. Inhibition of heparin-induced tau filament formation by phenothiazines, polyphenols, and porphyrins. J. Biol. Chem. 2005, 280, 7614–7623. [Google Scholar] [CrossRef]
- Breydo, L.; Uversky, V.N. Structural, morphological, and functional diversity of amyloid oligomers. FEBS Lett. 2015, 589, 2640–2648. [Google Scholar] [CrossRef]
- Sandberg, A.; Luheshi, L.M.; Söllvander, S.; Barros, T.P.; de Macao, B.; Knowles, T.P.J.; Biverstål, H.; Lendel, C.; Ekholm-Petterson, F.; Dubnovitsky, A.; et al. Stabilization of neurotoxic Alzheimer amyloid-β oligomers by protein engineering. Proc. Natl. Acad. Sci. USA 2010, 107, 15595–15600. [Google Scholar] [CrossRef]
- Bitan, G.; Kirkitadze, M.D.; Lomakin, A.; Vollers, S.S.; Benedek, G.B.; Teplow, D.B. Amyloid β-protein (Aβ) assembly: Aβ40 and Aβ42 oligomerize through distinct pathways. Proc. Natl. Acad. Sci. USA 2003, 100, 330–335. [Google Scholar] [CrossRef]
- Campioni, S.; Mannini, B.; Zampagni, M.; Pensalfini, A.; Parrini, C.; Evangelisti, E.; Relini, A.; Stefani, M.; Dobson, C.M.; Cecchi, C.; et al. A causative link between the structure of aberrant protein oligomers and their toxicity. Nat. Chem. Biol. 2010, 6, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Mannini, B.; Cascella, R.; Zampagni, M.; van Waarde-Verhagen, M.; Meehan, S.; Roodveldt, C.; Campioni, S.; Boninsegna, M.; Penco, A.; Relini, A.; et al. Molecular mechanisms used by chaperones to reduce the toxicity of aberrant protein oligomers. Proc. Natl. Acad. Sci. USA 2012, 109, 12479–12484. [Google Scholar] [CrossRef] [PubMed]
- Mannini, B.; Chiti, F. Chaperones as suppressors of protein misfolded oligomer toxicity. Front. Mol. Neurosci. 2017, 10, 98. [Google Scholar] [CrossRef] [PubMed]
- Bolognesi, B.; Kumita, J.R.; Barros, T.P.; Esbjorner, E.K.; Luheshi, L.M.; Crowther, D.C.; Wilson, M.R.; Dobson, C.M.; Favrin, G.; Yerbury, J.J. ANS binding reveals common features of cytotoxic amyloid species. ACS Chem. Biol. 2010, 5, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Ladiwala, A.R.A.; Litt, J.; Kane, R.S.; Aucoin, D.S.; Smith, S.O.; Ranjan, S.; Davis, J.; Nostrand, W.E.V.; Tessier, P.M. Conformational differences between two amyloid β oligomers of similar size and dissimilar toxicity. J. Biol. Chem. 2012, 287, 24765–24773. [Google Scholar] [CrossRef]
- Limbocker, R.; Mannini, B.; Cataldi, R.; Chhangur, S.; Wright, A.K.; Kreiser, R.P.; Albright, J.A.; Chia, S.; Habchi, J.; Sormanni, P.; et al. Rationally designed antibodies as research tools to study the structure–toxicity relationship of amyloid-β oligomers. Int. J. Mol. Sci. 2020, 21, 4542. [Google Scholar] [CrossRef]
- Cizas, P.; Budvytyte, R.; Morkuniene, R.; Moldovan, R.; Broccio, M.; Lösche, M.; Niaura, G.; Valincius, G.; Borutaite, V. Size-dependent neurotoxicity of beta-amyloid oligomers. Arch. Biochem. Biophys. 2010, 496, 84–92. [Google Scholar] [CrossRef]
- Cappelli, S.; Penco, A.; Mannini, B.; Cascella, R.; Wilson, M.R.; Ecroyd, H.; Li, X.; Buxbaum, J.N.; Dobson, C.M.; Cecchi, C.; et al. Effect of molecular chaperones on aberrant protein oligomers in vitro: Super-versus sub-stoichiometric chaperone concentrations. Biol. Chem. 2016, 397, 401–415. [Google Scholar] [CrossRef][Green Version]
- Ojha, J.; Masilamoni, G.; Dunlap, D.; Udoff, R.A.; Cashikar, A.G. Sequestration of toxic oligomers by HspB1 as a cytoprotective mechanism. Mol. Cell. Biol. 2011, 31, 3146–3157. [Google Scholar] [CrossRef]
- Benoit, M.E.; Hernandez, M.X.; Dinh, M.L.; Benavente, F.; Vasquez, O.; Tenner, A.J. C1q-induced LRP1B and GPR6 proteins expressed early in Alzheimer disease mouse models, are essential for the C1q-mediated protection against amyloid-β neurotoxicity. J. Biol. Chem. 2013, 288, 654–665. [Google Scholar] [CrossRef]
- Cheng, J.S.; Dubal, D.B.; Kim, D.H.; Legleiter, J.; Cheng, I.H.; Yu, G.-Q.; Tesseur, I.; Wyss-Coray, T.; Bonaldo, P.; Mucke, L. Collagen VI protects neurons against Aβ toxicity. Nat. Neurosci. 2009, 12, 119. [Google Scholar] [CrossRef] [PubMed]
- Hochberg, G.K.A.; Ecroyd, H.; Liu, C.; Cox, D.; Cascio, D.; Sawaya, M.R.; Collier, M.P.; Stroud, J.; Carver, J.A.; Baldwin, A.J.; et al. The structured core domain of αB-crystallin can prevent amyloid fibrillation and associated toxicity. Proc. Natl. Acad. Sci. USA 2014, 111, E1562–E1570. [Google Scholar] [CrossRef] [PubMed]
- Santhoshkumar, P.; Sharma, K.K. Inhibition of amyloid fibrillogenesis and toxicity by a peptide chaperone. Mol. Cell. Biochem. 2004, 267, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Robbins, J.P.; Perfect, L.; Ribe, E.M.; Maresca, M.; Dangla-Valls, A.; Foster, E.M.; Killick, R.; Nowosiad, P.; Reid, M.J.; Polit, L.D.; et al. Clusterin is required for β-amyloid toxicity in human iPSC-derived neurons. Front. Neurosci. 2018, 12, 504. [Google Scholar] [CrossRef]
- Chakraborty, K.; Georgescauld, F.; Hayer-Hartl, M.; Hartl, F.U. Role of molecular chaperones in protein folding. In Protein Misfolding Diseases; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2010; pp. 47–72. ISBN 978-0-470-57270-2. [Google Scholar]
- Rivera, I.; Capone, R.; Cauvi, D.M.; Arispe, N.; De Maio, A. Modulation of Alzheimer’s amyloid β peptide oligomerization and toxicity by extracellular Hsp70. Cell Stress Chaperones 2018, 23, 269–279. [Google Scholar] [CrossRef]
- Fernandez-Funez, P.; Sanchez-Garcia, J.; de Mena, L.; Zhang, Y.; Levites, Y.; Khare, S.; Golde, T.E.; Rincon-Limas, D.E. Holdase activity of secreted Hsp70 masks amyloid-β42 neurotoxicity in Drosophila. Proc. Natl. Acad. Sci. USA 2016, 113, E5212–E5221. [Google Scholar] [CrossRef]
- Ojha, J.; Karmegam, R.V.; Masilamoni, J.G.; Terry, A.V., Jr.; Cashikar, A.G. Behavioral defects in chaperone-deficient Alzheimer’s disease model mice. PLoS ONE 2011, 6, e16550. [Google Scholar] [CrossRef]
- Garai, K.; Posey, A.E.; Li, X.; Buxbaum, J.N.; Pappu, R.V. Inhibition of amyloid beta fibril formation by monomeric human transthyretin. Protein Sci. 2018, 27, 1252–1261. [Google Scholar] [CrossRef]
- Beeg, M.; Stravalaci, M.; Romeo, M.; Carrá, A.D.; Cagnotto, A.; Rossi, A.; Diomede, L.; Salmona, M.; Gobbi, M. Clusterin binds to Aβ1–42 oligomers with high affinity and interferes with peptide aggregation by inhibiting primary and secondary nucleation. J. Biol. Chem. 2016, 291, 6958–6966. [Google Scholar] [CrossRef]
- Lee, J.; Kim, Y.H.; Arce, F.T.; Gillman, A.L.; Jang, H.; Kagan, B.L.; Nussinov, R.; Yang, J.; Lal, R. Amyloid β ion channels in a membrane comprising brain total lipid extracts. ACS Chem. Neurosci. 2017, 8, 1348–1357. [Google Scholar] [CrossRef]
- Williams, T.L.; Johnson, B.R.G.; Urbanc, B.; Jenkins, A.T.A.; Connell, S.D.A.; Serpell, L.C. Aβ42 oligomers, but not fibrils, simultaneously bind to and cause damage to ganglioside-containing lipid membranes. Biochem. J. 2011, 439, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Hashemi, M.; Zagorski, K.; Lyubchenko, Y.L. Interaction of Aβ42 with membranes triggers the self-Assembly into oligomers. Int. J. Mol. Sci. 2020, 21, 1129. [Google Scholar] [CrossRef] [PubMed]
- Egawa, J.; Pearn, M.L.; Lemkuil, B.P.; Patel, P.M.; Head, B.P. Membrane lipid rafts and neurobiology: Age-related changes in membrane lipids and loss of neuronal function. J. Physiol. 2016, 594, 4565–4579. [Google Scholar] [CrossRef]
- Dinamarca, M.C.; Ríos, J.A.; Inestrosa, N.C. Postsynaptic receptors for amyloid-β oligomers as mediators of neuronal damage in Alzheimer’s disease. Front. Physiol. 2012, 3, 464. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Shang, Z.; Imai, Y.; Hoshino, T.; Tero, R.; Tanaka, M.; Yamamoto, N.; Yanagisawa, K.; Urisu, T. Surface-induced phase separation of a sphingomyelin/cholesterol/ganglioside GM1-planar bilayer on mica surfaces and microdomain molecular conformation that accelerates Aβ oligomerization. Biochim. Biophys. Acta BBA Biomembr. 2010, 1798, 1090–1099. [Google Scholar] [CrossRef]
- Fabiani, C.; Antollini, S.S. Alzheimer’s disease as a membrane disorder: Spatial cross-talk among beta-amyloid peptides, nicotinic acetylcholine receptors and lipid rafts. Front. Cell. Neurosci. 2019, 13, 309. [Google Scholar] [CrossRef]
- Lahdo, R.; de La Fournière-Bessueille, L. Insertion of the amyloid precursor protein into lipid monolayers: Effects of cholesterol and apolipoprotein E. Biochem. J. 2004, 382, 987–994. [Google Scholar] [CrossRef]
- Brown, D.A.; London, E. Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J. Biol. Chem. 2000, 275, 17221–17224. [Google Scholar] [CrossRef]
- Grimm, M.O.W.; Grimm, H.S.; Pätzold, A.J.; Zinser, E.G.; Halonen, R.; Duering, M.; Tschäpe, J.-A.; Strooper, B.D.; Müller, U.; Shen, J.; et al. Regulation of cholesterol and sphingomyelin metabolism by amyloid-β and presenilin. Nat. Cell Biol. 2005, 7, 1118–1123. [Google Scholar] [CrossRef]
- Cutler, R.G.; Kelly, J.; Storie, K.; Pedersen, W.A.; Tammara, A.; Hatanpaa, K.; Troncoso, J.C.; Mattson, M.P. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 2070–2075. [Google Scholar] [CrossRef]
- Brinkmalm, G.; Hong, W.; Wang, Z.; Liu, W.; O’Malley, T.T.; Sun, X.; Frosch, M.P.; Selkoe, D.J.; Portelius, E.; Zetterberg, H.; et al. Identification of neurotoxic cross-linked amyloid-β dimers in the Alzheimer’s brain. Brain J. Neurol. 2019, 142, 1441–1457. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Shepardson, N.; Yang, T.; Chen, G.; Walsh, D.; Selkoe, D.J. Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc. Natl. Acad. Sci. USA 2011, 108, 5819–5824. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Hafiz, L.; Müller-Schiffmann, A.; Korth, C.; Fazari, B.; Chao, O.Y.; Nikolaus, S.; Schäble, S.; Herring, A.; Keyvani, K.; Lamounier-Zepter, V.; et al. Aβ dimers induce behavioral and neurochemical deficits of relevance to early Alzheimer’s disease. Neurobiol. Aging 2018, 69, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Jana, M.K.; Cappai, R.; Pham, C.L.L.; Ciccotosto, G.D. Membrane-bound tetramer and trimer Aβ oligomeric species correlate with toxicity towards cultured neurons. J. Neurochem. 2016, 136, 594–608. [Google Scholar] [CrossRef] [PubMed]
- Townsend, M.; Shankar, G.M.; Mehta, T.; Walsh, D.M.; Selkoe, D.J. Effects of secreted oligomers of amyloid β-protein on hippocampal synaptic plasticity: A potent role for trimers. J. Physiol. 2006, 572, 477–492. [Google Scholar] [CrossRef] [PubMed]
- Lesné, S.E.; Sherman, M.A.; Grant, M.; Kuskowski, M.; Schneider, J.A.; Bennett, D.A.; Ashe, K.H. Brain amyloid-β oligomers in ageing and Alzheimer’s disease. Brain 2013, 136, 1383–1398. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Teng, Z.; Cui, C.; Wang, R.; Liu, M.; Zhang, Y. Amyloid beta-derived diffusible ligands (ADDLs) induce abnormal expression of insulin receptors in rat hippocampal meurons. J. Mol. Neurosci. 2014, 52, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, S.; Shinoda, K.; Yamada, M.; Yokojima, S.; Inoue, M.; Ohnishi, T.; Shimada, T.; Kikuchi, K.; Masui, D.; Hashimoto, S.; et al. Two distinct amyloid β-protein (Aβ) assembly pathways leading to oligomers and fibrils identified by combined fluorescence correlation spectroscopy, morphology, and toxicity analyses. J. Biol. Chem. 2011, 286, 11555–11562. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Liu, R. The toxicity and polymorphism of β-amyloid oligomers. Int. J. Mol. Sci. 2020, 21, 4477. [Google Scholar] [CrossRef]
- Calamai, M.; Pavone, F.S. Single molecule tracking analysis reveals that the surface mobility of amyloid oligomers is driven by their conformational structure. J. Am. Chem. Soc. 2011, 133, 12001–12008. [Google Scholar] [CrossRef]
- Johnson, R.D.; Steel, D.G.; Gafni, A. Structural evolution and membrane interactions of Alzheimer’s amyloid-beta peptide oligomers: New knowledge from single-molecule fluorescence studies. Protein Sci. 2014, 23, 869–883. [Google Scholar] [CrossRef] [PubMed]
- Demuro, A.; Mina, E.; Kayed, R.; Milton, S.C.; Parker, I.; Glabe, C.G. Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J. Biol. Chem. 2005, 280, 17294–17300. [Google Scholar] [CrossRef]
- Galvagnion, C.; Brown, J.W.P.; Ouberai, M.M.; Flagmeier, P.; Vendruscolo, M.; Buell, A.K.; Sparr, E.; Dobson, C.M. Chemical properties of lipids strongly affect the kinetics of the membrane-induced aggregation of α-synuclein. Proc. Natl. Acad. Sci. USA 2016, 113, 7065–7070. [Google Scholar] [CrossRef] [PubMed]
- Lantz, K.A.; Hart, S.G.E.; Planey, S.L.; Roitman, M.F.; Ruiz-White, I.A.; Wolfe, H.R.; McLane, M.P. Inhibition of PTP1B by trodusquemine (MSI-1436) causes fat-specific weight loss in diet-induced obese mice. Obesity 2010, 18, 1516–1523. [Google Scholar] [CrossRef] [PubMed]
- Yip, C.M.; Elton, E.A.; Darabie, A.A.; Morrison, M.R.; McLaurin, J. Cholesterol, a modulator of membrane-associated aβ-fibrillogenesis and neurotoxicity. J. Mol. Biol. 2001, 311, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Evangelisti, E.; Cascella, R.; Becatti, M.; Marrazza, G.; Dobson, C.M.; Chiti, F.; Stefani, M.; Cecchi, C. Binding affinity of amyloid oligomers to cellular membranes is a generic indicator of cellular dysfunction in protein misfolding diseases. Sci. Rep. 2016, 6, 32721. [Google Scholar] [CrossRef]
- Martinez Hernandez, A.; Urbanke, H.; Gillman, A.L.; Lee, J.; Ryazanov, S.; Agbemenyah, H.Y.; Benito, E.; Jain, G.; Kaurani, L.; Grigorian, G.; et al. The diphenylpyrazole compound anle138b blocks Aβ channels and rescues disease phenotypes in a mouse model for amyloid pathology. EMBO Mol. Med. 2018, 10, 32–47. [Google Scholar] [CrossRef]
- Sala, A.J.; Bott, L.C.; Morimoto, R.I. Shaping proteostasis at the cellular, tissue, and organismal level. J. Cell Biol. 2017, 216, 1231–1241. [Google Scholar] [CrossRef]
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435. [Google Scholar] [CrossRef]
- Klaips, C.L.; Jayaraj, G.G.; Hartl, F.U. Pathways of cellular proteostasis in aging and disease. J. Cell Biol. 2018, 217, 51–63. [Google Scholar] [CrossRef]
- Sweeney, P.; Park, H.; Baumann, M.; Dunlop, J.; Frydman, J.; Kopito, R.; McCampbell, A.; Leblanc, G.; Venkateswaran, A.; Nurmi, A.; et al. Protein misfolding in neurodegenerative diseases: Implications and strategies. Transl. Neurodegener. 2017, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Brehme, M.; Voisine, C.; Rolland, T.; Wachi, S.; Soper, J.H.; Zhu, Y.; Orton, K.; Villella, A.; Garza, D.; Vidal, M.; et al. A chaperome subnetwork safeguards proteostasis in aging and neurodegenerative disease. Cell Rep. 2014, 9, 1135–1150. [Google Scholar] [CrossRef] [PubMed]
- Labbadia, J.; Morimoto, R.I. The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 2015, 84, 435–464. [Google Scholar] [CrossRef] [PubMed]
- Balchin, D.; Hayer-Hartl, M.; Hartl, F.U. In vivo aspects of protein folding and quality control. Science 2016, 353, aac4354. [Google Scholar] [CrossRef] [PubMed]
- Sinnige, T.; Yu, A.; Morimoto, R.I. Challenging proteostasis: Role of the chaperone network to control aggregation-prone proteins in human disease. In HSF1 and Molecular Chaperones in Biology and Cancer; Advances in Experimental Medicine and Biology; Mendillo, M.L., Pincus, D., Scherz-Shouval, R., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 53–68. ISBN 978-3-030-40204-4. [Google Scholar]
- Smith, H.L.; Li, W.; Cheetham, M.E. Molecular chaperones and neuronal proteostasis. Semin. Cell Dev. Biol. 2015, 40, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Cristóvão, J.S.; Gomes, C.M. S100 Proteins in Alzheimer’s disease. Front. Neurosci. 2019, 13, 463. [Google Scholar] [CrossRef]
- Cristóvão, J.S.; Morris, V.K.; Cardoso, I.; Leal, S.S.; Martínez, J.; Botelho, H.M.; Göbl, C.; David, R.; Kierdorf, K.; Alemi, M.; et al. The neuronal S100B protein is a calcium-tuned suppressor of amyloid-β aggregation. Sci. Adv. 2018, 4, eaaq1702. [Google Scholar] [CrossRef]
- Wang, B.; Liu, Y.; Huang, L.; Chen, J.; Li, J.J.; Wang, R.; Kim, E.; Chen, Y.; Justicia, C.; Sakata, K.; et al. A CNS-permeable Hsp90 inhibitor rescues synaptic dysfunction and memory loss in APP-overexpressing Alzheimer’s mouse model via an HSF1-mediated mechanism. Mol. Psychiatry 2017, 22, 990–1001. [Google Scholar] [CrossRef]
- Martin, M.D.; Baker, J.D.; Suntharalingam, A.; Nordhues, B.A.; Shelton, L.B.; Zheng, D.; Sabbagh, J.J.; Haystead, T.A.J.; Gestwicki, J.E.; Dickey, C.A. Inhibition of both Hsp70 activity and tau aggregation in vitro best predicts tau lowering activity of small molecules. ACS Chem. Biol. 2016, 11, 2041–2048. [Google Scholar] [CrossRef]
- Kalia, S.K.; Kalia, L.V.; McLean, P.J. Molecular chaperones as rational drug targets for Parkinson’s disease therapeutics. CNS Neurol. Disord. Drug Targets 2010, 9, 741–753. [Google Scholar] [CrossRef]
- Husna Ibrahim, N.; Yahaya, M.F.; Mohamed, W.; Teoh, S.L.; Hui, C.K.; Kumar, J. Pharmacotherapy of Alzheimer’s disease: Seeking clarity in a time of uncertainty. Front. Pharmacol. 2020, 11, 261. [Google Scholar] [CrossRef] [PubMed]
- Kalmar, B.; Novoselov, S.; Gray, A.; Cheetham, M.E.; Margulis, B.; Greensmith, L. Late stage treatment with arimoclomol delays disease progression and prevents protein aggregation in the SOD1 mouse model of ALS. J. Neurochem. 2008, 107, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, S.N.; Martin, M.D.; Dickey, C.A. Neurodegeneration and the heat shock protein 70 machinery: Implications for therapeutic development. Curr. Top. Med. Chem. 2016, 16, 2741–2752. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Zheng, W.; Wang, X.; Chen, Z. Proteostasis of α-synuclein and its role in the pathogenesis of Parkinson’s disease. Front. Cell. Neurosci. 2020, 14, 45. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Leidal, A.M.; Levine, B.; Debnath, J. Autophagy and the cell biology of age-related disease. Nat. Cell Biol. 2018, 20, 1338–1348. [Google Scholar] [CrossRef]
- Galluzzi, L.; Pietrocola, F.; Levine, B.; Kroemer, G. Metabolic control of autophagy. Cell 2014, 159, 1263–1276. [Google Scholar] [CrossRef]
- Guglielmotto, M.; Monteleone, D.; Piras, A.; Valsecchi, V.; Tropiano, M.; Ariano, S.; Fornaro, M.; Vercelli, A.; Puyal, J.; Arancio, O.; et al. Aβ1-42 monomers or oligomers have different effects on autophagy and apoptosis. Autophagy 2014, 10, 1827–1843. [Google Scholar] [CrossRef]
- Majumder, S.; Richardson, A.; Strong, R.; Oddo, S. Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS ONE 2011, 6, e25416. [Google Scholar] [CrossRef]
- Spilman, P.; Podlutskaya, N.; Hart, M.J.; Debnath, J.; Gorostiza, O.; Bredesen, D.; Richardson, A.; Strong, R.; Galvan, V. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. PLoS ONE 2010, 5, e9979. [Google Scholar] [CrossRef]
- Liu, J.; Li, L. Targeting autophagy for the treatment of Alzheimer’s disease: Challenges and opportunities. Front. Mol. Neurosci. 2019, 12, 203. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A. Intracellular protein degradation: From a vague idea thru the lysosome and the ubiquitin–proteasome system and onto human diseases and drug targeting. Cell Death Differ. 2005, 12, 1178–1190. [Google Scholar] [CrossRef] [PubMed]
- Arndt, V.; Rogon, C.; Höhfeld, J. To be, or not to be--molecular chaperones in protein degradation. Cell. Mol. Life Sci. 2007, 64, 2525–2541. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D. β-secretase inhibitors for Alzheimer’s disease: Heading in the wrong direction? Lancet Neurol. 2019, 18, 624–626. [Google Scholar] [CrossRef]
- Liu, C.-C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms, and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef]
- Giau, V.V.; Bagyinszky, E.; An, S.S.A.; Kim, S.Y. Role of apolipoprotein E in neurodegenerative diseases. Neuropsychiatr. Dis. Treat. 2015, 11, 1723–1737. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Zhao, N.; Caulfield, T.R.; Liu, C.-C.; Bu, G. Apolipoprotein E and Alzheimer disease: Pathobiology and targeting strategies. Nat. Rev. Neurol. 2019, 15, 501–518. [Google Scholar] [CrossRef]
- Aprile, F.A.; Sormanni, P.; Podpolny, M.; Chhangur, S.; Needham, L.-M.; Ruggeri, F.S.; Perni, M.; Limbocker, R.; Heller, G.T.; Sneideris, T.; et al. Rational design of a conformation-specific antibody for the quantification of Aβ oligomers. Proc. Natl. Acad. Sci. USA 2020, 117, 13509–13518. [Google Scholar] [CrossRef]
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef]
- Falcon, B.; Zhang, W.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Vidal, R.; Crowther, R.A.; Ghetti, B.; Scheres, S.H.W.; Goedert, M. Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature 2018, 561, 137. [Google Scholar] [CrossRef]
- Falcon, B.; Zivanov, J.; Zhang, W.; Murzin, A.G.; Garringer, H.J.; Vidal, R.; Crowther, R.A.; Newell, K.L.; Ghetti, B.; Goedert, M.; et al. Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature 2019, 568, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Tarutani, A.; Newell, K.L.; Murzin, A.G.; Matsubara, T.; Falcon, B.; Vidal, R.; Garringer, H.J.; Shi, Y.; Ikeuchi, T.; et al. Novel tau filament fold in corticobasal degeneration. Nature 2020, 580, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Arakhamia, T.; Lee, C.E.; Carlomagno, Y.; Duong, D.M.; Kundinger, S.R.; Wang, K.; Williams, D.; DeTure, M.; Dickson, D.W.; Cook, C.N.; et al. Posttranslational modifications mediate the structural diversity of tauopathy strains. Cell 2020, 180, 633–644.e12. [Google Scholar] [CrossRef] [PubMed]
- Schweighauser, M.; Shi, Y.; Tarutani, A.; Kametani, F.; Murzin, A.G.; Ghetti, B.; Matsubara, T.; Tomita, T.; Ando, T.; Hasegawa, K.; et al. Structures of α-synuclein filaments from multiple system atrophy. Nature 2020, 585, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Nakane, T.; Kotecha, A.; Sente, A.; McMullan, G.; Masiulis, S.; Brown, P.M.G.E.; Grigoras, I.T.; Malinauskaite, L.; Malinauskas, T.; Miehling, J.; et al. Single-particle cryo-EM at atomic resolution. Nature 2020, 587, 152–156. [Google Scholar] [CrossRef]
- Gustafsson, G.; Lindström, V.; Rostami, J.; Nordström, E.; Lannfelt, L.; Bergström, J.; Ingelsson, M.; Erlandsson, A. Alpha-synuclein oligomer-selective antibodies reduce intracellular accumulation and mitochondrial impairment in alpha-synuclein exposed astrocytes. J. Neuroinflamm. 2017, 14, 241. [Google Scholar] [CrossRef]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- Söllvander, S.; Nikitidou, E.; Gallasch, L.; Zyśk, M.; Söderberg, L.; Sehlin, D.; Lannfelt, L.; Erlandsson, A. The Aβ protofibril selective antibody mAb158 prevents accumulation of Aβ in astrocytes and rescues neurons from Aβ-induced cell death. J. Neuroinflammation 2018, 28, 98. [Google Scholar] [CrossRef]
- Salloway, S.; Sperling, R.; Fox, N.C.; Blennow, K.; Klunk, W.; Raskind, M.; Sabbagh, M.; Honig, L.S.; Porsteinsson, A.P.; Ferris, S.; et al. Two phase 3 trials of Bapineuzumab in mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 2014, 370, 322–333. [Google Scholar] [CrossRef]
- Weihofen, A.; Liu, Y.; Arndt, J.W.; Huy, C.; Quan, C.; Smith, B.A.; Baeriswyl, J.-L.; Cavegn, N.; Senn, L.; Su, L.; et al. Development of an aggregate-selective, human-derived α-synuclein antibody BIIB054 that ameliorates disease phenotypes in Parkinson’s disease models. Neurobiol. Dis. 2019, 124, 276–288. [Google Scholar] [CrossRef]
- Tolar, M.; Abushakra, S.; Hey, J.A.; Porsteinsson, A.; Sabbagh, M. Aducanumab, gantenerumab, BAN2401, and ALZ-801—The first wave of amyloid-targeting drugs for Alzheimer’s disease with potential for near term approval. Alzheimers Res. Ther. 2020, 12, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Dawson, V.L.; Dawson, T.M. Promising disease-modifying therapies for Parkinson’s disease. Sci. Transl. Med. 2019, 11, eaba1659. [Google Scholar] [CrossRef] [PubMed]
- Schofield, D.J.; Irving, L.; Calo, L.; Bogstedt, A.; Rees, G.; Nuccitelli, A.; Narwal, R.; Petrone, M.; Roberts, J.; Brown, L.; et al. Preclinical development of a high affinity α-synuclein antibody, MEDI1341, that can enter the brain, sequester extracellular α-synuclein and attenuate α-synuclein spreading in vivo. Neurobiol. Dis. 2019, 132, 104582. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, E.; Silverman, J.M.; Zhao, B.; Peng, X.; Wang, J.; Wellington, C.L.; Mackenzie, I.R.; Plotkin, S.S.; Kaplan, J.M.; Cashman, N.R. A rationally designed humanized antibody selective for amyloid beta oligomers in Alzheimer’s disease. Sci. Rep. 2019, 9, 9870. [Google Scholar] [CrossRef] [PubMed]
- Schenk, D.B.; Koller, M.; Ness, D.K.; Griffith, S.G.; Grundman, M.; Zago, W.; Soto, J.; Atiee, G.; Ostrowitzki, S.; Kinney, G.G. First-in-human assessment of PRX002, an anti-α-synuclein monoclonal antibody, in healthy volunteers. Mov. Disord. 2017, 32, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Doody, R.S.; Thomas, R.G.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; Raman, R.; Sun, X.; Aisen, P.S.; et al. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 2014, 370, 311–321. [Google Scholar] [CrossRef]
- Kiyota, T.; Machhi, J.; Lu, Y.; Dyavarshetty, B.; Nemati, M.; Yokoyama, I.; Mosley, R.L.; Gendelman, H.E. Granulocyte-macrophage colony-stimulating factor neuroprotective activities in Alzheimer’s disease mice. J. Neuroimmunol. 2018, 319, 80–92. [Google Scholar] [CrossRef]
- Grimm, M.O.W.; Kuchenbecker, J.; Grösgen, S.; Burg, V.K.; Hundsdörfer, B.; Rothhaar, T.L.; Friess, P.; de Wilde, M.C.; Broersen, L.M.; Penke, B.; et al. Docosahexaenoic acid reduces amyloid β production via multiple pleiotropic mechanisms. J. Biol. Chem. 2011, 286, 14028–14039. [Google Scholar] [CrossRef]
- Habchi, J.; Chia, S.; Galvagnion, C.; Michaels, T.C.T.; Bellaiche, M.M.J.; Ruggeri, F.S.; Sanguanini, M.; Idini, I.; Kumita, J.R.; Sparr, E.; et al. Cholesterol catalyses Aβ42 aggregation through a heterogeneous nucleation pathway in the presence of lipid membranes. Nat. Chem. 2018, 10, 673–683. [Google Scholar] [CrossRef]
- Michaels, T.C.T.; Šarić, A.; Meisl, G.; Heller, G.T.; Curk, S.; Arosio, P.; Linse, S.; Dobson, C.M.; Vendruscolo, M.; Knowles, T.P.J. Thermodynamic and kinetic design principles for amyloid-aggregation inhibitors. Proc. Natl. Acad. Sci. USA 2020, 117, 24251–24257. [Google Scholar] [CrossRef]
- Villard, V.; Espallergues, J.; Keller, E.; Vamvakides, A.; Maurice, T. Anti-amnesic and neuroprotective potentials of the mixed muscarinic receptor/sigma1 (σ1) ligand ANAVEX2-73, a novel aminotetrahydrofuran derivative. J. Psychopharmacol. Oxf. 2011, 25, 1101–1117. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.; Ryazanov, S.; Leonov, A.; Levin, J.; Shi, S.; Schmidt, F.; Prix, C.; Pan-Montojo, F.; Bertsch, U.; Mitteregger-Kretzschmar, G.; et al. Anle138b: A novel oligomer modulator for disease-modifying therapy of neurodegenerative diseases such as prion and Parkinson’s disease. Acta Neuropathol. Berl. 2013, 125, 795–813. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Wang, J.; Ho, L.; Ono, K.; Teplow, D.B.; Pasinetti, G.M. Identification of antihypertensive drugs which inhibit amyloid-β protein oligomerization. J. Alzheimers Dis. 2009, 16, 49–57. [Google Scholar] [CrossRef]
- Ono, K.; Tsuji, M. Pharmacological potential of Cilostazol for Alzheimer’s disease. Front. Pharmacol. 2019, 10, 599. [Google Scholar] [CrossRef]
- Shozawa, H.; Oguchi, T.; Tsuji, M.; Yano, S.; Kiuchi, Y.; Ono, K. Supratherapeutic concentrations of cilostazol inhibits β-amyloid oligomerization in vitro. Neurosci. Lett. 2018, 677, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Neumann, U.; Ufer, M.; Jacobson, L.H.; Rouzade-Dominguez, M.-L.; Huledal, G.; Kolly, C.; Lüönd, R.M.; Machauer, R.; Veenstra, S.J.; Hurth, K.; et al. The BACE-1 inhibitor CNP520 for prevention trials in Alzheimer’s disease. EMBO Mol. Med. 2018, 10, e9316. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y.; Takeda, S.; Cho, H.; Wegmann, S.; Shoup, T.M.; Takahashi, K.; Irimia, D.; Elmaleh, D.R.; Hyman, B.T.; Hudry, E. A Food and Drug Administration-approved asthma therapeutic agent impacts amyloid β in the brain in a transgenic model of Alzheimer disease. J. Biol. Chem. 2015, 290, 1966–1978. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.; Congdon, E.E.; Honson, N.S.; Duff, K.E.; Kuret, J. Structure−activity relationship of cyanine tau aggregation inhibitors. J. Med. Chem. 2009, 52, 3539–3547. [Google Scholar] [CrossRef] [PubMed]
- Lai, R.; Albala, B.; Kaplow, J.M.; Aluri, J.; Yen, M.; Satlin, A. O1-06-05: First-in-human study of E2609, a novel BACE1 inhibitor, demonstrates prolonged reductions in plasma beta-amyloid levels after single dosing. Alzheimers Dement. 2012, 8, P96. [Google Scholar] [CrossRef]
- Grundman, M.; Morgan, R.; Lickliter, J.D.; Schneider, L.S.; DeKosky, S.; Izzo, N.J.; Guttendorf, R.; Higgin, M.; Pribyl, J.; Mozzoni, K.; et al. A phase 1 clinical trial of the sigma-2 receptor complex allosteric antagonist CT1812, a novel therapeutic candidate for Alzheimer’s disease. Alzheimers Dement. Transl. Res. Clin. Interv. 2019, 5, 20–26. [Google Scholar] [CrossRef]
- Izzo, N.J.; Staniszewski, A.; To, L.; Fa, M.; Teich, A.F.; Saeed, F.; Wostein, H.; Walko, T.; Vaswani, A.; Wardius, M.; et al. Alzheimer’s therapeutics targeting amyloid beta 1-42 oligomers I: Abeta 42 oligomer binding to specific neuronal receptors is displaced by drug candidates that improve cognitive deficits. PLoS ONE 2014, 9, e111898. [Google Scholar] [CrossRef] [PubMed]
- Izzo, N.J.; Xu, J.; Zeng, C.; Kirk, M.J.; Mozzoni, K.; Silky, C.; Rehak, C.; Yurko, R.; Look, G.; Rishton, G.; et al. Alzheimer’s therapeutics targeting amyloid beta 1-42 oligomers II: Sigma-2/PGRMC1 receptors mediate Abeta 42 oligomer binding and synaptotoxicity. PLoS ONE 2014, 9, e111899. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Carver, J.A.; Calabrese, A.N.; Pukala, T.L. Gallic acid interacts with α-synuclein to prevent the structural collapse necessary for its aggregation. Biochim. Biophys. Acta 2014, 1844, 1481–1485. [Google Scholar] [CrossRef] [PubMed]
- Cho, W.; Park, J.-C.; Kim, D.; Kim, M.; Han, J.-S. ID1201, the ethanolic extract of the fruit of Melia toosendan ameliorates impairments in spatial learning and reduces levels of amyloid beta in 5XFAD mice. Neurosci. Lett. 2014, 583, 170–175. [Google Scholar] [CrossRef]
- Park, H.; Yoo, J.-S.; Kim, J.-Y.; Hwang, B.Y.; Han, J.-S.; Yeon, S.-W.; Kang, J.-H. Anti-amyloidogenic effects of ID1201, the ethanolic extract of the fruits of Melia toosendan, through activation of the phosphatidylinositol 3-kinase/Akt pathway. Environ. Toxicol. Pharmacol. 2014, 37, 513–520. [Google Scholar] [CrossRef]
- Congdon, E.E.; Wu, J.W.; Myeku, N.; Figueroa, Y.H.; Herman, M.; Marinec, P.S.; Gestwicki, J.E.; Dickey, C.A.; Yu, W.H.; Duff, K.E. Methylthioninium chloride (methylene blue) induces autophagy and attenuates tauopathy in vitro and in vivo. Autophagy 2012, 8, 609–622. [Google Scholar] [CrossRef]
- Hebron, M.L.; Lonskaya, I.; Moussa, C.E.-H. Nilotinib reverses loss of dopamine neurons and improves motor behavior via autophagic degradation of α-synuclein in Parkinson’s disease models. Hum. Mol. Genet. 2013, 22, 3315–3328. [Google Scholar] [CrossRef]
- Pagan, F.L.; Hebron, M.L.; Wilmarth, B.; Torres-Yaghi, Y.; Lawler, A.; Mundel, E.E.; Yusuf, N.; Starr, N.J.; Anjum, M.; Arellano, J.; et al. Nilotinib effects on safety, tolerability, and potential biomarkers in Parkinson disease: A phase 2 randomized clinical trial. JAMA Neurol. 2020, 77, 309–317. [Google Scholar] [CrossRef]
- Price, D.L.; Koike, M.A.; Khan, A.; Wrasidlo, W.; Rockenstein, E.; Masliah, E.; Bonhaus, D. The small molecule alpha-synuclein misfolding inhibitor, NPT200-11, produces multiple benefits in an animal model of Parkinson’s disease. Sci. Rep. 2018, 8, 16165. [Google Scholar] [CrossRef]
- Finkelstein, D.I.; Billings, J.L.; Adlard, P.A.; Ayton, S.; Sedjahtera, A.; Masters, C.L.; Wilkins, S.; Shackleford, D.M.; Charman, S.A.; Bal, W.; et al. The novel compound PBT434 prevents iron mediated neurodegeneration and alpha-synuclein toxicity in multiple models of Parkinson’s disease. Acta Neuropathol. Commun. 2017, 5, 53. [Google Scholar] [CrossRef]
- Lahiri, D.K.; Chen, D.; Maloney, B.; Holloway, H.W.; Yu, Q.; Utsuki, T.; Giordano, T.; Sambamurti, K.; Greig, N.H. The experimental Alzheimer’s disease drug posiphen [(+)-phenserine] lowers amyloid-beta peptide levels in cell culture and mice. J. Pharmacol. Exp. Ther. 2007, 320, 386–396. [Google Scholar] [CrossRef] [PubMed]
- Teich, A.F.; Sharma, E.; Barnwell, E.; Zhang, H.; Staniszewski, A.; Utsuki, T.; Padmaraju, V.; Mazell, C.; Tzekou, A.; Sambamurti, K.; et al. Translational inhibition of APP by Posiphen: Efficacy, pharmacodynamics, and pharmacokinetics in the APP/PS1 mouse. Alzheimers Dement. 2018, 4, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Kuo, Y.-M.; Nwankwo, E.I.; Nussbaum, R.L.; Rogers, J.; Maccecchini, M.L. Translational inhibition of α-synuclein by Posiphen normalizes distal colon motility in transgenic Parkinson mice. Am. J. Neurodegener. Dis. 2019, 8, 1–15. [Google Scholar] [PubMed]
- Yu, Q.-S.; Reale, M.; Kamal, M.A.; Holloway, H.W.; Luo, W.; Sambamurti, K.; Ray, B.; Lahiri, D.K.; Rogers, J.T.; Greig, N.H. Synthesis of the Alzheimer drug Posiphen into its primary metabolic products (+)-N1-norPosiphen, (+)-N8-norPosiphen and (+)-N1, N8-bisnorPosiphen, their inhibition of amyloid precursor protein, α-Synuclein synthesis, interleukin-1β release, and cholinergic action. Anti-Inflamm. Anti-Allergy Agents Med. Chem. 2013, 12, 117–128. [Google Scholar] [CrossRef]
- Wang, H.-Y.; Lee, K.-C.; Pei, Z.; Khan, A.; Bakshi, K.; Burns, L.H. PTI-125 binds and reverses an altered conformation of filamin A to reduce Alzheimer’s disease pathogenesis. Neurobiol. Aging 2017, 55, 99–114. [Google Scholar] [CrossRef]
- Kennedy, M.E.; Stamford, A.W.; Chen, X.; Cox, K.; Cumming, J.N.; Dockendorf, M.F.; Egan, M.; Ereshefsky, L.; Hodgson, R.A.; Hyde, L.A.; et al. The BACE1 inhibitor verubecestat (MK-8931) reduces CNS β-amyloid in animal models and in Alzheimer’s disease patients. Sci. Transl. Med. 2016, 8, 363ra150. [Google Scholar] [CrossRef]
- Hamaguchi, T.; Ono, K.; Murase, A.; Yamada, M. Phenolic compounds prevent Alzheimer’s pathology through different effects on the amyloid-β aggregation pathway. Am. J. Pathol. 2009, 175, 2557–2565. [Google Scholar] [CrossRef]
- Zelus, C.; Fox, A.; Calciano, A.; Faridian, B.S.; Nogaj, L.A.; Moffet, D.A. Myricetin inhibits Islet Amyloid Polypeptide (IAPP) aggregation and rescues living mammalian cells from IAPP toxicity. Open Biochem. J. 2012, 6, 66–70. [Google Scholar] [CrossRef]
- Kim, J.-Y.; Kim, D.H.; Kim, J.H.; Lee, D.; Jeon, H.B.; Kwon, S.-J.; Kim, S.M.; Yoo, Y.J.; Lee, E.H.; Choi, S.J.; et al. Soluble intracellular adhesion molecule-1 secreted by human umbilical cord blood-derived mesenchymal stem cell reduces amyloid- β plaques. Cell Death Differ. 2012, 19, 680–691. [Google Scholar] [CrossRef]
- Kontsekova, E.; Zilka, N.; Kovacech, B.; Novak, P.; Novak, M. First-in-man tau vaccine targeting structural determinants essential for pathological tau–tau interaction reduces tau oligomerisation and neurofibrillary degeneration in an Alzheimer’s disease model. Alzheimers Res. Ther. 2014, 6, 44. [Google Scholar] [CrossRef]
- West, T.; Hu, Y.; Verghese, P.B.; Bateman, R.J.; Braunstein, J.B.; Fogelman, I.; Budur, K.; Florian, H.; Mendonca, N.; Holtzman, D.M. Preclinical and clinical development of ABBV-8E12, a humanized anti-tau antibody, for treatment of Alzheimer’s disease and other tauopathies. J. Prev. Alzheimers Dis. 2017, 4, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Wiessner, C.; Wiederhold, K.-H.; Tissot, A.C.; Frey, P.; Danner, S.; Jacobson, L.H.; Jennings, G.T.; Lüönd, R.; Ortmann, R.; Reichwald, J.; et al. The second-generation active Aβ immunotherapy CAD106 reduces amyloid accumulation in APP transgenic mice while minimizing potential side effects. J. Neurosci. 2011, 31, 9323–9331. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Wang, P.-N.; Chiu, M.-J.; Finstad, C.L.; Lin, F.; Lynn, S.; Tai, Y.-H.; De Fang, X.; Zhao, K.; Hung, C.-H.; et al. UB-311, a novel UBITh® amyloid β peptide vaccine for mild Alzheimer’s disease. Alzheimers Dement. Transl. Res. Clin. Interv. 2017, 3, 262–272. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).