1. Introduction
The last decade has witnessed unprecedented progress in understanding the genetic complexity of amyotrophic lateral sclerosis (ALS), the most common motor neuron disease, and it is now clear that regardless of the gene mutation, ALS results from both cell autonomous degeneration of cortical and/or spinal motor neurons as well as non-cell autonomous cell damage [
1].
Evidence from ALS mouse models, in particular the superoxide dismutase 1 (SOD1) mutant transgenic mice, together with findings from ALS patients, indicate that multiple mechanisms are involved in the death of the motor neurons (MNs) and the loss of muscular innervation. They include a mix of molecular processes (e.g., protein aggregation, impaired RNA processing and metabolism, mitochondrial dysfunction, oxidative stress, cytoskeletal alterations and impairment of axonal transport) that cause cell-autonomous motor neuron dysfunction [
2]. Other mechanisms involve the participation of different cell types of the central nervous system (CNS) such as reactive astroglia and microglia [
3], Schwann cells of the peripheral nervous system (PNS) [
4] and macrophages and T-cells of the peripheral immune system [
5,
6] that may also elicit neuroinflammatory processes contributing actively to motor neuron degeneration. However, motor neurons become vulnerable to these mechanisms in adult life through activation of signaling pathways promoting cell death [
7] or loss of compensatory pro-survival mechanisms such as Protein kinase B (AKT) or the extracellular signal-related kinase (ERK) signaling pathway [
8,
9] and/or the loss of an anti-inflammatory protective environment around damaged motor neurons [
10].
The hepatocyte growth factor/scatter factor (HGF/SF), through the activation of its receptor MET, is one of the most potent survival-promoting factors for sensory and motor neurons during the development and in adulthood in the event of tissue damage [
11,
12]. Interestingly, certain residual anterior horn cells from post-mortem ALS patients overexpressed both HGF/SF and MET in comparison with those of normal subjects [
13]. Most importantly, transgenic overexpression or intrathecal delivery of the growth factor markedly delayed the progression of the disease in SOD1G93A transgenic mice and rats [
14,
15]. Hence, early clinical trials involving intramuscular administration of plasmid HGF/SF DNA [
16] or intrathecal delivery of HGF/SF protein [
17] in ALS patients have displayed adequate safety profiles and are now progressing to efficacy studies. However, HGF/SF has a complex multidomain structure consisting of a 69 kDa alpha-chain comprising an N terminal domain (N) and four kringle domains (K) linked via a disulfide bond to a 34 kDa beta-chain consisting of an inactive serine protease homology domain (SPH) (
Figure S1) and may be rapidly degraded in the proteinase-rich microenvironment of degenerating motor neurons and activated glial cells typical of ALS, limiting its efficacy. Therefore, fragments of HGF/SF engineered for superior stability, potency and ability to cross the blood–brain barrier may be more suitable for therapy than the natural molecule.
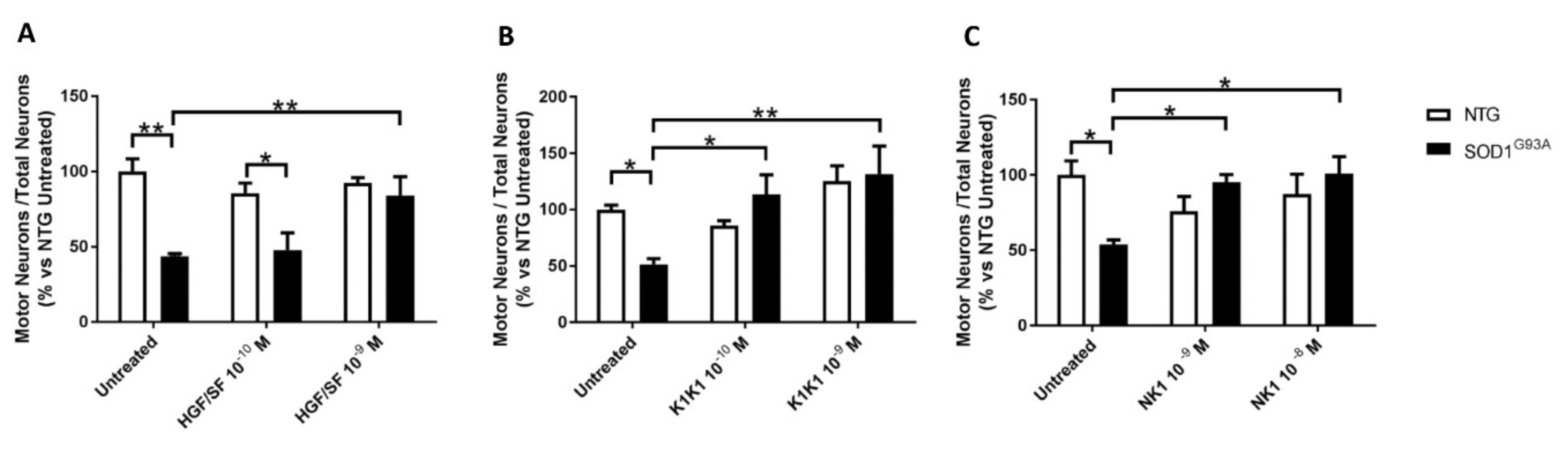
We recently developed and tested in vitro stable and potent recombinant MET agonists based on HGF/SF. They include NK1, a small natural fragment of the growth factor consisting of the N and first kringle domain (K1) of HGF/SF and encompassing the high-affinity MET binding site [
18] (
Supplementary Figure S1), and a novel dimeric form of the K1 domain, designated K1K1 [
19]. After preliminary studies in vitro, we selected the most potent protein, K1K1, as the molecule of choice to assess the neuroprotective effect in in vitro and in vivo models of ALS. To overcome potential immunogenicity of the human proteins, a mouse version of K1K1 was generated and produced for the in vivo experiments in SOD1G93A transgenic mice. We observed a delay in the disease course of mice treated intraperitoneally with K1K1 and this effect was accompanied by a partial protection of motor neurons and neuromuscular junctions as well as alterations in inflammatory and immune response in the spinal cord and skeletal muscle. The molecular mechanisms involved in these effects of K1K1 were examined.
3. Discussion
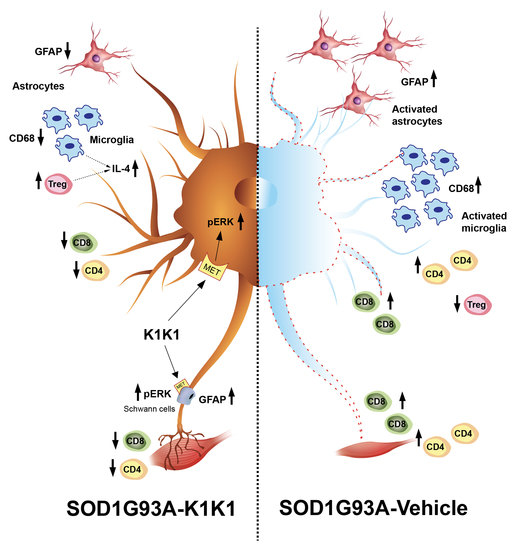
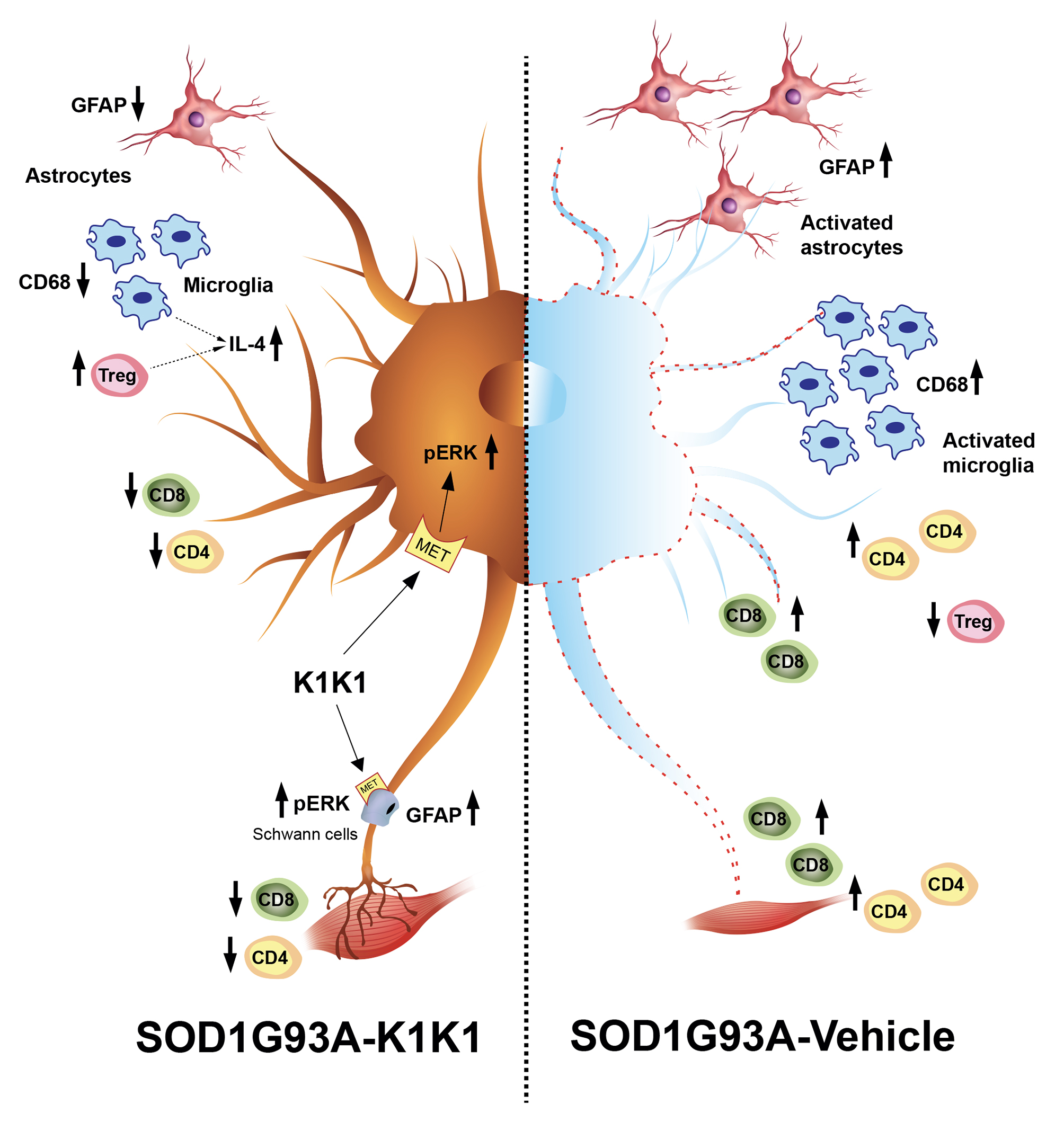
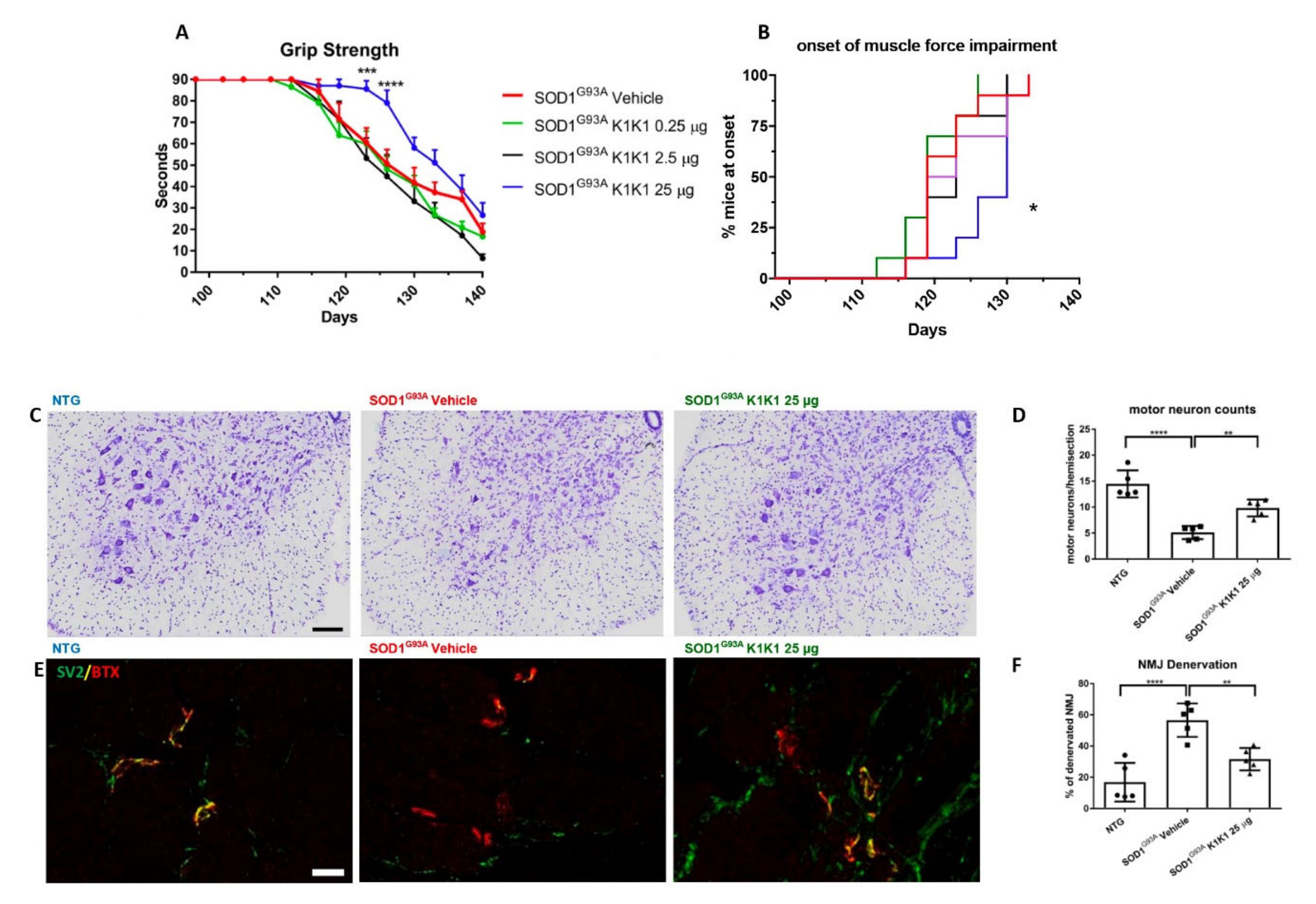
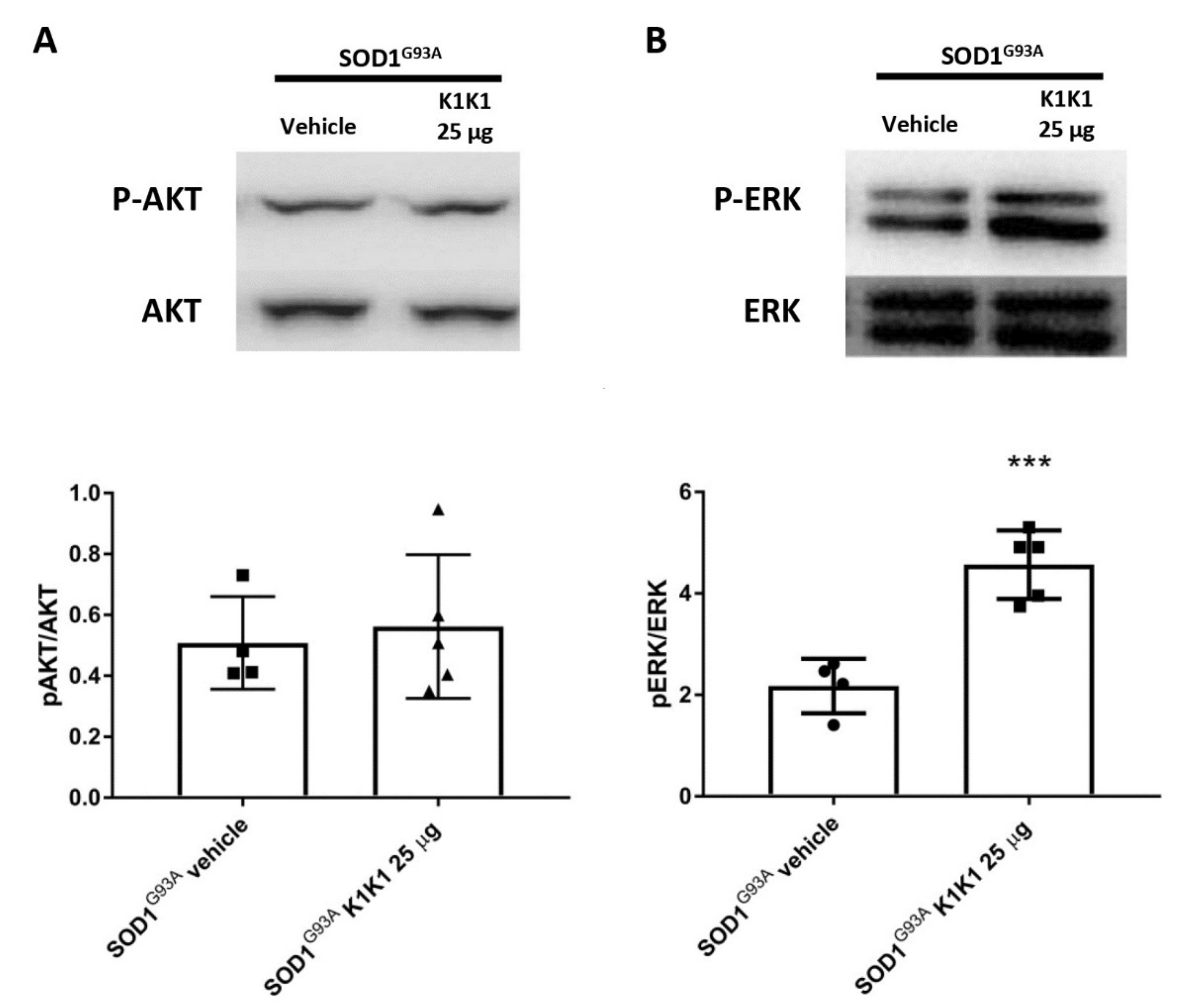
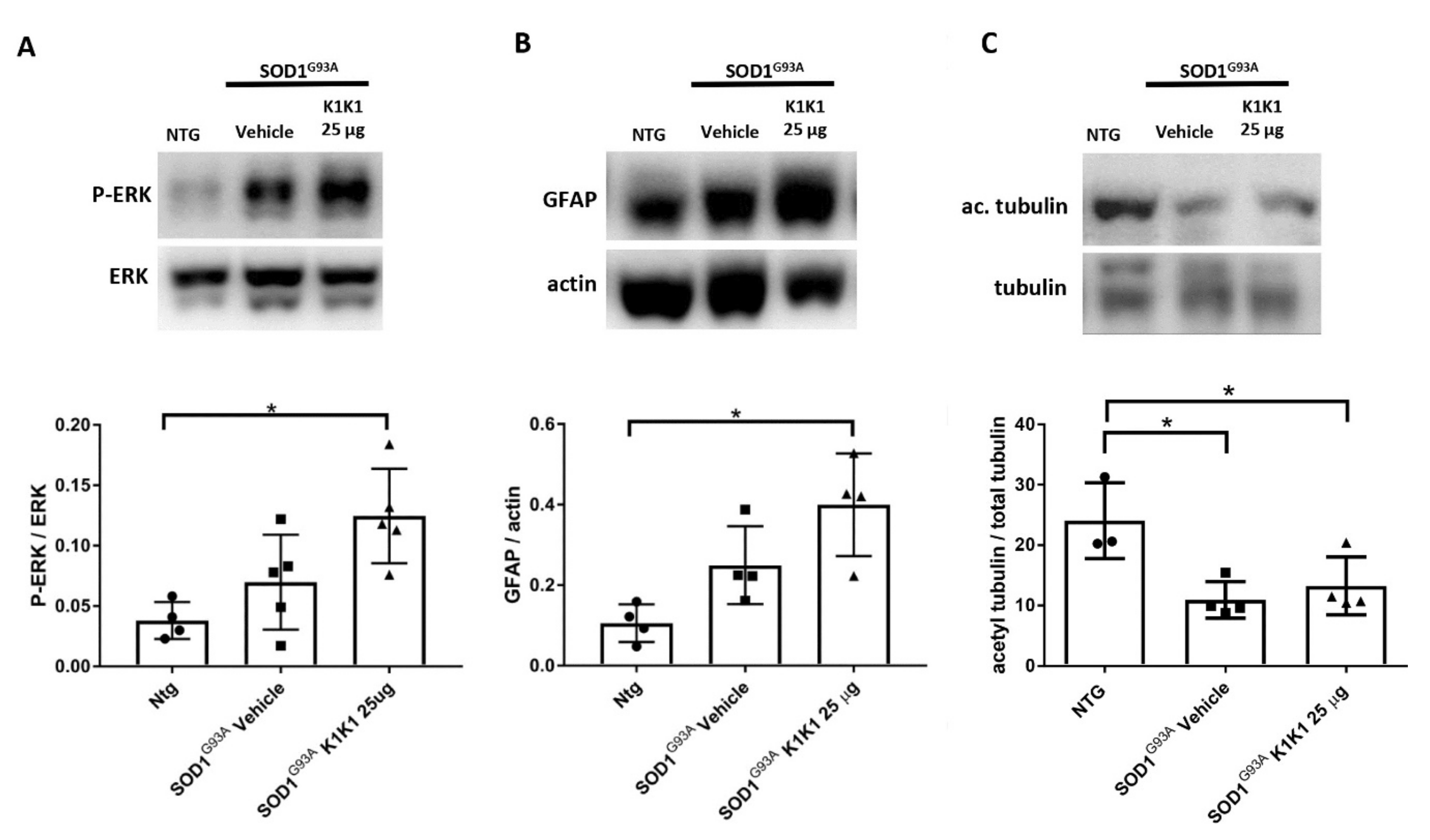
The present study strengthens the evidence that activation of the MET receptor by engineered derivatives of HGF/SF results in a strong neuroprotective activity on the motor neurons of SOD1G93A transgenic mice and demonstrates for the first time—to the best of our knowledge—that this result can be accomplished through the systemic administration of a potent MET ligand. Treatment with K1K1 resulted in the rescue of motor neurons from death in spinal-neuron astrocyte co-cultures as well as in the lumbar spinal cord of SOD1G93A transgenic mice accompanied by a transient amelioration of the muscle force impairment, although it did not prolong the mouse survival. This improved outcome of K1K1 treated ALS mice was characterized by the protection of motor neurons both at the level of their soma in the spinal cord and of the motor axons terminals as demonstrated by the reduced neuromuscular junction denervation. In the spinal cord, we found that the neuroprotection was accompanied by the activation (phosphorylation) of ERK but not AKT. This result complies with a recent study showing that intrathecal delivery of recombinant AAV1 encoding HGF/SF protected spinal motor neurons in SOD1G93A mice through an increase in phosphorylated ERK but no other signaling molecules of the HGF/SF-MET pathway such as STAT3, cJUN and GSK3b [
15]. This highlights the importance of ERK activation in the rescue of motor neurons induced by HGF/MET signaling. Interestingly, ERK was remarkably activated also in the sciatic nerves of SOD1G93A mice treated with K1K1 together with the increased expression of GFAP. Activation of ERK and GFAP in the sciatic nerve is essential to stimulate the de-differentiation and promote the proliferation of Schwann cells, respectively, during re-innervation [
25]. This is consistent with the recent evidence demonstrating that HGF/SF increased the migration and proliferation of cultured Schwann cells by activating the ERK pathway and accelerated the nerve regeneration process after nerve crush [
29]. Interestingly, we previously demonstrated that the expression of GFAP and p-ERK were higher in the sciatic nerves of SOD1G93A mice showing a delayed disease onset and progression compared to those with fast disease progression and this was associated with higher levels of acetyl-tubulin, a marker of axonal function [
24]. With K1K1 treatment, we did not find a rescue of acetyl-tubulin reduction in symptomatic SOD1G93A mice, however it should be noted that while in the previous study the analysis had been performed at the disease onset, here the mice were examined at the symptomatic stage. We cannot rule out that a rescue of acetylated tubulin occurred at an earlier time point after K1K1 treatment. Noteworthily, the prolonged treatment with K1K1 until the end stage of the disease lost its efficacy in activating p-ERK/ERK in LSC and sciatic nerve of SOD1G93A mice (
Supplementary Figure S5) suggesting a mechanism of “drug tolerance” which could explain the transiency of the amelioration of the neuromuscular deficit of SOD1G93A mice.
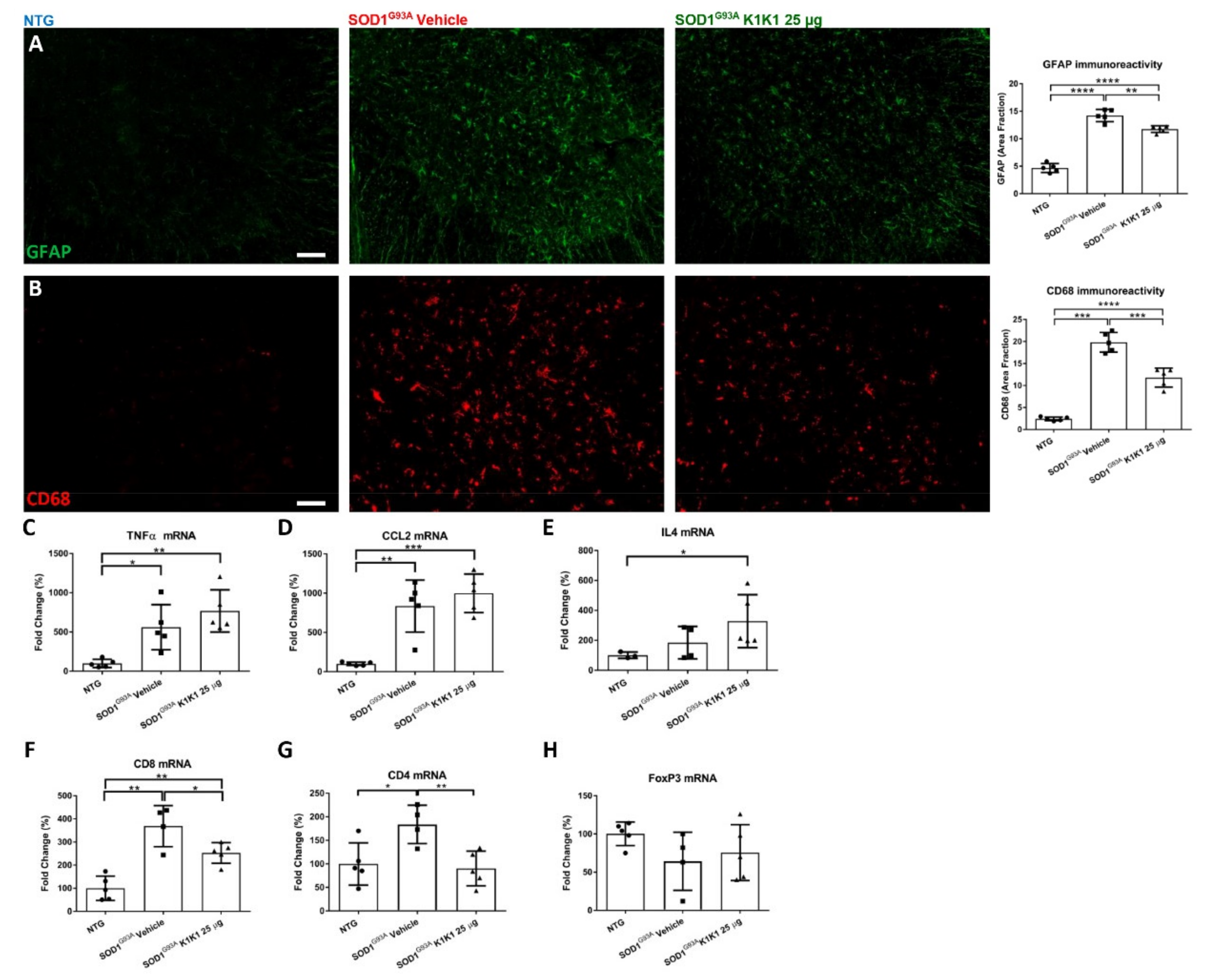
K1K1 treatment also produced a significant reduction in the reactive astrogliosis and microgliosis in the LSC of SOD1G93A mice. We do not know whether this is a response to the partial rescue of MNs or is causative of the neuroprotection. Unexpectedly, the reduction in reactive gliosis did not match with a reduction in proinflammatory cytokines TNFalpha and CCL2 that remained higher in SOD1G93A mice after treatment with K1K1. However, we detected a concomitant increase in anti-inflammatory IL-4 in the LSC of K1K1 treated mice, suggesting that reactive microglia, besides being reduced in number, might have adopted a partial anti-inflammatory phenotype in response to K1K1 treatment [
30]. An association between motor neuron protection, reduction in microgliosis and increased IL-4 was previously observed with other treatments in SOD1G93A mice sustaining the protective role of this cytokine [
22,
31]. Interestingly, the intracerebroventricular delivery of IL-4 in SOD1G93A mice, via a lentiviral-mediated gene therapy strategy, was able to modulate microglia and to delay the disease onset in these mice but did not prolong their survival [
32].
Another potential mechanism through which K1K1 treatment may exert it protective effect on neuromuscular system relates to the modulation of the immune response in both the spinal cord and the skeletal muscle. The MET signaling pathways have been implicated in the modulation of different immune–inflammatory responses [
33]. For example, in a multiple sclerosis mouse model HGF/SF has been shown to modulate both CD4
+ and CD8
+ T cell effector responses [
26,
27]. Here, we show that the increased recruitment of CD8
+ and CD4
+ T cells in the spinal cord of symptomatic SOD1G93A mice was significantly counteracted by treatment with K1K1 during the early progressing phase of the disease, although this effect disappeared on the long-term treatment. Whether this effect may depend on a direct action of K1K1 on the migratory activity of T cells or being a consequence of the protective effect on the MNs needs to be investigated. We and other groups have recently demonstrated that the ablation of CD8
+ T cells in SOD1G93A mice protected spinal motor neurons from death at the early symptomatic stage [
24,
34] and this effect was associated with the reduction in macrophagic microglia hyperactivation [
24]. On the contrary, the lack of CD4
+ T cells in SOD1G93A mice showed a detrimental effect on disease progression, in the presence of attenuated microglia and astrocyte reactivity in the spinal cord [
35]. The CD4
+ T lymphocytes in the spinal cord of SOD1G93A mice are probably a mixture of Th1 and Th2 effector T cells (Teffs) and Th2 lymphocytes possibly provide the increased, although not significant, levels of IL-4 mRNA found in vehicle treated SOD1G93A mice, compared to NTG. However, IL-4 mRNA levels were further increased by the treatment with K1K1 despite the reduction in CD4
+ T cells mRNA in the spinal cord. IL-4 is also produced by the Treg, a CD4
+ T lymphocyte subtype with marked neuroprotective effect in SOD1G93A mice [
32]. Through the analysis of FoxP3, a transcription factor typical of the Treg, we showed that the proportion of Tregs present in the CD4
+ T cell population of SOD1G93A mice treated with K1K1 is higher than that present in vehicle treated mice. This is consistent with the protective action of K1K1 in SOD1G93A mice. In fact, growing evidence suggest that the expansion of Tregs plays a significant role in the modulation of disease progression in ALS patients and mouse models [
6,
10]. Passive transfer of T-cell populations enriched in Tregs were shown to sustain IL-4 levels and M2 microglia, lengthen disease duration, and prolong survival of SOD1G93A mice [
35]. Moreover, treatment of transgenic mice with interleukin 2 complex (IL-2c) with rapamycin, which is known to induced Treg expansion, significantly increased the levels of FOXP3 mRNA in the spinal cord of SOD1G93A mice and this effect was associated to a rescue of motor neurons and reduced glial activation [
36]. It has been reported that dendritic cells exposed to HGF/SF develop tolerogenic properties and facilitate the expansion of Tregs [
37]. Dendritic cells (DCs) were detected early in the spinal cord of SOD1G93A mice [
38] as well as in post-mortem ALS patient spinal cord [
39]. Thus, we can speculate that infiltrated DCs could have been activated by K1K1 in the spinal cord of SOD1G93A mice in an attempt to preserve the Treg population.
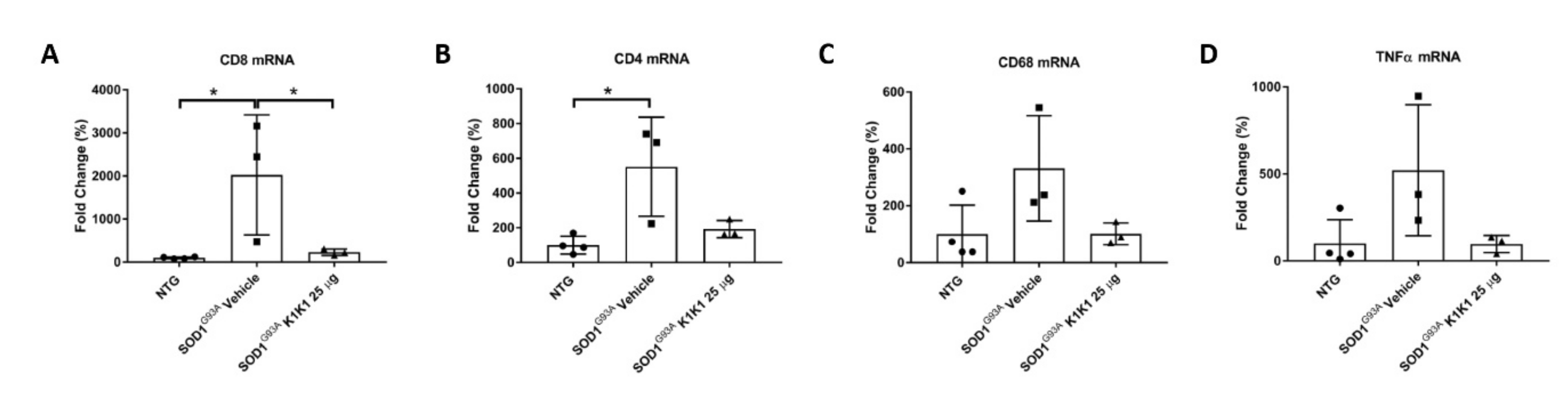
The immunomodulatory effect by K1K1 in SOD1G93A mice was detected at the skeletal muscle level as well. In fact, the overexpression of macrophages and T lymphocytes found in the TAM of symptomatic SOD1G93A mice were markedly reduced in K1K1 treated mice with consequent partial reduction in the proinflammatory cytokine, TNFalpha. There is a consolidated hypothesis that, following damage, the skeletal muscle activates a series of immune mediators and proinflammatory cytokines that allow the recruitment of macrophages, neutrophils and adaptive immune T cells important for the improvement and modulation of the muscle growth and regeneration [
28]. We recently reported that a depletion of CD8
+ T cells in SOD1G93A mice lacking beta2-microglobulin, while protecting the soma of MNs in spinal cord, accelerated the NMJs denervation in the TAM and this was accompanied by an anticipated onset of hind limb impairment [
24]. However, the effect was transient as at the later symptomatic stage there was no difference in TAM NMJs denervation between SOD1G93A mice with or without CD8
+ T cells while the onset of disability was delayed [
24]. Apparently, this result is at variance with those obtained in the present study showing that the decrease in CD8
+ T induced by K1K1 was accompanied by reduced NMJs denervation. Nevertheless, unlike the study on counteracting roles of MHCI and CD8
+ T cells [
24], with K1K1 we induced a massive immune suppression in the skeletal muscle that include both the macrophages and the CD4
+ T cells and reduced the proinflammatory cytokine TNFalpha that may have contributed to the delay of symptoms. Activated macrophages have been reported to infiltrate and accumulate in the skeletal muscles of SOD1G93A mice beginning from disease onset [
39] and its reduction by ablation of complement signaling was reported to reduce NMJs denervation and to improve hind limb grip strength in mice [
40] similarly to what we observed with K1K1. Noteworthily, although we found that K1K1 treatment partially rescued the denervation of NMJs in TAM, the decrease in muscle mass was not counteracted by the treatment. Since there is evidence that the recruitment of CD4
+ T cells in damaged skeletal muscles is important for the repair and the regeneration of the muscle [
28], we cannot exclude that their reduction by K1K1 may have interfered with this process.
4. Materials and Methods
The wild-type human and mouse HGF/SF were produced in the myeloma line NS0 (MRC, UK) and purified as previously described [
18]. The natural splice-variant NK1 was produced recombinantly in the yeast expression system
Pichia pastoris (Invitrogen, Carlsbad, CA, USA) [
41]. The engineered recombinant K1K1 was produced as described in Leclercq et al., 2020 [
19]. Briefly, K1K1 was produced in bacterial inclusion bodies using the
E. coli strain BL21 (Invitrogen) giving yields of around 10 mg/litre. The protein was extracted from the inclusion bodies using a Tris buffered 2 M arginine solution and was subsequently diluted in a Tris pH 7.4 buffer and purified by Heparin Sepharose affinity and gel filtration chromatography. Each batch of MET agonist was quantified by absorbance measurement at 280 nm or standard BCA assay (Pierce, Walthman, MA, USA).
4.1. Madin-Darby Canine Kidney Cell Colony Scatter Assay
For the testing of biological activity, mouse and human variants of HGF/SF and K1K1 were tested in a sensitive colony dispersal (“scatter”) assay [
18]. MDCK cells (ICRF, UK) were maintained in DMEM (Gibco, Life Technologies, Grovemont, MD, USA) supplemented with 10% FCS (Gibco) in a humidified incubator at 37 °C with 5% CO
2. Small and compact colonies appear overnight after seeding 1000 cells per well in a 96-well plate. The medium was replaced with DMEM + 10% FCS supplemented with the ligands at different concentrations ranging from 10 µM down to 0.1 pM using serial half-log dilutions. The next day, colony dispersal was observed at low magnification under an inverted microscope and the endpoint of observable scattering was determined for each protein. Using low magnification, three photos were taken of individual colonies at each concentration for all ligands.
4.2. Quantification of Protein Degradation
Purified HGF/SF and K1K1 was diluted to a final concentration of 10 µM in 200 µL Phosphate Buffer Saline (PBS) at pH 7.4 and in 200 µL RPMI medium (Gibco, Life Technology) supplemented with 10% FCS (Gibco). The proteins were incubated at 37 °C for 4 weeks. Samples of 40 µl were collected each week, flash frozen and stored at −80 °C for further analysis by SDS-PAGE and Thermal shift assay. An amount of 5 µg HGF/SF and 3.5 µg K1K1 sample was loaded on a 12% SDS-PAGE gel and subsequently stained with Coomassie Blue. A thermal shift assay was performed using the Tycho NT.6 (NanoTemper Technologies, München, Germany) measuring changes in total fluorescence intensity (“brightness”) at 330 nm and 350 nm with a 30 °C/min ramp (from 35 to 95 °C). The total sample brightness was used for calculating the percentage reduction in soluble protein in the samples using Excel (Microsoft) and the bar chart was produced using Prism (Graphpad, San Diego, CS, USA).
4.3. SOD1G93A Mice
Transgenic mice (C57B6.CgTg SOD1.G93A1Gur/J) were originally obtained from Jackson Laboratories (Bar Harbor, ME, USA) and then maintained on a C57BL6/J background at the Istituto di Ricerche Farmacologiche Mario Negri IRCCS, Milan, Italy (IRFMN). The animals were housed under specific pathogen free (SPF) standard conditions (22 °C ± 1, 55% ± 10 relative humidity and 12 h light/dark schedule), 3–4 per cage, with free access to food (standard pellet, Altromin, MT, Rieper) and water. Procedures involving animals and their care were conducted in conformity with the institutional guidelines of the Mario Negri Institute for Pharmacological Research IRCCS, Milan, Italy, which are in compliance with national (D.lgs 26/2014; Authorization n.783/2016-PR issued on 8 August 2016 by Ministry of Health) and Mario Negri Institutional regulations and Policies providing internal authorization for persons conducting animal experiments (Quality Management System certificate—UNI EN ISO 9001:2008–reg. N° 6121), the NIH Guide for the Care and Use of Laboratory Animals (2011 edition) and EU directives and guidelines (EEC Council Directive 2010/63/UE).
4.4. Primary Astrocyte-Spinal Neuron Co-Cultures
SOD1G93A and non-transgenic co-cultures were prepared as previously described [
21,
22]. Briefly, astrocytes were obtained dissecting cortices of E13-E14 embryos from SOD1G93A mice or their non-transgenic littermates and mechanically dissociation in Hanks’ balanced salt solution (HBSS) containing 33 mM glucose. After centrifugation, the pellet was suspended in culture medium (Dulbecco’s modified Eagle’s medium/F12, 2 mM L-glutamine, 33 mM glucose, 5 µg/mL gentamycin, 10% horse serum) and seeded (500,000 cells/mL) onto 48- or 6-well plates coated with 1.5 µg/mL poly-L-ornithine. Repeated washing with HBSS, 12 h of orbital shaking at 200 rpm and treatment with 10 M AraC once they reached confluence, rendered astrocyte cultures free of microglia, oligodendrocytes, and neurons. Spinal cords of E13-E14 embryos were dissected and mechanically dissociated in HBSS, 33 mM glucose. The cells were centrifuged onto a 4% bovine serum albumin cushion at 201 rcf for 10 min and the pellet resuspended in neuron culture medium: Neurobasal (Gibco), 2 mM L-glutamine, 33 mM glucose, 5 µg/mL gentamycin, 1 ng/mL brain-derived neurotrophic factor, 25 µg/mL insulin, 10 µg/mL putrescine, 30 nM sodium selenite, 2 µM progesterone, 100 µg/mL apo-transferrin, 10% heat-inactivated horse serum, 10 µM AraC. Cells were seeded (1,000,000 cells/mL) onto 48- or 6-well plates coated with 15 µg/mL poly-L-ornithine and 2 µg/mL laminin or onto a pre-established astrocyte confluent layer to obtain spinal neuron-cortical astrocyte co-cultures. Non-transgenic and SOD1G93A co-culture were obtained from non-transgenic and SOD1G93A neurons seeded on non-transgenic and SOD1G93A astrocytes, respectively. Motor neurons obtained from E13–14 embryos are fully differentiated and express the specific transcription factors Hb9 and Islet-1/2. A few days after plating, they show adult characteristics such as profuse dendrite and axon outgrowth and synapse formation [
21,
22]. Co-cultures were treated with HGF/SF, NK1 and K1K1. The molecules were administered at different doses every other day starting from the plating of the neurons, days in vitro (DIV) 0, 2 and 4. Co-cultures were fixed and analyzed at 6 DIV.
4.5. Immunocytochemistry and Evaluation of Motor Neuron Survival In Vitro
Immunocytochemistry was performed as previously described [
21,
22,
31]. After an incubation with blocking solution containing 10% normal goat serum (NGS) and 0.1% Triton in phosphate-buffered saline (PBS) 0.01 M, the cells were incubated overnight at 4 °C with the primary antibodies mouse monoclonal anti-SMI32 (1:1000, Covance, Cambridge, MA, USA) and mouse monoclonal anti-NeuN (1:250, Chemicon, Burlington, MA, USA), diluted in a solution containing 1% NGS and 0.1% Triton in PBS 0.01M. Cells were then washed and incubated with the appropriate secondary fluorescent antibody (1:500, Alexa Fluor Dyes, Life Technologies) or secondary biotinylated antibody (1:500, Vector Laboratories, Burlingame, CA, USA) for tyramide signal amplification (TSA, Perkin Elmer, Walthman, MA, USA) following the manufacturer’s instructions. Images were acquired with an Olympus BX41 fluorescence microscope. Motor neuron survival was evaluated as previously described [
21,
22]. The labelling with anti-SMI32 antibody highlights motor neurons with typical morphology and large cell bodies (diameter ≥ 20 μm) and anti-NeuN antibody was used to identify all neurons in twelve adjacent frames per well at 10X-magnification. Data were calculated as the ratio of the number of motor neurons to the total neurons and expressed as percentage of untreated non-transgenic co-culture to compare different experiments.
4.6. MET Agonist K1K1 In Vivo Treatment
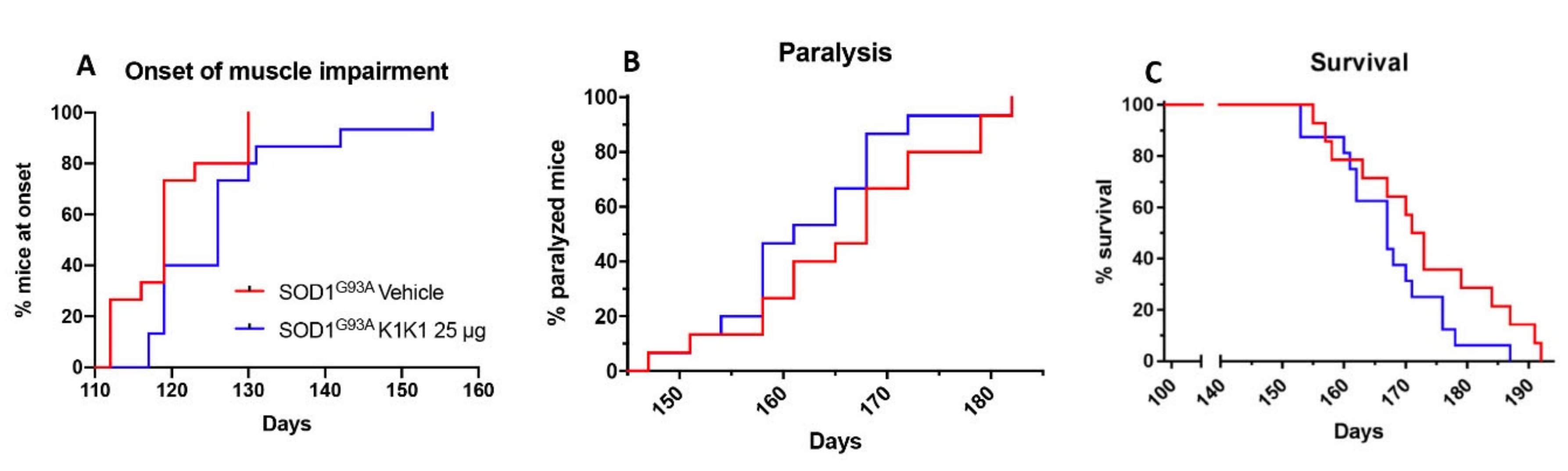
For the first in vivo treatment SOD1G93A female mice were treated intraperitoneally (IP) with PBS vehicle or mouse K1K1 (0.25, 2.5, and 25 μg/injection) every day, five days a week, starting from 98 days of age, when the mice show the first signs of symptoms (hind limb tremors and reduced limb abduction), and until the overt symptomatic phase (140 days of age). At 140 days of age, ten mice per group were sacrificed to perform histopathological, biochemical and molecular analyses. For the second in vivo treatment, 15 female SOD1G93A mice per group were treated IP with vehicle or K1K1 (25 μg) every day, five days a week, starting at the onset of symptoms (98 days of age) and until the end stage of the disease to assess the effect on survival.
4.7. Behavioural Analysis and Survival
Behavioral analyses were performed in all mice two times a week from the onset of the disease, by the same investigator blinded to the treatment. The grip strength test was used to measure disease progression by evaluation the limb resistance as previously described [
21,
22,
31]. Mice were placed on a horizontal metallic grid which was then gently inverted. The latency to fall of each mouse was recorded. The test ended after 90 s. The measurement was repeated three times in case the mice fell off before 90 s and the best performance holding on the grid was considered for the statistical analysis. The onset of neuromuscular deficit was evaluated at the age when the mouse exhibits the first failure in the paw grip strength test at two consecutive time points. The age at which the mice were no longer able to perform the grip strength test was considered as time of paralysis. The mice were sacrificed when they were unable to right themselves within 10 s after being placed on either side. This time was considered the end stage of the disease and was used to calculate the survival.
4.8. Immunohistochemistry
Spinal cords were processed as previously described [
22,
31]. Briefly, under deep anesthesia, mice were transcardially perfused with PBS followed by 4% paraformaldehyde (PFA) solution. The spinal cords were quickly removed, post fixed for 24 h, and cryopreserved in 30% sucrose solution at 4 °C until they sank, embedded in Tissue-tek OCT (Sakura, AJ Alphen aan den Rijn, The Netherlands), frozen in n-pentane at −45 °C and stored at −80 °C until analysis. Spinal cord immunohistochemistry was performed on free floating sections (30 μm). The number of motor neurons was determined on serial sections (20, one every 10th) from lumbar spinal cord (LSC) segments L2-L5 of 5 mice per group as previously described [
22]. The sections were stained with cresyl violet to detect the Nissl substance of neuronal cells. Motor neuron counting was performed at 10× magnification using the free software ImageJ (Windows v8.0,
http://imagej.nih.gov/ij/), previously calibrated. Intense Nissl labelled neurons with clear nucleus and nucleolus and an area of the cell body higher than 400 μm
2 were identified as motor neurons. The number of motor neurons was calculated for each hemisection and the means used for statistical analysis. Immunofluorescence was evaluated on 5–7 coronal spinal cord slices (1 every 10) from LSC per animal. After an incubation with blocking solution containing 3% NGS and 0.1% Triton in 0.01 M PBS, primary antibodies
mouse anti-GFAP (1:2500, Millipore, Burlington, MA, USA) and rat anti-CD68 (1:200, AbDSerotec, Hercules, CA, USA) and appropriate fluoro-conjugated secondary antibodies (1:500 dilution), Alexa 647 and Alexa 488 (Alexa Fluor
® Dyes, Life Technologies) were used. The sections were analyzed under Olympus Fluoview confocal microscope. The quantification of GFAP and CD68 intensity in the ventral horns was carried out using the free software ImageJ by determining the area fraction of fluorescent signal after setting a threshold value within a grey-scale (corresponding to the maximum level of an unstained section background) and considering all the pixels falling over this value as positive. These analyses were executed by the same operator blinded to treatment.
4.9. Neuromuscular Junctions (NMJs)
NMJ denervation was detected according to the previously described protocol [
22]. Briefly, tibialis anterior muscles (TAM) were dissected from mice transcardially perfused with PBS under deep anesthesia and snap-frozen in isopentane cooled on dry ice. Serial 20 µm cryostat longitudinal muscle sections were collected on poly-Lysine objective slides (VWR International), fixed in chilled acetone for 10 min, incubated in a blocking solution (0.3% Triton, 10% NGS in 0.01 M PBS) for 1 h at 22 °C and left overnight at 4 °C with anti-SV2 primary antibody (1:100, DSHB) in 0.15% Triton, 5% NGS, 0.01 M PBS. Then the sections were incubated with goat anti-rabbit 647 (1:500, Alexa Fluor
® Dyes, Life Technologies) secondary antibody and with bungarotoxin (1:500, Invitrogen) conjugated with Alexa Fluor
® 488 (Life Technologies). Innervated neuromuscular junctions were identified by the bungarotoxin labelling, totally or partially co-localized with synaptophysin labelling. Endplates marked with bungarotoxin only were considered denervated. Data were expressed as the percentage of the denervated plaques over the total ones counted in 8 adjacent frames per section. Five sections at 20× magnification were analyzed for each mouse. Fluorescence images along the z axis were taken by Olympus confocal microscopy using a 20× objective and z-stacking was performed by using ImageJ software.
4.10. Western Blot
Mice were sacrificed according to institutional ethical procedures by decapitation and the spinal cord and sciatic nerve were rapidly dissected. The spinal cord was immediately frozen on dry ice and stored at −80 °C. For each mouse, LSC was longitudinally transected at 50 µm in a cryostat with ventral and dorsal spinal cord sections as separate samples. The resulting cryostat ventral material was homogenized by sonication in ice-cold homogenization buffer (20 mM Tris-HCl pH 7.4, 2% Triton X-100, 150 mM NaCl, 1 mM EDTA, 5 mM MgCl
2, 10% anhydrous glycerol, protease and phosphates inhibitor cocktail by Roche), centrifuged at 15,700 rcf for 30 min at 4 °C and the supernatants were collected and stored at −80 °C. The sciatic nerves were processed as previously described [
24]. Briefly, tissues were powdered in liquid nitrogen, next homogenized by sonication in ice-cold homogenization buffer (20 mM Tris-HCl pH 7.4, 1% Triton X-100, 150 mM NaCl, 1 mM EDTA, 5 mM MgCl
2, 10% anhydrous glycerol, protease and phosphates from Roche) and centrifuged at 15,700 rcf for 15 min at 4 °C. Equal amounts of total protein homogenates were loaded on polyacrylamide gels and electroblotted onto PVDF membrane (Millipore) as previously described [
31]. Membranes were first blocked with 5% BSA (Sigma, St. Luis, MO, USA) in TBS with additional 0.1% Tween (TBS-T) for 1 h at room temperature and then incubated over night at 4 °C with one of the following primary antibodies: mouse monoclonal anti-GFAP (1:1000, Millipore), mouse monoclonal anti β-actin (1:15,000, Chemicon), rabbit monoclonal anti pAKT (1:750, Cell Signaling, Danvers, MA, USA), rabbit anti AKT (1:1000, Cell Signaling), mouse anti pERK (1:1000, Santa Cruz Biotechnology, Dallas, Tx, USA ), rabbit anti ERK (1:1000 Promega), rabbit anti human SOD1 (1:1000, Upstate, Burlington, MA, USA), mouse anti CNPase (1:1000 Chemicon), mouse anti β- III Tubulin (1:1000, Millipore), mouse monoclonal anti-Acetylated Tubulin (1:1000, Sigma Aldrich). Membranes were then washed and incubated with horseradish peroxidase-conjugated anti-rabbit, anti-mouse or anti-rat secondary antibodies (Santa Cruz) and developed by Luminata Forte Western Chemiluminescent horse radish peroxidase (HRP) Substrate (Millipore) on the Chemi-Doc XRS system (Bio-Rad, Hercules, CA, USA). Densitometry analysis was performed with ImageLab (Bio-Rad) software.
4.11. Real-Time PCR
Spinal cords and muscles were freshly collected from mice transcardially perfused with PBS under deep anesthesia. All tissues were immediately frozen on dry-ice. Tissue was homogenized and total RNA was extracted using the Trizol method (Invitrogen) and purified with PureLink RNA columns (Life Technologies). RNA samples were treated with DNase I and reverse transcription was performed with High Capacity cDNA Reverse Transcription Kit (Life Technologies). Real-time PCR was performed using the Taq Man Gene expression assay (Applied Biosystems, Foster City, CA, USA) following the manufacturer’s instructions, on cDNA specimens in triplicate, using 1× Universal PCR master mix (Life technologies) and 1× mix containing specific probes for Tumor Necrosis Factor (TNFα, Mm00443258_m1), chemokine (C-C motif) ligand 2 (CCL2, Mm00441242_m1), interleukin 4 (IL4, Mm00445259_m1), interleukin 10 (IL-10, Mm00439614_m1), forkhead box P3 (FoxP3, Mm00475162_m1), cluster of differentiation 4 (CD4, Mm00442754_m1), cluster of differentiation 8 (CD8, Mm01182107_g1), cluster of differentiation 68 (CD68, Mm03047343_m1) and β-actin (Hs01060665_g1) all from Life technologies. Relative quantification was calculated from the ratio between the cycle number (Ct) at which the signal crossed a threshold set within the logarithmic phase of the given gene and that of the reference gene (β actin). Mean values of the triplicate results for each animal were used as individual data for 2-ΔΔCt statistical analysis.
4.12. Statistical Analysis
One-way ANOVA was used to compare differences between more than two groups, followed by post hoc Fisher’s least significant difference (LSD) while Student’s “t” test was used for the analysis of two groups. Two-way ANOVA was used for the analysis of dose response effect in NTG and SOD1G93A cell cultures and for the analysis of repeated measures of grip strength test in mice. The Mantel–Cox log rank test was used for comparing motor deficit onset, paralysis and survival between groups.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






