PPARs and Microbiota in Skeletal Muscle Health and Wasting




{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Peroxisome Proliferator-Activated Receptors (PPARs)
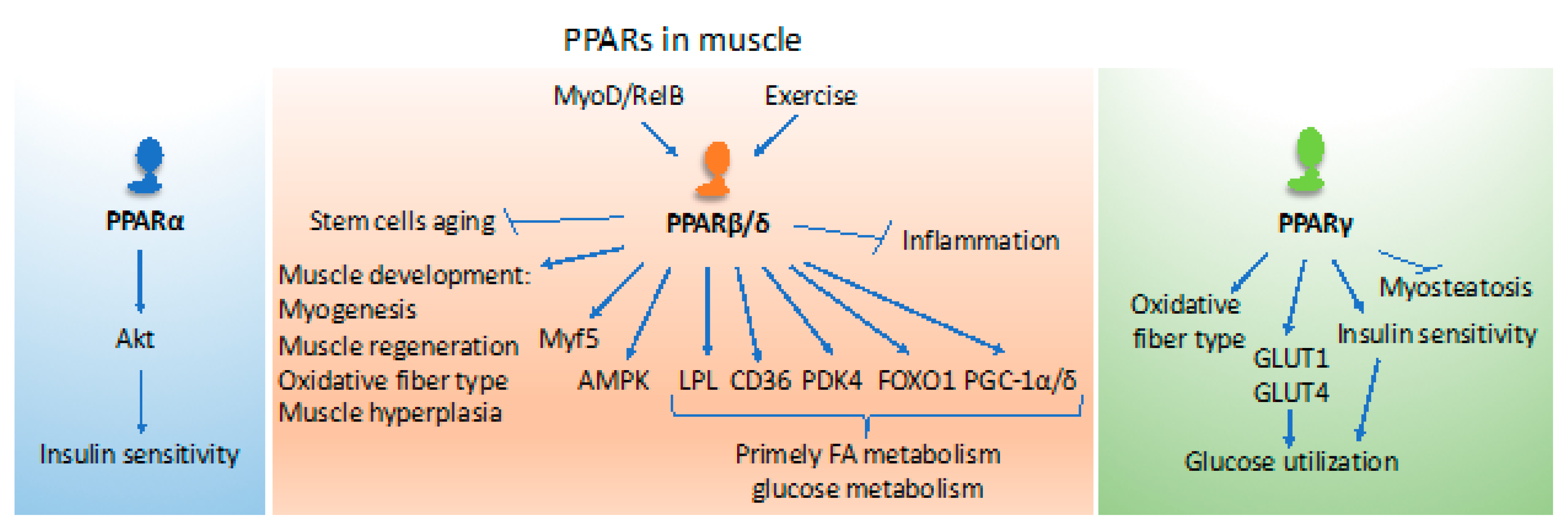
3. Roles of PPARs in Muscle
4. PPARs in Muscle Wasting
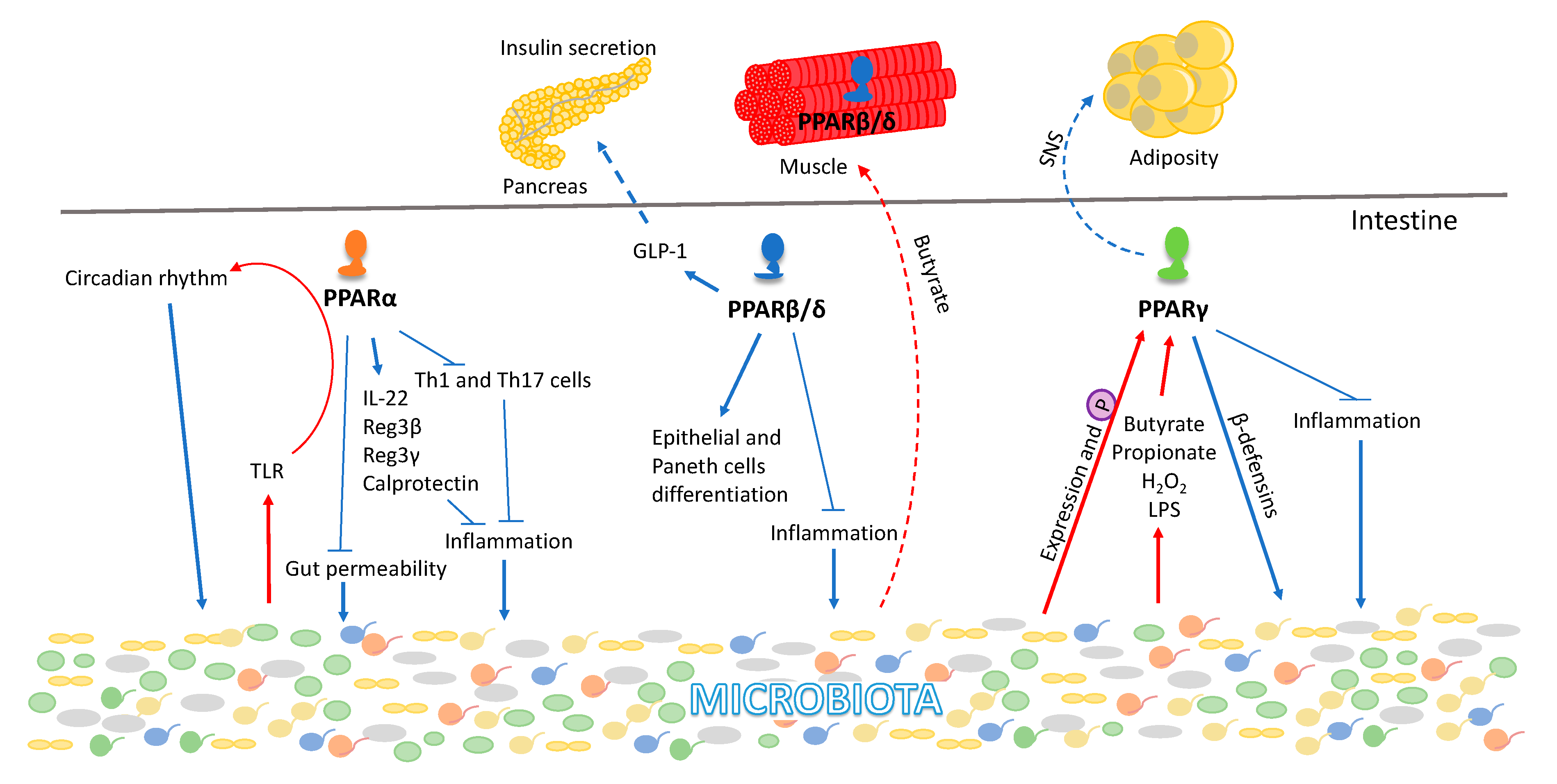
5. PPAR Interactions with the Gut Microbiota
6. PPARs and the Gut-Muscle Axis
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AICAR | 5-Aminiimidazole-4-carboxamide ribonucleotide |
| Akt | Protein kinase B |
| Ampk | AMP-activated protein kinase |
| Angptl4 | Angiopoietin-like 4 |
| ApoC3 | Apolipoprotein C3 |
| ATP | Adenosine Triphosphate |
| CD36 | Cluster of differentiation 36 |
| C/EBP | CCAAT/enhancer binding protein |
| Cox10 | Cytochrome oxidase 10 |
| CPT1/2 | Carnitine palmitoyltransferase½ |
| Cry1/2 | Cryptochrome ½ |
| DMD | Duchenne muscular dystrophy |
| FA | Fatty acid |
| FABP3 | Fatty acid binding protein 3 |
| FoxO1 | Forkhead box O1 |
| GLP-1 | Glucagon-like peptide-1 |
| GLUT1/4 | Glucose transporter ¼ |
| HGF | Hepatocyte growth factor |
| H2O2 | Hydrogen peroxide |
| IGF1 | Insulin-like growth factor |
| IBD | Inflammatory bowel disease |
| IBS | Irritable bowel syndrome |
| IL-22 | Interleukin-22 |
| iNOS | Inducible nitric oxide synthase |
| KO | Knockout |
| LPL | Lipoprotein lipase |
| LPS | Lipopolysaccharide |
| mdx | Muscular dystrophy X-linked |
| Mef2 | Myocyte enhancer factor 2 |
| MRF4 | Myogenic regulatory factor 4 |
| MyoD | Myogenic differentiation |
| Myf5 | Myogenic factor 5 |
| NAFLD | Nonalcoholic fatty liver disease |
| NFAT | Nuclear factor of activated T-cells |
| NR1C1-3 | Nuclear receptor 1C-1-3 |
| Pax3/7 | Paired box 3/7 |
| PDK4 | Pyruvate dehydrogenase kinase 4 |
| PGC1 | PPAR gamma coactivator 1 |
| PI3K | Phosphoinositide 3-kinase |
| PPAR | Peroxisome proliferator-activated receptor |
| PPRE | Peroxisome proliferator response element |
| Reg3 | Regenerating islet-derived protein 3 |
| SDH | Succinate dehydrogenase |
| SF | Scatter factor |
| SNS | Sympathetic nervous system |
| T2D | Type 2 diabetes |
| Th1/17 | T-helper cells 1/17 |
| TLR | Toll-like receptor |
| VLCAD | Very long-chain acyl-CoA dehydrogenase |
| Wnt | Wingless-type MMTV integration site family |
References
- Janssen, I.; Heymsfield, S.B.; Baumgartner, R.N.; Ross, R. Estimation of skeletal muscle mass by bioelectrical impedance analysis. J. Appl. Physiol. 2000, 89, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Dressel, U.; Allen, T.L.; Pippal, J.B.; Rohde, P.R.; Lau, P.; Muscat, G.E.O. The Peroxisome Proliferator-Activated Receptor β/δ Agonist, GW501516, regulates the expression of genes involved in lipid catabolism and energy uncoupling in skeletal muscle cells. Mol. Endocrinol. 2003, 17, 2477–2493. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Jacot, E.; Jequier, E.; Maeder, E.; Wahren, J.; Felber, J.P. The effect of insulin on the disposal of intravenous glucose: Results from indirect calorimetry and hepatic and femoral venous catheterization. Diabetes 1981, 30, 1000–1007. [Google Scholar] [CrossRef] [PubMed]
- Shulman, G.I.; Rothman, D.L.; Jue, T.; Stein, P.; DeFronzo, R.A.; Shulman, R.G. Quantitation of muscle glycogen synthesis in normal subjects and subjects with non-insulin-dependent diabetes by 13C nuclear magnetic resonance spectroscopy. N. Engl. J. Med. 1990, 322, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Luquet, S.; Gaudel, C.; Holst, D.; Lopez-Soriano, J.; Jehl-Pietri, C.; Fredenrich, A.; Grimaldi, P.A. Roles of PPAR delta in lipid absorption and metabolism: A new target for the treatment of type 2 diabetes. Biochim. Biophys. Acta 2005, 1740, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Luquet, S.; Lopez-Soriano, J.; Holst, D.; Gaudel, C.; Jehl-Pietri, C.; Fredenrich, A.; Grimaldi, P.A. Roles of peroxisome proliferator-activated receptor delta (PPARdelta) in the control of fatty acid catabolism. A new target for the treatment of metabolic syndrome. Biochime 2004, 86, 833–837. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S.; Reggiani, C. Fiber types in mammalian skeletal muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef]
- Manickam, R.; Wahli, W. Roles of peroxisome proliferator-activated receptorβ/δ in skeletal muscle physiology. Biochime 2017, 136, 42–48. [Google Scholar] [CrossRef]
- Buckingham, M.; Montarras, D. Skeletal muscle stem cells. Curr. Opin. Genet. Dev. 2008, 18, 330–336. [Google Scholar] [CrossRef]
- Denetclaw, W.F.; Christ, B.; Ordahl, C.P. Location and growth of epaxial myotome precursor cells. Development 1997, 124, 1601–1610. [Google Scholar]
- Gros, J.; Manceau, M.; Thome, V.; Marcelle, C. A common somitic origin for embryonic muscle progenitors and satellite cells. Nat. Cell Biol. 2005, 435, 954–958. [Google Scholar] [CrossRef] [PubMed]
- Chal, J.; Pourquie, O. Making muscle: Skeletal myogenesis in vivo and in vitro. Development 2017, 144, 2104–2122. [Google Scholar] [CrossRef] [PubMed]
- Christ, B.; Brand-Saberi, B. Limb muscle development. Int. J. Dev. Biol. 2002, 46, 905–914. [Google Scholar] [PubMed]
- Buckingham, M.; Bajard, L.; Chang, T.; Daubas, P.; Hadchouel, J.; Meilhac, S.; Montarras, D.; Rocancourt, D.; Relaix, F. The formation of skeletal muscle: From somite to limb. J. Anat. 2003, 202, 59–68. [Google Scholar] [CrossRef]
- Bober, E.; Franz, T.; Arnold, H.H.; Gruss, P.; Tremblay, P. Pax-3 is required for the development of limb muscles: A possible role for the migration of dermomyotomal muscle progenitor cells. Development 1994, 120, 603–612. [Google Scholar]
- Kahane, N.; Cinnamon, Y.; Kalcheim, C. The cellular mechanism by which the dermomyotome contributes to the second wave of myotome development. Development 1998, 125, 4259–4271. [Google Scholar]
- Brand-Saberi, B.; Christ, B. Genetic and epigenetic control of muscle development in vertebrates. Cell Tissue Res. 1999, 296, 199–212. [Google Scholar] [CrossRef]
- Zhao, P.; Hoffman, E.P. Embryonic myogenesis pathways in muscle regeneration. Dev. Dyn. 2004, 229, 380–392. [Google Scholar] [CrossRef]
- Buckingham, M. Early stages of myogenesis as seen through the action of the myf-5 gene. C. R. Seances. Sco. Biol. Fil. 1997, 191, 43–54. [Google Scholar]
- Tajbakhsh, S.; Rocancourt, D.; Buckingham, M. Muscle progenitor cells failing to respond to positional cues adopt non-myogenic fates in myf-5 null mice. Nature 1996, 384, 266–270. [Google Scholar] [CrossRef]
- Olguin, H.C.; Olwin, B.B. Pax-7 up-regulation inhibits myogenesis and cell cycle progression in satellite cells: A potential mechanism for self-renewal. Dev. Biol. 2004, 275, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Mauro, A. Satellite cell of skeletal muscle fibers. J. Biophys. Biochem. Cytol. 1961, 9, 493–495. [Google Scholar] [CrossRef] [PubMed]
- Biressi, S.; Rando, T.A. Heterogeneity in the muscle satellite cell population. Semin. Cell Dev. Biol. 2010, 21, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Collins, C.A.; Olsen, I.; Zammit, P.S.; Heslop, L.; Petrie, A.; Patridge, T.A.; Morgan, J.E. Stem cell function, self-renewal, and behavioral heterogeneity of cells from the adult muscle satellite cell niche. Cell 2005, 122, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Bachman, J.F.; Klose, A.; Liu, W.; Paris, N.D.; Blanc, R.S.; Schmalz, M.; Knapp, E.; Chakkalakal, J. Prepubertal skeletal muscle growth requires Pax7-expressing satellite cell-derived myonuclear contribution. Development 2018, 145, dev167197. [Google Scholar] [CrossRef] [PubMed]
- Yamakawa, H.; Kusumoto, D.; Hashimoto, H.; Yuasa, S. Stem cell aging in skeletal muscle regeneration and disease. Int. J. Mol. Sci. 2020, 21, 1830. [Google Scholar] [CrossRef]
- Rai, M.; Nongthomba, U.; Grounds, M.D. Skeletal muscle degeneration and regeneration in mice and flies. Curr. Top.Dev. Biol. 2014, 108, 247–281. [Google Scholar] [CrossRef]
- Pallafacchina, G.; Blaauw, B.; Schiaffino, S. Role of satellite cells in muscle growth and maintenance of muscle mass. Nutr. Metab. Cardiovasc. Dis. 2013, 23, S12–S18. [Google Scholar] [CrossRef]
- Kang, J.-S.; Krauss, R.S. Muscle stem cells in developmental and regenerative myogenesis. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 243–248. [Google Scholar] [CrossRef]
- Tajbakhsh, S. Skeletal muscle stem cells in developmental versus regenerative myogenesis. J. Intern. Med. 2009, 266, 372–389. [Google Scholar] [CrossRef]
- Grefte, S.; Kuijpers-Jagtman, A.M.; Torensma, R.; Von den Hoff, J.W. Skeletal muscle development and regeneration. Stem Cells Dev. 2007, 16, 857–868. [Google Scholar] [CrossRef] [PubMed]
- McKinnell, I.W.; Parise, G.; Rudnicki, M.A. Muscle stem cells and regenerative myogenesis. Curr. Top. Dev. Biol. 2005, 71, 113–130. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, J.; Rando, T.A. Stem cells in postnatal myogenesis: Molecular mechanisms of satellite cell quiescence, activation and replenishment. Trends Cell Biol. 2005, 15, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Snow, M.H. The effects of aging on satellite cells in skeletal muscles of mice and rats. Cell Tissue Res. 1977, 185, 399–408. [Google Scholar] [CrossRef]
- Sajko, S.; Kubínová, L.; Cvetko, E.; Kreft, M.; Wernig, A.; Erzen, I. Frequency of M-cadherin-stained satellite cells declines in human muscles during aging. J. Histochem. Cytochem. 2004, 52, 179–185. [Google Scholar] [CrossRef]
- Gibson, M.C.; Schultz, E. Age-related differences in absolute numbers of skeletal muscle satellite cells. Muscle Nerve 1983, 6, 574–580. [Google Scholar] [CrossRef]
- Day, K.; Shefer, G.; Shearer, A.; Yablonka-Reuveni, Z. The depletion of skeletal muscle satellite cells with age is concomitant with reduced capacity of single progenitors to produce reserve progeny. Dev. Biol. 2010, 340, 330–343. [Google Scholar] [CrossRef]
- Asfour, H.A.; Allouh, M.Z.; Said, R.S. Myogenic regulatory factors: The orchestrators of myogenesis after 30 years of discovery. Exp. Biol. Med. 2018, 243, 118–128. [Google Scholar] [CrossRef]
- Punch, V.G.; Jones, A.E.; Rudnicki, M.A. Transcriptional networks that regulate muscle stem cell function. Wiley Interdiscip. Rev. Syst. Biol. Med. 2009, 1, 128–140. [Google Scholar] [CrossRef]
- Rudnicki, M.A.; Grand, F.L.; Mckinnell, I.; Kuang, S. The molecular regulation of muscle stem cell function. Cold Spring Harb. Symp. Quant. Biol. 2008, 73, 323–331. [Google Scholar] [CrossRef]
- White, T.P.; Esser, K.A. Satellite cell and growth factor involvement in skeletal muscle growth. Med. Sci. Sports Exerc. 1989, 21, S158–S163. [Google Scholar] [CrossRef] [PubMed]
- Relaix, F.; Rocancourt, D.; Mansouri, A.; Buckingham, M. A Pax3/Pax7-dependent population of skeletal muscle progenitor cells. Nature 2005, 435, 948–953. [Google Scholar] [CrossRef] [PubMed]
- Kuang, S.; Chargé, S.B.; Seale, P.; Huh, M.; Rudnicki, M.A. Distinct roles for Pax7 and Pax3 in adult regenerative myogenesis. J. Cell Biol. 2006, 172, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Seale, P.; Sabourin, L.A.; Girgis-Gabardo, A.; Mansouri, A.; Gruss, P.; Rudnicki, M.A. Pax7 is required for the specification of myogenic satellite cells. Cell 2000, 102, 777–786. [Google Scholar] [CrossRef]
- Von Maltzahn, J.; Jones, A.E.; Parks, R.J.; Rudnicki, M.A. Pax7 is critical for the normal function of satellite cells in adult skeletal muscle. Proc. Natl. Acad. Sci. USA 2013, 110, 16474–16479. [Google Scholar] [CrossRef]
- Shi, X.; Garry, D.J. Muscle stem cells in development, regeneration, and disease. Genes Dev. 2006, 20, 1692–1708. [Google Scholar] [CrossRef]
- Collins, K.H.; Herzog, W.; MacDonald, G.Z.; Reimer, R.A.; Rios, J.L.; Smith, I.C.; Zernicke, R.F.; Hart, D.A. Obesity, metabolic syndrome, and musculoskeletal disease: Common inflammatory pathways suggest a central role for loss of muscle integrity. Front. Physiol. 2018, 9, 112. [Google Scholar] [CrossRef]
- Anker, S.D.; Coats, A.J.S.; Morley, J.E.; Rosano, G.; Bernabei, R.; Von Haehling, S.; Kalantar-Zadeh, K. Muscle wasting disease: A proposal for a new disease classification. J. Cachexia Sarcopenia Muscle 2014, 5, 1–3. [Google Scholar] [CrossRef]
- Greco, E.A.; Pietschmann, P.; Migliaccio, S. Osteoporosis and sarcopenia increase frailty syndrome in the elderly. Front. Endocrinol. 2019, 10, 255. [Google Scholar] [CrossRef]
- Desvergne, B.; Michalik, L.; Wahli, W. Transcriptional regulation of metabolism. Physiol. Rev. 2006, 86, 465–514. [Google Scholar] [CrossRef]
- Chawla, A.; Repa, J.J.; Evans, R.M.; Mangelsdorf, D.J. Nuclear receptors and lipid physiology: Opening the X-files. Science 2001, 294, 1866–1870. [Google Scholar] [CrossRef] [PubMed]
- Burris, T.P.; Solt, L.A.; Wang, Y.; Crumbley, C.; Banerjee, S.; Griffett, K.; Lundasen, T.; Hughes, T.; Kojetin, D.J. Nuclear receptors and their selective pharmacologic modulators. Pharmacol. Rev. 2013, 65, 710–778. [Google Scholar] [CrossRef] [PubMed]
- Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptors: Nuclear control of metabolism. Endocr. Rev. 1999, 20, 649–688. [Google Scholar] [CrossRef]
- Duszka, K.; Wahli, W. Enteric microbiota–gut–brain axis from the perspective of nuclear receptors. Int. J. Mol. Sci. 2018, 19, 2210. [Google Scholar] [CrossRef] [PubMed]
- Dreyer, C.; Krey, G.; Keller, H.; Givel, F.; Helftenbein, G.; Wahli, W. Control of the peroxisomal beta-oxidation pathway by a novel family of nuclear hormone receptors. Cell 1992, 68, 879–887. [Google Scholar] [CrossRef]
- Kliewer, S.A.; Forman, B.M.; Blumberg, B.; Ong, E.S.; Borgmeyer, U.; Mangelsdorf, D.J.; Umesono, K.; Evans, R.M. Differential expression and activation of a family of murine peroxisome proliferator-activated receptors. Proc. Natl. Acad. Sci. USA 1994, 91, 7355–7359. [Google Scholar] [CrossRef]
- Issemann, I.; Green, S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature Cell Biol. 1990, 347, 645–650. [Google Scholar] [CrossRef]
- Tontonoz, P.; Hu, E.; Graves, R.A.; Budavari, A.I.; Spiegelman, B.M. mPPAR gamma 2: Tissue-specific regulator of an adipocyte enhancer. Genes Dev. 1994, 8, 1224–1234. [Google Scholar] [CrossRef]
- Ijpenberg, A.; Tan, N.S.; Gelman, L.; Kersten, S.; Seydoux, J.; Xu, J.; Metzger, D.; Canaple, L.; Chambon, P.; Wahli, W.; et al. In vivo activation of PPAR target genes by RXR homodimers. EMBO J. 2004, 23, 2083–2091. [Google Scholar] [CrossRef]
- Krey, G.; Keller, H.; Mahfoudi, A.; Medin, J.; Ozato, K.; Dreyer, C.; Wahli, W. Xenopus peroxisome proliferator activated receptors: Genomic organization, response element recognition, heterodimer formation with retinoid X receptor and activation by fatty acids. J. Steroid Biochem. Mol. Biol. 1993, 47, 65–73. [Google Scholar] [CrossRef]
- Tugwood, J.D.; Issemann, I.; Anderson, R.G.; Bundell, K.R.; McPheat, W.L.; Green, S. The mouse peroxisome proliferator activated receptor recognizes a response element in the 5′ flanking sequence of the rat acyl CoA oxidase gene. EMBO J. 1992, 11, 433–439. [Google Scholar] [CrossRef]
- Nolte, R.T.; Wisely, G.B.; Westin, S.; Cobb, J.E.; Lambert, M.H.; Kurokawa, R.; Rosenfeld, M.G.; Willson, T.M.; Glass, C.K.; Milburn, M.V. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature 1998, 395, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Schulman, I.G.; Shao, G.; Heyman, R.A. Transactivation by retinoid X receptor peroxisome proliferator-activated receptor gamma (PPARgamma) heterodimers: Intermolecular synergy requires only the PPARgamma hormone dependent activation function. Mol. Cell Biol. 1998, 18, 3483–3494. [Google Scholar] [CrossRef] [PubMed]
- Gearing, K.L.; Göttlicher, M.; Teboul, M.; Widmark, E.; Gustafsson, J.A. Interaction of the peroxisome-proliferator-activated receptor and retinoid X receptor. Proc. Natl. Acad. Sci. USA 1993, 90, 1440–1444. [Google Scholar] [CrossRef] [PubMed]
- IJpenberg, A.; Jeannin, E.; Wahli, W.; Desvergne, B. Polarity and specific sequence requirements of peroxisome proliferator-activated receptor (PPAR)/retinoid X receptor heterodimer binding to DNA. A functional analysis of the malic enzyme gene PPAR response element. J. Biol. Chem. 1997, 272, 20108–20117. [Google Scholar] [CrossRef]
- Feige, J.N.; Gelman, L.; Michalik, L.; Desvergne, B.; Wahli, W. From molecular action to physiological outputs: Peroxisome proliferator-activated receptors are nuclear receptors at the crossroads of key cellular functions. Prog. Lipid Res. 2006, 45, 120–159. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Downes, M.; Yu, R.T.; Bookout, A.L.; He, W.; Straume, M.; Mangelsdorf, D.J.; Evans, R.M. Nuclear receptor expression links the circadian clock to metabolism. Cell 2006, 126, 801–810. [Google Scholar] [CrossRef]
- Braissant, O.; Foufelle, F.; Scotto, C.; Dauca, M.; Wahli, W. Differential expression of peroxisome proliferator activated receptors (PPARs): Tissue distribution of PPAR-alpha,-beta, and -gamma in the adult rat. Endocrinology 1996, 137, 354–366. [Google Scholar] [CrossRef]
- Braissant, O.; Wahli, W. Differential expression of peroxisome proliferator-activated receptor-α, -β, and -γ during rat embryonic development. Endocrinology 1998, 139, 2748–2754. [Google Scholar] [CrossRef]
- Muoio, D.M.; MacLean, P.S.; Lang, D.B.; Li, S.; Haumard, J.A.; Way, J.M.; Winegar, D.A.; Corton, J.C.; Dohm, G.L.; Kraus, W.E. Fatty acid homeostasis and induction of lipid regulatory genes in skeletal muscles of peroxisome proliferator-activated receptor (PPAR) alpha knock-out mice. J. Biol. Chem. 2002, 277, 26089–26097. [Google Scholar] [CrossRef]
- Palmer, C.N.; Hsu, M.H.; Griffin, K.J.; Raucy, J.L.; Johnson, E.F. Peroxisome proliferator activated receptor-alpha expression in human liver. Mol. Pharmacol. 1998, 53, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Misra, P.; Reddy, J.K. Peroxisome proliferator-activated receptor-α activation and excess energy burning in hepatocarcinogenesis. Biochimie 2014, 98, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Pyper, S.R.; Viswakarma, N.; Yu, S.; Reddy, J.K. PPARalpha: Energy combustion, hypolipidemia, inflammation and cancer. Nucl. Recept. Signal 2010, 8, e002. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Jia, Y.; Fu, T.; Viwakarma, N.; Bai, L.; Rao, M.S.; Zhu, Y.; Borensztajn, J.; Reddy, J.K. Sustained activation of PPARα by endogenous ligands increases hepatic fatty acid oxidation and prevents obesity in ob/ob mice. FASEB J. 2011, 26, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Neels, J.G.; Grimaldi, P.A. Physiological functions of peroxisome proliferator activated receptor beta. Physiol. Rev. 2014, 94, 795–858. [Google Scholar] [CrossRef] [PubMed]
- Girroir, E.E.; Hollingshead, H.E.; He, P.; Zhu, B.; Perdew, G.H.; Peters, J.M. Quantitative expression patterns of peroxisome proliferator activated receptor-beta/delta (PPARbeta/delta) protein in mice. Biochem. Biophys. Res. Commun. 2008, 371, 456–461. [Google Scholar] [CrossRef]
- Lahiri, S.; Wahli, W. Peroxisome proliferator-activated receptor beta/delta: A master regulator of metabolic pathways in skeletal muscle. Horm. Mol. Biol. Clin. Investig. 2010, 4, 565–573. [Google Scholar] [CrossRef]
- Tan, N.S.; Vazquez-Carrera, M.; Montagner, A.; Sng, M.K.; Guillou, H.; Wahli, W. Transcriptional control of physiological and pathological processes by the nuclear receptor PPARbeta/delta. Prog. Lipid Res. 2016, 64, 98–122. [Google Scholar] [CrossRef]
- Zhu, Y.; Qi, C.; Korenberg, J.R.; Chen, X.N.; Noya, D.; Rao, M.S.; Reddy, J.K. Structural organization of mouse peroxisome proliferator-activated receptor gamma (mPPAR gamma) gene: Alternative promoter use and different splicing yield two mPPAR gamma isoforms. Proc. Natl. Acad. Sci. USA 1995, 92, 7921–7925. [Google Scholar] [CrossRef]
- Zhu, Y.; Alvares, K.; Huang, Q.; Rao, M.S.; Reddy, J.K. Cloning of a new member of the peroxisome proliferator-activated receptor gene family from mouse liver. J. Biol. Chem. 1993, 268, 26817–26820. [Google Scholar]
- Lazar, M.A. PPAR gamma, 10 years later. Biochimie 2005, 87, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Zhang, Y.; Zhu, S.; Luo, Y.; Xu, P.; Huang, Z. PPAR-ediated toxicology and applied pharmacology. Cells 2020, 9, 352. [Google Scholar] [CrossRef]
- Wagner, N.; Wagner, N. PPAR beta/delta and the hallmarks of cancer. Cells 2020, 9, 1133. [Google Scholar] [CrossRef] [PubMed]
- Fougerat, A.; Montagner, A.; Loiseau, N.; Guillou, H.; Wahli, W. Peroxisome proliferator-activated receptors and their novel ligands as candidates for the treatment of non-alcoholic fatty liver disease. Cells 2020, 9, 1638. [Google Scholar] [CrossRef] [PubMed]
- Wagner, N.; Wagner, K.-D. PPARs and angiogenesis—Implications in pathology. Int. J. Mol. Sci. 2020, 21, 5723. [Google Scholar] [CrossRef]
- Cheng, H.S.; Tan, W.R.; Low, Z.S.; Marvalim, C.; Lee, J.Y.H.; Tan, N.S. Exploration and development of PPAR modulators in health and disease: An update of clinical evidence. Int. J. Mol. Sci. 2019, 20, 5055. [Google Scholar] [CrossRef]
- Takada, I.; Makishima, M. Peroxisome proliferator-activated receptor agonists and antagonists: A patent review (2014-present). Expert Opin. Ther. Pat. 2019, 30, 1–13. [Google Scholar] [CrossRef]
- Kersten, S.; Desvergne, B.; Wahli, W. Roles of PPARs in health and disease. Nature 2000, 405, 421–424. [Google Scholar] [CrossRef]
- Phua, W.W.T.; Wong, M.X.Y.; Liao, Z.; Tan, N.S. An aPPARent functional consequence in skeletal muscle physiology via peroxisome proliferator-activated receptors. Int. J. Mol. Sci. 2018, 19, 1425. [Google Scholar] [CrossRef]
- Finck, B.N.; Bernal-Mizrachi, C.; Han, D.H.; Coleman, T.; Sambandam, N.; LaRiviere, L.L.; Holloszy, J.O.; Semenkovich, C.F.; Kelly, D.P. A potential link between muscle peroxisome proliferator- activated receptor-α signaling and obesity-related diabetes. Cell Metab. 2005, 1, 133–144. [Google Scholar] [CrossRef]
- Franko, A.; Huypens, P.; Neschen, S.; Irmler, M.; Rozman, J.; Rathkolb, B.; Neff, F.; Prehn, C.; Dubois, G.; Baumann, M.; et al. Bezafibrate improves insulin sensitivity and metabolic flexibility in STZ-induced diabetic mice. Diabetes 2016, 65, 2540–2552. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, M.D.; Hwang, S.-K.; Pauli, C.; Murphy, C.J.; Cheng, Z.; Hopkins, B.D.; Wu, D.; Loughran, R.M.; Emerling, B.M.; Zhang, G.; et al. Fenofibrate prevents skeletal muscle loss in mice with lung cancer. Proc. Natl. Acad. Sci. USA 2018, 115, E743–E752. [Google Scholar] [CrossRef]
- Russell, A.P.; Feilchenfeldt, J.; Schreiber, S.; Praz, M.; Crettenand, A.; Gobelet, C.; Meier, C.A.; Bell, D.R.; Kralli, A.; Giacobino, J.-P.; et al. Endurance training in humans leads to fiber type-specific increases in levels of peroxisome proliferator-activated receptor-gamma coactivator-1 and peroxisome proliferator-activated receptor-alpha in skeletal muscle. Diabetes 2003, 52, 2874–2881. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Leon, S.; Tuvblad, C.; Forero, D.A. Sports genetics: The PPARA gene and athletes’ high ability in endurance sports. A systematic review and meta-analysis. Biol. Sport 2015, 33, 3–6. [Google Scholar] [CrossRef]
- Nahle, Z.; Hsieh, M.; Pietka, T.; Coburn, C.T.; Grimaldi, P.A.; Zhang, M.Q.; Das, D.; Abumrad, N.A. CD36-dependent regulation of muscle FoxO1 and PDK4 in the PPAR delta/beta-mediated adaptation to metabolic stress. J. Biol. Chem. 2008, 283, 14317–14326. [Google Scholar] [CrossRef]
- Peters, S.J.; Harris, R.A.; Heigenhauser, G.J.; Spriet, L.L. Muscle fiber type comparison of PDH kinase activity and isoform expression in fed and fasted rats. Am. J. Physiol. Integr. Comp. Physiol. 2001, 280, R661–R668. [Google Scholar] [CrossRef] [PubMed]
- Spriet, L.L.; Tunstall, R.J.; Watt, M.J.; Mehan, K.A.; Hargreaves, M.; Cameron-Smith, D. Pyruvate dehydrogenase activation and kinase expression in human skeletal muscle during fasting. J. Appl. Physiol. 2004, 96, 2082–2087. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Gao, R.; Xie, X.; Zheng, Z.; Li, H.; Li, S.; Dong, F.; Wang, L. A metabolomic study of the PPARδ agonist GW501516 for enhancing running endurance in Kunming mice. Sci. Rep. 2015, 5, 9884. [Google Scholar] [CrossRef]
- Luquet, S.; Lopez-Soriano, J.; Holst, D.; Fredenrich, A.; Melki, J.; Rassoulzadegan, M.; Grimaldi, P.A. Peroxisome proliferator-activated receptor delta controls muscle development and oxidative capability. FASEB J. 2003, 17, 2299–2301. [Google Scholar] [CrossRef]
- Wang, Y.-X.; Zhang, C.-L.; Yu, R.T.; Cho, H.K.; Nelson, M.C.; Bayuga-Ocampo, C.R.; Ham, J.; Kang, H.; Evans, R.M. Regulation of muscle fiber type and running endurance by PPARdelta. PLoS Biol. 2004, 2, e294. [Google Scholar] [CrossRef]
- Wang, Y.-X.; Lee, C.-H.; Tiep, S.; Yu, R.T.; Ham, J.; Kang, H.; Evans, R.M. Peroxisome-proliferator-activated receptor delta activates fat metabolism to prevent obesity. Cell 2003, 113, 159–170. [Google Scholar] [CrossRef]
- Schuler, M.; Ali, F.; Chambon, C.; Duteil, D.; Bornert, J.-M.; Tardivel, A.; Desvergne, B.; Wahli, W.; Chambon, P.; Metzger, D. PGC1alpha expression is controlled in skeletal muscles by PPARbeta, whose ablation results in fiber-type switching, obesity, and type 2 diabetes. Cell Metab. 2006, 4, 407–414. [Google Scholar] [CrossRef]
- Kleiner, S.; Nguyen-Tran, V.; Bare, O.; Huang, X.; Spiegelman, B.; Wu, Z. PPAR{delta} agonism activates fatty acid oxidation via PGC-1{alpha} but does not increase mitochondrial gene expression and function. J. Biol. Chem. 2009, 284, 18624–18633. [Google Scholar] [CrossRef]
- Koh, J.-H.; Hancock, C.R.; Terada, S.; Higashida, K.; Holloszy, J.O.; Han, D.-H. PPARβ is essential for maintaining Nnrmal levels of PGC-1α and mitochondria and for the increase in muscle mitochondria induced by exercise. Cell Metab. 2017, 25, 1176–1185.e5. [Google Scholar] [CrossRef] [PubMed]
- Arany, Z. PGC-1 coactivators and skeletal muscle adaptations in health and disease. Curr. Opin. Genet. Dev. 2008, 18, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Handschin, C.; Rhee, J.; Lin, J.; Tarr, P.T.; Spiegelman, B.M. An autoregulatory loop controls peroxisome proliferator-activated receptor gamma coactivators 1 alpha expression in muscle. Proc. Natl. Acad. Sci. USA 2003, 100, 7111–7116. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Wu, H.; Tarr, P.T.; Zhang, C.-Y.; Wu, Z.; Boss, O.; Michael, L.F.; Puigserver, P.; Isotani, E.; Olson, E.N.; et al. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibers. Nature 2002, 418, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Arany, Z.; Lebrasseur, N.; Morris, C.; Smith, E.; Yang, W.; Ma, Y.; Chin, S.; Spiegelman, B.M. The transcriptional coactivators PGC-1 beta drives the formation of oxidative type IIx fibers in skeletal muscle. Cell Metab. 2007, 5, 35–46. [Google Scholar] [CrossRef]
- Chandrashekar, P.; Manickam, R.; Ge, X.; Bonala, S.; McFarlane, C.; Sharma, M.; Wahli, W.; Kambadur, R. Inactivation of PPARbeta/delta adversely affects satellite cells and reduces postnatal myogenesis. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E122–E131. [Google Scholar] [CrossRef]
- Angione, A.R.; Jiang, C.; Pan, D.; Wang, Y.-X.; Kuang, S. PPARδ regulates satellite cell proliferation and skeletal muscle regeneration. Skelet. Muscle 2011, 1, 33. [Google Scholar] [CrossRef]
- Gaudel, C.; Schwartz, C.; Giordana, C.; Abumrad, N.A.; Grimaldi, P.A. Pharmacological activation of PPARbeta promotes rapid and calcineurin-dependent fiber remodeling and angiogenesis in mouse skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E297–E304. [Google Scholar] [CrossRef] [PubMed]
- Giordano, C.; Rousseau, A.; Wagner, N.; Gaudel, C.; Murdaca, J.; Jehl-Pietri, C.; Sibille, B.; Grimaldi, P.A.; Lopez, P. Peroxisome proliferator-activated receptor β activation promotes myonuclear accretion in skeletal muscle of adult and aged mice. Pflügers Arch. 2009, 458, 901–913. [Google Scholar] [CrossRef] [PubMed]
- Shintaku, J.; Peterson, J.M.; Talbert, E.E.; Gu, J.-M.; Ladner, K.J.; Williams, D.R.; Mousavi, K.; Wang, R.; Sartorelli, V.; Guttridge, D.C. MyoD regulates skeletal muscle oxidative metabolism cooperatively with alternative NF-κB. Cell Rep. 2016, 17, 514–526. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, B.L.; Wachtmann, T.S.; Cosgrove, P.G.; Kuhn, M.; Opsahl, A.C.; Judkins, K.M.; Freeman, T.B.; Hadcock, J.R.; LeBrasseur, N.K. Postnatal PPARdelta activation and myostatin inhibition exert distinct yet complimentary effects on the metabolic profile of obese insulin-resistant mice. PLoS ONE 2010, 5, e11307. [Google Scholar] [CrossRef]
- Manio, M.C.C.; Inoue, K.; Fujitani, M.; Matsumara, S.; Fushiki, T. Combined pharmacological activation of AMPK and PPARdelta potentiates the effects of exercise in trained mice. Physiol. Rep. 2016, 4, e12625. [Google Scholar] [CrossRef]
- Salvado, L.; Barroso, E.; Gomez-Foix, A.M.; Palomer, X.; Michalik, L.; Wahli, W.; Vazquez-Carrera, M. PPARbeta/delta prevents endoplasmic reticulum stress associated inflammation and insulin resistance in skeletal muscle cells through an AMPK-dependent mechanism. Diabetologia 2014, 57, 2126–2135. [Google Scholar] [CrossRef]
- Klingler, C.; Zhao, X.; Adhikary, T.; Li, J.; Xu, G.; Häring, H.-U.; Schleicher, E.; Lehmann, R.; Weigert, C. Lysophosphatidylcholines activate PPARδ and protect human skeletal muscle cells from lipotoxicity. Biochim. Biophys. Acta 2016, 1861, 1980–1992. [Google Scholar] [CrossRef]
- Amin, R.H.; Mathews, S.T.; Camp, H.S.; Ding, L.; Leff, T. Selective activation of PPAR gamma in skeletal muscle induces endogenous production of adiponectin and protects mice from diet-induced insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2010, 298, 28–37. [Google Scholar] [CrossRef]
- Hevener, A.L.; He, W.; Barak, Y.; Le, J.; Bandyopadhyay, G.; Olson, P.; Wilkes, J.; Evans, R.M.; Olefsky, J. Muscle-specific Pparg deletion causes insulin resistance. Nat. Med. 2003, 9, 1491–1497. [Google Scholar] [CrossRef]
- Norris, A.W.; Chen, L.; Fisher, S.J.; Szanto, I.; Ristow, M.; Jozsi, A.C.; Hirshman, M.F.; Rosen, E.D.; Goodyear, L.J.; Gonzalezz, F.J.; et al. Muscle-specific PPAR gamma-deficient mice develop increased adiposity and insulin resistance but respond to thiazolidinediones. J. Clin. Investig. 2003, 112, 608–618. [Google Scholar] [CrossRef]
- He, W.; Barak, Y.; Hevener, A.; Olson, P.; Liao, D.; Le, J.; Nelson, M.; Ong, E.; Olefsky, J.M.; Evans, R.M. Adipose-specific peroxisome proliferator-activated receptor gamma knockout causes insulin resistance in fat and liver but not in muscle. Proc. Natl. Acad. Sci. USA 2003, 100, 15712–15717. [Google Scholar] [CrossRef] [PubMed]
- Marín-Juez, R.; Diaz, M.; Morata, J.; Planas, J.V. Mechanisms regulating GLUT4 transcription in skeletal muscle cells are highly conserved across vertebrates. PLoS ONE 2013, 8, e80628. [Google Scholar] [CrossRef]
- Djouadi, F.; Bonnefont, J.-P.; Thuillier, L.; Droin, V.; Khadom, N.; Munnich, A.; Bastin, J. Correction of fatty acid oxidation in carnitine palmitoyl transferase 2–deficient cultured skin fibroblasts by bezafibrate. Pediatr. Res. 2003, 54, 446–451. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Djouadi, F.; Aubey, F.; Schlemmer, D.; Ruiter, J.; Wanders, R.; Strauss, A.; Bastin, J. Bezafibrate increases very-long-chain acyl-CoA dehydrogenase protein and mRNA expression in deficient fibroblasts and is a potential therapy for fatty acid oxidation disorders. Hum. Mol. Genet. 2005, 14, 2695–2703. [Google Scholar] [CrossRef]
- Miura, P.; Chakkalakal, J.V.; Boudreault, L.; Bélanger, G.; Hébert, R.L.; Renaud, J.-M.; Jasmin, B.J. Pharmacological activation of PPAR/stimulates utrophin A expression in skeletal muscle fibers and restores sarcolemmal integrity in mature mdx mice. Hum. Mol. Genet. 2009, 18, 4640–4649. [Google Scholar] [CrossRef] [PubMed]
- Friday, B.B.; Pavlath, G.K. A calcineurin- and NFAT-dependent pathway regulates Myf5 gene expression in skeletal muscle reserve cells. J. Cell Sci. 2001, 114, 303–310. [Google Scholar] [PubMed]
- Chakkalakal, J.V.; Stocksley, M.A.; Harrison, M.-A.; Angus, L.M.; Deschênes-Furry, J.; St-Pierre, S.; Megeney, L.A.; Chin, E.R.; Michel, R.N.; Jasmin, B.J. Expression of utrophin A mRNA correlates with the oxidative capacity of skeletal muscle fiber types and is regulated by calcineurin/NFAT signaling. Proc. Natl. Acad. Sci. USA 2003, 100, 7791–7796. [Google Scholar] [CrossRef]
- Wagner, N.; Jehl-Pietri, C.; Lopez, P.; Murdaca, J.; Giordano, C.; Schwartz, C.; Gounon, P.; Hatem, S.N.; Grimaldi, P.; Wagner, K.-D. Peroxisome proliferator-activated receptor beta stimulation induces rapid cardiac growth and angiogenesis via direct activation of calcineurin. Cardiovasc. Res. 2009, 83, 61–71. [Google Scholar] [CrossRef]
- Gong, L.; Jin, H.; Li, Y.; Quan, Y.; Yang, J.; Tang, Q.; Zou, Z. Rosiglitazone ameliorates skeletal muscle insulin resistance by decreasing free fatty acids release from adipocytes. Biochem. Biophys. Res. Commun. 2020, S0006-291X, 31904–31905. [Google Scholar] [CrossRef]
- Cha, B.S.; Ciaraldi, T.P.; Carter, L.; Nikoulina, S.E.; Mudaliar, S.; Mukherjee, R.; Paterniti, J.R.; Henry, R.R. Peroxisome proliferator-activated receptor (PPAR) gamma and retinoid X receptor (RXR) agonists have complementary effects on glucose and lipid metabolism in human skeletal muscle. Diabetologia 2001, 44, 444–452. [Google Scholar] [CrossRef]
- Shang, Y.C.; Zhang, C.; Wang, S.H.; Xiong, F.; Zhao, C.P.; Peng, F.N.; Yu, M.J.; Li, M.S.; Zhang, Y.N. Activated beta-catenin induces myogenesis and inhibits adipogenesis in BM-derived mesenchymal stromal cells. Cytotherapy 2007, 9, 667–681. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Serrano, R.M.; González-Dávalos, M.L.; Lozano-Flores, C.; Shimada, A.; Antaramian, A.; Varela-Echavarría, A.; Mora, O. PPAR agonists promote the differentiation of porcine bone marrow mesenchymal Ssem cells into the adipogenic and myogenic lineages. Cells Tissues Organs 2016, 203, 153–172. [Google Scholar] [CrossRef] [PubMed]
- Barberi, T.; Willis, L.M.; Socci, N.D.; Studer, L. Derivation of multipotent mesenchymal precursors from human embryonic stem cells. PLoS Med. 2005, 2, e161. [Google Scholar] [CrossRef]
- Wan, Y.; Chong, L.-W.; Evans, R.M. PPAR-γ regulates osteoclastogenesis in mice. Nat. Med. 2007, 13, 1496–1503. [Google Scholar] [CrossRef] [PubMed]
- Wahli, W. PPAR gamma: Ally and foe in bone metabolism. Cell Metab. 2008, 7, 188–190. [Google Scholar] [CrossRef][Green Version]
- Muruganandan, S.; Ionescu, A.M.; Sinal, C.J. At the crossroads of the adipocyte and osteoclast differentiation programs: Future therapeutic perspectives. Int. J. Mol. Sci. 2020, 21, 2277. [Google Scholar] [CrossRef]
- Gupta, V.K.; Paul, S.; Dutta, C. Geography, ethnicity or subsistence-specific variations in human microbiome composition and diversity. Front. Microbiol. 2017, 8, 1162. [Google Scholar] [CrossRef]
- Ianiro, G.; Tilg, H.; Gasbarrini, A. Antibiotics as deep modulators of gut microbiota: Between good and evil. Gut 2016, 65, 1906–1915. [Google Scholar] [CrossRef]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef]
- Kim, H.; Sitarik, A.R.; Woodcroft, K.; Johnson, C.C.; Zoratti, E. Birth mode, breastfeeding, pet exposure, and antibiotic use: Associations with the gut microbiome and sensitization in children. Curr. Allergy Asthma Rep. 2019, 19, 22. [Google Scholar] [CrossRef]
- Heggie, A.D.; Wyrick, P.B.; Chase, P.A.; Sorensen, R.U. Cell-mediated immune responses to chlamydia trachomatis in mothers and infants. Exp. Biol. Med. 1986, 181, 586–595. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.Y.P.; Visvalingam, V.; Wahli, W. The PPAR–microbiota–metabolic organ trilogy to fine-tune physiology. FASEB J. 2019, 33, 9706–9730. [Google Scholar] [CrossRef] [PubMed]
- Manoharan, I.; Suryawanshi, A.; Hong, Y.; Ranganathan, P.; Shanmugam, A.; Ahmad, S.; Swafford, D.; Manicassamy, B.; Ramesh, G.; Koni, P.A.; et al. Homeostatic PPARα signaling limits inflammatory responses to commensal microbiota in the intestine. J. Immunol. 2016, 196, 4739–4749. [Google Scholar] [CrossRef] [PubMed]
- Mazzon, E.; Cuzzocrea, S. Absence of functional peroxisome proliferator-activated receptor enhanced ileum permeability during experimental colitis. Shock 2007, 28, 192–201. [Google Scholar] [CrossRef]
- Riccardi, L.; Mazzon, E.; Bruscoli, S.; Esposito, E.; Crisafulli, C.; Di Paola, R.; Caminiti, R.; Riccardi, C.; Cuzzocrea, S. Peroxisome proliferator-activated receptor-alpha modulates the anti-inflammatory effect of glucocorticoids in a model of inflammatory bowel disease in mice. Shock 2009, 31, 308–316. [Google Scholar] [CrossRef]
- Esposito, E.; Mazzon, E.; Paterniti, I.; Toso, R.D.; Pressi, G.; Caminiti, R.; Cuzzocrea, S. PPAR-alphacontributes to the anti-inflammatory activity of verbascoside in a model of inflammatory bowel disease in mice. PPAR Res. 2010, 2010, 917312. [Google Scholar] [CrossRef]
- Cuzzocrea, S.; Di Paola, R.; Mazzon, E.; Genovese, T.; Muià, C.; Centorrino, T.; Caputi, A.P. Role of endogenous and exogenous ligands for the peroxisome proliferators activated receptors alpha (PPAR-α) in the development of inflammatory bowel disease in mice. Lab. Investig. 2004, 84, 1643–1654. [Google Scholar] [CrossRef]
- Canaple, L.; Rambaud, J.; Dkhissi-Benyahya, O.; Rayet, B.; Tan, N.S.; Michalik, L.; Delaunay, F.; Wahli, W.; Laudet, V. Reciprocal regulation of brain and muscle Arnt-like protein 1 and peroxisome proliferator-activated receptor alpha defines a novel positive feedback loop in the rodent liver circadian clock. Mol. Endocrinol. 2006, 20, 1715–1727. [Google Scholar] [CrossRef]
- Montagner, A.; Rando, G.; Degueurce, G.; Leuenberger, N.; Michalik, L.; Wahli, W. New insights into the role of PPARs. Prostaglandins Leukot. Essent. Fatty Acids 2011, 85, 235–243. [Google Scholar] [CrossRef]
- Oishi, K.; Shirai, H.; Ishida, N. Clock is involved in the circadian transactivation of peroxisome-proliferator-activated receptor alpha (PPARalpha) in mice. Biochem. J. 2005, 386, 575–581. [Google Scholar] [CrossRef]
- Mukherji, A.; Kobiita, A.; Ye, T.; Chambon, P. Homeostasis in intestinal epithelium is orchestrated by the circadian clock and microbiota cues transduced by TLRs. Cell 2013, 153, 812–827. [Google Scholar] [CrossRef] [PubMed]
- Varnat, F.; Heggeler, B.B.-T.; Grisel, P.; Boucard, N.; Corthesy-Theulaz, I.; Wahli, W.; Desvergne, B. PPARbeta/delta regulates paneth cell differentiation via controlling the hedgehog signaling pathway. Gastroenterology 2006, 131, 538–553. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.M.; Hollingshead, H.E.; Gonzalez, F.J. Role of peroxisome-proliferator-activated receptor beta/delta (PPARbeta/delta) in gastrointestinal tract function and disease. Clin. Sci. 2008, 115, 107–127. [Google Scholar] [CrossRef] [PubMed]
- Hollingshead, H.E.; Morimura, K.; Adachi, M.; Kennett, M.J.; Billin, A.N.; Willson, T.M.; Gonzalez, F.J.; Peter, J.M. PPARbeta/delta protects against experimental colitis through a ligand-independent mechanism. Dig. Dis. Sci. 2007, 52, 2912–2919. [Google Scholar] [CrossRef]
- Annese, V.; Rogai, F.; Settesoldi, A.; Bagnoli, S. PPARγ in inflammatory bowel disease. PPAR Res. 2012, 2012, 620839. [Google Scholar] [CrossRef]
- Daoudi, M.; Hennuyer, N.; Borland, M.G.; Touche, V.; Duhem, C.; Gross, B.; Caiazzo, R.; Kerr-Conte, J.; Pattou, F.; Peters, J.M.; et al. PPARβ/δ activation induces enteroendocrine L cell GLP-1 production. Gasteroenterology 2011, 140, 1564–1574. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, S.; Kim, H.; Garcia-Perez, I.; Reza, M.M.; Martin, K.A.; Kundu, P.; Cox, L.M.; Selkrig, J.; Posma, J.M.; Zhang, H.; et al. The gut microbiota influences skeletal muscle mass and function in mice. Sci. Transl. Med. 2019, 11, eaan5662. [Google Scholar] [CrossRef]
- Fielding, R.A.; Reeves, A.R.; Jasuja, R.; Liu, C.; Barrett, B.B.; Lustgarten, M.S. Muscle strength is increased in mice that are colonized with microbiota from high-functioning older adults. Exp. Gerontol. 2019, 127, 110722. [Google Scholar] [CrossRef]
- Manickam, R.; Oh, H.Y.P.; Tan, C.K.; Paramalingam, E.; Wahli, W. Metronidazole causes skeletal muscle atrophy and modulates muscle chronometabolism. Int. J. Mol. Sci. 2018, 19, 2418. [Google Scholar] [CrossRef]
- Nay, K.; Jollet, M.; Goustard, B.; Baati, N.; Vernus, B.; Pontones, M.; Lefeuvre-Orfila, L.; Bendavid, C.; Rué, O.; Mariadassou, M.; et al. Gut bacteria are critical for optimal muscle function: A potential link with glucose homeostasis. Am. J. Physiol. Metab. 2019, 317, E158–E171. [Google Scholar] [CrossRef]
- Okamoto, T.; Morino, K.; Ugi, S.; Nakagawa, F.; Lemecha, M.; Ida, S.; Ohashi, N.; Sato, D.; Fujita, Y.; Maegawa, H. Microbiome potentiates endurance exercise through intestinal acetate production. Am. J. Physiol. Metab. 2019, 316, E956–E966. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Yin, J.; Zhang, J.; Ward, R.E.; Martin, R.J.; Lefevre, M.; Cefalu, W.T.; Ye, J. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 2009, 58, 1509–1517. [Google Scholar] [CrossRef]
- Mansén, A.; Guardiola-Diaz, H.; Rafter, J.; Branting, C.; Gustafsson, J.A. Expression of the peroxisome proliferator-activated receptor (PPAR) in the mouse colonic mucosa. Biochem. Biophys. Res. Commun. 1996, 222, 844–851. [Google Scholar] [CrossRef] [PubMed]
- Martinasso, G.; Oraldi, M.; Trombetta, A.; Maggiora, M.; Bertetto, O.; Canuto, R.A.; Muzio, G. Involvement of PPARs in cell proliferation and apoptosis in human colon cancer specimens and in normal and cancer cell lines. PPAR Res. 2007, 2007, 93416. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.G.; Lee, J.F.Y.; Wang, S.H.; Chan, U.P.F.; Ip, P.C.; Lau, W.Y. Apoptosis induced by activation of peroxisome-proliferator activated receptor-gamma is associated with Bcl-2 and NF-kappaB in human colon cancer. Life Sci. 2002, 70, 2631–2646. [Google Scholar] [CrossRef]
- Chen, G.G.; Xu, H.; Lee, J.F.; Subramaniam, M.; Leung, K.L.; Wang, S.H.; Chan, U.P.F.; Spelsberg, T.C. 15-hydroxy-eicosatetraenoic acid arrests growth of colorectal cancer cells via a peroxisome proliferator-activated receptor gamma-dependent pathway. Int. J. Cancer 2003, 107, 837–843. [Google Scholar] [CrossRef]
- Peyrin-Biroulet, L.; Beisner, J.; Wang, G.; Nuding, S.; Oommen, S.T.; Kelly, D.; Parmentier-Decrucq, E.; Dessein, R.; Merour, E.; Chavatte, P.; et al. Peroxisome proliferator-activated receptor gamma activation is required for maintenance of innate antimicrobial immunity in the colon. Proc. Natl. Acad. Sci. USA 2010, 107, 8772–8777. [Google Scholar] [CrossRef]
- Lewis, J.D.; Lichtenstein, G.R.; Deren, J.J.; Sands, B.E.; Hanauer, S.B.; Katz, J.A.; Lashner, B.; Present, D.H.; Chuai, S.; Ellenberg, J.H.; et al. Rosiglitazone for active ulcerative colitis: A randomized placebo-controlled trial. Gastroenterology 2008, 134, 688–695. [Google Scholar] [CrossRef]
- Bassaganya-Riera, J.; Hontecillas, R. CLA and n-3 PUFA differentially modulate clinical activity and colonic PPAR-responsive gene expression in a pig model of experimental IBD. Clin. Nutr. 2006, 25, 454–465. [Google Scholar] [CrossRef]
- Kundu, P.; Ling, T.W.; Korecka, A.; Li, Y.; D’Arienzo, R.; Bunte, R.M.; Berger, T.; Arulampalam, V.; Chambon, P.; Mak, T.W.; et al. Absence of intestinal PPARγ aggravates acute infectious colitis in mice through a lipocalin-2–dependent pathway. PLoS Pathog. 2014, 10, e1003887. [Google Scholar] [CrossRef]
- Su, C.G.; Wen, X.; Bailey, S.T.; Jiang, W.; Rangwala, S.M.; Keilbaugh, S.A.; Flanigan, A.; Murthy, S.; Lazar, M.A.; Wu, G.D. A novel therapy for colitis utilizing PPAR-γ ligands to inhibit the epithelial inflammatory response. J. Clin. Investig. 1999, 104, 383–389. [Google Scholar] [CrossRef]
- Byndloss, M.X.; Olsan, E.E.; Rivera-Chávez, F.; Tiffany, C.R.; Cevallos, S.A.; Lokken, K.L.; Torres, T.P.; Byndloss, A.J.; Faber, F.; Gao, Y.; et al. Microbiota-activated PPAR-γ signaling inhibits dysbiotic Enterobacteriaceae expansion. Science 2017, 357, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Nepelska, M.; de Wouters, T.; Jacouton, E.; Béguet-Crespel, F.; Lapaque, N.; Doré, J.; Arulampalam, V.; Blottière, H.M. Commensal gut bacteria modulate phosphorylation-dependent PPARγ transcriptional activity in human intestinal epithelial cells. Sci. Rep. 2017, 7, 43199. [Google Scholar] [CrossRef] [PubMed]
- Schwab, M.; Reynders, V.; Loitsch, S.; Steinhilber, D.; Stein, J.; Schroder, O. Involvement of different nuclear hormone receptors in butyrate-mediated inhibition of inducible NF kappa B signaling. Mol. Immunol. 2007, 44, 3625–3632. [Google Scholar] [CrossRef]
- Wächtershäuser, A.; Loitsch, S.M.; Stein, J. PPAR-γ is selectively upregulated in Caco-2 cells by butyrate. Biochem. Biophys. Res. Commun. 2000, 272, 380–385. [Google Scholar] [CrossRef]
- Voltan, S.; Martines, D.; Elli, M.; Brun, P.; Longo, S.; Porzionato, A.; Macchi, V.; D’Inca, R.; Scarpa, M.; Palu, G.; et al. Lactobacillus crispatus M247-derived H2O2 acts as a signal transducing molecule activating peroxisome proliferator activated receptor-gamma in the intestinal mucosa. Gasteroenterology 2008, 135, 1216–1227. [Google Scholar] [CrossRef] [PubMed]
- Are, A.; Aronsson, L.; Wang, S.; Greicius, G.; Lee, Y.K.; Gustafsson, J.-A.; Pettersson, S.; Arulampalam, V. Enterococcus faecalis from newborn babies regulate endogenous PPARgamma activity and IL-10 levels in colonic epithelial cells. Proc. Natl. Acad. Sci. USA 2008, 105, 1943–1948. [Google Scholar] [CrossRef]
- Couvigny, B.; de Wouters, T.; Kaci, G.; Jacouton, E.; Delorme, C.; Doré, J.; Renault, P.; Blottière, H.M.; Guédon, E.; Lapaque, N. Commensal Streptococcus salivarius modulates PPARγ transcriptional activity in human intestinal epithelial cells. PLoS ONE 2015, 10, e0125371. [Google Scholar] [CrossRef]
- Lukovac, S.; Belzer, C.; Pellis, L.; Keijser, B.J.; de Vos, W.M.; Montijn, R.C.; Roeselers, G. Differential modulation by Akkermansia muciniphila and Faecali bacterium prausnitzii of host peripheral lipid metabolism and histone acetylation in mouse gut organoids. mBio 2014, 5, e01438. [Google Scholar] [CrossRef]
- Duszka, K.; Picard, A.; Ellero-Simatos, S.; Chen, J.; Defernez, M.; Paramalingam, E.; Pigram, A.; Vanoaica, L.; Canlet, C.; Parini, P.; et al. Intestinal PPARγ signaling is required for sympathetic nervous system activation in response to calorie restriction. Sci. Rep. 2016, 6, 36937. [Google Scholar] [CrossRef]
- Lustgarten, M.S. The role of the gut microbiota on skeletal muscle mass and physical function: 2019 update. Front. Physiol. 2019, 10, 1435. [Google Scholar] [CrossRef]
- Ticinesi, A.; Nouvenne, A.; Cerundolo, N.; Catania, P.; Prati, B.; Tana, C.; Meschi, T. Gut icrobiota, muscle mass and function in aging: A focus on physical frailty and sarcopenia. Nutrients 2019, 11, 1633. [Google Scholar] [CrossRef] [PubMed]
- Ticinesi, A.; Lauretani, F.; Milani, C.; Nouvenne, A.; Tana, C.; Del Rio, D.; Maggio, M.; Ventura, M.; Meschi, T. Aging gut microbiota at the cross-road between nutrition, physical frailty, and sarcopenia: Is there a gut–muscle axis? Nutrients 2017, 9, 1303. [Google Scholar] [CrossRef]
- Lochlainn, M.N.; Bowyer, R.C.E.; Steves, C.J. Dietary protein and muscle in aging people: The potential role of the gut microbiome. Nutrients 2018, 10, 929. [Google Scholar] [CrossRef] [PubMed]
- Ticinesi, A.; Lauretani, F.; Tana, C.; Nouvenne, A.; Ridolo, E.; Meschi, T. Exercise and immune system as modulators of intestinal microbiome: Implications for the gut-muscle axis hypothesis. Exerc. Immunol. Rev. 2019, 25, 84–95. [Google Scholar] [PubMed]
- De Sire, R.; Rizzatti, G.; Ingravalle, F.; Pizzoferrato, M.; Petito, V.; Lopetuso, L.; Graziani, C.; de Sire, A.; Mentella, M.C.; Mele, M.C.; et al. Skeletal muscle-gut axis: Emerging mechanisms of sarcopenia for intestinal and extra intestinal diseases. Minerva Gastroenterol. Dietol. 2018, 64, 351–362. [Google Scholar] [CrossRef]
- Grosicki, G.J.; Fielding, R.A.; Lustgarten, M.S. Gut microbiota contribute to age-related changes in skeletal muscle size, composition, and function: Biological basis for a gut-muscle axis. Calcif. Tissue Int. 2017, 102, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Fanelli, F.; Calvani, R.; Mulè, G.; Pesce, V.; Sisto, A.; Pantanelli, C.; Bernabei, R.; Landi, F.; Marzetti, E. Gut dysbiosis and muscle aging: Searching for novel targets against sarcopenia. Mediators Inflamm. 2018, 2018, 7026198. [Google Scholar] [CrossRef]
- Bäckhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef]
- Nobel, Y.R.; Cox, L.M.; Kirigin, F.F.; Bokulich, N.A.; Yamanishi, S.; Teitler, I.; Chung, J.; Sohn, J.; Barber, C.M.; Goldfarb, D.S.; et al. Metabolic and metagenomic outcomes from early-life pulsed antibiotic treatment. Nat. Commun. 2015, 6, 7486. [Google Scholar] [CrossRef]
- Bischoff, S.C. Microbiota and aging. Curr. Opin. Clin. Nutr. Metab. Care 2016, 19, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Yang, X.; Zheng, L.; Wang, Z.; Wu, L.; Jiang, J.; Yang, T.; Ma, L.; Fu, Z. Lactobacillus and Bifidobacterium improves physiological function and cognitive ability in aged mice by the regulation of gut microbiota. Mol. Nutr. Food Res. 2019, 63, e1900603. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.-C.; Chen, Y.-H.; Chuang, H.-L.; Chiu, C.-C.; Huang, C.-C. Investigation of the effects of microbiota on exercise physiological adaptation, performance, and energy utilization using a gnotobiotic animal model. Front. Microbiol. 2019, 10, 1906. [Google Scholar] [CrossRef]
- Scheiman, J.; Luber, J.M.; Chavkin, T.A.; MacDonald, T.; Tung, A.; Pham, L.-D.; Wibowo, M.C.; Wurth, R.C.; Punthambaker, S.; Tierney, B.T.; et al. Meta-omics analysis of elite athletes identifies a performance-enhancing microbe that functions via lactate metabolism. Nat. Med. 2019, 25, 1104–1109. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Diao, H.; Xiao, Y.; Li, W.; Yu, B.; He, J.; Yu, J.; Zheng, P.; Mao, X.; Luo, Y.; et al. Gut microbiota can transfer fiber characteristics and lipid metabolic profiles of skeletal muscle from pigs to germ-free mice. Sci. Rep. 2016, 6, 31786. [Google Scholar] [CrossRef]
- Houghton, M.J.; Kerimi, A.; Mouly, V.; Tumova, S.; Williamson, G. Gut microbiome catabolites as novel modulators of muscle cell glucose metabolism. FASEB J. 2018, 33, 1887–1898. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manickam, R.; Duszka, K.; Wahli, W. PPARs and Microbiota in Skeletal Muscle Health and Wasting. Int. J. Mol. Sci. 2020, 21, 8056. https://doi.org/10.3390/ijms21218056
Manickam R, Duszka K, Wahli W. PPARs and Microbiota in Skeletal Muscle Health and Wasting. International Journal of Molecular Sciences. 2020; 21(21):8056. https://doi.org/10.3390/ijms21218056
Chicago/Turabian StyleManickam, Ravikumar, Kalina Duszka, and Walter Wahli. 2020. "PPARs and Microbiota in Skeletal Muscle Health and Wasting" International Journal of Molecular Sciences 21, no. 21: 8056. https://doi.org/10.3390/ijms21218056
APA StyleManickam, R., Duszka, K., & Wahli, W. (2020). PPARs and Microbiota in Skeletal Muscle Health and Wasting. International Journal of Molecular Sciences, 21(21), 8056. https://doi.org/10.3390/ijms21218056