Herpes Simplex Virus Oncolytic Immunovirotherapy: The Blossoming Branch of Multimodal Therapy

Abstract
:1. Introduction
2. Attenuated oHSVs
2.1. Conditionally Replicating oHSVs with Single or Multiple Mutations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
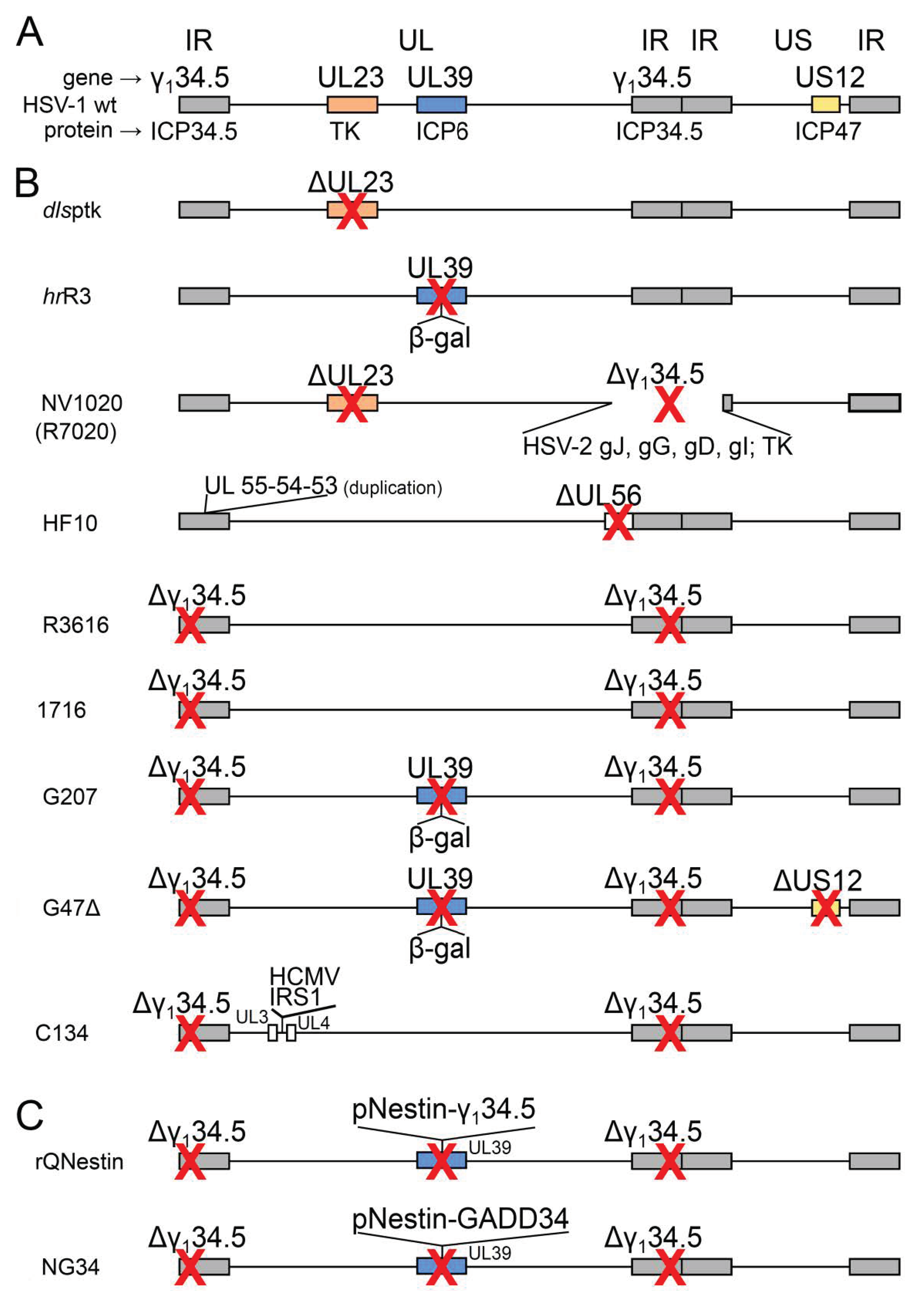
| oHSV Name (Alternative Name) | Genetic Modification | Diagram in Figure 1, Line | Clinical Trial Identifier (Status) | Ref. |
|---|---|---|---|---|
| Conditionally replicating oHSVs with single or multiple mutations | ||||
| dlsptk | deletion of UL23 (encodes TK) | a | – | [28] |
| hrR3 | inactivation of UL39 (encodes ICP6, large subunit of RR) by lacZ insertion | b | – | [29] |
| NV1020 (R7020) | deletion of one copy of γ134.5 (encodes ICP34.5 neurovirulence factor, anti-PKR) + HSV-2 US2-US8 genes + α4-tk | c | NCT00149396 (C) | [31] |
| HF10 | duplications of UL53, UL54, UL55; deletion of UL56 | d | NCT02428036 (C) | [41] |
| R3616 | deletion of two copies of γ134.5 (encodes ICP34.5 neurovirulence factor, anti-PKR) | e | – | [32] |
| 1716 | deletion of two copies of γ134.5 (encodes ICP34.5 neurovirulence factor, anti-PKR) | f | NCT00931931 (C) | [33] |
| G207 | deletion of two copies of γ134.5 (encodes ICP34.5 neurovirulence factor, anti-PKR); inactivation of UL39 (encodes ICP6, large subunit of RR) by lacZ insertion | g | NCT00028158 (C) | [30] |
| G47Δ | deletion of two copies of γ134.5 (encodes ICP34.5 neurovirulence factor, anti-PKR); deletion of US12 (encodes ICP47, immune evasion protein); increased expression of US11 (encodes anti-PKR factor) | h | UMIN000015995 (C) | [46] |
| C134 | deletion of two copies of γ134.5 (encodes ICP34.5 neurovirulence factor, anti-PKR); insertion of HCMV IRS1 gene (inhibits antiviral state in the host cell) | i | NCT03657576 (A) | [47] |
| Tumor-specific transcriptionally targeted oHSVs | ||||
| rQNestin (rQNestin34.5v.2) | γ134.5 (encodes ICP34.5 neurovirulence factor, anti-PKR) under control of nestin promoter | j | NCT03152318 (R) | [48] |
| NG34 | GADD34 (human counterpart of γ134.5) under control of nestin promoter | k | – | [49] |
2.2. Armed oHSVs
2.3. Talimogene Laherparepvec (T-VEC)
3. Tropism Retargeted, Unattenuated, oHSVs
4. oHSV Delivery
5. oHSV Combination Therapies and Immunotherapies
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 3-OS HS | 3-O-sulphated heparan sulphate |
| BAC | bacterial artificial chromosome |
| BIRC5 | baculoviral inhibitor of apoptosis repeat-containing 5 |
| CEA | carcinoembryonic antigen |
| CTL | cytotoxic T lymphocyte |
| CTLA-4 | CTL antigen-4 |
| CYP2B1 | cytochrome P450, family 2, subfamily b, polypeptide 1 |
| EGFP | enhanced green fluorescent protein |
| EGFR | epidermal growth factor receptor |
| EGFRvIII | epidermal growth factor receptor variant III |
| EMA | European Medicines Agency |
| FDA | Food and Drug Administration (USA) |
| GALV | gibbon ape leukemia virus |
| GBM | glioblastoma multiforme |
| GM-CSF | granulocyte macrophage colony-stimulating factor (GM-CSF) |
| GMP | Good Manufacturing Practice |
| GSCs | glioblastoma stem-like cells |
| HCMV | human cytomegalovirus |
| HER2 | human epidermal growth factor receptor 2 |
| HS | heparan sulphate |
| HveA | herpesvirus entry mediator A |
| HveC | herpesvirus entry mediator C |
| HVEM | herpesvirus entry mediator |
| ICI | immune-checkpoint inhibitor |
| ICP | infected cell protein |
| IL | interleukin |
| MAG | myelin-associated glycoprotein |
| NMHC-II | non-muscle myosin heavy chain II |
| N-ter | amino terminus |
| OPTiM | OncoVEXGM-CSF Pivotal Trial in Melanoma |
| OV | oncolytic virus |
| PD-1 | programmed cell death 1 |
| PD-L1 | programmed cell death 1 ligand 1 |
| PILRα | paired immunoglobulin-like type 2 receptor-α |
| PKR | protein kinase R |
| PSMA | prostate-specific membrane antigen |
| RR | ribonucleotide reductase |
| scFv | single chain antibody variable fragment |
| ScGCs | serum-cultured GBM cells |
| TK | thymidine kinase |
| UL | unique long |
| uPA | urokinase-type plasminogen activator |
| uPAR | urokinase-type plasminogen activator receptor |
| US | unique short |
| wt | wild type |
| yCD | yeast cytosine deaminase |
| Δ | deletion |
References
- Roizman, B.; Knipe, D.M.; Whitley, R.J. Herpes simplex viruses. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Cohen, J.I., Griffin, D.E., Lamb, R.A., Martin, M.A., Racaniello, V.R., Roizman, B., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; Volume 2, pp. 1823–1897. [Google Scholar]
- Kato, A.; Adachi, S.; Kawano, S.; Takeshima, K.; Watanabe, M.; Kitazume, S.; Sato, R.; Kusano, H.; Koyanagi, N.; Maruzuru, Y.; et al. Identification of a herpes simplex virus 1 gene encoding neurovirulence factor by chemical proteomics. Nat. Commun. 2020, 11, 4894. [Google Scholar] [CrossRef] [PubMed]
- Campadelli-Fiume, G.; Amasio, M.; Avitabile, E.; Cerretani, A.; Forghieri, C.; Gianni, T.; Menotti, L. The multipartite system that mediates entry of herpes simplex virus into the cell. Rev. Med Virol. 2007, 17, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Campadelli-Fiume, G.; Menotti, L.; Avitabile, E.; Gianni, T. Viral and cellular contributions to herpes simplex virus entry into the cell. Curr. Opin. Virol. 2012, 2, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Sathiyamoorthy, K.; Chen, J.; Longnecker, R.; Jardetzky, T.S. The COMPLEXity in herpesvirus entry. Curr. Opin. Virol. 2017, 24, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Messerle, M.; Crnkovic, I.; Hammerschmidt, W.; Ziegler, H.; Koszinowski, U.H. Cloning and mutagenesis of a herpesvirus genome as an infectious bacterial artificial chromosome. Proc. Natl. Acad. Sci. USA 1997, 94, 14759–14763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borst, E.M.; Hahn, G.; Koszinowski, U.H.; Messerle, M. Cloning of the human cytomegalovirus (HCMV) genome as an infectious bacterial artificial chromosome in Escherichia coli: A new approach for construction of HCMV mutants. J. Virol. 1999, 73, 8320–8329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brune, W.; Messerle, M.; Koszinowski, U.H. Forward with BACs: New tools for herpesvirus genomics. Trends Genet. Tig. 2000, 16, 254–259. [Google Scholar] [CrossRef]
- Tanaka, M.; Kagawa, H.; Yamanashi, Y.; Sata, T.; Kawaguchi, Y. Construction of an excisable bacterial artificial chromosome containing a full-length infectious clone of herpes simplex virus type 1: Viruses reconstituted from the clone exhibit wild-type properties in vitro and in vivo. J. Virol. 2003, 77, 1382–1391. [Google Scholar] [CrossRef] [Green Version]
- Warming, S.; Costantino, N.; Court, D.L.; Jenkins, N.A.; Copeland, N.G. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res. 2005, 33, e36. [Google Scholar] [CrossRef]
- Terada, K.; Wakimoto, H.; Tyminski, E.; Chiocca, E.A.; Saeki, Y. Development of a rapid method to generate multiple oncolytic HSV vectors and their in vivo evaluation using syngeneic mouse tumor models. Gene Ther. 2006, 13, 705–714. [Google Scholar] [CrossRef]
- Suenaga, T.; Kohyama, M.; Hirayasu, K.; Arase, H. Engineering large viral DNA genomes using the CRISPR-Cas9 system. Microbiol. Immunol. 2014, 58, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Menotti, L.; Leoni, V.; Gatta, V.; Petrovic, B.; Vannini, A.; Pepe, S.; Gianni, T.; Campadelli-Fiume, G. oHSV Genome Editing by Means of galK Recombineering. Methods Mol. Biol. 2020, 2060, 131–151. [Google Scholar] [CrossRef]
- Vannini, A.; Petrovic, B.; Gatta, V.; Leoni, V.; Pepe, S.; Menotti, L.; Campadelli-Fiume, G.; Gianni, T. Rescue, Purification, and Characterization of a Recombinant HSV Expressing a Transgenic Protein. Methods Mol. Biol. 2020, 2060, 153–168. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Li, H.; Hao, M.; Xiong, D.; Luo, Y.; Huang, C.; Yuan, Q.; Zhang, J.; Xia, N. Increasing the Efficiency of CRISPR/Cas9-mediated Precise Genome Editing of HSV-1 Virus in Human Cells. Sci. Rep. 2016, 6, 34531. [Google Scholar] [CrossRef] [PubMed]
- Pearl, T.M.; Markert, J.M.; Cassady, K.A.; Ghonime, M.G. Oncolytic Virus-Based Cytokine Expression to Improve Immune Activity in Brain and Solid Tumors. Mol. Ther. Oncolytics 2019, 13, 14–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraefel, C.; Epstein, A.L. Preparation of Herpes Simplex Virus Type 1 (HSV-1)-Based Amplicon Vectors. Methods Mol. Biol. 2020, 2060, 91–109. [Google Scholar] [CrossRef]
- Goins, W.F.; Huang, S.; Hall, B.; Marzulli, M.; Cohen, J.B.; Glorioso, J.C. Engineering HSV-1 Vectors for Gene Therapy. Methods Mol. Biol. 2020, 2060, 73–90. [Google Scholar] [CrossRef] [PubMed]
- Campadelli-Fiume, G.; De Giovanni, C.; Gatta, V.; Nanni, P.; Lollini, P.L.; Menotti, L. Rethinking herpes simplex virus: The way to oncolytic agents. Rev. Med Virol. 2011, 21, 213–226. [Google Scholar] [CrossRef]
- Peters, C.; Rabkin, S.D. Designing Herpes Viruses as Oncolytics. Mol. Ther. Oncolytics 2015, 2, 15010. [Google Scholar] [CrossRef]
- Sokolowski, N.A.; Rizos, H.; Diefenbach, R.J. Oncolytic virotherapy using herpes simplex virus: How far have we come? Oncolytic Virotherapy 2015, 4, 207–219. [Google Scholar] [CrossRef] [Green Version]
- Sanchala, D.S.; Bhatt, L.K.; Prabhavalkar, K.S. Oncolytic Herpes Simplex Viral Therapy: A Stride toward Selective Targeting of Cancer Cells. Front. Pharmacol. 2017, 8, 270. [Google Scholar] [CrossRef]
- Watanabe, D.; Goshima, F. Oncolytic Virotherapy by HSV. Adv. Exp. Med. Biol. 2018, 1045, 63–84. [Google Scholar] [CrossRef] [PubMed]
- Varghese, S.; Rabkin, S.D. Oncolytic herpes simplex virus vectors for cancer virotherapy. Cancer Gene Ther. 2002, 9, 967–978. [Google Scholar] [CrossRef] [Green Version]
- Fukuhara, H.; Ino, Y.; Todo, T. Oncolytic virus therapy: A new era of cancer treatment at dawn. Cancer Sci. 2016, 107, 1373–1379. [Google Scholar] [CrossRef]
- Cripe, T.P.; Chen, C.Y.; Denton, N.L.; Haworth, K.B.; Hutzen, B.; Leddon, J.L.; Streby, K.A.; Wang, P.Y.; Markert, J.M.; Waters, A.M.; et al. Pediatric cancer gone viral. Part I: Strategies for utilizing oncolytic herpes simplex virus-1 in children. Mol. Ther. Oncolytics 2015, 2. [Google Scholar] [CrossRef] [PubMed]
- Totsch, S.K.; Schlappi, C.; Kang, K.D.; Ishizuka, A.S.; Lynn, G.M.; Fox, B.; Beierle, E.A.; Whitley, R.J.; Markert, J.M.; Gillespie, G.Y.; et al. Oncolytic herpes simplex virus immunotherapy for brain tumors: Current pitfalls and emerging strategies to overcome therapeutic resistance. Oncogene 2019, 38, 6159–6171. [Google Scholar] [CrossRef]
- Martuza, R.L.; Malick, A.; Markert, J.M.; Ruffner, K.L.; Coen, D.M. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science 1991, 252, 854–856. [Google Scholar] [CrossRef]
- Goldstein, D.J.; Weller, S.K. Herpes simplex virus type 1-induced ribonucleotide reductase activity is dispensable for virus growth and DNA synthesis: Isolation and characterization of an ICP6 lacZ insertion mutant. J. Virol. 1988, 62, 196–205. [Google Scholar] [CrossRef] [Green Version]
- Mineta, T.; Rabkin, S.D.; Yazaki, T.; Hunter, W.D.; Martuza, R.L. Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nat. Med. 1995, 1, 938–943. [Google Scholar] [CrossRef] [PubMed]
- Meignier, B.; Longnecker, R.; Roizman, B. In vivo behavior of genetically engineered herpes simplex viruses R7017 and R7020: Construction and evaluation in rodents. J. Infect. Dis. 1988, 158, 602–614. [Google Scholar] [CrossRef] [PubMed]
- Chou, J.; Kern, E.R.; Whitley, R.J.; Roizman, B. Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science 1990, 250, 1262–1266. [Google Scholar] [CrossRef]
- MacLean, A.R.; ul-Fareed, M.; Robertson, L.; Harland, J.; Brown, S.M. Herpes simplex virus type 1 deletion variants 1714 and 1716 pinpoint neurovirulence-related sequences in Glasgow strain 17+ between immediate early gene 1 and the ’a’ sequence. J. Gen. Virol. 1991, 72 Pt 3, 631–639. [Google Scholar] [CrossRef] [PubMed]
- McKie, E.A.; MacLean, A.R.; Lewis, A.D.; Cruickshank, G.; Rampling, R.; Barnett, S.C.; Kennedy, P.G.; Brown, S.M. Selective in vitro replication of herpes simplex virus type 1 (HSV-1) ICP34.5 null mutants in primary human CNS tumours--evaluation of a potentially effective clinical therapy. Br. J. Cancer 1996, 74, 745–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markert, J.M.; Liechty, P.G.; Wang, W.; Gaston, S.; Braz, E.; Karrasch, M.; Nabors, L.B.; Markiewicz, M.; Lakeman, A.D.; Palmer, C.A.; et al. Phase Ib trial of mutant herpes simplex virus G207 inoculated pre-and post-tumor resection for recurrent GBM. Mol. Ther. J. Am. Soc. Gene Ther. 2009, 17, 199–207. [Google Scholar] [CrossRef]
- Harrow, S.; Papanastassiou, V.; Harland, J.; Mabbs, R.; Petty, R.; Fraser, M.; Hadley, D.; Patterson, J.; Brown, S.M.; Rampling, R. HSV1716 injection into the brain adjacent to tumour following surgical resection of high-grade glioma: Safety data and long-term survival. Gene Ther. 2004, 11, 1648–1658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacKie, R.M.; Stewart, B.; Brown, S.M. Intralesional injection of herpes simplex virus 1716 in metastatic melanoma. Lancet 2001, 357, 525–526. [Google Scholar] [CrossRef]
- Streby, K.A.; Geller, J.I.; Currier, M.A.; Warren, P.S.; Racadio, J.M.; Towbin, A.J.; Vaughan, M.R.; Triplet, M.; Ott-Napier, K.; Dishman, D.J.; et al. Intratumoral Injection of HSV1716, an Oncolytic Herpes Virus, Is Safe and Shows Evidence of Immune Response and Viral Replication in Young Cancer Patients. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 3566–3574. [Google Scholar] [CrossRef] [Green Version]
- Kemeny, N.; Brown, K.; Covey, A.; Kim, T.; Bhargava, A.; Brody, L.; Guilfoyle, B.; Haag, N.P.; Karrasch, M.; Glasschroeder, B.; et al. Phase I, open-label, dose-escalating study of a genetically engineered herpes simplex virus, NV1020, in subjects with metastatic colorectal carcinoma to the liver. Hum. Gene Ther. 2006, 17, 1214–1224. [Google Scholar] [CrossRef]
- Geevarghese, S.K.; Geller, D.A.; de Haan, H.A.; Horer, M.; Knoll, A.E.; Mescheder, A.; Nemunaitis, J.; Reid, T.R.; Sze, D.Y.; Tanabe, K.K.; et al. Phase I/II study of oncolytic herpes simplex virus NV1020 in patients with extensively pretreated refractory colorectal cancer metastatic to the liver. Hum. Gene Ther. 2010, 21, 1119–1128. [Google Scholar] [CrossRef] [Green Version]
- Nishiyama, Y.; Kimura, H.; Daikoku, T. Complementary lethal invasion of the central nervous system by nonneuroinvasive herpes simplex virus types 1 and 2. J. Virol. 1991, 65, 4520–4524. [Google Scholar] [CrossRef] [Green Version]
- Ushijima, Y.; Luo, C.; Goshima, F.; Yamauchi, Y.; Kimura, H.; Nishiyama, Y. Determination and analysis of the DNA sequence of highly attenuated herpes simplex virus type 1 mutant HF10, a potential oncolytic virus. Microbes Infect. 2007, 9, 142–149. [Google Scholar] [CrossRef]
- Kasuya, H.; Kodera, Y.; Nakao, A.; Yamamura, K.; Gewen, T.; Zhiwen, W.; Hotta, Y.; Yamada, S.; Fujii, T.; Fukuda, S.; et al. Phase I Dose-escalation Clinical Trial of HF10 Oncolytic Herpes Virus in 17 Japanese Patients with Advanced Cancer. Hepato-gastroenterology 2014, 61, 599–605. [Google Scholar]
- Hirooka, Y.; Kasuya, H.; Ishikawa, T.; Kawashima, H.; Ohno, E.; Villalobos, I.B.; Naoe, Y.; Ichinose, T.; Koyama, N.; Tanaka, M.; et al. A Phase I clinical trial of EUS-guided intratumoral injection of the oncolytic virus, HF10 for unresectable locally advanced pancreatic cancer. BMC Cancer 2018, 18, 596. [Google Scholar] [CrossRef]
- Hamada, M.; Yura, Y. Efficient Delivery and Replication of Oncolytic Virus for Successful Treatment of Head and Neck Cancer. Int. J. Mol. Sci. 2020, 21, 7073. [Google Scholar] [CrossRef]
- Todo, T.; Martuza, R.L.; Rabkin, S.D.; Johnson, P.A. Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc. Natl. Acad. Sci. USA 2001, 98, 6396–6401. [Google Scholar] [CrossRef] [Green Version]
- Cassady, K.A. Human cytomegalovirus TRS1 and IRS1 gene products block the double-stranded-RNA-activated host protein shutoff response induced by herpes simplex virus type 1 infection. J. Virol. 2005, 79, 8707–8715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiocca, E.A.; Nakashima, H.; Kasai, K.; Fernandez, S.A.; Oglesbee, M. Preclinical Toxicology of rQNestin34.5v.2: An Oncolytic Herpes Virus with Transcriptional Regulation of the ICP34.5 Neurovirulence Gene. Mol. Therapy. Methods Clin. Dev. 2020, 17, 871–893. [Google Scholar] [CrossRef]
- Nakashima, H.; Nguyen, T.; Kasai, K.; Passaro, C.; Ito, H.; Goins, W.F.; Shaikh, I.; Erdelyi, R.; Nishihara, R.; Nakano, I.; et al. Toxicity and Efficacy of a Novel GADD34-expressing Oncolytic HSV-1 for the Treatment of Experimental Glioblastoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 2574–2584. [Google Scholar] [CrossRef] [Green Version]
- Cassady, K.A.; Gross, M.; Roizman, B. The second-site mutation in the herpes simplex virus recombinants lacking the gamma134.5 genes precludes shutoff of protein synthesis by blocking the phosphorylation of eIF-2alpha. J. Virol. 1998, 72, 7005–7011. [Google Scholar] [CrossRef] [Green Version]
- Mohr, I.; Sternberg, D.; Ward, S.; Leib, D.; Mulvey, M.; Gluzman, Y. A herpes simplex virus type 1 gamma34.5 second-site suppressor mutant that exhibits enhanced growth in cultured glioblastoma cells is severely attenuated in animals. J. Virol. 2001, 75, 5189–5196. [Google Scholar] [CrossRef] [Green Version]
- Nakatake, R.; Kaibori, M.; Nakamura, Y.; Tanaka, Y.; Matushima, H.; Okumura, T.; Murakami, T.; Ino, Y.; Todo, T.; Kon, M. Third-generation oncolytic herpes simplex virus inhibits the growth of liver tumors in mice. Cancer Sci. 2018, 109, 600–610. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, H.; Kaibori, M.; Hatta, M.; Ishizaki, M.; Nakatake, R.; Okumura, T.; Yoshii, K.; Todo, T. Efficacy of a third-generation oncolytic herpes simplex virus in neuroendocrine tumor xenograft models. Oncotarget 2019, 10, 7132–7141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugawara, K.; Iwai, M.; Yajima, S.; Tanaka, M.; Yanagihara, K.; Seto, Y.; Todo, T. Efficacy of a Third-Generation Oncolytic Herpes Virus G47Delta in Advanced Stage Models of Human Gastric Cancer. Mol. Ther. Oncolytics 2020, 17, 205–215. [Google Scholar] [CrossRef]
- Wakimoto, H.; Kesari, S.; Farrell, C.J.; Curry, W.T., Jr.; Zaupa, C.; Aghi, M.; Kuroda, T.; Stemmer-Rachamimov, A.; Shah, K.; Liu, T.C.; et al. Human glioblastoma-derived cancer stem cells: Establishment of invasive glioma models and treatment with oncolytic herpes simplex virus vectors. Cancer Res. 2009, 69, 3472–3481. [Google Scholar] [CrossRef] [Green Version]
- Peters, C.; Paget, M.; Tshilenge, K.T.; Saha, D.; Antoszczyk, S.; Baars, A.; Frost, T.; Martuza, R.L.; Wakimoto, H.; Rabkin, S.D. restriction of gamma34.5-deleted oncolytic herpes simplex virus replication in glioblastoma stem-like cells. J. Virol. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taguchi, S.; Fukuhara, H.; Todo, T. Oncolytic virus therapy in Japan: Progress in clinical trials and future perspectives. Jpn. J. Clin. Oncol. 2019, 49, 201–209. [Google Scholar] [CrossRef]
- Kambara, H.; Okano, H.; Chiocca, E.A.; Saeki, Y. An oncolytic HSV-1 mutant expressing ICP34.5 under control of a nestin promoter increases survival of animals even when symptomatic from a brain tumor. Cancer Res. 2005, 65, 2832–2839. [Google Scholar] [CrossRef] [Green Version]
- Friedman, G.K.; Nan, L.; Haas, M.C.; Kelly, V.M.; Moore, B.P.; Langford, C.P.; Xu, H.; Han, X.; Beierle, E.A.; Markert, J.M.; et al. gamma(1)34.5-deleted HSV-1-expressing human cytomegalovirus IRS1 gene kills human glioblastoma cells as efficiently as wild-type HSV-1 in normoxia or hypoxia. Gene Ther. 2015, 22, 348–355. [Google Scholar] [CrossRef]
- Todo, T. “Armed” oncolytic herpes simplex viruses for brain tumor therapy. Cell Adhes. Migr. 2008, 2, 208–213. [Google Scholar] [CrossRef] [Green Version]
- Burton, C.; Bartee, E. Syncytia Formation in Oncolytic Virotherapy. Mol. Ther. Oncolytics 2019, 15, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Fu, X.; Tao, L.; Jin, A.; Vile, R.; Brenner, M.K.; Zhang, X. Expression of a fusogenic membrane glycoprotein by an oncolytic herpes simplex virus potentiates the viral antitumor effect. Mol. Ther. J. Am. Soc. Gene Ther. 2003, 7, 748–754. [Google Scholar] [CrossRef]
- Chase, M.; Chung, R.Y.; Chiocca, E.A. An oncolytic viral mutant that delivers the CYP2B1 transgene and augments cyclophosphamide chemotherapy. Nat. Biotechnol. 1998, 16, 444–448. [Google Scholar] [CrossRef]
- Nakamura, H.; Mullen, J.T.; Chandrasekhar, S.; Pawlik, T.M.; Yoon, S.S.; Tanabe, K.K. Multimodality therapy with a replication-conditional herpes simplex virus 1 mutant that expresses yeast cytosine deaminase for intratumoral conversion of 5-fluorocytosine to 5-fluorouracil. Cancer Res. 2001, 61, 5447–5452. [Google Scholar]
- Simpson, G.R.; Han, Z.; Liu, B.; Wang, Y.; Campbell, G.; Coffin, R.S. Combination of a fusogenic glycoprotein, prodrug activation, and oncolytic herpes simplex virus for enhanced local tumor control. Cancer Res. 2006, 66, 4835–4842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.C.; Zhang, T.; Fukuhara, H.; Kuroda, T.; Todo, T.; Martuza, R.L.; Rabkin, S.D.; Kurtz, A. Oncolytic HSV armed with platelet factor 4, an antiangiogenic agent, shows enhanced efficacy. Mol. Ther. J. Am. Soc. Gene Ther. 2006, 14, 789–797. [Google Scholar] [CrossRef]
- Han, Z.Q.; Assenberg, M.; Liu, B.L.; Wang, Y.B.; Simpson, G.; Thomas, S.; Coffin, R.S. Development of a second-generation oncolytic Herpes simplex virus expressing TNFalpha for cancer therapy. J. Gene Med. 2007, 9, 99–106. [Google Scholar] [CrossRef]
- Xu, B.; Ma, R.; Russell, L.; Yoo, J.Y.; Han, J.; Cui, H.; Yi, P.; Zhang, J.; Nakashima, H.; Dai, H.; et al. An oncolytic herpesvirus expressing E-cadherin improves survival in mouse models of glioblastoma. Nat. Biotechnol. 2018. [Google Scholar] [CrossRef]
- Andreansky, S.; He, B.; van Cott, J.; McGhee, J.; Markert, J.M.; Gillespie, G.Y.; Roizman, B.; Whitley, R.J. Treatment of intracranial gliomas in immunocompetent mice using herpes simplex viruses that express murine interleukins. Gene Ther. 1998, 5, 121–130. [Google Scholar] [CrossRef] [Green Version]
- Parker, J.N.; Gillespie, G.Y.; Love, C.E.; Randall, S.; Whitley, R.J.; Markert, J.M. Engineered herpes simplex virus expressing IL-12 in the treatment of experimental murine brain tumors. Proc. Natl. Acad. Sci. USA 2000, 97, 2208–2213. [Google Scholar] [CrossRef] [Green Version]
- Patel, D.M.; Foreman, P.M.; Nabors, L.B.; Riley, K.O.; Gillespie, G.Y.; Markert, J.M. Design of a Phase I Clinical Trial to Evaluate M032, a Genetically Engineered HSV-1 Expressing IL-12, in Patients with Recurrent/Progressive Glioblastoma Multiforme, Anaplastic Astrocytoma, or Gliosarcoma. Hum. Gene Ther. Clin. Dev. 2016, 27, 69–78. [Google Scholar] [CrossRef]
- Wong, R.J.; Patel, S.G.; Kim, S.; DeMatteo, R.P.; Malhotra, S.; Bennett, J.J.; St-Louis, M.; Shah, J.P.; Johnson, P.A.; Fong, Y. Cytokine gene transfer enhances herpes oncolytic therapy in murine squamous cell carcinoma. Hum. Gene Ther. 2001, 12, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.L.; Robinson, M.; Han, Z.Q.; Branston, R.H.; English, C.; Reay, P.; McGrath, Y.; Thomas, S.K.; Thornton, M.; Bullock, P.; et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003, 10, 292–303. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.C.; Coffin, R.S.; Davis, C.J.; Graham, N.J.; Groves, N.; Guest, P.J.; Harrington, K.J.; James, N.D.; Love, C.A.; McNeish, I.; et al. A phase I study of OncoVEXGM-CSF, a second-generation oncolytic herpes simplex virus expressing granulocyte macrophage colony-stimulating factor. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 6737–6747. [Google Scholar] [CrossRef] [Green Version]
- Martin, N.T.; Bell, J.C. Oncolytic Virus Combination Therapy: Killing One Bird with Two Stones. Mol. Ther. J. Am. Soc. Gene Ther. 2018, 26, 1414–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todo, T.; Rabkin, S.D.; Sundaresan, P.; Wu, A.; Meehan, K.R.; Herscowitz, H.B.; Martuza, R.L. Systemic antitumor immunity in experimental brain tumor therapy using a multimutated, replication-competent herpes simplex virus. Hum. Gene Ther. 1999, 10, 2741–2755. [Google Scholar] [CrossRef]
- Hellums, E.K.; Markert, J.M.; Parker, J.N.; He, B.; Perbal, B.; Roizman, B.; Whitley, R.J.; Langford, C.P.; Bharara, S.; Gillespie, G.Y. Increased efficacy of an interleukin-12-secreting herpes simplex virus in a syngeneic intracranial murine glioma model. Neuro-oncology 2005, 7, 213–224. [Google Scholar] [CrossRef]
- Parker, J.N.; Meleth, S.; Hughes, K.B.; Gillespie, G.Y.; Whitley, R.J.; Markert, J.M. Enhanced inhibition of syngeneic murine tumors by combinatorial therapy with genetically engineered HSV-1 expressing CCL2 and IL-12. Cancer Gene Ther. 2005, 12, 359–368. [Google Scholar] [CrossRef] [Green Version]
- Markert, J.M.; Cody, J.J.; Parker, J.N.; Coleman, J.M.; Price, K.H.; Kern, E.R.; Quenelle, D.C.; Lakeman, A.D.; Schoeb, T.R.; Palmer, C.A.; et al. Preclinical evaluation of a genetically engineered herpes simplex virus expressing interleukin-12. J. Virol. 2012, 86, 5304–5313. [Google Scholar] [CrossRef] [Green Version]
- Friedman, G.K.; Moore, B.P.; Nan, L.; Kelly, V.M.; Etminan, T.; Langford, C.P.; Xu, H.; Han, X.; Markert, J.M.; Beierle, E.A.; et al. Pediatric medulloblastoma xenografts including molecular subgroup 3 and CD133+ and CD15+ cells are sensitive to killing by oncolytic herpes simplex viruses. Neuro-oncology 2016, 18, 227–235. [Google Scholar] [CrossRef]
- Bennett, J.J.; Malhotra, S.; Wong, R.J.; Delman, K.; Zager, J.; St-Louis, M.; Johnson, P.; Fong, Y. Interleukin 12 secretion enhances antitumor efficacy of oncolytic herpes simplex viral therapy for colorectal cancer. Ann. Surg. 2001, 233, 819–826. [Google Scholar] [CrossRef]
- Varghese, S.; Rabkin, S.D.; Nielsen, P.G.; Wang, W.; Martuza, R.L. Systemic oncolytic herpes virus therapy of poorly immunogenic prostate cancer metastatic to lung. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 2919–2927. [Google Scholar] [CrossRef] [Green Version]
- Varghese, S.; Rabkin, S.D.; Nielsen, G.P.; MacGarvey, U.; Liu, R.; Martuza, R.L. Systemic therapy of spontaneous prostate cancer in transgenic mice with oncolytic herpes simplex viruses. Cancer Res. 2007, 67, 9371–9379. [Google Scholar] [CrossRef] [Green Version]
- Roth, J.C.; Cassady, K.A.; Cody, J.J.; Parker, J.N.; Price, K.H.; Coleman, J.M.; Peggins, J.O.; Noker, P.E.; Powers, N.W.; Grimes, S.D.; et al. Evaluation of the safety and biodistribution of M032, an attenuated herpes simplex virus type 1 expressing hIL-12, after intracerebral administration to aotus nonhuman primates. Hum. Gene Ther. Clin. Dev. 2014, 25, 16–27. [Google Scholar] [CrossRef] [Green Version]
- Chambers, M.R.; Bentley, R.T.; Crossman, D.K.; Foote, J.B.; Koehler, J.W.; Markert, J.M.; Omar, N.B.; Platt, S.R.; Self, D.M.; Shores, A.; et al. The One Health Consortium: Design of a Phase I Clinical Trial to Evaluate M032, a Genetically Engineered HSV-1 Expressing IL-12, in Combination With a Checkpoint Inhibitor in Canine Patients With Sporadic High Grade Gliomas. Front. Surg. 2020, 7, 59. [Google Scholar] [CrossRef]
- Ino, Y.; Saeki, Y.; Fukuhara, H.; Todo, T. Triple combination of oncolytic herpes simplex virus-1 vectors armed with interleukin-12, interleukin-18, or soluble B7-1 results in enhanced antitumor efficacy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 643–652. [Google Scholar] [CrossRef] [Green Version]
- Zhan, Y.; Lew, A.M.; Chopin, M. The Pleiotropic Effects of the GM-CSF Rheostat on Myeloid Cell Differentiation and Function: More Than a Numbers Game. Front. Immunol. 2019, 10, 2679. [Google Scholar] [CrossRef]
- Varghese, S.; Rabkin, S.D.; Liu, R.; Nielsen, P.G.; Ipe, T.; Martuza, R.L. Enhanced therapeutic efficacy of IL-12, but not GM-CSF, expressing oncolytic herpes simplex virus for transgenic mouse derived prostate cancers. Cancer Gene Ther. 2006, 13, 253–265. [Google Scholar] [CrossRef] [Green Version]
- Toda, M.; Martuza, R.L.; Kojima, H.; Rabkin, S.D. In situ cancer vaccination: An IL-12 defective vector/replication-competent herpes simplex virus combination induces local and systemic antitumor activity. J. Immunol. 1998, 160, 4457–4464. [Google Scholar]
- Toda, M.; Martuza, R.L.; Rabkin, S.D. Tumor growth inhibition by intratumoral inoculation of defective herpes simplex virus vectors expressing granulocyte-macrophage colony-stimulating factor. Mol. Ther. J. Am. Soc. Gene Ther. 2000, 2, 324–329. [Google Scholar] [CrossRef]
- Senzer, N.N.; Kaufman, H.L.; Amatruda, T.; Nemunaitis, M.; Reid, T.; Daniels, G.; Gonzalez, R.; Glaspy, J.; Whitman, E.; Harrington, K.; et al. Phase II clinical trial of a granulocyte-macrophage colony-stimulating factor-encoding, second-generation oncolytic herpesvirus in patients with unresectable metastatic melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 5763–5771. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Bines, S.D. OPTIM trial: A Phase III trial of an oncolytic herpes virus encoding GM-CSF for unresectable stage III or IV melanoma. Future Oncol. 2010, 6, 941–949. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Kim, D.W.; DeRaffele, G.; Mitcham, J.; Coffin, R.S.; Kim-Schulze, S. Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann. Surg. Oncol. 2010, 17, 718–730. [Google Scholar] [CrossRef]
- Harrington, K.J.; Hingorani, M.; Tanay, M.A.; Hickey, J.; Bhide, S.A.; Clarke, P.M.; Renouf, L.C.; Thway, K.; Sibtain, A.; McNeish, I.A.; et al. Phase I/II study of oncolytic HSV GM-CSF in combination with radiotherapy and cisplatin in untreated stage III/IV squamous cell cancer of the head and neck. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 4005–4015. [Google Scholar] [CrossRef] [Green Version]
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef]
- Kohlhapp, F.J.; Kaufman, H.L. Molecular Pathways: Mechanism of Action for Talimogene Laherparepvec, a New Oncolytic Virus Immunotherapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 1048–1054. [Google Scholar] [CrossRef] [Green Version]
- Pol, J.; Buque, A.; Aranda, F.; Bloy, N.; Cremer, I.; Eggermont, A.; Erbs, P.; Fucikova, J.; Galon, J.; Limacher, J.M.; et al. Trial Watch-Oncolytic viruses and cancer therapy. Oncoimmunology 2016, 5, e1117740. [Google Scholar] [CrossRef] [Green Version]
- Dummer, R.; Hoeller, C.; Gruter, I.P.; Michielin, O. Combining talimogene laherparepvec with immunotherapies in melanoma and other solid tumors. Cancer Immunol. Immunother. 2017, 66, 683–695. [Google Scholar] [CrossRef] [Green Version]
- Conry, R.M.; Westbrook, B.; McKee, S.; Norwood, T.G. Talimogene laherparepvec: First in class oncolytic virotherapy. Hum. Vaccines Immunother. 2018, 14, 839–846. [Google Scholar] [CrossRef]
- Pol, J.G.; Levesque, S.; Workenhe, S.T.; Gujar, S.; Le Boeuf, F.; Clements, D.R.; Fahrner, J.E.; Fend, L.; Bell, J.C.; Mossman, K.L.; et al. Trial Watch: Oncolytic viro-immunotherapy of hematologic and solid tumors. Oncoimmunology 2018, 7, e1503032. [Google Scholar] [CrossRef] [Green Version]
- Reale, A.; Calistri, A.; Palu, G. A clinical trial investigating biodistribution and shedding of an oncolytic virus. EBioMedicine 2019, 47, 4–5. [Google Scholar] [CrossRef] [Green Version]
- Andtbacka, R.H.I.; Amatruda, T.; Nemunaitis, J.; Zager, J.S.; Walker, J.; Chesney, J.A.; Liu, K.; Hsu, C.P.; Pickett, C.A.; Mehnert, J.M. Biodistribution, shedding, and transmissibility of the oncolytic virus talimogene laherparepvec in patients with melanoma. EBioMedicine 2019, 47, 89–97. [Google Scholar] [CrossRef] [Green Version]
- Coffin, R.S. From virotherapy to oncolytic immunotherapy: Where are we now? Curr. Opin. Virol. 2015, 13, 93–100. [Google Scholar] [CrossRef]
- Coffin, R. Interview with Robert Coffin, inventor of T-VEC: The first oncolytic immunotherapy approved for the treatment of cancer. Immunotherapy 2016, 8, 103–106. [Google Scholar] [CrossRef] [Green Version]
- Campadelli-Fiume, G.; Petrovic, B.; Leoni, V.; Gianni, T.; Avitabile, E.; Casiraghi, C.; Gatta, V. Retargeting Strategies for Oncolytic Herpes Simplex Viruses. Viruses 2016, 8, 63. [Google Scholar] [CrossRef] [Green Version]
- Rey, F.A. Molecular gymnastics at the herpesvirus surface. Embo Rep. 2006, 7, 1000–1005. [Google Scholar] [CrossRef]
- Campadelli-Fiume, G.; Menotti, L. Entry of alphaherpesviruses into the cell. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Campadelli-Fiume, G.; Gianni, T. HSV Glycoproteins and Their Roles in Virus Entry and Egress; Caister Academic Press: Wymondham, UK, 2007; Volume 1, pp. 135–156. [Google Scholar]
- Arii, J.; Kawaguchi, Y. The Role of HSV Glycoproteins in Mediating Cell Entry. Adv. Exp. Med. Biol. 2018, 1045, 3–21. [Google Scholar] [CrossRef]
- Liu, X.Q.; Xin, H.Y.; Lyu, Y.N.; Ma, Z.W.; Peng, X.C.; Xiang, Y.; Wang, Y.Y.; Wu, Z.J.; Cheng, J.T.; Ji, J.F.; et al. Oncolytic herpes simplex virus tumor targeting and neutralization escape by engineering viral envelope glycoproteins. Drug Deliv. 2018, 25, 1950–1962. [Google Scholar] [CrossRef]
- Oliver, S.L.; Xing, Y.; Chen, D.H.; Roh, S.H.; Pintilie, G.D.; Bushnell, D.A.; Sommer, M.H.; Yang, E.; Carfi, A.; Chiu, W.; et al. A glycoprotein B-neutralizing antibody structure at 2.8 A uncovers a critical domain for herpesvirus fusion initiation. Nat. Commun. 2020, 11, 4141. [Google Scholar] [CrossRef]
- Eisenberg, R.J.; Atanasiu, D.; Cairns, T.M.; Gallagher, J.R.; Krummenacher, C.; Cohen, G.H. Herpes virus fusion and entry: A story with many characters. Viruses 2012, 4, 800–832. [Google Scholar] [CrossRef] [PubMed]
- Atanasiu, D.; Cairns, T.M.; Whitbeck, J.C.; Saw, W.T.; Rao, S.; Eisenberg, R.J.; Cohen, G.H. Regulation of herpes simplex virus gB-induced cell-cell fusion by mutant forms of gH/gL in the absence of gD and cellular receptors. MBio 2013, 4. [Google Scholar] [CrossRef] [Green Version]
- Campadelli-Fiume, G.; Collins-McMillen, D.; Gianni, T.; Yurochko, A.D. Integrins as Herpesvirus Receptors and Mediators of the Host Signalosome. Annu. Rev. Virol. 2016, 3, 215–236. [Google Scholar] [CrossRef] [Green Version]
- Heldwein, E.E. gH/gL supercomplexes at early stages of herpesvirus entry. Curr. Opin. Virol. 2016, 18, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azab, W.; Lehmann, M.J.; Osterrieder, N. Glycoprotein H and alpha4beta1 integrins determine the entry pathway of alphaherpesviruses. J. Virol. 2013, 87, 5937–5948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicola, A.V. Herpesvirus Entry into Host Cells Mediated by Endosomal Low pH. Traffic 2016, 17, 965–975. [Google Scholar] [CrossRef] [Green Version]
- Azab, W.; Osterrieder, K. Initial Contact: The First Steps in Herpesvirus Entry. Adv. Anat. Embryol. Cell Biol. 2017, 223, 1–27. [Google Scholar] [CrossRef]
- Goins, W.F.; Hall, B.; Cohen, J.B.; Glorioso, J.C. Retargeting of herpes simplex virus (HSV) vectors. Curr. Opin. Virol. 2016, 21, 93–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, G.; Ye, G.J.; Debinski, W.; Roizman, B. Engineered herpes simplex virus 1 is dependent on IL13Ralpha 2 receptor for cell entry and independent of glycoprotein D receptor interaction. Proc. Natl. Acad. Sci. USA 2002, 99, 15124–15129. [Google Scholar] [CrossRef] [Green Version]
- Kamiyama, H.; Zhou, G.; Roizman, B. Herpes simplex virus 1 recombinant virions exhibiting the amino terminal fragment of urokinase-type plasminogen activator can enter cells via the cognate receptor. Gene Ther. 2006, 13, 621–629. [Google Scholar] [CrossRef] [Green Version]
- Zhou, G.; Roizman, B. Construction and properties of a herpes simplex virus 1 designed to enter cells solely via the IL-13alpha2 receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 5508–5513. [Google Scholar] [CrossRef] [Green Version]
- Menotti, L.; Cerretani, A.; Campadelli-Fiume, G. A herpes simplex virus recombinant that exhibits a single-chain antibody to HER2/neu enters cells through the mammary tumor receptor, independently of the gD receptors. J. Virol. 2006, 80, 5531–5539. [Google Scholar] [CrossRef] [Green Version]
- Menotti, L.; Cerretani, A.; Hengel, H.; Campadelli-Fiume, G. Construction of a fully retargeted herpes simplex virus 1 recombinant capable of entering cells solely via human epidermal growth factor receptor 2. J. Virol. 2008, 82, 10153–10161. [Google Scholar] [CrossRef] [Green Version]
- Menotti, L.; Nicoletti, G.; Gatta, V.; Croci, S.; Landuzzi, L.; De Giovanni, C.; Nanni, P.; Lollini, P.L.; Campadelli-Fiume, G. Inhibition of human tumor growth in mice by an oncolytic herpes simplex virus designed to target solely HER-2-positive cells. Proc. Natl. Acad. Sci. USA 2009, 106, 9039–9044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchida, H.; Marzulli, M.; Nakano, K.; Goins, W.F.; Chan, J.; Hong, C.S.; Mazzacurati, L.; Yoo, J.Y.; Haseley, A.; Nakashima, H.; et al. Effective treatment of an orthotopic xenograft model of human glioblastoma using an EGFR-retargeted oncolytic herpes simplex virus. Mol. Ther. J. Am. Soc. Gene Ther. 2013, 21, 561–569. [Google Scholar] [CrossRef] [Green Version]
- Menotti, L.; Avitabile, E.; Gatta, V.; Malatesta, P.; Petrovic, B.; Campadelli-Fiume, G. HSV as A Platform for the Generation of Retargeted, Armed, and Reporter-Expressing Oncolytic Viruses. Viruses 2018, 10, 352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leoni, V.; Petrovic, B.; Gianni, T.; Gatta, V.; Campadelli-Fiume, G. Simultaneous Insertion of Two Ligands in gD for Cultivation of Oncolytic Herpes Simplex Viruses in Noncancer Cells and Retargeting to Cancer Receptors. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Gatta, V.; Petrovic, B.; Campadelli-Fiume, G. The Engineering of a Novel Ligand in gH Confers to HSV an Expanded Tropism Independent of gD Activation by Its Receptors. PLoS Pathog. 2015, 11, e1004907. [Google Scholar] [CrossRef] [Green Version]
- Leoni, V.; Gatta, V.; Casiraghi, C.; Nicosia, A.; Petrovic, B.; Campadelli-Fiume, G. A Strategy for Cultivation of Retargeted Oncolytic Herpes Simplex Viruses in Non-cancer Cells. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Petrovic, B.; Gianni, T.; Gatta, V.; Campadelli-Fiume, G. Insertion of a ligand to HER2 in gB retargets HSV tropism and obviates the need for activation of the other entry glycoproteins. PLoS Pathog. 2017, 13, e1006352. [Google Scholar] [CrossRef]
- Nanni, P.; Gatta, V.; Menotti, L.; De Giovanni, C.; Ianzano, M.; Palladini, A.; Grosso, V.; Dall’ora, M.; Croci, S.; Nicoletti, G.; et al. Preclinical therapy of disseminated HER-2(+) ovarian and breast carcinomas with a HER-2-retargeted oncolytic herpesvirus. PLoS Pathog. 2013, 9, e1003155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gambini, E.; Reisoli, E.; Appolloni, I.; Gatta, V.; Campadelli-Fiume, G.; Menotti, L.; Malatesta, P. Replication-competent herpes simplex virus retargeted to HER2 as therapy for high-grade glioma. Mol. Ther. J. Am. Soc. Gene Ther. 2012, 20, 994–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reisoli, E.; Gambini, E.; Appolloni, I.; Gatta, V.; Barilari, M.; Menotti, L.; Malatesta, P. Efficacy of HER2 retargeted herpes simplex virus as therapy for high-grade glioma in immunocompetent mice. Cancer Gene Ther. 2012, 19, 788–795. [Google Scholar] [CrossRef]
- Alessandrini, F.; Ceresa, D.; Appolloni, I.; Marubbi, D.; Malatesta, P. Noninvasive Monitoring of Glioma Growth in the Mouse. J. Cancer 2016, 7, 1791–1797. [Google Scholar] [CrossRef] [Green Version]
- Leoni, V.; Vannini, A.; Gatta, V.; Rambaldi, J.; Sanapo, M.; Barboni, C.; Zaghini, A.; Nanni, P.; Lollini, P.L.; Casiraghi, C.; et al. A fully-virulent retargeted oncolytic HSV armed with IL-12 elicits local immunity and vaccine therapy towards distant tumors. PLoS Pathog. 2018, 14, e1007209. [Google Scholar] [CrossRef] [Green Version]
- Alessandrini, F.; Menotti, L.; Avitabile, E.; Appolloni, I.; Ceresa, D.; Marubbi, D.; Campadelli-Fiume, G.; Malatesta, P. Eradication of glioblastoma by immuno-virotherapy with a retargeted oncolytic HSV in a preclinical model. Oncogene 2019, 38, 4467–4479. [Google Scholar] [CrossRef]
- Uchida, H.; Chan, J.; Goins, W.F.; Grandi, P.; Kumagai, I.; Cohen, J.B.; Glorioso, J.C. A double mutation in glycoprotein gB compensates for ineffective gD-dependent initiation of herpes simplex virus type 1 infection. J. Virol. 2010, 84, 12200–12209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sette, P.; Amankulor, N.; Li, A.; Marzulli, M.; Leronni, D.; Zhang, M.; Goins, W.F.; Kaur, B.; Bolyard, C.; Cripe, T.P.; et al. GBM-Targeted oHSV Armed with Matrix Metalloproteinase 9 Enhances Anti-tumor Activity and Animal Survival. Mol. Ther. Oncolytics 2019, 15, 214–222. [Google Scholar] [CrossRef] [Green Version]
- Mazzacurati, L.; Marzulli, M.; Reinhart, B.; Miyagawa, Y.; Uchida, H.; Goins, W.F.; Li, A.; Kaur, B.; Caligiuri, M.; Cripe, T.; et al. Use of miRNA response sequences to block off-target replication and increase the safety of an unattenuated, glioblastoma-targeted oncolytic HSV. Mol. Ther. J. Am. Soc. Gene Ther. 2015, 23, 99–107. [Google Scholar] [CrossRef] [Green Version]
- Uchida, H.; Hamada, H.; Nakano, K.; Kwon, H.; Tahara, H.; Cohen, J.B.; Glorioso, J.C. Oncolytic Herpes Simplex Virus Vectors Fully Retargeted to Tumor- Associated Antigens. Curr. Cancer Drug Targets 2018, 18, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Sasso, E.; Froechlich, G.; Cotugno, G.; D’Alise, A.M.; Gentile, C.; Bignone, V.; De Lucia, M.; Petrovic, B.; Campadelli-Fiume, G.; Scarselli, E.; et al. Replicative conditioning of Herpes simplex type 1 virus by Survivin promoter, combined to ERBB2 retargeting, improves tumour cell-restricted oncolysis. Sci. Rep. 2020, 10, 4307. [Google Scholar] [CrossRef] [Green Version]
- Di Giovine, P.; Settembre, E.C.; Bhargava, A.K.; Luftig, M.A.; Lou, H.; Cohen, G.H.; Eisenberg, R.J.; Krummenacher, C.; Carfi, A. Structure of herpes simplex virus glycoprotein D bound to the human receptor nectin-1. PLoS Pathog. 2011, 7, e1002277. [Google Scholar] [CrossRef] [Green Version]
- Tuzmen, C.; Cairns, T.M.; Atanasiu, D.; Lou, H.; Saw, W.T.; Hall, B.L.; Cohen, J.B.; Cohen, G.H.; Glorioso, J.C. Point Mutations in Retargeted gD Eliminate the Sensitivity of EGFR/EGFRvIII-Targeted HSV to Key Neutralizing Antibodies. Mol. Therapy. Methods Clin. Dev. 2020, 16, 145–154. [Google Scholar] [CrossRef] [Green Version]
- Wong, R.J.; Chan, M.K.; Yu, Z.; Kim, T.H.; Bhargava, A.; Stiles, B.M.; Horsburgh, B.C.; Shah, J.P.; Ghossein, R.A.; Singh, B.; et al. Effective intravenous therapy of murine pulmonary metastases with an oncolytic herpes virus expressing interleukin 12. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2004, 10, 251–259. [Google Scholar] [CrossRef] [Green Version]
- Ungerechts, G.; Bossow, S.; Leuchs, B.; Holm, P.S.; Rommelaere, J.; Coffey, M.; Coffin, R.; Bell, J.; Nettelbeck, D.M. Moving oncolytic viruses into the clinic: Clinical-grade production, purification, and characterization of diverse oncolytic viruses. Mol. Ther. Methods Clin. Dev. 2016, 3, 16018. [Google Scholar] [CrossRef] [Green Version]
- Cattaneo, R.; Miest, T.; Shashkova, E.V.; Barry, M.A. Reprogrammed viruses as cancer therapeutics: Targeted, armed and shielded. Nat. Reviews. Microbiol. 2008, 6, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Leoni, V.; Gatta, V.; Palladini, A.; Nicoletti, G.; Ranieri, D.; Dall’Ora, M.; Grosso, V.; Rossi, M.; Alviano, F.; Bonsi, L.; et al. Systemic delivery of HER2-retargeted oncolytic-HSV by mesenchymal stromal cells protects from lung and brain metastases. Oncotarget 2015, 6, 34774–34787. [Google Scholar] [CrossRef] [Green Version]
- Fu, X.; Tao, L.; Zhang, X. Genetically coating oncolytic herpes simplex virus with CD47 allows efficient systemic delivery and prolongs virus persistence at tumor site. Oncotarget 2018, 9, 34543–34553. [Google Scholar] [CrossRef] [Green Version]
- Kuruppu, D.; Tanabe, K.K. HSV-1 as a novel therapy for breast cancer meningeal metastases. Cancer Gene Ther. 2015, 22, 506–508. [Google Scholar] [CrossRef] [Green Version]
- Senzer, N.N.; Kaufman, H.; Amatruda, A.; Nemunaitis, M.; Daniels, G.; Glaspy, J.; Goldsweig, H.; Coffin, R.S.; Nemunaitis, J. Phase II clinical trial with a second generation, GM-CSF encoding, oncolytic herpesvirus in unresectable metastatic melanoma. J. Clin. Oncol. (Asco Meet. Abstr.) 2009, 27, 9035. [Google Scholar] [CrossRef]
- Simpson, G.R.; Relph, K.; Harrington, K.; Melcher, A.; Pandha, H. Cancer immunotherapy via combining oncolytic virotherapy with chemotherapy: Recent advances. Oncolytic Virotherapy 2016, 5, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alayo, Q.A.; Ito, H.; Passaro, C.; Zdioruk, M.; Mahmoud, A.B.; Grauwet, K.; Zhang, X.; Lawler, S.E.; Reardon, D.A.; Goins, W.F.; et al. Glioblastoma infiltration of both tumor- and virus-antigen specific cytotoxic T cells correlates with experimental virotherapy responses. Sci. Rep. 2020, 10, 5095. [Google Scholar] [CrossRef] [Green Version]
- Currier, M.A.; Gillespie, R.A.; Sawtell, N.M.; Mahller, Y.Y.; Stroup, G.; Collins, M.H.; Kambara, H.; Chiocca, E.A.; Cripe, T.P. Efficacy and safety of the oncolytic herpes simplex virus rRp450 alone and combined with cyclophosphamide. Mol. Ther. J. Am. Soc. Gene Ther. 2008, 16, 879–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghonime, M.G.; Cassady, K.A. Combination Therapy Using Ruxolitinib and Oncolytic HSV Renders Resistant MPNSTs Susceptible to Virotherapy. Cancer Immunol. Res. 2018, 6, 1499–1510. [Google Scholar] [CrossRef] [Green Version]
- Yoo, J.Y.; Swanner, J.; Otani, Y.; Nair, M.; Park, F.; Banasavadi-Siddegowda, Y.; Liu, J.; Jaime-Ramirez, A.C.; Hong, B.; Geng, F.; et al. oHSV therapy increases trametinib access to brain tumors and sensitizes them in vivo. Neuro-oncology 2019. [Google Scholar] [CrossRef]
- Currier, M.A.; Eshun, F.K.; Sholl, A.; Chernoguz, A.; Crawford, K.; Divanovic, S.; Boon, L.; Goins, W.F.; Frischer, J.S.; Collins, M.H.; et al. VEGF blockade enables oncolytic cancer virotherapy in part by modulating intratumoral myeloid cells. Mol. Ther. J. Am. Soc. Gene Ther. 2013, 21, 1014–1023. [Google Scholar] [CrossRef] [Green Version]
- Advani, S.J.; Mezhir, J.J.; Roizman, B.; Weichselbaum, R.R. ReVOLT: Radiation-enhanced viral oncolytic therapy. Int. J. Radiat. Oncol. Biol. Phys. 2006, 66, 637–646. [Google Scholar] [CrossRef]
- Markert, J.M.; Razdan, S.N.; Kuo, H.C.; Cantor, A.; Knoll, A.; Karrasch, M.; Nabors, L.B.; Markiewicz, M.; Agee, B.S.; Coleman, J.M.; et al. A phase 1 trial of oncolytic HSV-1, G207, given in combination with radiation for recurrent GBM demonstrates safety and radiographic responses. Mol. Ther. J. Am. Soc. Gene Ther. 2014, 22, 1048–1055. [Google Scholar] [CrossRef] [Green Version]
- Foreman, P.M.; Friedman, G.K.; Cassady, K.A.; Markert, J.M. Oncolytic Virotherapy for the Treatment of Malignant Glioma. Neurother. J. Am. Soc. Exp. Neurother. 2017, 14, 333–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, S.C.; Levine, J.H.; Cogdill, A.P.; Zhao, Y.; Anang, N.A.S.; Andrews, M.C.; Sharma, P.; Wang, J.; Wargo, J.A.; Pe’er, D.; et al. Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell 2017, 170, 1120–1133. [Google Scholar] [CrossRef] [Green Version]
- Filley, A.C.; Dey, M. Immune System, Friend or Foe of Oncolytic Virotherapy? Front. Oncol. 2017, 7, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.S.; Liu, Z.; Kowalsky, S.; Feist, M.; Kalinski, P.; Lu, B.; Storkus, W.J.; Bartlett, D.L. Oncolytic Immunotherapy: Conceptual Evolution, Current Strategies, and Future Perspectives. Front. Immunol. 2017, 8, 555. [Google Scholar] [CrossRef] [Green Version]
- Haanen, J. Converting Cold into Hot Tumors by Combining Immunotherapies. Cell 2017, 170, 1055–1056. [Google Scholar] [CrossRef] [Green Version]
- Maroun, J.; Munoz-Alia, M.; Ammayappan, A.; Schulze, A.; Peng, K.W.; Russell, S. Designing and building oncolytic viruses. Future Virol. 2017, 12, 193–213. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.; Markert, J.M.; Leavenworth, J.W. Modulation of the Intratumoral Immune Landscape by Oncolytic Herpes Simplex Virus Virotherapy. Front. Oncol. 2017, 7, 136. [Google Scholar] [CrossRef] [Green Version]
- Ma, W.; He, H.; Wang, H. Oncolytic herpes simplex virus and immunotherapy. BMC Immunol. 2018, 19, 40. [Google Scholar] [CrossRef] [Green Version]
- Sivanandam, V.; LaRocca, C.J.; Chen, N.G.; Fong, Y.; Warner, S.G. Oncolytic Viruses and Immune Checkpoint Inhibition: The Best of Both Worlds. Mol. Ther. Oncolytics 2019, 13, 93–106. [Google Scholar] [CrossRef] [Green Version]
- Chiocca, E.A.; Rabkin, S.D. Oncolytic viruses and their application to cancer immunotherapy. Cancer Immunol. Res. 2014, 2, 295–300. [Google Scholar] [CrossRef] [Green Version]
- Breitbach, C.J.; Lichty, B.D.; Bell, J.C. Oncolytic Viruses: Therapeutics With an Identity Crisis. EBioMedicine 2016, 9, 31–36. [Google Scholar] [CrossRef] [Green Version]
- Friedman, G.K.; Markert, J.M.; Gillespie, G.Y. Combination strategies enhance oncolytic virotherapy. Oncotarget 2017, 8, 34020–34021. [Google Scholar] [CrossRef]
- Lawler, S.E.; Speranza, M.C.; Cho, C.F.; Chiocca, E.A. Oncolytic Viruses in Cancer Treatment: A Review. Jama Oncol. 2017, 3, 841–849. [Google Scholar] [CrossRef] [Green Version]
- Meyers, D.E.; Wang, A.A.; Thirukkumaran, C.M.; Morris, D.G. Current Immunotherapeutic Strategies to Enhance Oncolytic Virotherapy. Front. Oncol. 2017, 7, 114. [Google Scholar] [CrossRef] [Green Version]
- Puzanov, I.; Milhem, M.M.; Minor, D.; Hamid, O.; Li, A.; Chen, L.; Chastain, M.; Gorski, K.S.; Anderson, A.; Chou, J.; et al. Talimogene Laherparepvec in Combination With Ipilimumab in Previously Untreated, Unresectable Stage IIIB-IV Melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 2619–2626. [Google Scholar] [CrossRef] [Green Version]
- Chesney, J.; Puzanov, I.; Collichio, F.; Singh, P.; Milhem, M.M.; Glaspy, J.; Hamid, O.; Ross, M.; Friedlander, P.; Garbe, C.; et al. Randomized, Open-Label Phase II Study Evaluating the Efficacy and Safety of Talimogene Laherparepvec in Combination With Ipilimumab Versus Ipilimumab Alone in Patients With Advanced, Unresectable Melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 1658–1667. [Google Scholar] [CrossRef]
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.I.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2017, 170, 1109–1119. [Google Scholar] [CrossRef] [Green Version]
- Middleton, M.R.; Aroldi, F.; Sacco, J.; Milhem, M.M.; Curti, B.D.; Vanderwalde, A.M.; Baum, S.; Samson, A.; Pavlick, A.C.; Chesney, J.A.; et al. An open-label, single-arm, phase II clinical trial of RP1, an enhanced potency oncolytic herpes virus, combined with nivolumab in four solid tumor types: Initial results from the skin cancer cohorts. J. Clin. Oncol. 2020, 38, e22050. [Google Scholar] [CrossRef]
- Saha, D.; Martuza, R.L.; Rabkin, S.D. Macrophage Polarization Contributes to Glioblastoma Eradication by Combination Immunovirotherapy and Immune Checkpoint Blockade. Cancer Cell 2017, 32, 253–267. [Google Scholar] [CrossRef] [Green Version]
- Saha, D.; Martuza, R.L.; Rabkin, S.D. Oncolytic herpes simplex virus immunovirotherapy in combination with immune checkpoint blockade to treat glioblastoma. Immunotherapy 2018, 10, 779–786. [Google Scholar] [CrossRef]
- Passaro, C.; Alayo, Q.; De Laura, I.; McNulty, J.; Grauwet, K.; Ito, H.; Bhaskaran, V.; Mineo, M.; Lawler, S.E.; Shah, K.; et al. Arming an Oncolytic Herpes Simplex Virus Type 1 with a Single-chain Fragment Variable Antibody against PD-1 for Experimental Glioblastoma Therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 290–299. [Google Scholar] [CrossRef] [Green Version]
- Cook, M.; Chauhan, A. Clinical Application of Oncolytic Viruses: A Systematic Review. Int. J. Mol. Sci. 2020, 21, 7505. [Google Scholar] [CrossRef] [PubMed]
- Macedo, N.; Miller, D.M.; Haq, R.; Kaufman, H.L. Clinical landscape of oncolytic virus research in 2020. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Cassidy, T.; Craig, M. Determinants of combination GM-CSF immunotherapy and oncolytic virotherapy success identified through in silico treatment personalization. PLoS Comput. Biol. 2019, 15, e1007495. [Google Scholar] [CrossRef]
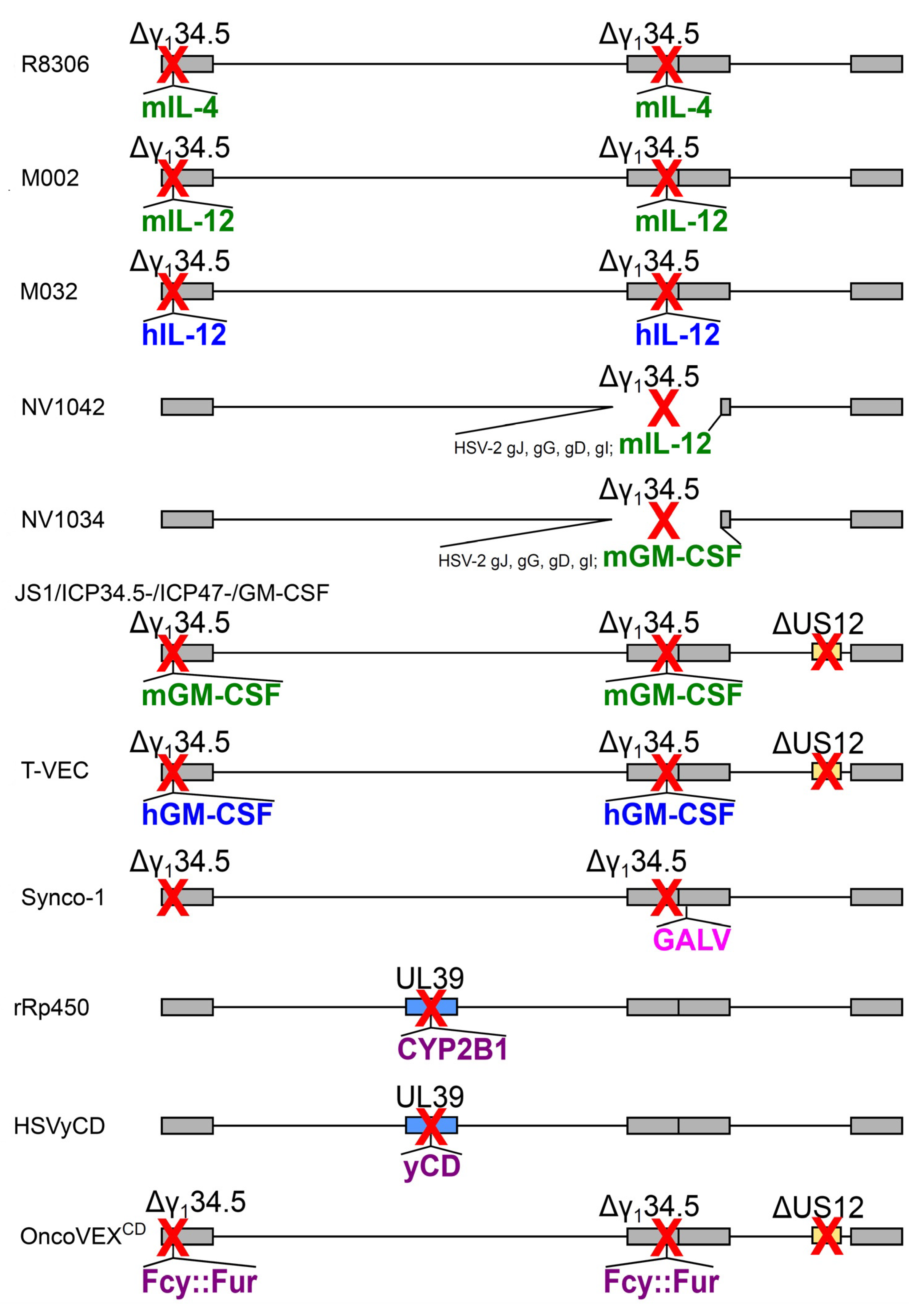
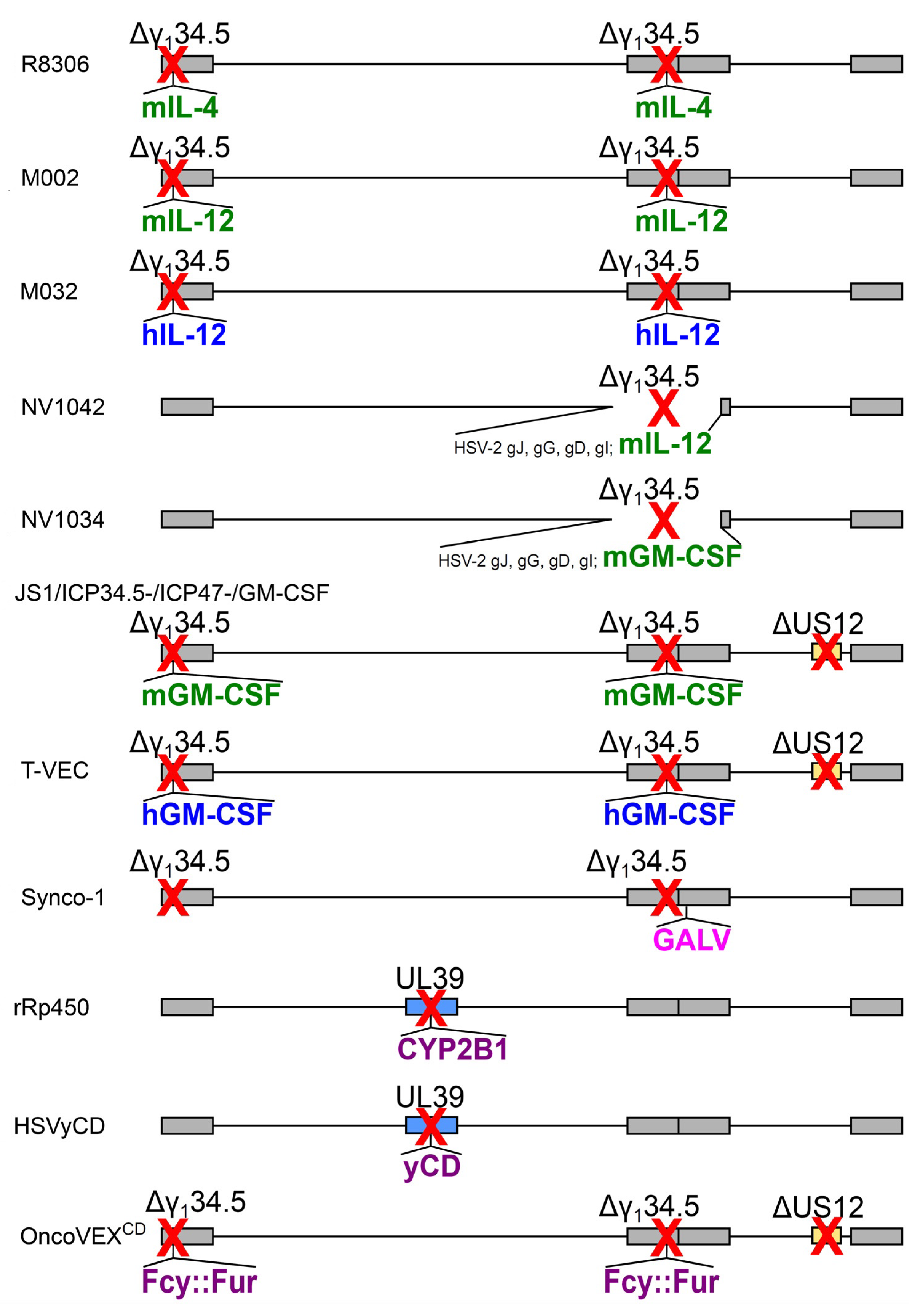
| oHSV Name | Expressed Transgene @ Viral Locus | Parental Virus | Diagram in Figure 2, Line | Clinical Trial Identifier (Status) | Ref. |
|---|---|---|---|---|---|
| R8306 | murine IL-4 @ γ134.5 loci | HSV-1(F), Δ 2 copies of γ134.5 | a | – | [69] |
| M002 | murine IL-12 @ γ134.5 loci | HSV-1(F), Δ 2 copies of γ134.5 | b | – | [70] |
| M032 | human IL-12 @ γ134.5 loci | HSV-1(F), Δ 2 copies of γ134.5 | c | NCT02062827 (R) | [71] |
| NV1042 | murine IL-12 @ γ134.5 locus | NV1020 (Δ 1 copy of γ134.5) | d | – | [72] |
| NV1034 | murine GM-CSF @ γ134.5 locus | NV1020 (Δ 1 copy of γ134.5) | e | – | [72] |
| JS1/ICP34.5-/ICP47-/GM-CSF | murine GM-CSF @ γ134.5 loci | JS-1 1, Δ 2 copies of γ134.5, ΔUS12 | f | – | [73] |
| OncoVEXGM-CSF, T-VEC, talimogene laherparepvec | human GM-CSF @ γ134.5 loci | JS-1 1, Δ 2 copies of γ134.5, ΔUS12 | g | NCT00769704 (C) | [74] |
| Synco-1 | GALV fusogenic protein @ packaging signal | HSV-1, Δ 2 copies of γ134.5 | h | – | [62] |
| rRp450 | rat CYP2B1 @ UL39 | HSV-1 (KOS) + inactivated UL39 | i | NCT01071941 (R) | [63] |
| HSVyCD | yCD @ UL39 | HSV-1 (KOS) + inactivated UL39 | j | – | [64] |
| OncoVEXCD | Fcy::Fur fusion @ γ134.5 locus | JS-1 1, Δ 2 copies of γ134.5, ΔUS12 | k | – | [65] |
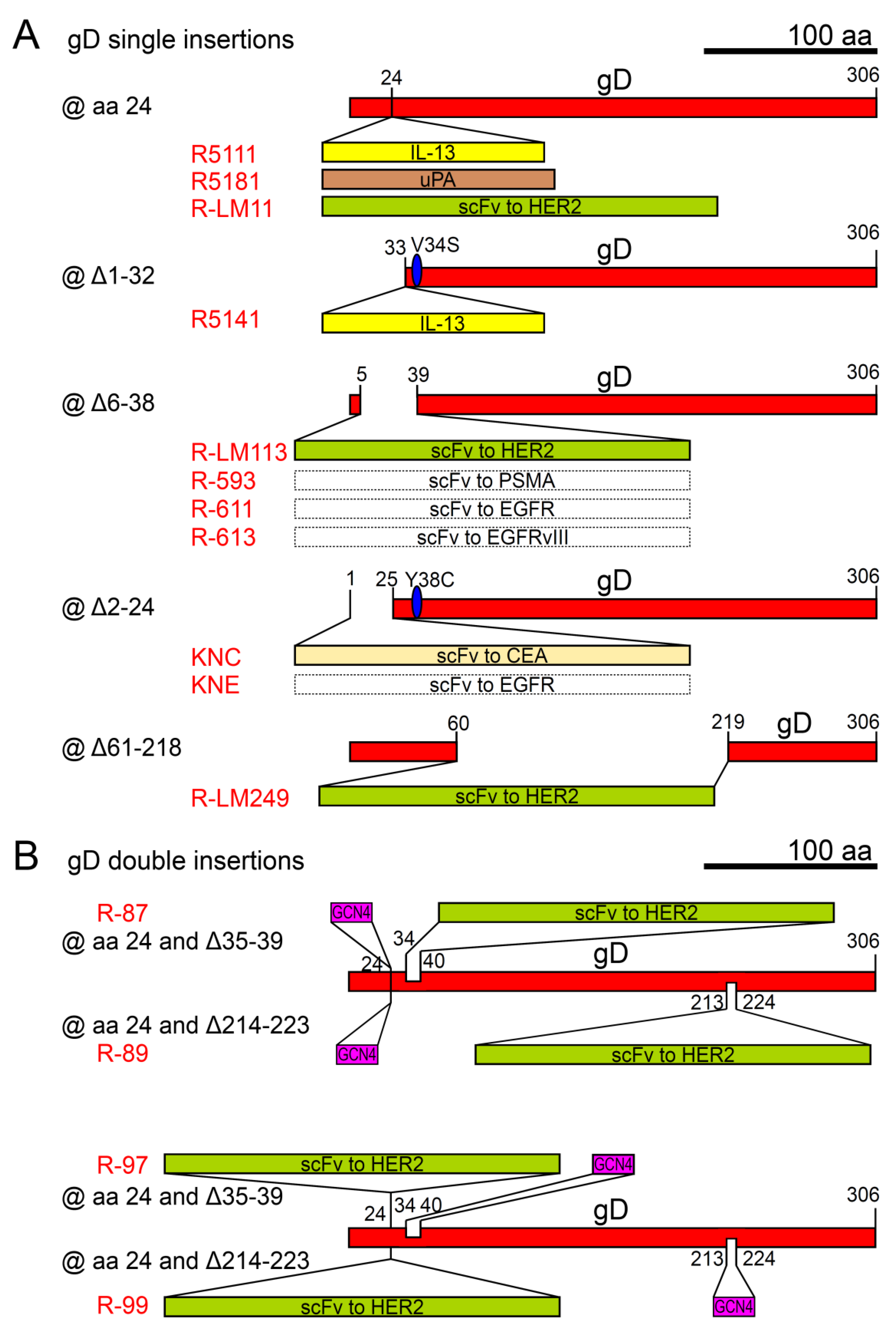
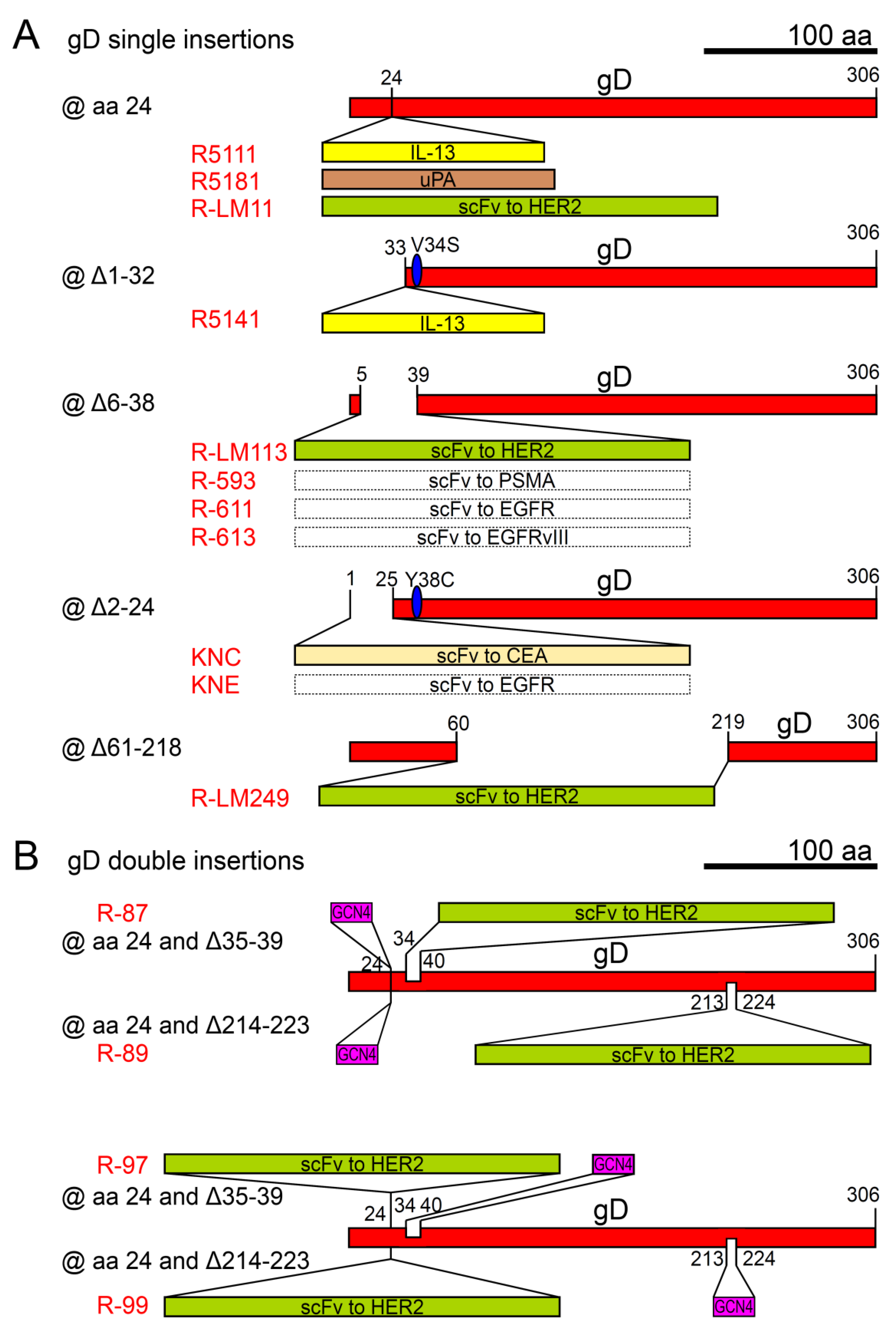
| Retargeting Ligand(s) @ Viral Glycoprotein | Target Heterologous Receptor | oHSV Name | Parental Strain | Ref. |
|---|---|---|---|---|
| @ gD | ||||
| IL-13 @ gD aa 24 | IL-13Rα2 | R5111 | HSV-1(F) | [120] |
| uPA @gD aa 24 | uPAR | R5181 | HSV-1(F) | [121] |
| IL-13 @ gD Δ1-32 | IL-13Rα2 | R5141 | HSV-1(F)+gDV34S | [122] |
| scFv to HER2 @ gD aa24 | HER2 | R-LM11 | HSV-1(F)BAC+lacZ | [123] |
| scFv to HER2 @ gDΔ6-38 | HER2 | R-LM113 1 | HSV-1(F)BAC+EGFP | [124] |
| scFv to HER2 @ gDΔ61-218 | HER2 | R-LM249 1 | HSV-1(F)BAC+EGFP | [125] |
| scFv to CEA @ gDΔ2-24 | CEA | KNC 1 | HSV-1(KOS)+gDY38C+gB:NT allele | [126] |
| scFv to EGFR @ gDΔ2-24 | EGFR, EGFRvIII | KNE 1 | HSV-1(KOS)+gDY38C+gB:NT allele | [126] |
| scFv to EGFR @ gDΔ6-38 | EGFR | R-611 1 | HSV-1(F)BAC+EGFP | [127] |
| scFv to PSMA @gDΔ6-38 | PSMA | R-593 1 | HSV-1(F)BAC+EGFP | [127] |
| scFv to EGFRvIII @ gDΔ6-38 | EGFRvIII | R-613 1 | HSV-1(F)BAC+EGFP | [127] |
| @ gD (double engineering) | ||||
| scFv to HER2 and GCN4 peptide @ gD aa 24, or @ gDΔ35-39, or @ gDΔ214-223 | HER2 and GCN4R | R-87 1, R-89 1, R-97 1, R-99 1 | HSV-1(F)BAC+EGFP | [128] |
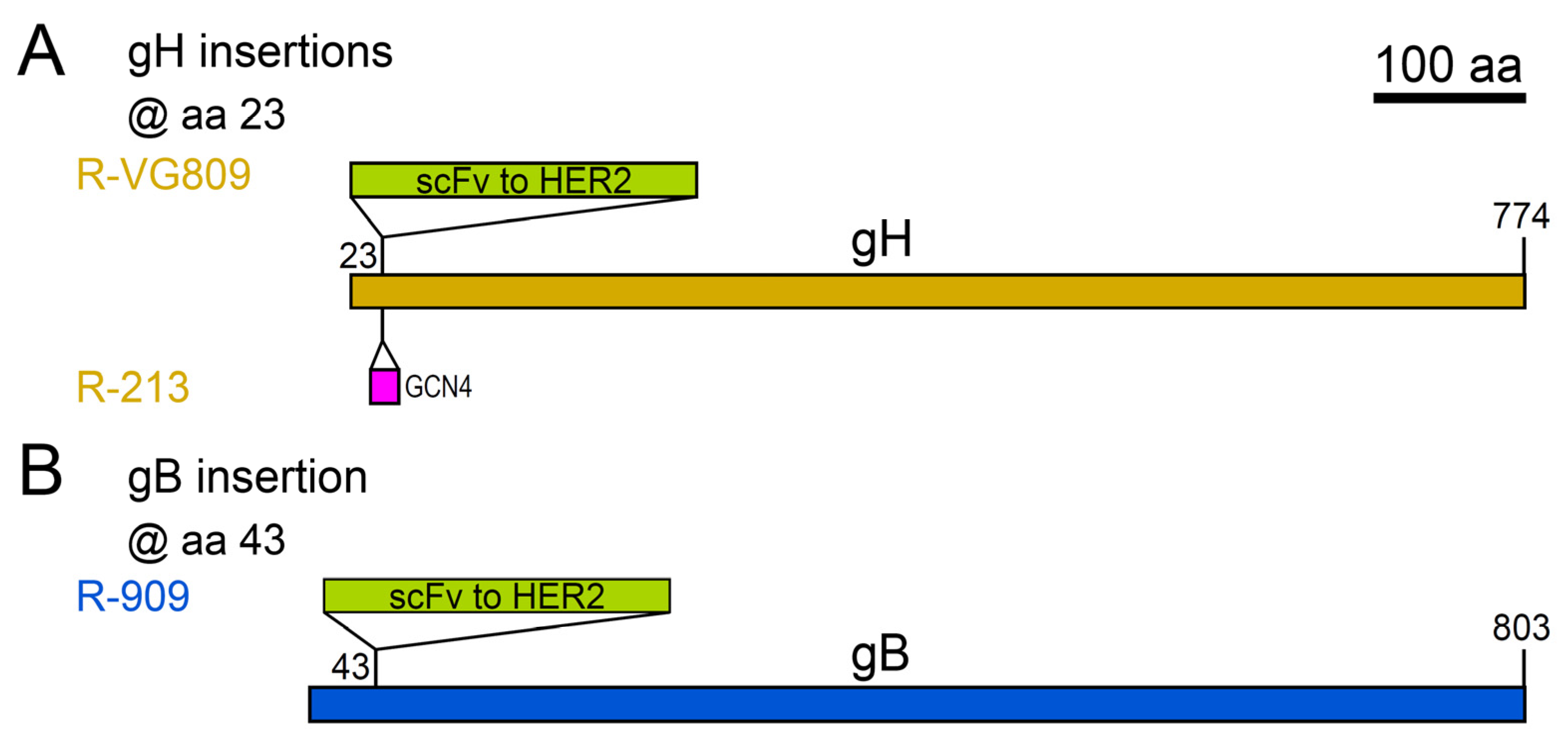
| @ gH | ||||
| scFv to HER2 @ gH aa 23 | HER2 | R-VG809 2 | HSV-1(F)BAC+mCherry+gDΔ6-38 | [129] |
| GCN4 peptide @ gH aa 23 | HER2 and GCN4R | R-213 2 | R-LM113 | [130] |
| @ gB | ||||
| scFv to HER2 @ gB aa 43 | HER2 | R-909 2 | HSV-1(F)BAC+EGFP+gDΔ6-38 | [131] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Menotti, L.; Avitabile, E. Herpes Simplex Virus Oncolytic Immunovirotherapy: The Blossoming Branch of Multimodal Therapy. Int. J. Mol. Sci. 2020, 21, 8310. https://doi.org/10.3390/ijms21218310
Menotti L, Avitabile E. Herpes Simplex Virus Oncolytic Immunovirotherapy: The Blossoming Branch of Multimodal Therapy. International Journal of Molecular Sciences. 2020; 21(21):8310. https://doi.org/10.3390/ijms21218310
Chicago/Turabian StyleMenotti, Laura, and Elisa Avitabile. 2020. "Herpes Simplex Virus Oncolytic Immunovirotherapy: The Blossoming Branch of Multimodal Therapy" International Journal of Molecular Sciences 21, no. 21: 8310. https://doi.org/10.3390/ijms21218310
APA StyleMenotti, L., & Avitabile, E. (2020). Herpes Simplex Virus Oncolytic Immunovirotherapy: The Blossoming Branch of Multimodal Therapy. International Journal of Molecular Sciences, 21(21), 8310. https://doi.org/10.3390/ijms21218310