In Vivo Pre-Instructed HSCs Robustly Execute Asymmetric Cell Divisions In Vitro


Abstract
:1. Introduction
2. Results
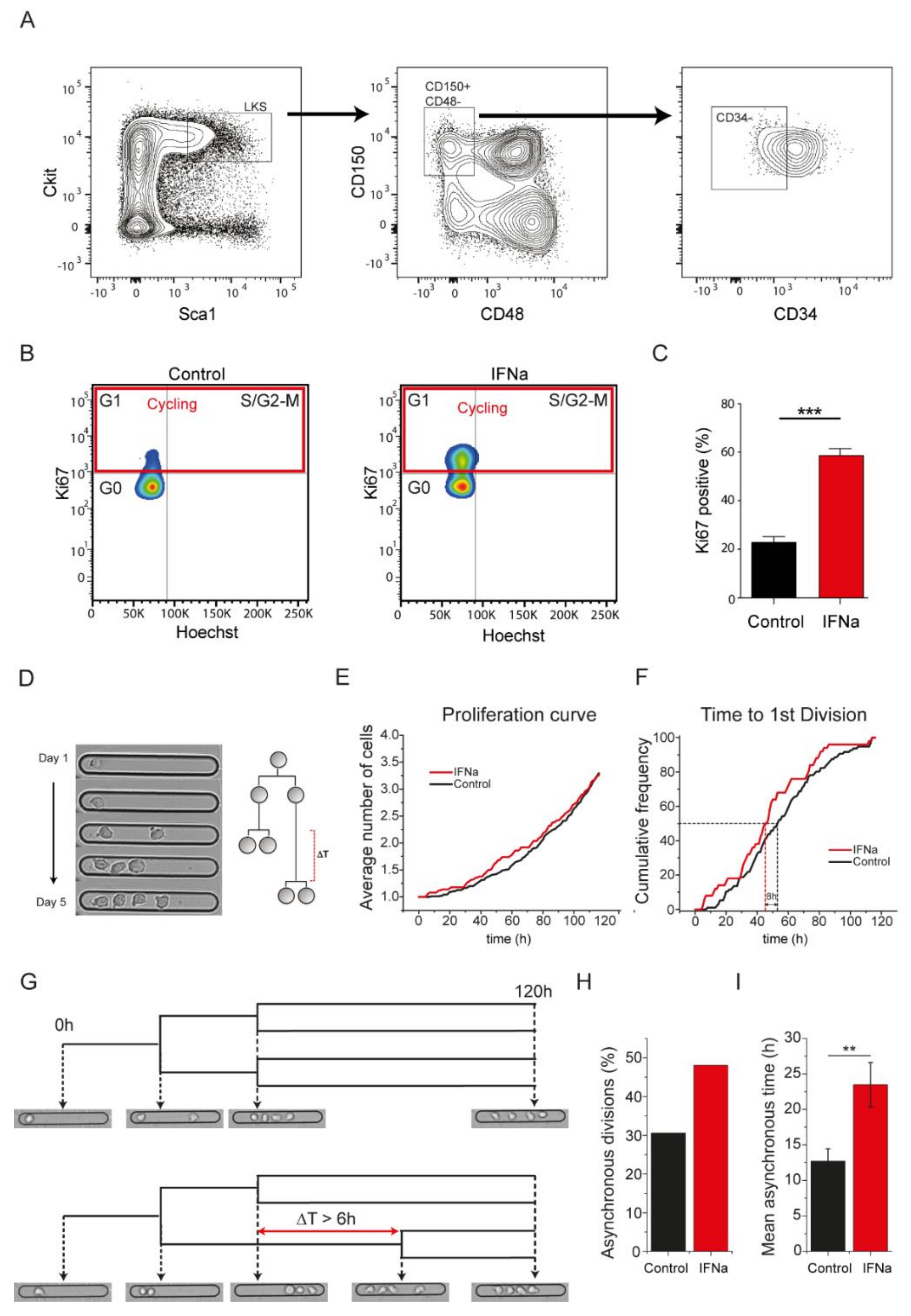
2.1. In Vivo Activation of Hematopoietic Stem Cells (HSCs) by IFN Alpha Increases Asynchronous Cell Divisions
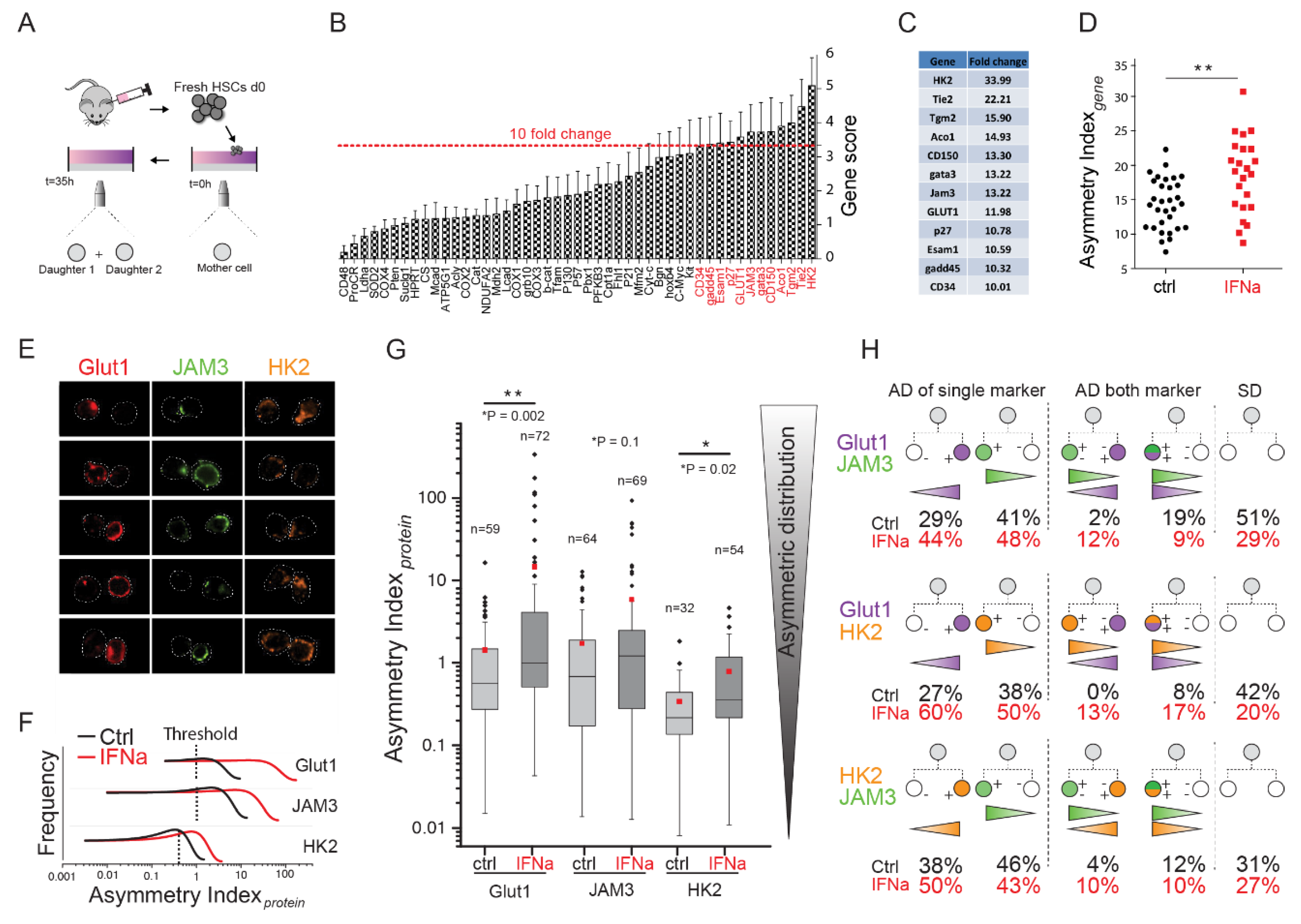
2.2. Identification of Asymmetrically Expressed Genes in Paired Daughter Cells (PDCs)
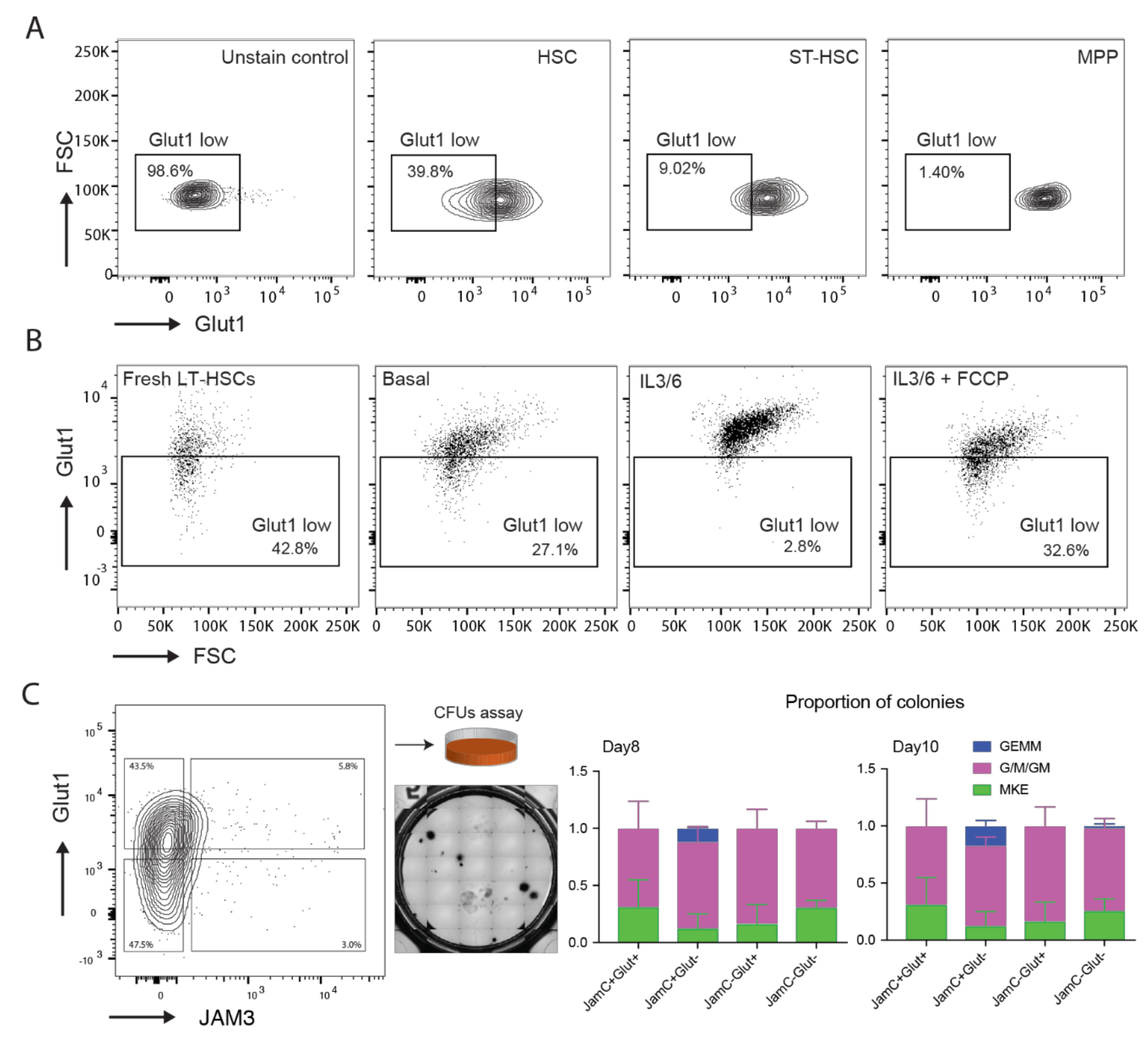
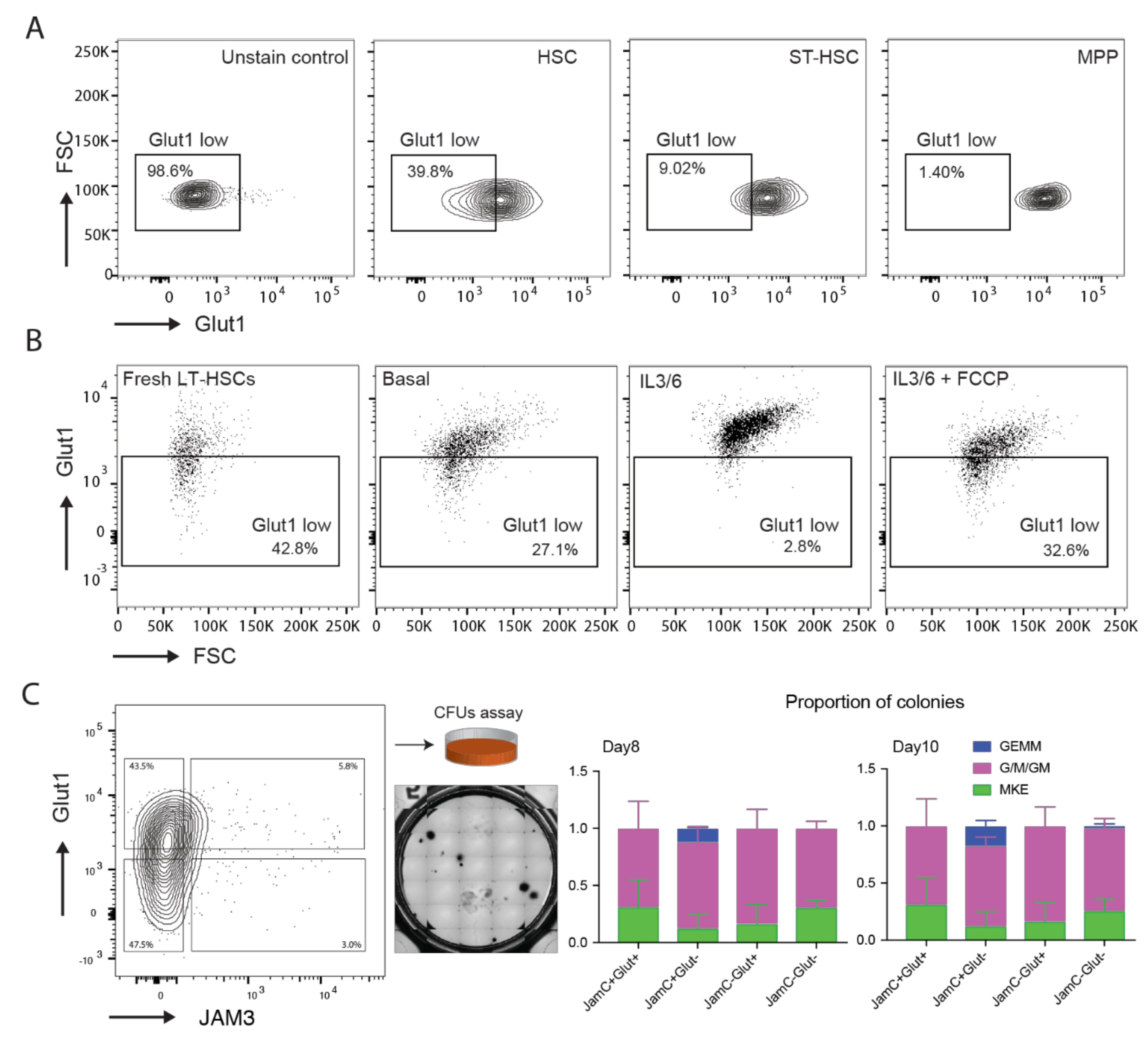
2.3. Analysis of the Protein Expression of Glut1, JAM3, and HK2
2.4. Characterization of Glut1 and JAM3 as Asymmetric Cell Division (ACD) Markers
3. Discussion
4. Materials and Methods
4.1. In Vivo Activation of HSCs
4.2. Antibodies
4.3. Flow Cytometry and Cell Sorting
4.4. Cell Cycle Analysis
4.5. Micro-Groove Array Platform Fabrication
4.6. PEG Microwell Array Fabrication
4.7. Single-Cell Proliferation Analysis by Time-Lapse Microscopy
4.8. HSC Cultures
4.9. Single-Cell Proliferation Analysis
4.10. Micromanipulation of Paired Daughter Cells for Single-Cell Analysis
4.11. Selection of Candidate Genes
4.12. Single-Cell qRT-PCR
4.13. Immunostaining and Images Analysis in the Multigroove Platform
4.14. Glut1 Staining for FACS Analysis
4.15. CFU Assay
4.16. Statistics
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Morrison, S.J.; Kimble, J. Asymmetric and symmetric stem-cell divisions in development and cancer. Nature 2006, 441, 1068–1074. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.; Laurenti, E.; Oser, G.; van der Wath, R.C.; Blanco-Bose, W.; Jaworski, M.; Offner, S.; Dunant, C.F.; Eshkind, L.; Bockamp, E.; et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell 2008, 135, 1118–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, L.; Lin, K.K.; Boles, N.C.; Yang, L.; King, K.Y.; Jeong, M.; Mayle, A.; Goodell, M.A. Less is more: Unveiling the functional core of hematopoietic stem cells through knockout mice. Cell Stem Cell 2012, 11, 302–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutolf, M.P.; Blau, H.M. Artificial stem cell niches. Adv. Mater. 2009, 21, 3255–3268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.D.; Wagers, A.J. Dynamic niches in the origination and differentiation of haematopoietic stem cells. Nat. Rev. Mol. Cell Biol. 2011, 12, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Arai, F.; Hirao, A.; Ohmura, M.; Sato, H.; Matsuoka, S.; Takubo, K.; Ito, K.; Koh, G.Y.; Suda, T. Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell 2004, 118, 149–161. [Google Scholar] [CrossRef] [Green Version]
- Calvi, L.M.; Adams, G.B.; Weibrecht, K.W.; Weber, J.M.; Olson, D.P.; Knight, M.C.; Martin, R.P.; Schipani, E.; Divieti, P.; Bringhurst, F.R.; et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 2003, 425, 841–846. [Google Scholar] [CrossRef]
- Fleming, H.E.; Janzen, V.; Lo Celso, C.; Guo, J.; Leahy, K.M.; Kronenberg, H.M.; Scadden, D.T. Wnt signaling in the niche enforces hematopoietic stem cell quiescence and is necessary to preserve self-renewal in vivo. Cell Stem Cell 2008, 2, 274–283. [Google Scholar] [CrossRef] [Green Version]
- Hosokawa, K.; Arai, F.; Yoshihara, H.; Iwasaki, H.; Hembree, M.; Yin, T.; Nakamura, Y.; Gomei, Y.; Takubo, K.; Shiama, H.; et al. Cadherin-based adhesion is a potential target for niche manipulation to protect hematopoietic stem cells in adult bone marrow. Cell Stem Cell 2010, 6, 194–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosokawa, K.; Arai, F.; Yoshihara, H.; Iwasaki, H.; Nakamura, Y.; Gomei, Y.; Suda, T. Knockdown of N-cadherin suppresses the long-term engraftment of hematopoietic stem cells. Blood 2010, 116, 554–563. [Google Scholar] [CrossRef] [Green Version]
- Qian, H.; Buza-Vidas, N.; Hyland, C.D.; Jensen, C.T.; Antonchuk, J.; Mansson, R.; Thoren, L.A.; Ekblom, M.; Alexander, W.S.; Jacobsen, S.E. Critical role of thrombopoietin in maintaining adult quiescent hematopoietic stem cells. Cell Stem Cell 2007, 1, 671–684. [Google Scholar] [CrossRef] [Green Version]
- Kiel, M.J.; Yilmaz, O.H.; Iwashita, T.; Yilmaz, O.H.; Terhorst, C.; Morrison, S.J. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 2005, 121, 1109–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendez-Ferrer, S.; Michurina, T.V.; Ferraro, F.; Mazloom, A.R.; Macarthur, B.D.; Lira, S.A.; Scadden, D.T.; Ma’ayan, A.; Enikolopov, G.N.; Frenette, P.S. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 2010, 466, 829–834. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Wada, M.; Minamikawa, H.; Kameta, N.; Masuda, M.; Shimizu, T. Cisplatin-encapsulated organic nanotubes by endo-complexation in the hollow cylinder. Chem. Commun. 2012, 48, 8625–8627. [Google Scholar] [CrossRef]
- Omatsu, Y.; Sugiyama, T.; Kohara, H.; Kondoh, G.; Fujii, N.; Kohno, K.; Nagasawa, T. The essential functions of adipo-osteogenic progenitors as the hematopoietic stem and progenitor cell niche. Immunity 2010, 33, 387–399. [Google Scholar] [CrossRef] [Green Version]
- Naveiras, O.; Nardi, V.; Wenzel, P.L.; Hauschka, P.V.; Fahey, F.; Daley, G.Q. Bone-marrow adipocytes as negative regulators of the haematopoietic microenvironment. Nature 2009, 460, 259–263. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.O.; Yu, H.; Yue, R.; Zhao, Z.; Rios, J.J.; Naveiras, O.; Morrison, S.J. Bone marrow adipocytes promote the regeneration of stem cells and haematopoiesis by secreting SCF. Nat. Cell Biol. 2017, 19, 891–903. [Google Scholar] [CrossRef] [PubMed]
- Spagnuolo, G.; Codispoti, B.; Marrelli, M.; Rengo, C.; Rengo, S.; Tatullo, M. Commitment of Oral-Derived Stem Cells in Dental and Maxillofacial Applications. Dent. J. 2018, 6, 72. [Google Scholar] [CrossRef] [Green Version]
- Giebel, B.; Zhang, T.; Beckmann, J.; Spanholtz, J.; Wernet, P.; Ho, A.D.; Punzel, M. Primitive human hematopoietic cells give rise to differentially specified daughter cells upon their initial cell division. Blood 2006, 107, 2146–2152. [Google Scholar] [CrossRef] [Green Version]
- Takano, H.; Ema, H.; Sudo, K.; Nakauchi, H. Asymmetric division and lineage commitment at the level of hematopoietic stem cells: Inference from differentiation in daughter cell and granddaughter cell pairs. J. Exp. Med. 2004, 199, 295–302. [Google Scholar] [CrossRef] [Green Version]
- Ema, H.; Takano, H.; Sudo, K.; Nakauchi, H. In vitro self-renewal division of hematopoietic stem cells. J. Exp. Med. 2000, 192, 1281–1288. [Google Scholar] [CrossRef]
- Yamamoto, R.; Morita, Y.; Ooehara, J.; Hamanaka, S.; Onodera, M.; Rudolph, K.L.; Ema, H.; Nakauchi, H. Clonal analysis unveils self-renewing lineage-restricted progenitors generated directly from hematopoietic stem cells. Cell 2013, 154, 1112–1126. [Google Scholar] [CrossRef] [Green Version]
- Ting, S.B.; Deneault, E.; Hope, K.; Cellot, S.; Chagraoui, J.; Mayotte, N.; Dorn, J.F.; Laverdure, J.P.; Harvey, M.; Hawkins, E.D.; et al. Asymmetric segregation and self-renewal of hematopoietic stem and progenitor cells with endocytic Ap2a2. Blood 2012, 119, 2510–2522. [Google Scholar] [CrossRef]
- Shin, J.W.; Buxboim, A.; Spinler, K.R.; Swift, J.; Christian, D.A.; Hunter, C.A.; Leon, C.; Gachet, C.; Dingal, P.C.; Ivanovska, I.L.; et al. Contractile forces sustain and polarize hematopoiesis from stem and progenitor cells. Cell Stem Cell 2014, 14, 81–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorgens, A.; Ludwig, A.K.; Mollmann, M.; Krawczyk, A.; Durig, J.; Hanenberg, H.; Horn, P.A.; Giebel, B. Multipotent hematopoietic progenitors divide asymmetrically to create progenitors of the lymphomyeloid and erythromyeloid lineages. Stem Cell Rep. 2014, 3, 1058–1072. [Google Scholar] [CrossRef] [Green Version]
- Loeffler, D.; Wehling, A.; Schneiter, F.; Zhang, Y.; Muller-Botticher, N.; Hoppe, P.S.; Hilsenbeck, O.; Kokkaliaris, K.D.; Endele, M.; Schroeder, T. Asymmetric lysosome inheritance predicts activation of haematopoietic stem cells. Nature 2019, 573, 426–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, R.; Arif, T.; Kalmykova, S.; Kasianov, A.; Lin, M.; Menon, V.; Qiu, J.; Bernitz, J.M.; Moore, K.; Lin, F.; et al. Restraining Lysosomal Activity Preserves Hematopoietic Stem Cell Quiescence and Potency. Cell Stem Cell 2020, 26, 359–376.e7. [Google Scholar] [CrossRef]
- Hinge, A.; He, J.; Bartram, J.; Javier, J.; Xu, J.; Fjellman, E.; Sesaki, H.; Li, T.; Yu, J.; Wunderlich, M.; et al. Asymmetrically Segregated Mitochondria Provide Cellular Memory of Hematopoietic Stem Cell Replicative History and Drive HSC Attrition. Cell Stem Cell 2020, 26, 420–430.e6. [Google Scholar] [CrossRef]
- Vannini, N.; Campos, V.; Girotra, M.; Trachsel, V.; Rojas-Sutterlin, S.; Tratwal, J.; Ragusa, S.; Stefanidis, E.; Ryu, D.; Rainer, P.Y.; et al. The NAD-Booster Nicotinamide Riboside Potently Stimulates Hematopoiesis through Increased Mitochondrial Clearance. Cell Stem Cell 2019, 24, 405–418.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Essers, M.A.; Offner, S.; Blanco-Bose, W.E.; Waibler, Z.; Kalinke, U.; Duchosal, M.A.; Trumpp, A. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature 2009, 458, 904–908. [Google Scholar] [CrossRef]
- Vannini, N.; Girotra, M.; Naveiras, O.; Nikitin, G.; Campos, V.; Giger, S.; Roch, A.; Auwerx, J.; Lutolf, M.P. Specification of haematopoietic stem cell fate via modulation of mitochondrial activity. Nat. Commun. 2016, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Osawa, M.; Hanada, K.; Hamada, H.; Nakauchi, H. Long-term lymphohematopoietic reconstitution by a single CD34-low/negative hematopoietic stem cell. Science 1996, 273, 242–245. [Google Scholar] [CrossRef] [Green Version]
- Lutolf, M.P.; Doyonnas, R.; Havenstrite, K.; Koleckar, K.; Blau, H.M. Perturbation of single hematopoietic stem cell fates in artificial niches. Integr. Biol. 2009, 1, 59–69. [Google Scholar] [CrossRef]
- Roch, A.; Giger, S.; Girotra, M.; Campos, V.; Vannini, N.; Naveiras, O.; Gobaa, S.; Lutolf, M.P. Single-cell analyses identify bioengineered niches for enhanced maintenance of hematopoietic stem cells. Nat. Commun. 2017, 8, 221. [Google Scholar] [CrossRef] [Green Version]
- Vannini, N.; Roch, A.; Naveiras, O.; Griffa, A.; Kobel, S.; Lutolf, M.P. Identification of in vitro HSC fate regulators by differential lipid raft clustering. Cell Cycle 2012, 11, 1535–1543. [Google Scholar] [CrossRef]
- Nygren, J.M.; Bryder, D.; Jacobsen, S.E. Prolonged cell cycle transit is a defining and developmentally conserved hemopoietic stem cell property. J. Immunol. 2006, 177, 201–208. [Google Scholar] [CrossRef]
- Orford, K.W.; Scadden, D.T. Deconstructing stem cell self-renewal: Genetic insights into cell-cycle regulation. Nat. Rev. Genet. 2008, 9, 115–128. [Google Scholar] [CrossRef]
- Arcangeli, M.L.; Bardin, F.; Frontera, V.; Bidaut, G.; Obrados, E.; Adams, R.H.; Chabannon, C.; Aurrand-Lions, M. Function of Jam-B/Jam-C interaction in homing and mobilization of human and mouse hematopoietic stem and progenitor cells. Stem Cells 2014, 32, 1043–1054. [Google Scholar] [CrossRef]
- Arcangeli, M.L.; Frontera, V.; Bardin, F.; Obrados, E.; Adams, S.; Chabannon, C.; Schiff, C.; Mancini, S.J.; Adams, R.H.; Aurrand-Lions, M. JAM-B regulates maintenance of hematopoietic stem cells in the bone marrow. Blood 2011, 118, 4609–4619. [Google Scholar] [CrossRef] [Green Version]
- Sarrazy, V.; Viaud, M.; Westerterp, M.; Ivanov, S.; Giorgetti-Peraldi, S.; Guinamard, R.; Gautier, E.L.; Thorp, E.B.; De Vivo, D.C.; Yvan-Charvet, L. Disruption of Glut1 in Hematopoietic Stem Cells Prevents Myelopoiesis and Enhanced Glucose Flux in Atheromatous Plaques of ApoE(-/-) Mice. Circ. Res. 2016, 118, 1062–1077. [Google Scholar] [CrossRef] [Green Version]
- Ito, K.; Suda, T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat. Rev. Mol. Cell Biol. 2014, 15, 243–256. [Google Scholar] [CrossRef] [Green Version]
- Simsek, T.; Kocabas, F.; Zheng, J.; Deberardinis, R.J.; Mahmoud, A.I.; Olson, E.N.; Schneider, J.W.; Zhang, C.C.; Sadek, H.A. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell 2010, 7, 380–390. [Google Scholar] [CrossRef] [Green Version]
- Takubo, K.; Nagamatsu, G.; Kobayashi, C.I.; Nakamura-Ishizu, A.; Kobayashi, H.; Ikeda, E.; Goda, N.; Rahimi, Y.; Johnson, R.S.; Soga, T.; et al. Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell 2013, 12, 49–61. [Google Scholar] [CrossRef] [Green Version]
- Kiger, A.A.; Jones, D.L.; Schulz, C.; Rogers, M.B.; Fuller, M.T. Stem cell self-renewal specified by JAK-STAT activation in response to a support cell cue. Science 2001, 294, 2542–2545. [Google Scholar] [CrossRef]
- Song, X.; Wong, M.D.; Kawase, E.; Xi, R.; Ding, B.C.; McCarthy, J.J.; Xie, T. Bmp signals from niche cells directly repress transcription of a differentiation-promoting gene, bag of marbles, in germline stem cells in the Drosophila ovary. Development 2004, 131, 1353–1364. [Google Scholar] [CrossRef] [Green Version]
- Tulina, N.; Matunis, E. Control of stem cell self-renewal in Drosophila spermatogenesis by JAK-STAT signaling. Science 2001, 294, 2546–2549. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, Y.M.; Fuller, M.T.; Jones, D.L. Signaling in stem cell niches: Lessons from the Drosophila germline. J. Cell Sci. 2005, 118, 665–672. [Google Scholar] [CrossRef] [Green Version]
- Ito, K.; Carracedo, A.; Weiss, D.; Arai, F.; Ala, U.; Avigan, D.E.; Schafer, Z.T.; Evans, R.M.; Suda, T.; Lee, C.H.; et al. A PML-PPAR-delta pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat. Med. 2012, 18, 1350–1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, P.; Yoshihara, H.; Hosokawa, K.; Tai, I.; Shinmyozu, K.; Tsukahara, F.; Maru, Y.; Nakayama, K.; Nakayama, K.I.; Suda, T. p57(Kip2) and p27(Kip1) cooperate to maintain hematopoietic stem cell quiescence through interactions with Hsc70. Cell Stem Cell 2011, 9, 247–261. [Google Scholar] [CrossRef] [Green Version]
- Mathupala, S.P.; Rempel, A.; Pedersen, P.L. Glucose catabolism in cancer cells: Identification and characterization of a marked activation response of the type II hexokinase gene to hypoxic conditions. J. Biol. Chem. 2001, 276, 43407–43412. [Google Scholar] [CrossRef] [Green Version]
- Signer, R.A.; Magee, J.A.; Salic, A.; Morrison, S.J. Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature 2014, 509, 49–54. [Google Scholar] [CrossRef]
- Forsberg, E.C.; Prohaska, S.S.; Katzman, S.; Heffner, G.C.; Stuart, J.M.; Weissman, I.L. Differential expression of novel potential regulators in hematopoietic stem cells. PLoS Genet. 2005, 1, e28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forsberg, E.C.; Passegue, E.; Prohaska, S.S.; Wagers, A.J.; Koeva, M.; Stuart, J.M.; Weissman, I.L. Molecular signatures of quiescent, mobilized and leukemia-initiating hematopoietic stem cells. PLoS ONE 2010, 5, e8785. [Google Scholar] [CrossRef] [PubMed]
- Umemoto, T.; Yamato, M.; Nishida, K.; Yang, J.; Tano, Y.; Okano, T. p57Kip2 is expressed in quiescent mouse bone marrow side population cells. Biochem. Biophys. Res. Commun. 2005, 337, 14–21. [Google Scholar] [CrossRef]
- Cheng, T.; Rodrigues, N.; Shen, H.; Yang, Y.; Dombkowski, D.; Sykes, M.; Scadden, D.T. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science 2000, 287, 1804–1808. [Google Scholar] [CrossRef]
- Passegue, E.; Wagers, A.J.; Giuriato, S.; Anderson, W.C.; Weissman, I.L. Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates. J. Exp. Med. 2005, 202, 1599–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, J.M.; He, X.C.; Sugimura, R.; Grindley, J.C.; Haug, J.S.; Ding, S.; Li, L. Cooperation between both Wnt/β-catenin and PTEN/PI3K/Akt signaling promotes primitive hematopoietic stem cell self-renewal and expansion. Genes Dev. 2011, 25, 1928–1942. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Grindley, J.C.; Yin, T.; Jayasinghe, S.; He, X.C.; Ross, J.T.; Haug, J.S.; Rupp, D.; Porter-Westpfahl, K.S.; Wiedemann, L.M.; et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature 2006, 441, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Ku, C.J.; Hosoya, T.; Maillard, I.; Engel, J.D. GATA-3 regulates hematopoietic stem cell maintenance and cell-cycle entry. Blood 2012, 119, 2242–2251. [Google Scholar] [CrossRef] [Green Version]
- Krosl, J.; Austin, P.; Beslu, N.; Kroon, E.; Humphries, R.K.; Sauvageau, G. In vitro expansion of hematopoietic stem cells by recombinant TAT-HOXB4 protein. Nat. Med. 2003, 9, 1428–1432. [Google Scholar] [CrossRef]
- Satoh, Y.; Matsumura, I.; Tanaka, H.; Ezoe, S.; Sugahara, H.; Mizuki, M.; Shibayama, H.; Ishiko, E.; Ishiko, J.; Nakajima, K.; et al. Roles for c-Myc in self-renewal of hematopoietic stem cells. J. Biol. Chem. 2004, 279, 24986–24993. [Google Scholar] [CrossRef] [Green Version]
- Wilson, A.; Murphy, M.J.; Oskarsson, T.; Kaloulis, K.; Bettess, M.D.; Oser, G.M.; Pasche, A.C.; Knabenhans, C.; Macdonald, H.R.; Trumpp, A. c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. Genes Dev. 2004, 18, 2747–2763. [Google Scholar] [CrossRef] [Green Version]
- Takubo, K.; Goda, N.; Yamada, W.; Iriuchishima, H.; Ikeda, E.; Kubota, Y.; Shima, H.; Johnson, R.S.; Hirao, A.; Suematsu, M.; et al. Regulation of the HIF-1alpha level is essential for hematopoietic stem cells. Cell Stem Cell 2010, 7, 391–402. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Classification | Gene Name | Classification | Gene Name |
|---|---|---|---|
| Housekeeping gene | HPRT | Glycolysis | HK2 |
| ECM proteins | Tgm2 | Glycolysis | PFKFB3 |
| ECM proteins | Bgn | Glycolysis | Glut1 |
| Membrane proteins | Esam1 | Glycolysis | Ldha |
| Membrane proteins | Tie2 | TCA cycle | CS |
| Membrane proteins | JAM3 | TCA cycle | Acly |
| HSC markers | CD150 | TCA cycle | Aco1 |
| HSC markers | CD48 | TCA cycle | Suclg1 |
| HSC markers | CD34 | TCA cycle | Mdh2 |
| HSC markers | C-kit | Oxidative phosphorylation | Cyt-C |
| Intracellular adaptors | Grb10 | Oxidative phosphorylation | NDUFA2 |
| Intracellular adaptors | ProCR | Oxidative phosphorylation | COX2 (Sdhd) |
| Intracellular adaptors | Fhl1 | Oxidative phosphorylation | ATP5g1 |
| Intracellular adaptors | b-catenin | Oxidative phosphorylation | COX1 |
| Cell cycle regulators | P57 (cdkn1c) | Oxidative phosphorylation | COX3 |
| Cell cycle regulators | P27 (cdkn1b) | Oxidative phosphorylation | COX4 |
| Cell cycle regulators | P21 (cdkn1a) | Mitochondrial biogenesis | Mfn2 |
| Cell cycle regulators | P130 (rab3gap) | Mitochondrial biogenesis | Tfam |
| Cell cycle regulators | Pten | Antioxidants | SOD2 |
| Transcription factors | Pbx1 | Antioxidants | Catalase |
| Transcription factors | Gata3 | Fatty acid oxidation | MCad |
| Transcription factors | c-myc | Fatty acid oxidation | LCad |
| Transcription factors | Hoxb4 | Fatty acid oxidation | CPT1a |
| DNA repair | Gadd45 | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Girotra, M.; Trachsel, V.; Roch, A.; Lutolf, M.P. In Vivo Pre-Instructed HSCs Robustly Execute Asymmetric Cell Divisions In Vitro. Int. J. Mol. Sci. 2020, 21, 8225. https://doi.org/10.3390/ijms21218225
Girotra M, Trachsel V, Roch A, Lutolf MP. In Vivo Pre-Instructed HSCs Robustly Execute Asymmetric Cell Divisions In Vitro. International Journal of Molecular Sciences. 2020; 21(21):8225. https://doi.org/10.3390/ijms21218225
Chicago/Turabian StyleGirotra, Mukul, Vincent Trachsel, Aline Roch, and Matthias P. Lutolf. 2020. "In Vivo Pre-Instructed HSCs Robustly Execute Asymmetric Cell Divisions In Vitro" International Journal of Molecular Sciences 21, no. 21: 8225. https://doi.org/10.3390/ijms21218225
APA StyleGirotra, M., Trachsel, V., Roch, A., & Lutolf, M. P. (2020). In Vivo Pre-Instructed HSCs Robustly Execute Asymmetric Cell Divisions In Vitro. International Journal of Molecular Sciences, 21(21), 8225. https://doi.org/10.3390/ijms21218225