1. Introduction
Neurogenesis is the development of new neurons from neural stem/precursor cells (NSCs). In the adult human brain, the NSCs reside primarily in two regions: The subgranular zone in the dentate gyrus of the hippocampus, and the subventricular zone of the lateral ventricles [
1].
The NSCs are a diverse population of cells with the capacity to self-renew and differentiate into neurons in response to stimuli in order to maintain central nervous system homeostasis. Local stimuli from the niche where these cells are found, as well as extrinsic factors such as neurotransmitters, growth factors, cytokines, and adhesion molecules, can positively or negatively affect the NSCs’ state and differentiation potential, thereby modulating neurogenesis in the adult brain [
1]. For example, microglial activation and secretion of pro-inflammatory cytokines are processes involved with healthy aging in the brain, and this pro-inflammatory environment is deleterious to NSCs, thereby decreasing neurogenesis [
2]. Age-related changes in the cell cycle due to accumulation of damaged proteins can cause a reduction in the proliferation rate of NSCs [
3] and cause the cells to become quiescent, but they can be reactivated upon stimulation, such as by physical exercise or epileptic seizures, through Notch signalling activation [
4].
This form of circuit plasticity may be altered in different diseases and targeted with pharmacological therapies [
5]. Cognitive deficits, mood dysregulation, and declines in hippocampal volume have been correlated with mental disorders that present decreased neurogenesis, such as major depression, post-traumatic stress disorder, schizophrenia, and Alzheimer’s disease [
6]. Neurogenesis in the hippocampus is necessary for the therapeutic effects of antidepressants [
7], but in the subventricular zone of the lateral ventricles, for example, chronic treatment with fluoxetine decreased neurogenesis [
8]. However, if chronic stress was first induced (known to be both a contributor to the development of mood disorders [
9] and decreased neurogenesis), the treatment with fluoxetine and imipramine can revert the decline in neurogenesis by increasing the NSCs pool and survival [
10].
While the effects of antidepressants acting through serotonergic receptors is widely investigated and thus better understood, the effects and mechanisms of action of other psychoactive drugs in neurogenesis remain unknown and/or controversial, with studies in mice showing positive effects on neurogenesis. Chronic treatment with atypical antipsychotics (olanzapine, quetiapine, clozapine, risperidone, and aripiprazole) increased neurogenesis in the hippocampus of adult mice [
11]. In contrast, haloperidol treatment resulted in decreased neurogenesis [
11]. On the other hand, in humans, the use of atypical antipsychotics (risperidone, olanzapine, paliperidone, amisulpride, and aripiprazole) was associated with a reduction in grey matter volume in first-episode schizophrenia patients [
12], suggesting a reduction in neurogenesis.
The changes that occur in the NSCs in response to environmental cues such as stress, psychiatric disorders, and aging, together with the evidence of NSCs responsiveness to drugs used in treating psychiatric disorders, supports the idea of neurogenesis being a potential therapeutic target for these diseases. Therefore, this study aims to evaluate the effects of common psychoactive drugs, used in the treatment of affective disorders (bipolar disorder and schizophrenia), on the expression of genes involved in neurogenesis in a human neuronal model in cell culture.
3. Discussion
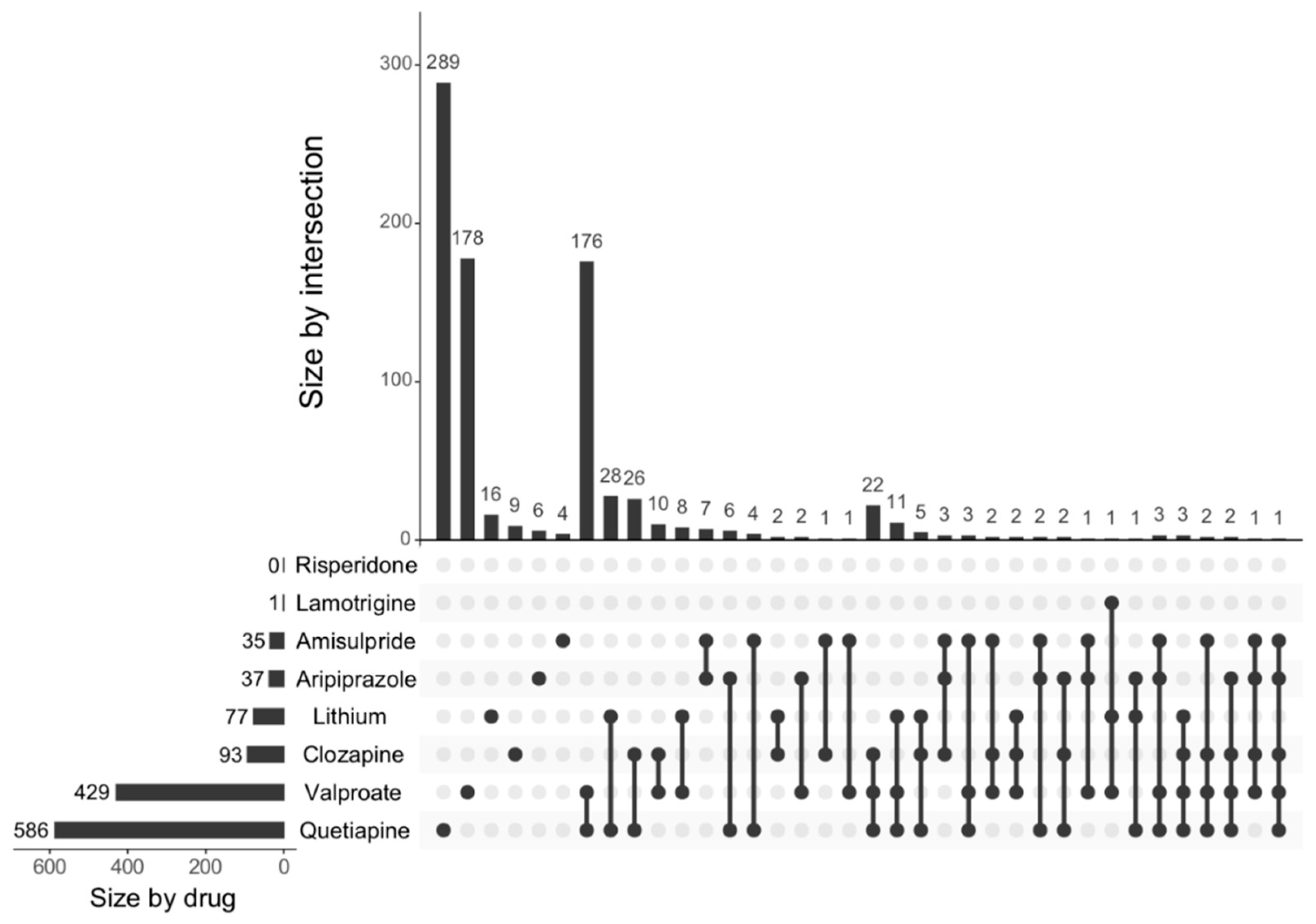
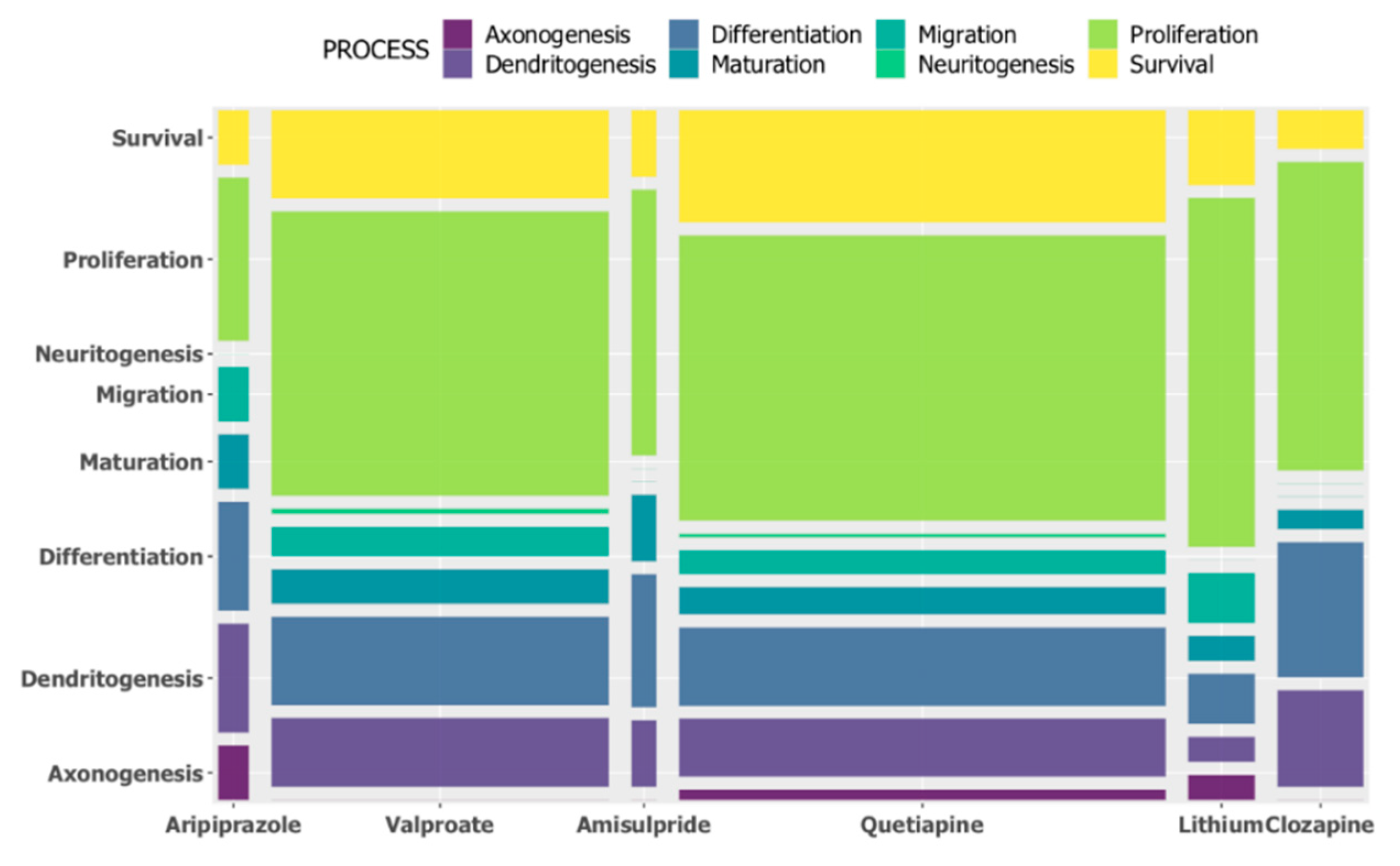
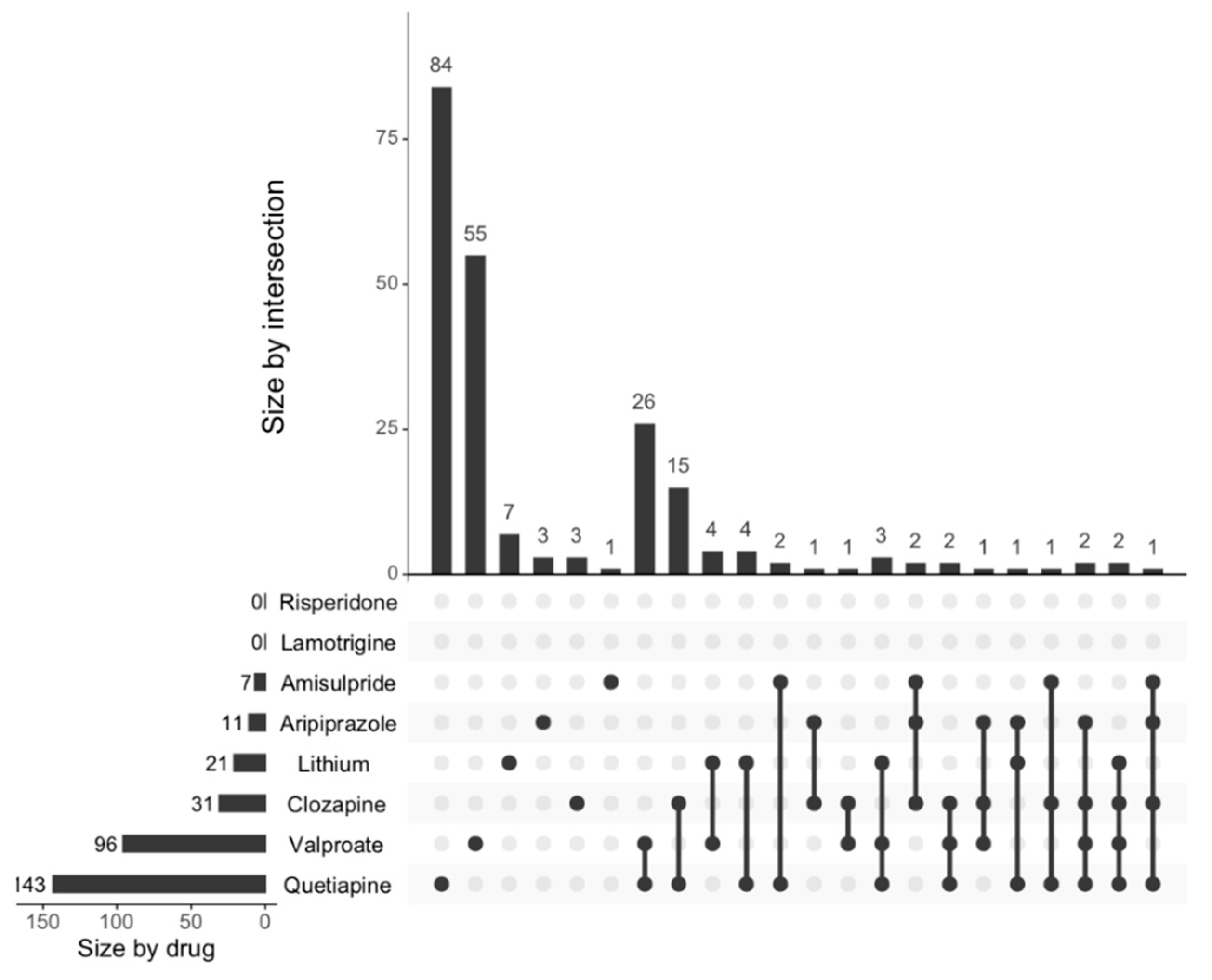
The generation of functional neurons from progenitor cells, neurogenesis, is a continuing process in the hippocampus throughout life, from embryonic development until adulthood [
13]. Altered neurogenesis is implicated in diverse neuropsychiatric disorders including depression, schizophrenia, and bipolar disorder [
14]. This study showed the effects of psychoactive drugs on genes involved in neurogenesis. Clozapine, amisulpride, aripiprazole, lithium, quetiapine, and lamotrigine all significantly decreased the expression of genes involved in neurogenesis, while risperidone significantly increased the expression of genes involved in neurogenesis. Apart from risperidone, the findings were confirmed when we investigated the effects of these drugs on the expression of neurogenesis genes using a more curated database (MANGO).
The literature around the effects of atypical antipsychotics in neurogenesis is controversial. In rodent models, chronic administration of aripiprazole, quetiapine, and clozapine significantly increased neurogenesis, cell proliferation, and survival [
11,
15,
16,
17]. Kim et al. (2015) suggested a reversal of depressive behaviours through preventing degeneration of dopaminergic neuronal cells and enhancing neurogenesis after treatment with aripiprazole [
18]. Aripiprazole treatment increased neurite branches in primary cortical neurons derived from mice with dopamine D2 receptor hyperactivity and disrupter in schizophrenia 1 (DISC1) [
19]. Clozapine treatment in male Wistar rats subjected to chronic mild stress upregulated adult neurogenesis and neuronal survival, reversing the behavioural effects of chronic stress [
20]. Quetiapine augmentation treatment increased hippocampal cell proliferation and neuronal differentiation and improved depressive-like behaviours in treated rats [
21].
In humans, some studies of antipsychotic treatment were associated with grey matter loss as shown in imaging studies in patients with schizophrenia, but studies have yielded inconsistent results as explored in a meta-analysis by Fusar-Poli [
22]. Wang et al. (2018) showed a significant decrease in grey matter volume in the left parahippocampal gyrus/hippocampus, right temporal pole mid/superior temporal gyrus, right parahippocampal/hippocampus, and right insula after four weeks of treatment with antipsychotics [
23]. Guo et al. (2019) also showed a decrease in grey matter volume in the bilateral frontal, temporal, and left parietal brain regions associated with antipsychotic treatment [
12]. In both studies, the majority of the treatment group were using several antipsychotics at the same time, and therefore conclusions on the specific effects of individuals drugs could not be made.
Antipsychotics are also prescribed for patients with bipolar disorder, together with mood stabilisers and antidepressants. These drugs have also been associated with structural brain differences in these patients, but data is still inconclusive and difficult to parse from illness effects. In bipolar disorder, total grey matter volume was reduced in individuals treated with atypical antipsychotics [
24]. The ENIGMA study reported significantly reduced cortical surface areas of large prefrontal areas in individuals with bipolar disorder taking atypical antipsychotics [
25]. However, lithium, considered the first line treatment for bipolar disorder, has been demonstrated to increase neurogenesis in the dentate gyrus of the rodent hippocampus [
26,
27]. Another study also demonstrated that lithium could promote neuronal differentiation of hippocampal neural progenitor cells both in vitro and in vivo [
28]. Yucel et al. (2008) also demonstrated a bilateral increase in hippocampal volume in bipolar disorder patients treated with lithium, but not lamotrigine [
29]. Similarly, in other human imaging studies, grey matter volume increased after four weeks of lithium treatment [
30] and appeared to be independent of long-term treatment response [
31]. Lithium increases brain-derived neurotrophic factor (BDNF) expression and genes associated with neuroprotection, such as
Bcl2 and
Bcl-XL, and also decreases the expression of pro-apoptotic genes
Bax,
Bad, and caspases in rat hippocampal neurons [
32]. This effect on apoptosis could, at least in part, be the mechanism involved in the increase of grey matter volume in vivo, rather than effects on neurogenesis.
Overall, drugs analysed in our study appear to increase neurogenesis in rodent models, which is in contrast to the findings from human imaging studies, where the drugs appear to decrease grey matter volume, suggestive of reduced neurogenesis, although it is unclear in some designs whether this relates to illness effects or other lifestyle or medical confounders. Our data is at transcriptional levels. The increases in grey matter demonstrated in imaging studies probably occurred because of neurotrophic effects and could explain the differences in results. To the best of our knowledge, there is no study with a similar approach to ours.
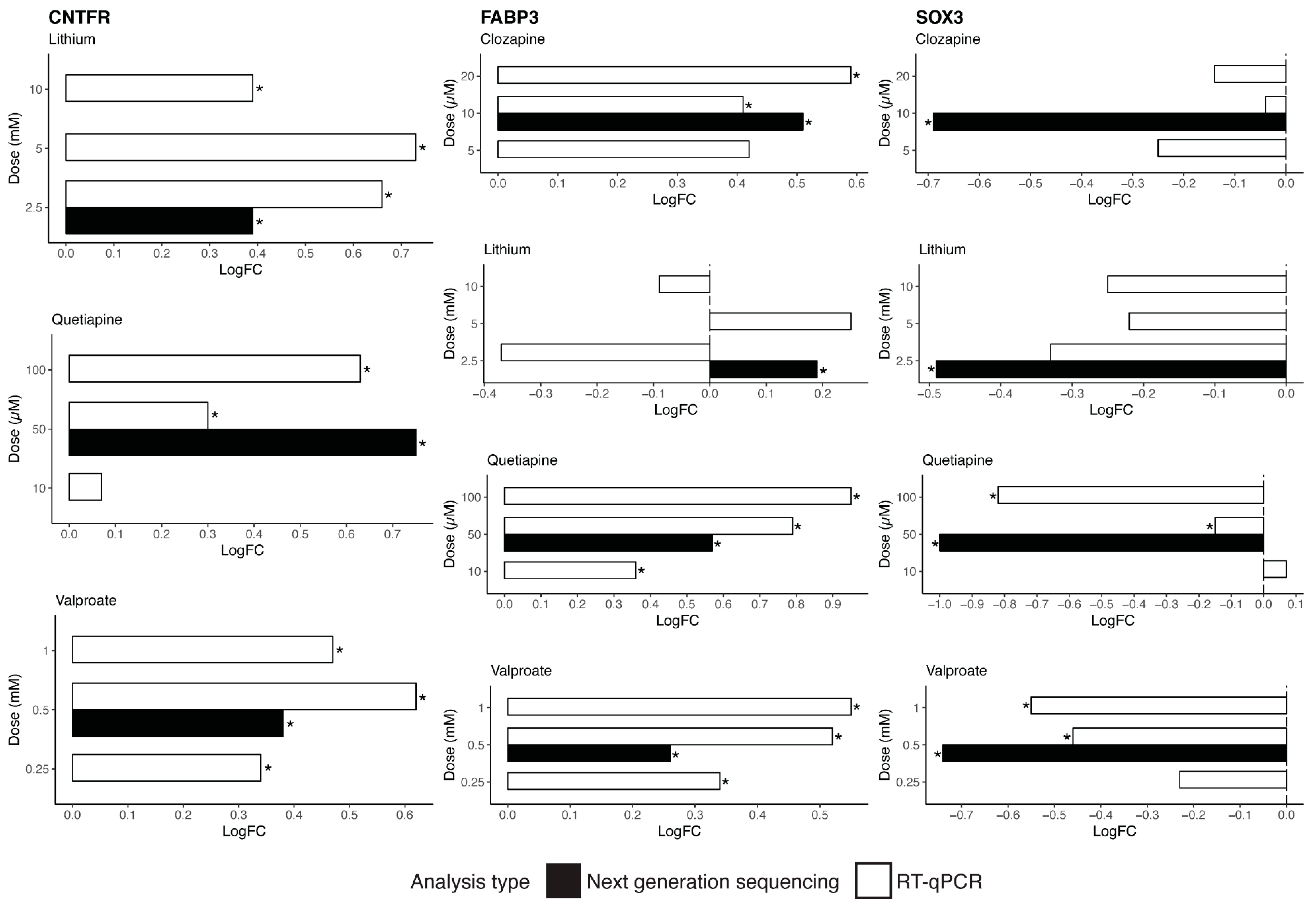
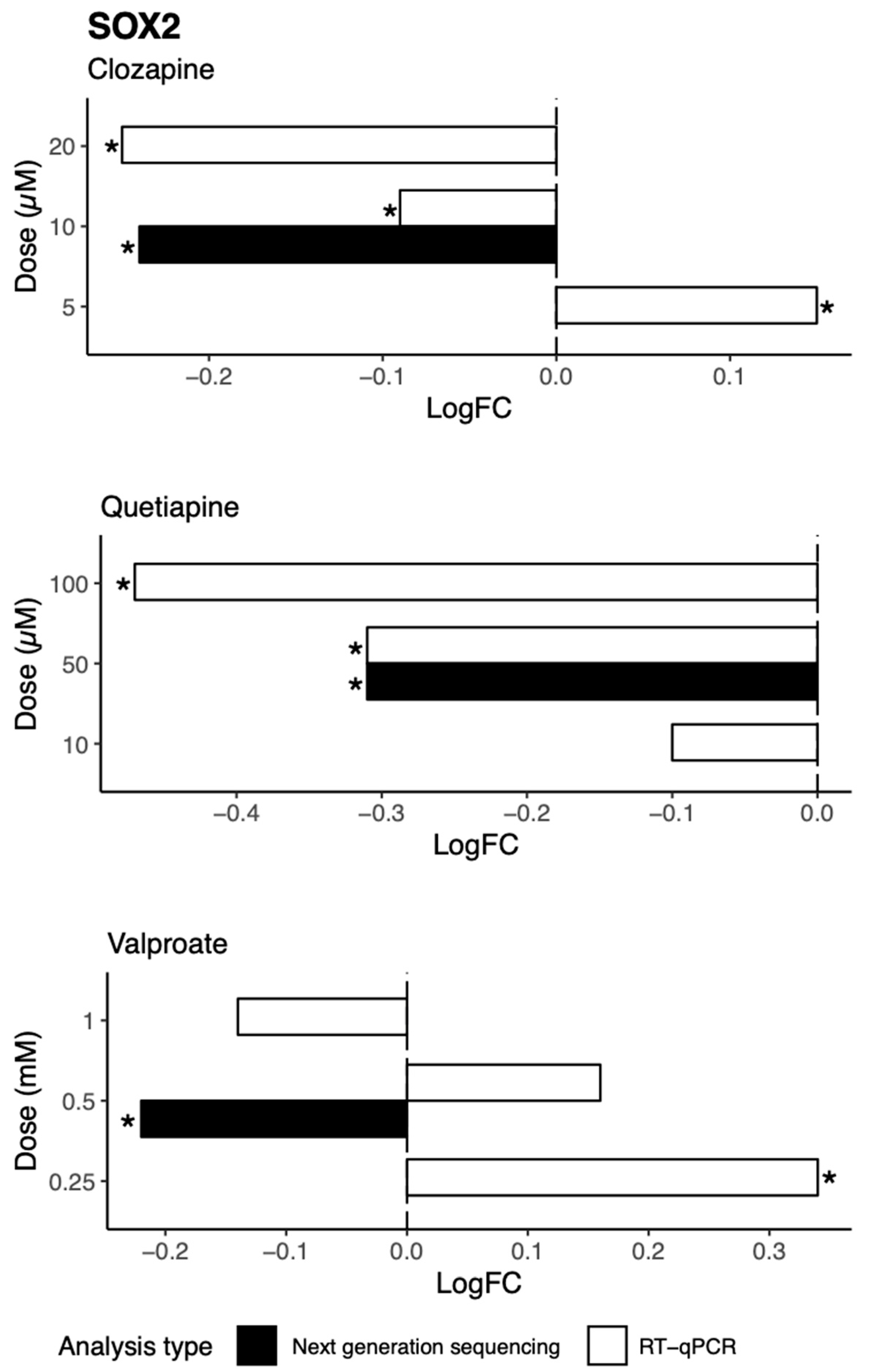
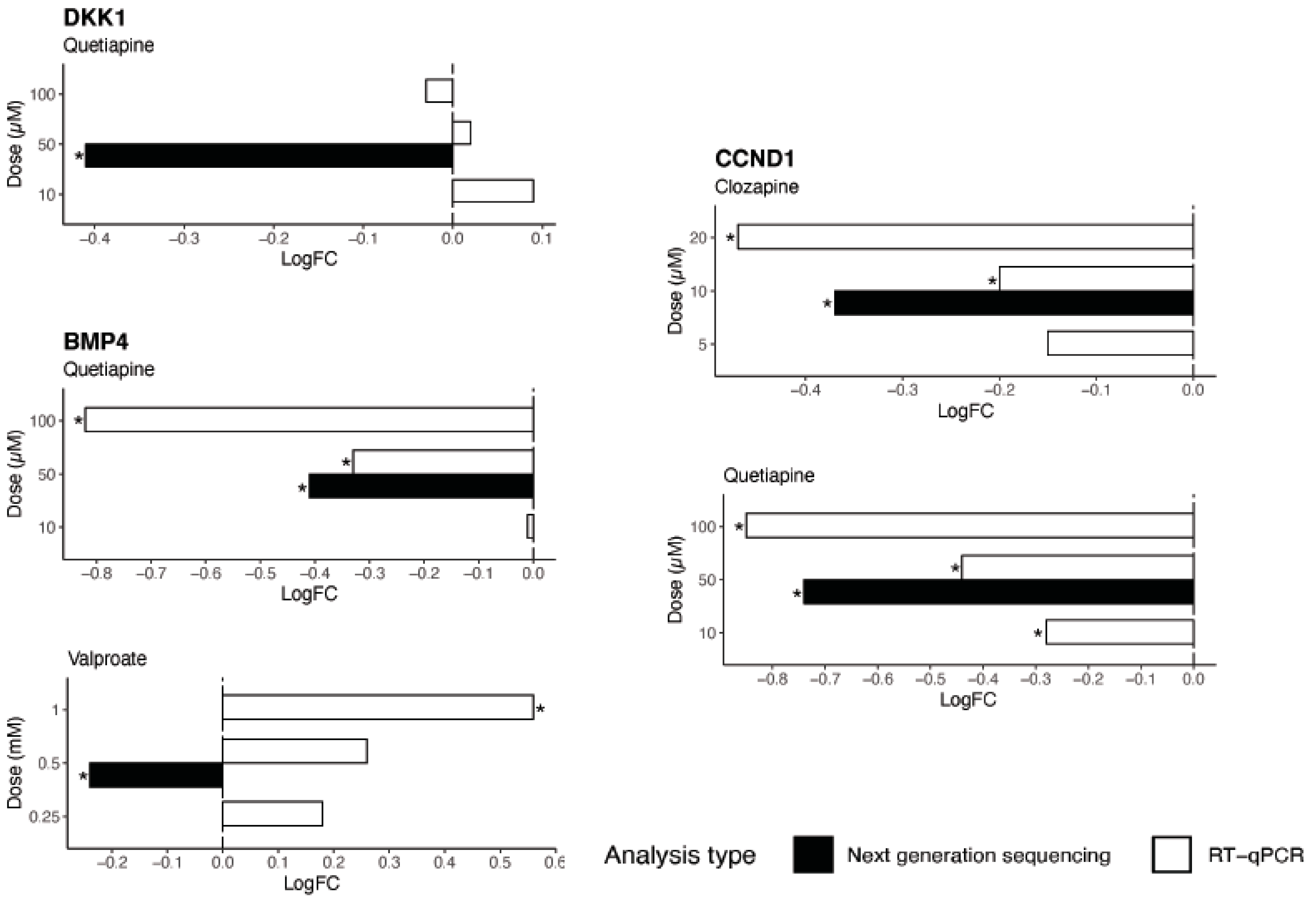
We further investigated possible mechanisms by which these drugs decrease genes involved in neurogenesis in vitro, identifying a candidate target molecule in
SOX2. SOX2 is a transcription factor and a marker of the nervous system from the beginning of development in many species, and acts to co-ordinate widespread transcriptional regulation of genes involved in neurogenesis.
SOX2 mutations in humans cause defects in the brain, particularly in the hippocampus, involving cognition, movement control, and vision [
33]. As identified by TRRUST analysis, three major gene targets of SOX2 in the neurogenesis pathway are
CCND1,
BMP4, and
DKK1. CCND1 promotes neurogenesis in vivo, a role that is not linked to its cell cycle function [
34]. Both in vivo and in vitro studies present evidence of the role of BMP4 in the modulation of adult neurogenesis in the hippocampus [
35]. DKK1 is a suppressor of neural stem cell proliferation, and studies demonstrate increased self-renewal of neural progenitors and increased generation of immature neurons after the deletion of
DKK1 in adult brains [
36]. The downregulation of
BMP4,
CCDN1, and
DKK1 by quetiapine could represent a molecular mechanism for the reduction in expression of neurogenesis genes observed in this study after treatment with quetiapine.
The drugs appeared to cause a general increase in a number of metabolites in NT2-N cells that have previously been suggested to be related to the process of neurogenesis. While it may appear that this suggests a positive effect on neurogenesis, the increased levels of these particular sets of metabolites are related to the presence of more mature neurons, with active neuritogenesis, and not the generation of new immature neurons.
For example, decreased levels of NAA is used as a marker of neuronal integrity and is reportedly decreased after whole-brain radiation in rodents, associated with changes in neurotransmission and loss of neuronal viability [
37]. The pathological pruning of dendrites has also been associated with a reduction of NAA, and this could contribute to the finding of reduced brain volume associated with reduced levels of NAA in schizophrenia, bipolar disorder, post-traumatic stress disorder, and obsessive-compulsive disorders [
38]. The effects seen in this study corroborate previous studies showing that antipsychotics and mood stabilizers increase the levels of NAA, suggesting a therapeutic response acting on increasing neuronal viability [
39].
GABA is another example, with four of the drugs increasing the levels of this metabolite in NT2-N cells. GABA agonists reportedly regulate synaptic integration by increasing the numbers and extension of neurites and promoting the survival of existing neurons [
40,
41].
In summary, the present study demonstrated that psychoactive drugs decrease the expression of genes involved in neurogenesis in neuronal cell culture after 24 h treatment, partially through inhibition of SOX2 and its targets genes BMP4, CCDN1, and DKK1. It also showed the alterations in the levels of metabolites involved in neuronal health, particularly NAA and GABA.
This study has some limitations that need to be acknowledged. Only one dose of each drug was tested. In addition, we explored acute effects of these drugs, and there is a possibility of a different effect during chronic administration. Our study did not evaluate the effects of these drugs in models of any particular disease state, therefore the nature of the pathophysiologic process of specific diseases might interfere in the effect of these drugs in patients.

 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}