Current Insights in Elucidation of Possible Molecular Mechanisms of the Juvenile Form of Batten Disease


Abstract
:
1. Introduction
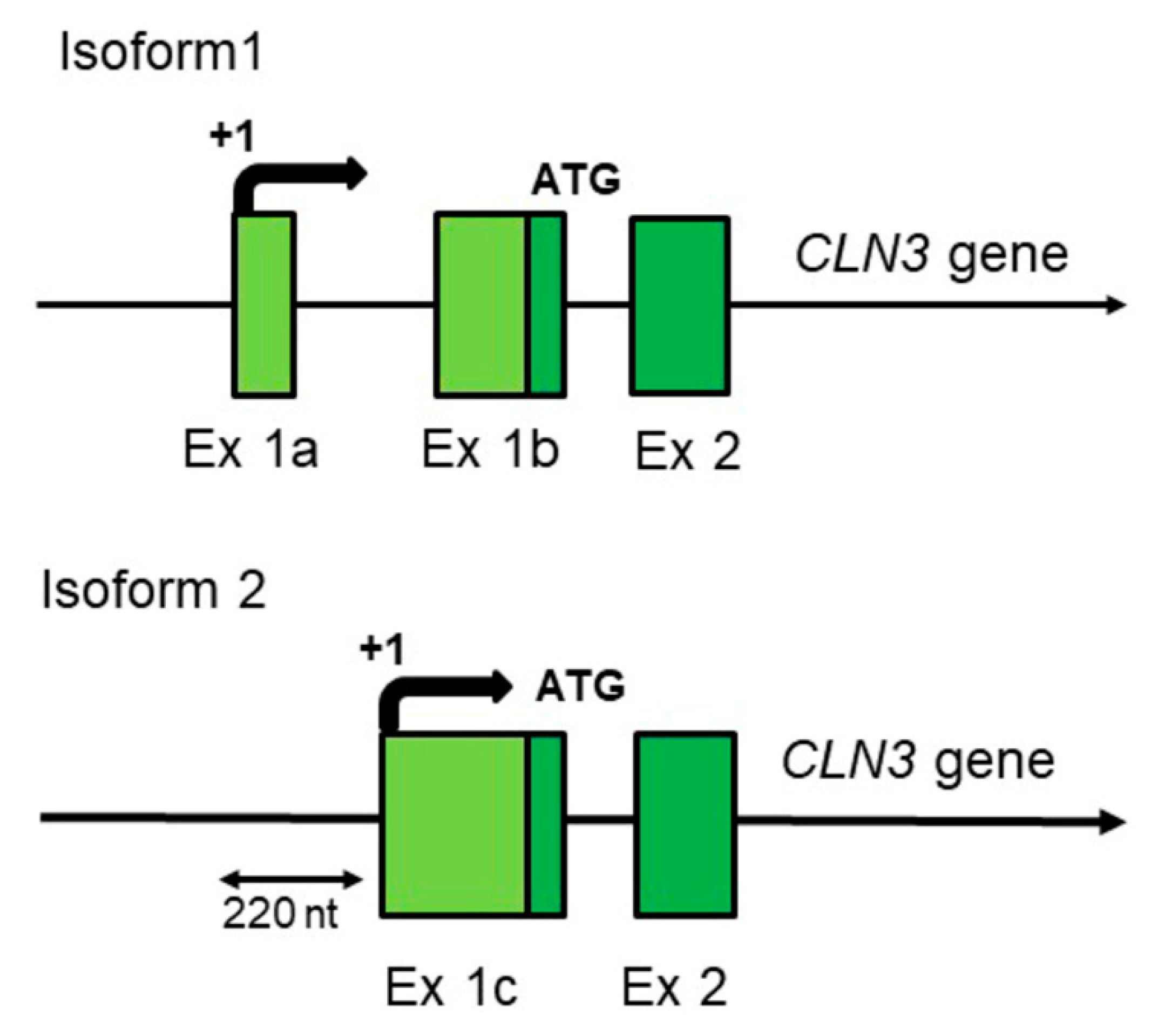
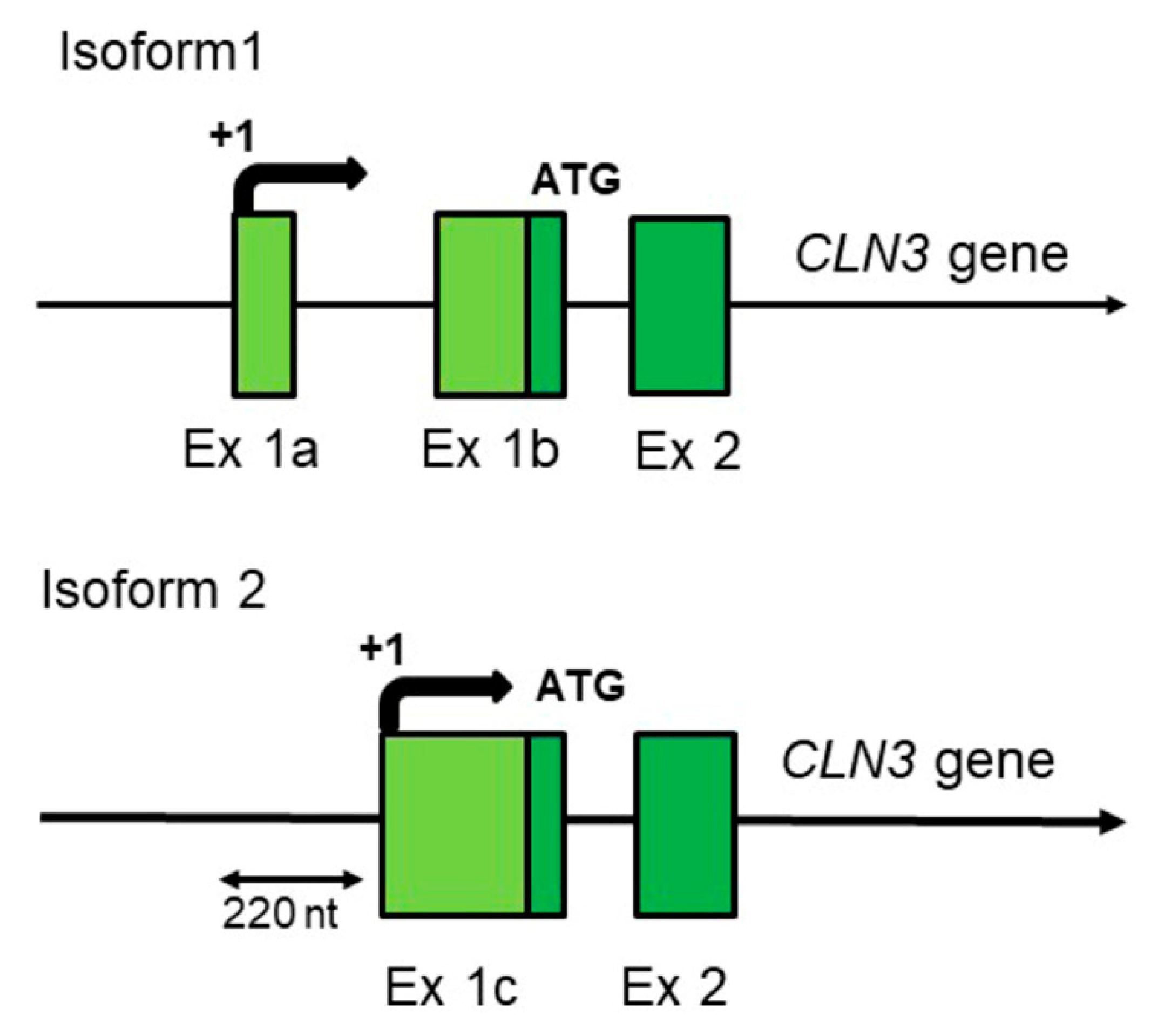
2. Human-Specific Aspects of the CLN3 Gene Structure and Expression Which Could Have Influence on the Disease Progression
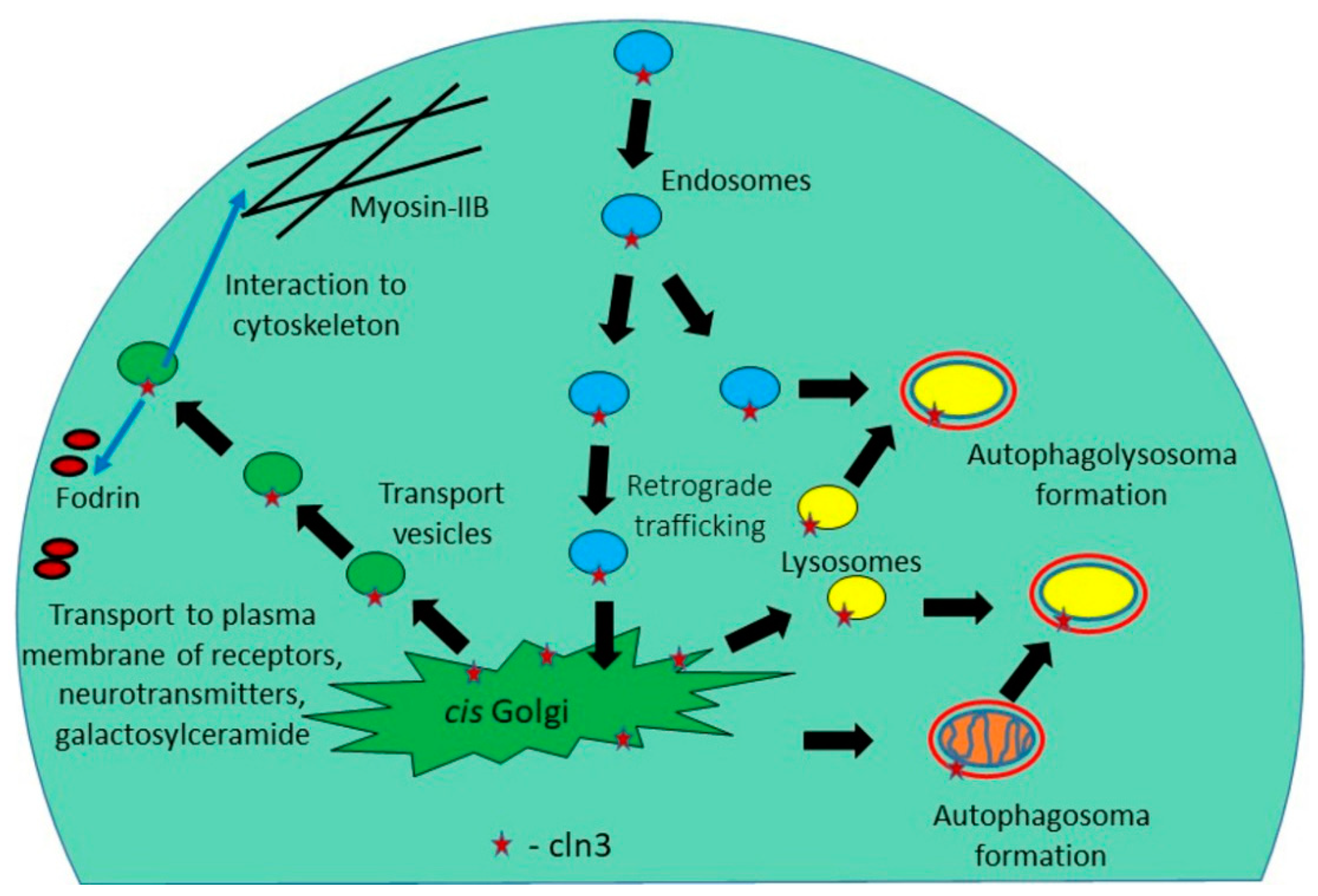
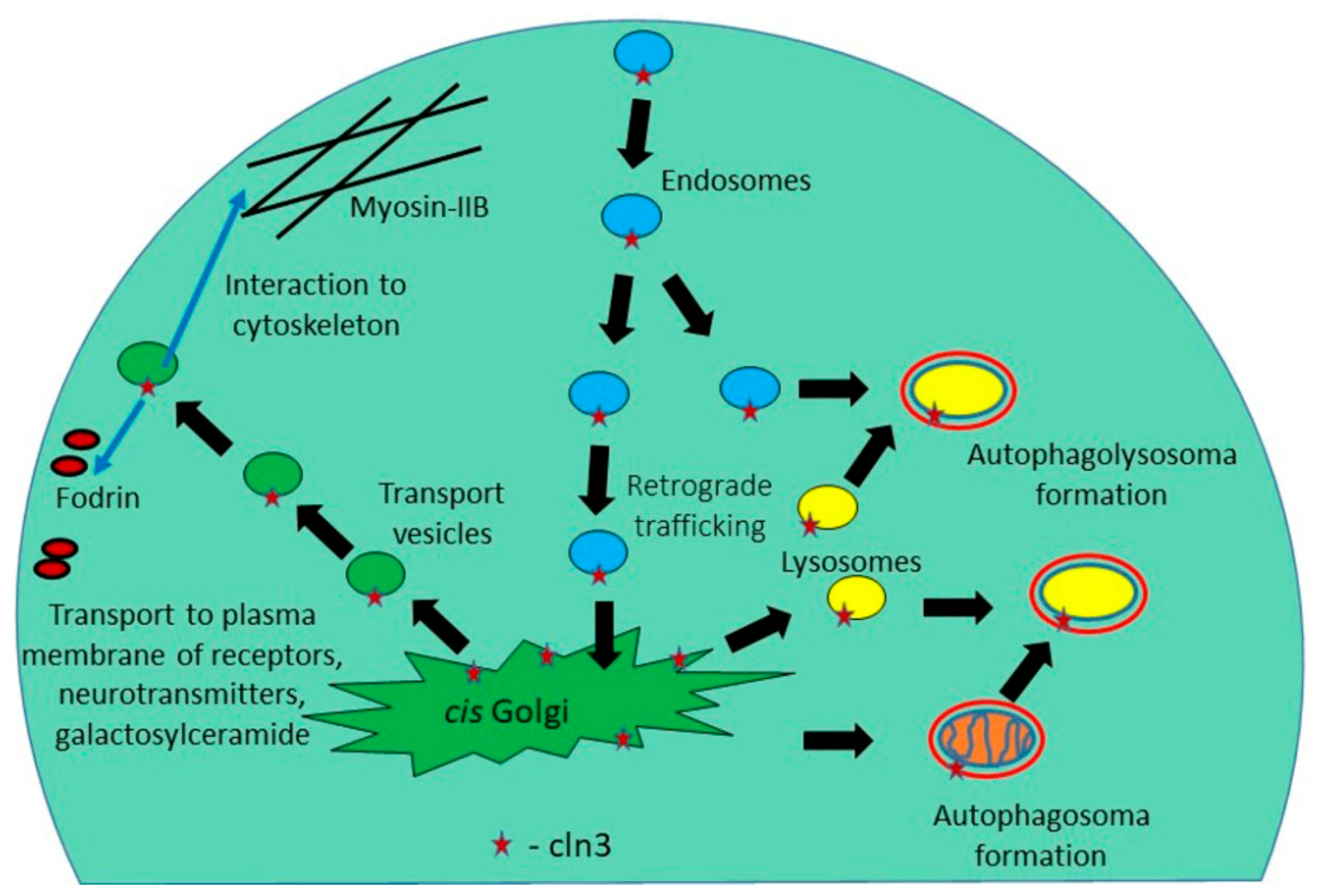
3. The CLN3 Protein Mediates Anterograde and Retrograde Trafficking of Proteins and Other Substances
4. Stress of the Endoplasmic Reticulum (ER Stress)
5. Dysfunction of Mitochondria
6. Reactive Oxygen Species (ROS)
7. pH Homeostasis and Osmoregulation
8. The Hyperactivation of Glial Cells and Astrocytes
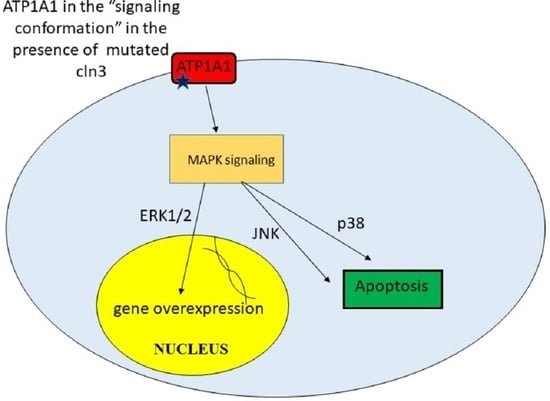
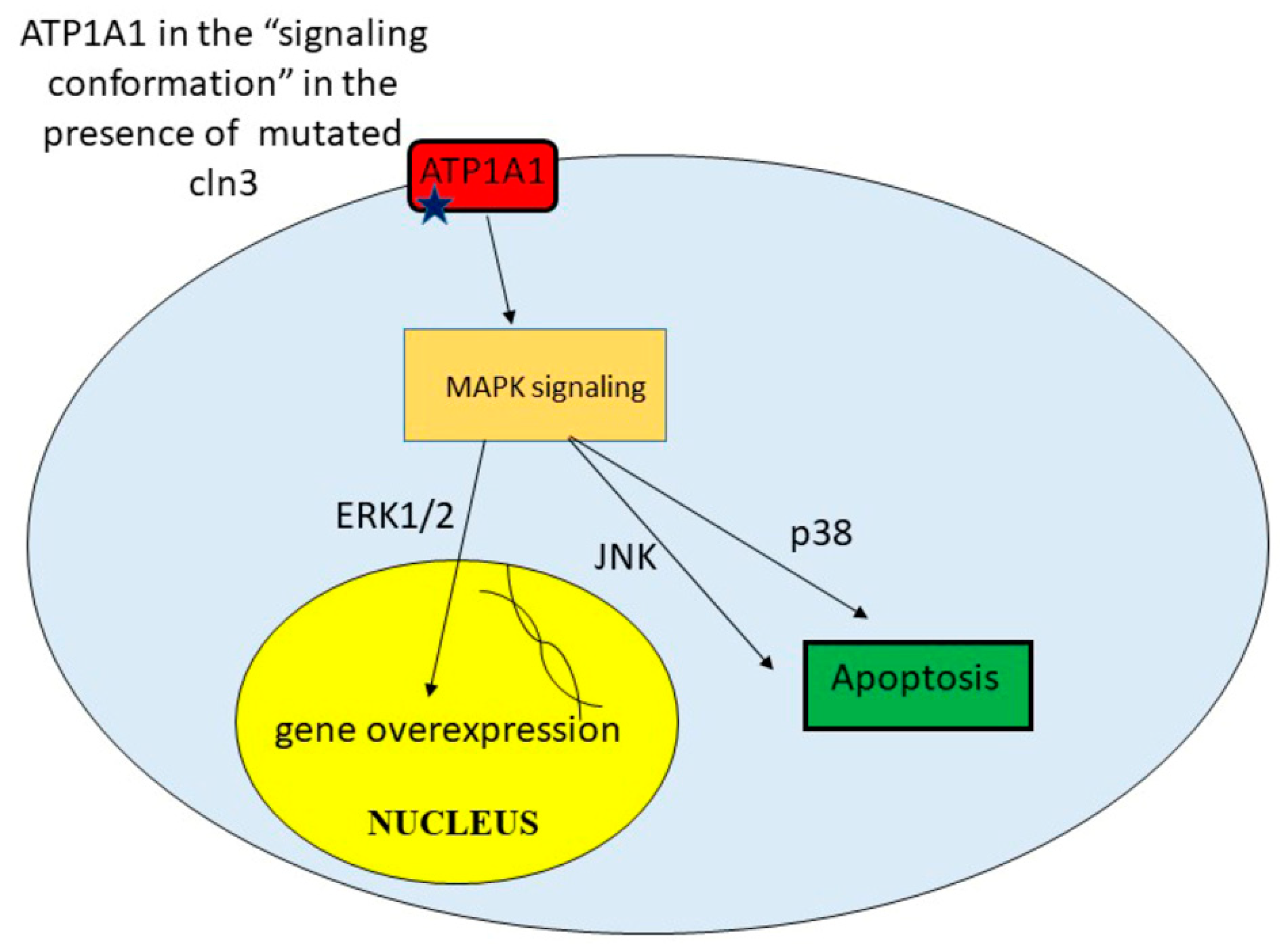
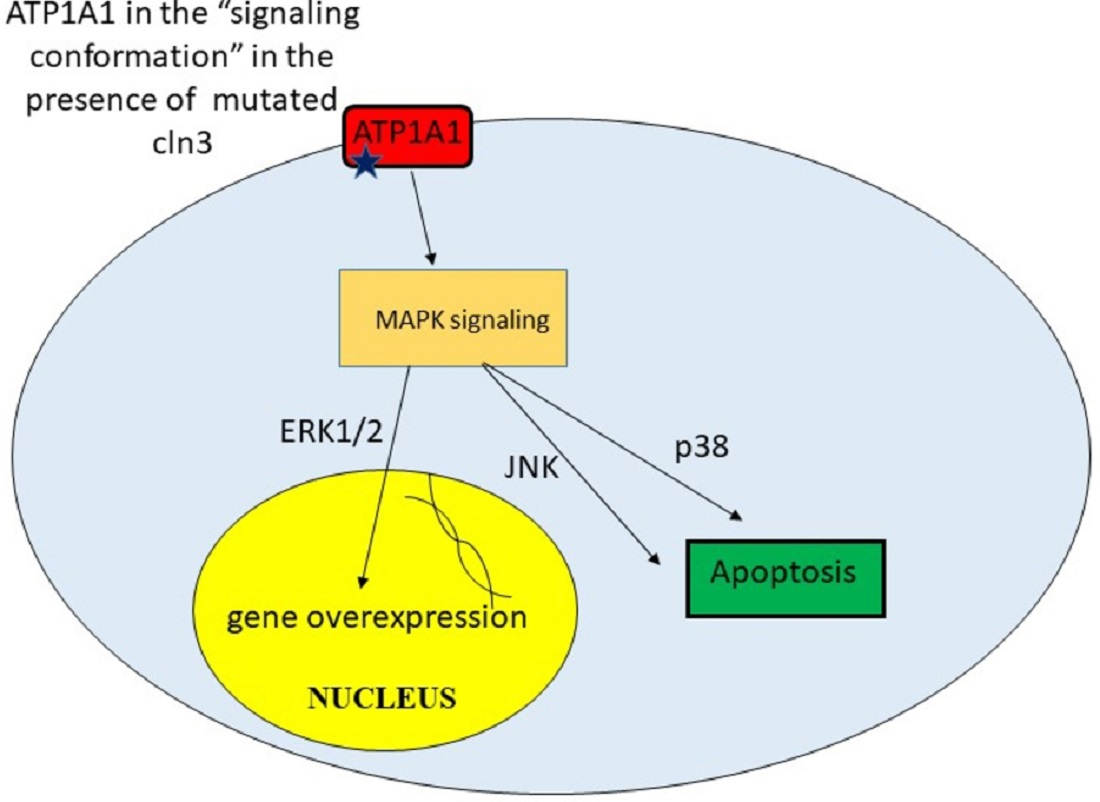
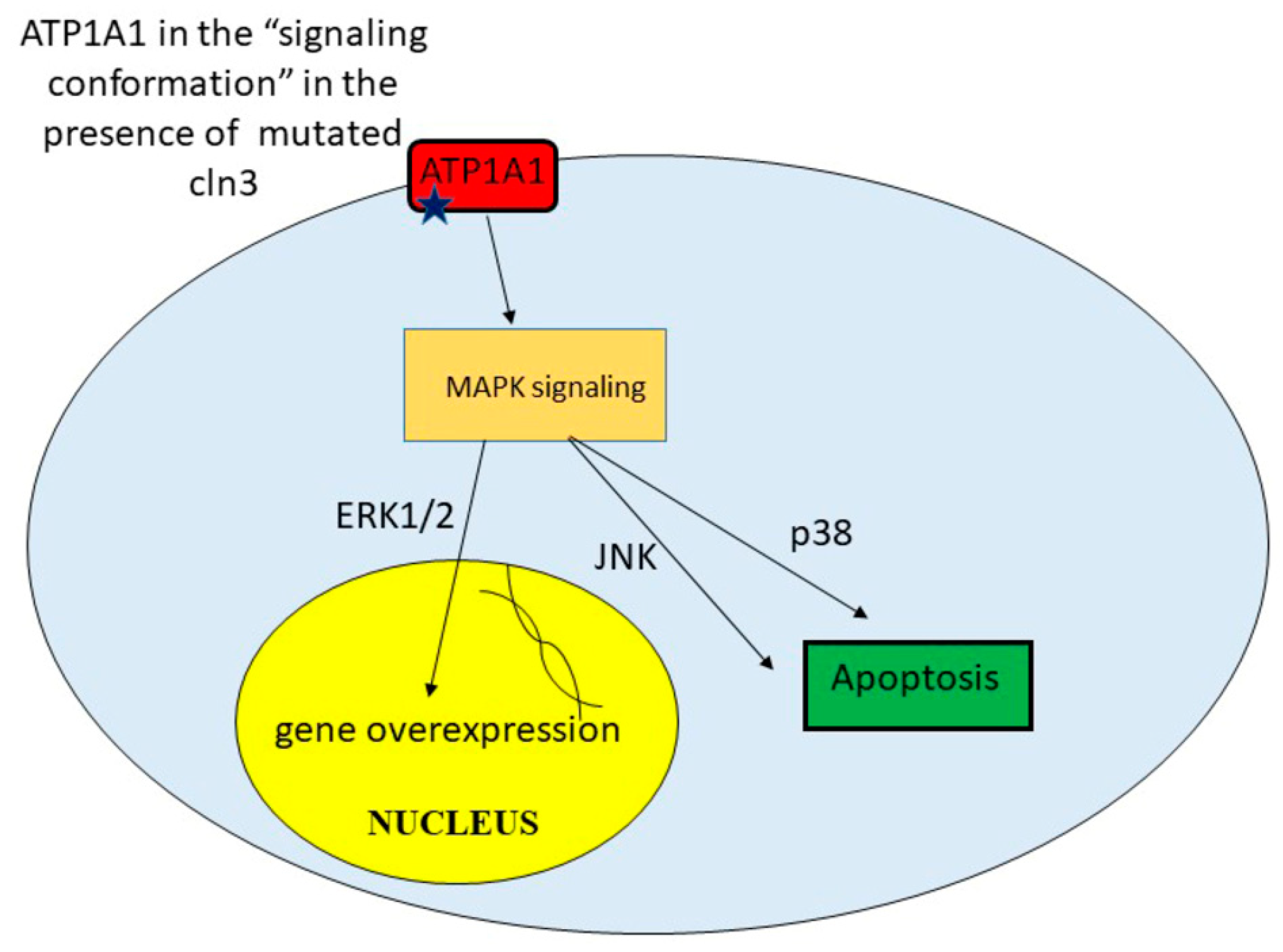
9. Unregulated Activation of Signal Cascades
10. Current Treatment Opportunities
11. Conclusions
Authors Contributions
Funding
Conflicts of Interest
Abbreviations
| ER | Endoplasmic Reticulum |
| JNCL | Juvenile Neuronal Ceroid Lipofuscinosis |
| NCLs | Neuronal Ceroid Lipofuscinoses |
| mPTP | Mitochondrial Permeability Transition Pore |
| ROS | Reactive Oxygen Species |
References
- Ostergaard, J.R. Juvenile neuronal ceroid lipofuscinosis (Batten disease): Current insights. Degener. Neurol. Neuromuscul. Dis. 2016, 6, 73–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mole, S.E.; Cotman, S.L. Genetics of the neuronal ceroid lipofuscinoses (Batten disease). Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 2237–2241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mole, S.E.; Williams, R.E.; Goebel, H.H. Correlations between genotype, ultrastructural morphology and clinical phenotype in the neuronal ceroid lipofuscinoses. Neurogenetics 2005, 6, 107–126. [Google Scholar] [CrossRef] [PubMed]
- Cárcel-Trullols, J.; Kovács, A.D.; Pearce, D.A. Cell biology of the NCL proteins: What they do and don’t do. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 2242–2255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirza, M.; Vainshtein, A.; di Ronza, A.; Chandrachud, U.; Haslett, L.J.; Palmieri, M.; Storch, S.; Groh, J.; Dobzinski, N.; Napolitano, G.; et al. The CLN3 gene and protein: What we know. Mol. Genet. Genom. Med. 2019, 7, e859. [Google Scholar] [CrossRef]
- Marshall, F.J.; de Blieck, E.A.; Mink, J.W.; Dure, L.; Adams, H.; Messing, S.; Rothberg, P.G.; Levy, E.; McDonough, T.; Deyoung, J.; et al. A clinical rating scale for Batten disease: Reliable and relevant for clinical trials. Neurolgy 2005, 65, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Collins, J. Batten disease: Features to facilitate early diagnosis. Br. J. Ophthalmol. 2006, 90, 1119–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouseph, M.M.; Kleinman, M.E.; Wang, Q.J. Vision loss in juvenile neuronal ceroid lipofuscinosis (CLN3 disease). Ann. New York Acad. Sci. 2016, 1371, 55–67. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, A.K.; Drack, A.V.; Ostergaard, J.R. Cataract and Glaucoma Development in Juvenile Neuronal Ceroid Lipofuscinosis (Batten Disease). Ophthalmic Genet. 2014, 36, 39–42. [Google Scholar] [CrossRef]
- Adams, H.R.; Mink, J.W.; University of Rochester Batten Center Study Group. Neurobehavioral Features and Natural History of Juvenile Neuronal Ceroid Lipofuscinosis (Batten Disease). J. Child Neurol. 2013, 28, 1128–1136. [Google Scholar] [CrossRef] [Green Version]
- Kwon, J.M.; Adams, H.; Rothberg, P.G.; Augustine, E.F.; Marshall, F.J.; Deblieck, E.A.; Vierhile, A.; Beck, C.A.; Newhouse, N.J.; Cialone, J.; et al. Quantifying physical decline in juvenile neuronal ceroid lipofuscinosis (Batten disease). Neurology 2011, 77, 1801–1807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claussen, M.; Heim, P.; Knispel, J.; Goebel, H.H.; Kohlschütter, A. Incidence of neuronal ceroid-lipofuscinoses in West Germany: Variation of a method for studying autosomal recessive disorders. Am. J. Med. Genet. 1992, 42, 536–538. [Google Scholar] [CrossRef] [PubMed]
- Mitchison, H.M.; O’Rawe, A.M.; Taschner, P.E.; Sandkuijl, L.A.; Santavuori, P.; de Vos, N.; Breuning, M.H.; Mole, S.E.; Gardiner, R.M.; Järvelä, I.E. Batten disease gene, CLN3: Linkage disequilibrium mapping in the Finnish population, and analysis of European haplotypes. Am. J. Hum. Genet. 1995, 56, 654–662. [Google Scholar] [PubMed]
- Munroe, P.B.; Mitchison, H.M.; O’Rawe, A.M.; Anderson, J.W.; Boustany, R.-M.; Lerner, T.J.; Taschner, P.E.; de Vos, N.; Breuning, M.H.; Gardiner, R.M.; et al. Spectrum of Mutations in the Batten Disease Gene, CLN3. Am. J. Hum. Genet. 1997, 61, 310–316. [Google Scholar] [CrossRef] [Green Version]
- Jarvela, I.; Mitchison, H.M.; Munroe, P.B.; O’Rawe, A.M.; Mole, S.E.; Syvanen, A.C. Rapid diagnostic test for the major mutation underlying Batten disease. J. Med. Genet. 1996, 33, 1041–1042. [Google Scholar] [CrossRef] [Green Version]
- Jilani, A.; Matviychuk, D.; Blaser, S.; Dyack, S.; Mathieu, J.; Prasad, A.N.; Prasad, C.; Kyriakopoulou, L.; Mercimek-Andrews, S. High diagnostic yield of direct Sanger sequencing in the diagnosis of neuronal ceroid lipofuscinoses. JIMD Rep. 2019, 50, 20–30. [Google Scholar] [CrossRef] [Green Version]
- Rietdorf, K.; Coode, E.E.; Schulz, A.; Wibbeler, E.; Bootman, M.D.; Ostergaard, J.R. Cardiac pathology in neuronal ceroid lipofuscinoses (NCL): More than a mere co-morbidity. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165643. [Google Scholar] [CrossRef]
- Radke, J.; Koll, R.; Gill, E.; Wiese, L.; Schulz, A.; Kohlschütter, A.; Schuelke, M.; Hagel, C.; Stenzel, W.; Goebel, H.H. Autophagic vacuolar myopathy is a common feature of CLN3 disease. Ann. Clin. Transl. Neurol. 2018, 5, 1385–1393. [Google Scholar] [CrossRef]
- Lerner, T.J.; Boustany, R.-M.N.; Anderson, J.W.; D’Arigo, K.L.; Schlumpf, K.; Buckler, A.J.; Gusella, J.F.; Haines, J.L. Isolation of a novel gene underlying batten disease, CLN3. Cell 1995, 82, 949–957. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A. The mutational constraint spectrum quantified from variation in 141,456 humans. Nat. Cell Biol. 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Kitzmüller, C.; Haines, R.L.; Codlin, S.; Cutler, D.F.; Mole, S.E. A function retained by the common mutant CLN3 protein is responsible for the late onset of juvenile neuronal ceroid lipofuscinosis. Hum. Mol. Genet. 2007, 17, 303–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janes, R.W.; Munroe, P.B.; Mitchison, H.M.; Gardiner, R.M.; Mole, S.E.; Wallace, B.A. A model for Batten disease protein CLN3: Functional implications from homology and mutations. FEBS Lett. 1996, 399, 75–77. [Google Scholar] [CrossRef] [Green Version]
- Kimura, K.; Wakamatsu, A.; Suzuki, Y.; Ota, T.; Nishikawa, T.; Yamashita, R.; Yamamoto, J.-I.; Sekine, M.; Tsuritani, K.; Wakaguri, H.; et al. Diversification of transcriptional modulation: Large-scale identification and characterization of putative alternative promoters of human genes. Genome Res. 2005, 16, 55–65. [Google Scholar] [CrossRef] [Green Version]
- Grandemange, S.; Schaller, S.; Yamano, S.; du Manoir, S.; Shpakovski, G.V.; Mattei, M.-G.; Kedinger, C.; Vigneron, M. A human RNA polymerase II subunit is encoded by a recently generated multigene family. BMC Mol. Biol. 2001, 2, 14. [Google Scholar] [CrossRef]
- Shpakovski, D.G.; Shematorova, E.K.; Shpakovski, G.V. New Genes on Human Chromosome 7: Bioinformation Analysis of a Cluster of Genes from the POLR2J Family. Russ. J. Bioorganic Chem. 2004, 30, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Benga, W.J.; Grandemange, S.; Shpakovski, G.V.; Shematorova, E.K.; Kedinger, C.; Vigneron, M. Distinct regions of RPB11 are required for heterodimerization with RPB3 in human and yeast RNA polymerase II. Nucleic Acids Res. 2005, 33, 3582–3590. [Google Scholar] [CrossRef] [Green Version]
- Shematorova, E.K.; Shpakovski, D.G.; Shpakovski, G.V. PMS2 and POLR2J gene families as molecular markers of the higher primate’s evolution. Russ. J. Genet. 2010, 46, 1112–1114. [Google Scholar] [CrossRef]
- Shematorova, E.K.; Shpakovski, D.G.; Shpakovski, G.V. Novel complexes of gene expression and their role in the appearance and evolution of the genus Homo. Cell Tissue Biol. 2013, 7, 314–319. [Google Scholar] [CrossRef]
- Lebrun, A.-H.; Moll-Khosrawi, P.; Pohl, S.; Makrypidi, G.; Storch, S.; Kilian, D.; Streichert, T.; Otto, B.; Mole, S.E.; Ullrich, K.; et al. Analysis of Potential Biomarkers and Modifier Genes Affecting the Clinical Course of CLN3 Disease. Mol. Med. 2011, 17, 1253–1261. [Google Scholar] [CrossRef] [PubMed]
- Proshkin, S.A.; Shematorova, E.K.; Souslova, E.A.; Proshkina, G.M.; Shpakovski, G.V. A minor isoform of the human RNA polymerase II subunit hRPB11 (POLR2J) interacts with several components of the translation initiation factor eIF3. Biochemistry 2011, 76, 976–980. [Google Scholar] [CrossRef]
- Proshkin, S.A.; Shematorova, E.K.; Shpakovski, G.V. The Human Isoform of RNA Polymerase II Subunit hRPB11bα Specifically Interacts with Transcription Factor ATF4. Int. J. Mol. Sci. 2019, 21, 135. [Google Scholar] [CrossRef] [Green Version]
- Huber, R.J.; Hughes, S.M.; Liu, W.; Morgan, A.; Tuxworth, R.I.; Russell, C. The contribution of multicellular model organisms to neuronal ceroid lipofuscinosis research. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165614. [Google Scholar] [CrossRef] [PubMed]
- Jolly, R.D.; Martinus, R.D.; Palmer, D.N. Sheep and other animals with ceroid-lipofuscinoses: Their relevance to Batten disease. Am. J. Med Genet. 1992, 42, 609–614. [Google Scholar] [CrossRef]
- Koppang, N. English setter model and juvenile ceroid-lipofuscinosis in man. Am. J. Med Genet. 1992, 42, 599–604. [Google Scholar] [CrossRef]
- Taylor, R.M.; Farrow, B.R.H. Ceroid lipofuscinosis in the Border Collie dog: Retinal lesions in an animal model of juvenile Batten disease. Am. J. Med Genet. 1992, 42, 622–627. [Google Scholar] [CrossRef]
- Johnson, T.B.; Sturdevant, D.A.; White, K.A.; Drack, A.V.; Bhattarai, S.; Rogers, C.; Cooper, J.D.; Pearce, D.A.; Weimer, J.M. Characterization of a novel porcine model of CLN3-Batten disease. Mol. Genet. Metab. 2019, 126, S81. [Google Scholar] [CrossRef]
- Lee, R.L.; Johnson, K.R.; Lerner, T.J. Isolation and Chromosomal Mapping of a Mouse Homolog of the Batten Disease GeneCLN3. Genomics 1996, 35, 617–619. [Google Scholar] [CrossRef] [PubMed]
- Cotman, S.L.; Vrbanac, V.; Lebel, L.-A.; Lee, R.L.; Johnson, K.A.; Donahue, L.-R.; Teed, A.M.; Antonellis, K.; Bronson, R.T.; Lerner, T.J.; et al. Cln3(Deltaex7/8) knock-in mice with the common JNCL mutation exhibit progressive neurologic disease that begins before birth. Hum. Mol. Genet. 2002, 11, 2709–2721. [Google Scholar] [CrossRef]
- Katz, M.L.; Shibuya, H.; Liu, P.C.; Kaur, S.; Gao, C.L.; Johnson, G.S. A mouse gene knockout model for juvenile ceroid-lipofuscinosis (Batten disease). J. Neurosci. Res. 1999, 57, 551–556. [Google Scholar] [CrossRef]
- Mitchison, H.M.; Bernard, D.J.; Greene, N.D.E.; Cooper, J.D.; Junaid, M.A.; Pullarkat, R.K.; de Vos, N.; Breuning, M.H.; Owens, J.W.; Mobley, W.C.; et al. Targeted Disruption of the Cln3 Gene Provides a Mouse Model for Batten Disease. Neurobiol. Dis. 1999, 6, 321–334. [Google Scholar] [CrossRef] [Green Version]
- Mohan, H.; Verhoog, M.B.; Doreswamy, K.K.; Eyal, G.; Aardse, R.; Lodder, B.N.; Goriounova, N.A.; Asamoah, B.; Brakspear, A.C.B.; Groot, C.; et al. Dendritic and Axonal Architecture of Individual Pyramidal Neurons across Layers of Adult Human Neocortex. Cereb. Cortex 2015, 25, 4839–4853. [Google Scholar] [CrossRef] [PubMed]
- Shematorova, E.K.; Shpakovski, D.G.; Chernysheva, A.D.; Shpakovski, G.V. Molecular mechanisms of the juvenile form of Batten disease: Important role of MAPK signaling pathways (ERK1/ERK2, JNK and p38) in pathogenesis of the malady. Biol. Direct. 2018, 13, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kida, E.; Kaczmarski, W.; Golabek, A.; Kaczmarski, A.; Michalewski, M.; Wisniewski, K. Analysis of Intracellular Distribution and Trafficking of the CLN3 Protein in Fusion with the Green Fluorescent Proteinin Vitro. Mol. Genet. Metab. 1999, 66, 265–271. [Google Scholar] [CrossRef]
- Fossale, E.; Wolf, P.; Espinola, J.A.; Lubicz-Nawrocka, T.; Teed, A.M.; Gao, H.; Rigamonti, D.; Cattaneo, E.; MacDonald, M.E.; Cotman, S.L. Membrane trafficking and mitochondrial abnormalities precede subunit c deposition in a cerebellar cell model of juvenile neuronal ceroid lipofuscinosis. BMC Neurosci. 2004, 5, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Ramirez-Montealegre, D.; Pearce, D.A. A role in vacuolar arginine transport for yeast Btn1p and for human CLN3, the protein defective in Batten disease. Proc. Natl. Acad. Sci. USA 2003, 100, 15458–15462. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Espinola, J.A.; Fossale, E.; Massey, A.C.; Cuervo, A.M.; Macdonald, M.E.; Cotman, S.L. Autophagy Is Disrupted in a Knock-in Mouse Model of Juvenile Neuronal Ceroid Lipofuscinosis. J. Biol. Chem. 2006, 281, 20483–20493. [Google Scholar] [CrossRef] [Green Version]
- Sleat, D.E.; Wiseman, J.A.; El-Banna, M.; Zheng, H.; Zhao, C.; Soherwardy, A.; Moore, D.F.; Lobel, P. Analysis of Brain and Cerebrospinal Fluid from Mouse Models of the Three Major Forms of Neuronal Ceroid Lipofuscinosis Reveals Changes in the Lysosomal Proteome. Mol. Cell. Proteom. 2019, 18, 2244–2261. [Google Scholar] [CrossRef]
- Bond, M.E.; Brown, R.; Rallis, C.; Bähler, J.; Mole, S.E. A central role for TOR signalling in a yeast model for juvenile CLN3 disease. Microb. Cell 2015, 2, 466–480. [Google Scholar] [CrossRef] [Green Version]
- Luiro, K.; Kopra, O.; Lehtovirta, M.; Jalanko, A. CLN3 protein is targeted to neuronal synapses but excluded from synaptic vesicles: New clues to Batten disease. Hum. Mol. Genet. 2001, 10, 2123–2131. [Google Scholar] [CrossRef] [Green Version]
- Uusi-Rauva, K.; Luiro, K.; Tanhuanpää, K.; Kopra, O.; Martín-Vasallo, P.; Kyttälä, A.; Jalanko, A. Novel interactions of CLN3 protein link batten disease to dysregulation of fodrin-Na+, K+ ATPase complex. Exp Cell Res. 2008, 314, 2895–2905. [Google Scholar] [CrossRef]
- Getty, A.; Benedict, J.; Pearce, D. A novel interaction of CLN3 with nonmuscle myosin-IIB and defects in cell motility of Cln3−/− cells. Exp. Cell Res. 2011, 317, 51–69. [Google Scholar] [CrossRef] [Green Version]
- Persaud-Sawin, D.-A.; McNamara, J.O.; Rylova, S.N.; Vandongen, A.M.J.; Boustany, R.-M.N. A Galactosylceramide Binding Domain Is Involved in Trafficking of CLN3 from Golgi to Rafts via Recycling Endosomes. Pediatr. Res. 2004, 56, 449–463. [Google Scholar] [CrossRef] [Green Version]
- Rakheja, D.; Narayan, S.B.; Pastor, J.V.; Bennett, M.J. CLN3P, the Batten disease protein, localizes to membrane lipid rafts (detergent-resistant membranes). Biochem. Biophys. Res. Commun. 2004, 317, 988–991. [Google Scholar] [CrossRef]
- Marotta, D.; Tinelli, E.; Mole, S.E. NCLs and ER: A stressful relationship. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1273–1281. [Google Scholar] [CrossRef]
- Wu, D.; Liu, J.; Wu, B.; Tu, B.; Zhu, W.-G.; Luo, J. The Batten disease gene CLN3 confers resistance to endoplasmic reticulum stress induced by tunicamycin. Biochem. Biophys. Res. Commun. 2014, 447, 115–120. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, M.; Jiang, J. Mitochondrial dysfunction in neurodegenerative diseases and drug targets via apoptotic signaling. Mitochondrion 2019, 49, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Zeman, W.; Donahue, S. Fine structure of the lipid bodies in juvenile amaurotic idiocy. Acta Neuropathol. 1963, 3, 144–149. [Google Scholar] [CrossRef]
- Das, A.; Jolly, R.; Kohlschütter, A. Anomalies of Mitochondrial ATP Synthase Regulation in Four Different Types of Neuronal Ceroid Lipofuscinosis. Mol. Genet. Metab. 1999, 66, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Das, A.M.; von Harlem, R.; Feist, M.; Lücke, T.; Kohlschütter, A.; Kohlschütter, A. Altered levels of high-energy phosphate compoundsin fibroblasts from different forms of neuronal ceroid lipofuscinoses: Further evidence for mitochondria) involvement. Eur. J. Paediatr. Neurol. 2001, 5, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Dawson, G.; Kilkus, J.; Siakotos, A.N.; Singh, I. Mitochondrial abnormalities in CLN2 and CLN3 forms of batten disease. Mol. Chem. Neuropathol. 1996, 29, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Palmer, D.N.; Fearnley, I.M.; Walker, J.E.; Hall, N.A.; Lake, B.D.; Wolfe, L.S.; Haltia, M.; Martinus, R.D.; Jolly, R.D. Mitochondrial ATP synthase subunitc storage in the ceroid-lipofuscinoses (Batten disease). Am. J. Med Genet. 1992, 42, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Goebel, H.H. Loss of pigment-laden stellate cells: A severe alteration of the isocortex in juvenile neuronal ceroid-lipofuscinosis. Acta Neuropathol. 1978, 42, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Donet, J.M.; Cárcel-Trullols, J.; Casanova, B.; Aguado, C.; Knecht, E. Alterations in ROS Activity and Lysosomal pH Account for Distinct Patterns of Macroautophagy in LINCL and JNCL Fibroblasts. PLoS ONE 2013, 8, e55526. [Google Scholar] [CrossRef] [Green Version]
- Tuxworth, R.I.; Chen, H.; Vivancos, V.; Carvajal, N.; Huang, X.; Tear, G. The Batten disease gene CLN3 is required for the response to oxidative stress. Hum. Mol. Genet. 2011, 20, 2037–2047. [Google Scholar] [CrossRef] [Green Version]
- Padilla-Lopez, S.; Pearce, D.A. Saccharomyces cerevisiae Lacking Btn1p Modulate Vacuolar ATPase Activity to Regulate pH Imbalance in the Vacuole. J. Biol. Chem. 2006, 281, 10273–10280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathavarajah, S.; McLaren, M.D.; Huber, R.J. Cln3 function is linked to osmoregulation in a Dictyostelium model of Batten disease. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3559–3573. [Google Scholar] [CrossRef] [PubMed]
- Getty, A.; Kovács, A.D.; Lengyel-Nelson, T.; Cardillo, A.; Hof, C.; Chan, C.-H.; Pearce, D.A. Osmotic Stress Changes the Expression and Subcellular Localization of the Batten Disease Protein CLN3. PLoS ONE 2013, 8, e66203. [Google Scholar] [CrossRef] [Green Version]
- Stein, C.S.; Yancey, P.H.; Martins, I.; Sigmund, R.D.; Stokes, J.B.; Davidson, B.L. Osmoregulation of ceroid neuronal lipofuscinosis type 3 in the renal medulla. Am. J. Physiol. Physiol. 2010, 298, C1388–C1400. [Google Scholar] [CrossRef]
- Tecedor, L.; Stein, C.S.; Schultz, M.L.; Farwanah, H.; Sandhoff, K.; Davidson, B.L. CLN3 Loss Disturbs Membrane Microdomain Properties and Protein Transport in Brain Endothelial Cells. J. Neurosci. 2013, 33, 18065–18079. [Google Scholar] [CrossRef] [Green Version]
- Xiong, J.; Kielian, T. Microglia in juvenile neuronal ceroid lipofuscinosis are primed toward a pro-inflammatory phenotype. J. Neurochem. 2013, 127, 245–258. [Google Scholar] [CrossRef]
- Lange, J.; Haslett, L.J.; Lloyd-Evans, E.; Pocock, J.M.; Sands, M.S.; Williams, B.P.; Cooper, J.D. Compromised astrocyte function and survival negatively impact neurons in infantile neuronal ceroid lipofuscinosis. Acta Neuropathol. Commun. 2018, 6, 74. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.-W.; Choi, H.; Kim, H.-J.; Jo, D.-G.; Jeon, Y.-J.; Noh, J.-Y.; Park, W.J.; Jung, Y.-K. Neuronal vulnerability of CLN3 deletion to calcium-induced cytotoxicity is mediated by calsenilin. Hum. Mol. Genet. 2006, 16, 317–326. [Google Scholar] [CrossRef] [Green Version]
- Cabral-Miranda, F.; Hetz, C. ER Stress and neurodegenerative disease: A cause or effect relationship? Curr. Top Microbiol. Immunol. 2018, 414, 131–157. [Google Scholar]
- di Ronza, A.; Bajaj, L.; Sharma, J.; Sanagasetti, D.; Lotfi, P.; Adamski, C.J.; Collette, J.; Palmieri, M.; Amawi, A.; Popp, L.; et al. CLN8 is an endoplasmic reticulum cargo receptor that regulates lysosome biogenesis. Nat. Cell Biol. 2018, 20, 1370–1377. [Google Scholar] [CrossRef]
- Huber, R.J.; Mathavarajah, S.; Yap, S.Q. Mfsd8 localizes to endocytic compartments and influences the secretion of Cln5 and cathepsin D in Dictyostelium. Cell. Signal. 2020, 70, 109572. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxidative Med. Cell. Longev. 2016, 2016, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Tejera, D.; Heneka, M.T. Microglia in Neurodegenerative Disorders. Pheromone Signal. 2019, 2034, 57–67. [Google Scholar] [CrossRef]
- Tian, J.; Cai, T.; Yuan, Z.; Wang, H.; Liu, L.; Haas, M.; Maksimova, E.; Huang, X.Y.; Xie, Z.J. Binding of Src to Na+/K+-ATPase forms a functional signaling complex. Mol. Biol Cell. 2006, 17, 317–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, D. Ammonium Activates Ouabain-Activated Signalling Pathway in Astrocytes: Therapeutic Potential of Ouabain Antagonist. Curr. Neuropharmacol. 2014, 12, 334–341. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Ye, Q.; Liu, C.; Xie, J.X.; Yan, Y.; Lai, F.; Duan, Q.; Li, X.; Tian, J.; Xie, Z. Involvement of Na/K-ATPase in hydrogen peroxide-induced activation of the Src/ERK pathway in LLC-PK1 cells. Free Radic. Biol. Med. 2014, 71, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Haas, M.; Wang, H.; Tian, J.; Xie, Z. Src-mediated inter-receptor cross-talk between the Na+/K+-ATPase and the epidermal growth factor receptor relays the signal from ouabain to mitogen-activated protein kinases. J. Biol. Chem. 2002, 277, 18694–18702. [Google Scholar] [CrossRef] [Green Version]
- Xia, Z.; Dickens, M.; Raingeaud, J.L.; Davis, R.J.; Greenberg, M.E. Opposing Effects of ERK and JNK-p38 MAP Kinases on Apoptosis. Science 1995, 270, 1326–1331. [Google Scholar] [CrossRef] [PubMed]
- Huber, R.J.; Mathavarajah, S. Comparative transcriptomics reveals mechanisms underlying cln3-deficiency phenotypes in Dictyostelium. Cell. Signal. 2019, 58, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Huber, R.J. Loss of Cln3 impacts protein secretion in the social amoeba Dictyostelium. Cell. Signal. 2017, 35, 61–72. [Google Scholar] [CrossRef]
- Cooper, J.D.; Mole, S.E. Future perspectives: What lies ahead for Neuronal Ceroid Lipofuscinosis research? Biochim. Biophys. Acta Mol. Basis Dis. 2020, 8, 165681. [Google Scholar] [CrossRef]
- Liu, W.; Kleine-Holthaus, S.-M.; Herranz-Martin, S.; Aristorena, M.; Mole, S.E.; Smith, A.J.; Ali, R.R.; Rahim, A.A. Experimental gene therapies for the NCLs. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165772. [Google Scholar] [CrossRef]
- Holthaus, S.-M.K.; Herranz-Martin, S.; Massaro, G.; Aristorena, M.; Hoke, J.; Hughes, M.P.; Maswood, R.; Semenyuk, O.; Basche, M.; Shah, A.Z.; et al. Neonatal brain-directed gene therapy rescues a mouse model of neurodegenerative CLN6 Batten disease. Hum. Mol. Genet. 2019, 28, 3867–3879. [Google Scholar] [CrossRef]
- Holthaus, S.-M.K.; Ribeiro, J.; Abelleira-Hervas, L.; Pearson, R.A.; Duran, Y.; Georgiadis, A.; Sampson, R.D.; Rizzi, M.; Hoke, J.; Maswood, R.; et al. Prevention of Photoreceptor Cell Loss in a Cln6 Mouse Model of Batten Disease Requires CLN6 Gene Transfer to Bipolar Cells. Mol. Ther. 2018, 26, 1343–1353. [Google Scholar] [CrossRef] [Green Version]
- Schulz, A.; Ajayi, T.; Specchio, N.; Reyes, E.D.L.; Gissen, P.; Ballon, D.J.; Dyke, J.P.; Cahan, H.; Slasor, P.; Jacoby, D.; et al. Study of Intraventricular Cerliponase Alfa for CLN2 Disease. New Engl. J. Med. 2018, 378, 1898–1907. [Google Scholar] [CrossRef]
- NCT03770572: Gene Transfer Study of AAV9-CLN3 for Treatment NCL Type 3. Available online: https://clinicaltrials.gov/ct2/show/NCT03770572?cond=CLN3&rank=1 (accessed on 22 November 2019).
- Berns, K.I.; Muzyczka, N. AAV: An Overview of Unanswered Questions. Hum. Gene Ther. 2017, 28, 308–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Hu, C.; el Achkar, C.M.; Black, L.E.; Douville, J.; Larson, A.; Pendergast, M.K.; Goldkind, S.F.; Lee, E.A.; Kuniholm, A.; et al. Patient-Customized Oligonucleotide Therapy for a Rare Genetic Disease. New Engl. J. Med. 2019, 381, 1644–1652. [Google Scholar] [CrossRef] [PubMed]
- Tarczyluk-Wells, M.A.; Salzlechner, C.; Najafi, A.R.; Lim, M.J.; Smith, D.; Platt, F.M.; Williams, B.P.; Cooper, J.D. Combined Anti-inflammatory and Neuroprotective Treatments Have the Potential to Impact Disease Phenotypes in Cln3−/− Mice. Front. Neurol. 2019, 10, 963. [Google Scholar] [CrossRef] [Green Version]
- Groh, J.; Berve, K.; Martini, R. Fingolimod and Teriflunomide Attenuate Neurodegeneration in Mouse Models of Neuronal Ceroid Lipofuscinosis. Mol. Ther. 2017, 25, 1889–1899. [Google Scholar] [CrossRef] [Green Version]
- Makoukji, J.; Saadeh, F.; Mansour, K.A.; Al Ali, J.; Kinarivala, N.; Trippier, P.C.; El-Sitt, S.; Boustany, R.-M. Flupirtine derivatives as potential treatment for the neuronal ceroid lipofuscinoses. Ann. Clin. Transl. Neurol. 2018, 5, 1089–1103. [Google Scholar] [CrossRef] [Green Version]
- Hong, M.; Song, K.D.; Lee, H.-K.; Yi, S.; Lee, Y.S.; Heo, T.-H.; Jun, H.S.; Kim, S.-J. Fibrates inhibit the apoptosis of Batten disease lymphoblast cells via autophagy recovery and regulation of mitochondrial membrane potential. Vitr. Cell. Dev. Biol. Anim. 2015, 52, 349–355. [Google Scholar] [CrossRef]
- Augustine, E.F.; Beck, C.A.; Adams, H.R.; Defendorf, S.; Vierhile, A.; Timm, D.; Weimer, J.M.; Mink, J.W.; Marshall, F.J. Short-Term Administration of Mycophenolate Is Well-Tolerated in CLN3 Disease (Juvenile Neuronal Ceroid Lipofuscinosis). JIMD Rep. 2018, 43, 117–124. [Google Scholar] [CrossRef]
- Margraf, L.R.; Boriack, R.L.; Routheut, A.A.; Cuppen, I.; AlHilali, L.; Bennett, C.J.; Bennett, M.J. Tissue Expression and Subcellular Localization of CLN3, the Batten Disease Protein. Mol. Genet. Metab. 1999, 66, 283–289. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Physiological Changes in cln3− Cells and/or the Cellular System Affected | Probable Cause of the Cell Death | References |
|---|---|---|
| Endosomal-lysosomal system, autophagy | Defects in traffic of proteins, cholesterol, sphingolipids, neurotransmitters. Accumulation of damaged organelles | [43,44,45,46,47,48,49,50,51,52,53] |
| Stress of endoplasmic reticulum (ER stress) | Accumulation of misfolded proteins and, as a consequence, apoptosis | [54,55] |
| Dysfunction of mitochondria | Deficiency in energy-rich phosphates ATP and ADP | [56,57,58,59,60,61,62] |
| Reactive oxygen species (ROS) | Damage of proteins, nucleic acids, lipids, membranes and organelles, as a consequence — apoptosis | [63,64] |
| pH homeostasis and osmoregulation | Сell destruction due to the inability to throw out excess of water | [65,66,67,68,69] |
| The hyperactivation of glial cells and astrocytes | Stimulation of inflammation | [70,71] |
| Upregulated activation of signal cascades | Excessive amount of some proteins leads to toxicity. Apoptosis | [42] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shematorova, E.K.; Shpakovski, G.V. Current Insights in Elucidation of Possible Molecular Mechanisms of the Juvenile Form of Batten Disease. Int. J. Mol. Sci. 2020, 21, 8055. https://doi.org/10.3390/ijms21218055
Shematorova EK, Shpakovski GV. Current Insights in Elucidation of Possible Molecular Mechanisms of the Juvenile Form of Batten Disease. International Journal of Molecular Sciences. 2020; 21(21):8055. https://doi.org/10.3390/ijms21218055
Chicago/Turabian StyleShematorova, Elena K., and George V. Shpakovski. 2020. "Current Insights in Elucidation of Possible Molecular Mechanisms of the Juvenile Form of Batten Disease" International Journal of Molecular Sciences 21, no. 21: 8055. https://doi.org/10.3390/ijms21218055
APA StyleShematorova, E. K., & Shpakovski, G. V. (2020). Current Insights in Elucidation of Possible Molecular Mechanisms of the Juvenile Form of Batten Disease. International Journal of Molecular Sciences, 21(21), 8055. https://doi.org/10.3390/ijms21218055