Treating Senescence like Cancer: Novel Perspectives in Senotherapy of Chronic Diseases

 , , ,
, , ,  ,
, 
Abstract
1. Introduction
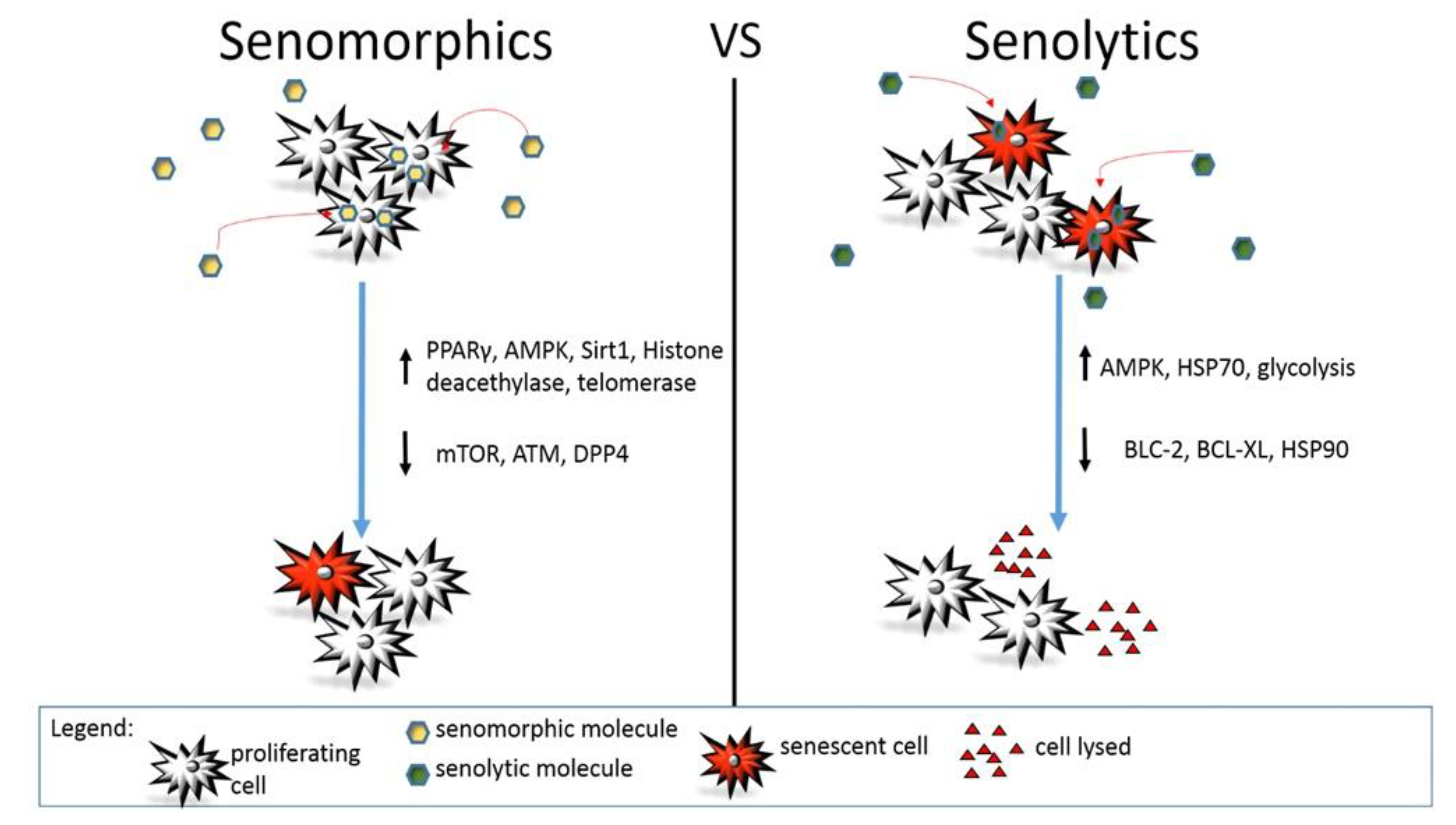
2. Senomorphics
3. Senolytics
4. Senotherapeutic Interventions in Models of Age-Related Diseases
4.1. Cardiovascular Diseases
4.2. Respiratory Diseases
4.3. Neurocognitive Diseases
4.4. Type 2 Diabetes
4.5. About Ongoing Clinical Trials
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L. The Cell Biology of Aging. J. Investig. Dermatol. 1979, 73, 8–14. [Google Scholar] [CrossRef][Green Version]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic Ras Provokes Premature Cell Senescence Associated with Accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Bornstein, R.; Gonzalez, B.; Johnson, S.C. Mitochondrial Pathways in Human Health and Aging. Mitochondrion 2020, 54, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Okuno, K.; Cicalese, S.; Elliott, K.J.; Kawai, T.; Hashimoto, T.; Eguchi, S. Targeting Molecular Mechanism of Vascular Smooth Muscle Senescence Induced by Angiotensin II, A Potential Therapy via Senolytics and Senomorphics. Int. J. Mol. Sci. 2020, 21, 6579. [Google Scholar] [CrossRef] [PubMed]
- Lozano-Torres, B.; Estepa-Fernández, A.; Rovira, M.; Orzáez, M.; Serrano, M.; Martínez-Máñez, R.; Sancenón, F. The Chemistry of Senescence. Nat. Rev. Chem. 2019, 3, 426–441. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. The DNA Damage Response and Cancer Therapy. Nat. Cell Biol. 2012, 481, 287–294. [Google Scholar] [CrossRef]
- Rossi, D.J.; Jamieson, C.H.; Weissman, I.L. Stems Cells and the Pathways to Aging and Cancer. Cell 2008, 132, 681–696. [Google Scholar] [CrossRef]
- Cao, K.; Blair, C.D.; Faddah, D.A.; Kieckhaefer, J.E.; Olive, M.; Erdos, M.R.; Nabel, E.G.; Collins, F.S. Progerin and Telomere Dysfunction Collaborate to Trigger Cellular Senescence in Normal Human Fibroblasts. J. Clin. Investig. 2011, 121, 2833–2844. [Google Scholar] [CrossRef]
- Vulliamy, T.; Marrone, A.; Goldman, F.; Dearlove, A.; Bessler, M.; Mason, P.J.; Dokal, I. The RNA component of telomerase is mutated in autosomal dominant dyskeratosis congenita. Nat. Cell Biol. 2001, 413, 432–435. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, J.; Liu, Y.; Zhang, S.; Wang, Y.; Liu, B.; Liu, H.; Li, R.; Lv, C.; Song, X. Regulation of TERRA on Telomeric and Mitochondrial Functions in IPF Pathogenesis. BMC Pulm. Med. 2017, 17, 163. [Google Scholar] [CrossRef] [PubMed]
- Alter, B.P.; Rosenberg, P.S.; Giri, N.; Baerlocher, G.M.; Lansdorp, P.M.; Savage, S.A. Telomere length is associated with disease severity and declines with age in dyskeratosis congenita. Haematologica 2011, 97, 353–359. [Google Scholar] [CrossRef]
- Han, S.; Brunet, A. Histone Methylation Makes its Mark on Longevity. Trends Cell Biol. 2012, 22, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Rondinelli, B.; Schwerer, H.; Antonini, E.; Gaviraghi, M.; Lupi, A.; Frenquelli, M.; Cittaro, D.; Segalla, S.; Lemaitre, J.-M.; Tonon, G. H3K4me3 Demethylation by the Histone Demethylase KDM5C/JARID1C Promotes DNA Replication Origin Firing. Nucleic Acids Res. 2015, 43, 2560–2574. [Google Scholar] [CrossRef]
- Chisholm, N.C.; Henderson, M.L.; Selvamani, A.; Park, M.J.; Dindot, S.; Miranda, R.C.; Sohrabji, F. Histone Methylation Patterns in Astrocytes are Influenced by Age Following Ischemia. Epigenetics 2015, 10, 142–152. [Google Scholar] [CrossRef]
- Bahar, R.; Hartmann, C.H.; Rodriguez, K.A.; Denny, A.D.; Busuttil, R.A.; Dollé, M.E.T.; Calder, R.B.; Chisholm, G.B.; Pollock, B.H.; Klein, C.A.; et al. Increased Cell-to-Cell Variation in Gene Expression in Ageing Mouse Heart. Nat. Cell Biol. 2006, 441, 1011–1014. [Google Scholar] [CrossRef]
- Iske, J.; Seyda, M.; Heinbokel, T.; Maenosono, R.; Minami, K.; Nian, Y.; Quante, M.; Falk, C.S.; Azuma, H.; Martin, F.; et al. Senolytics Prevent mt-DNA-Induced Inflammation and Promote the Survival of Aged Organs Following Transplantation. Nat. Commun. 2020, 11, 4289. [Google Scholar] [CrossRef]
- De Magalhães, J.P.; Curado, J.; Church, G.M. Meta-Analysis of Age-Related Gene Expression Profiles Identifies Common Signatures of Aging. Bioinformatics 2009, 25, 875–881. [Google Scholar] [CrossRef]
- Koga, H.; Kaushik, S.; Cuervo, A.M. Protein Homeostasis and Aging: The Importance of Exquisite Quality Control. Ageing Res. Rev. 2011, 10, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Fedintsev, A.; Moskalev, A. Stochastic Non-Enzymatic Modification of Long-Lived Macromolecules—A Missing Hallmark of Aging. Ageing Res. Rev. 2020, 62, 101097. [Google Scholar] [CrossRef] [PubMed]
- Pospich, S.; Stefan, R. The Molecular Basis of Alzheimer’s Plaques. Science 2017, 358, 45–46. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.-P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.N.; Nelson, P.S.; Desprez, P.-Y.; Campisi, J. Senescence-Associated Secretory Phenotypes Reveal Cell-Nonautonomous Functions of Oncogenic RAS and the p53 Tumor Suppressor. PLoS Biol. 2008, 6, e301–e368. [Google Scholar] [CrossRef] [PubMed]
- Rodier, F.; Campisi, J. Four Faces of Cellular Senescence. J. Cell Biol. 2011, 192, 547–556. [Google Scholar] [CrossRef]
- Panwar, P.; Butler, G.S.; Jamroz, A.; Azizi, P.; Overall, C.M.; Bromme, D. Aging-associated modifications of collagen affect its degradation by matrix metalloproteinases. J. Int. Soc. Matrix Biol. 2018, 65, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, S.D.; Endicott, S.K.; Province, M.A.; Pierce, J.A.; Campbell, E.J. Marked Longevity of Human Lung Parenchymal Elastic Fibers Deduced from Prevalence of D-Aspartate and Nuclear Weapons-Related Radiocarbon. J. Clin. Investig. 1991, 87, 1828–1834. [Google Scholar] [CrossRef]
- Ritz-Timme, S.; Laumeier, I.; Collins, M.J. Aspartic Acid Racemization: Evidence for Marked Longevity of Elastin in Human Skin. Br. J. Dermatol. 2003, 149, 951–959. [Google Scholar] [CrossRef]
- Imayama, S.; Braverman, I.M. A Hypothetical Explanation for the Aging of Skin. Chronologic Alteration of the Three-Dimensional Arrangement of Collagen and Elastic Fibers in Connective Tissue. Am. J. Pathol. 1989, 134, 1019–1025. [Google Scholar]
- Mongelli, A.; Gaetano, C. Controversial Impact of Sirtuins in Chronic Non-Transmissible Diseases and Rehabilitation Medicine. Int. J. Mol. Sci. 2018, 19, 3080. [Google Scholar] [CrossRef]
- Valenzano, D.R.; Terzibasi, E.; Genade, T.; Cattaneo, A.; Domenici, L.; Cellerino, A. Resveratrol Prolongs Lifespan and Retards the Onset of Age-Related Markers in a Short-Lived Vertebrate. Curr. Biol. 2006, 16, 296–300. [Google Scholar] [CrossRef]
- Ye, K.; Ji, C.-B.; Lu, X.-W.; Ni, Y.-H.; Gao, C.-L.; Chen, X.-H.; Zhao, Y.-P.; Gu, G.-X.; Guo, X. Resveratrol Attenuates Radiation Damage in Caenorhabditis elegans by Preventing Oxidative Stress. J. Radiat. Res. 2010, 51, 473–479. [Google Scholar] [CrossRef]
- Muhammad, M.H.; Allam, M.M. Resveratrol and/or Exercise Training Counteract Aging-Associated Decline of Physical Endurance in Aged Mice; Targeting Mitochondrial Biogenesis and Function. J. Physiol. Sci. 2017, 68, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Barger, J.L.; Kayo, T.; Vann, J.M.; Arias, E.B.; Wang, J.; Hacker, T.A.; Wang, Y.; Raederstorff, D.; Morrow, J.D.; Leeuwenburgh, C.; et al. A Low Dose of Dietary Resveratrol Partially Mimics Caloric Restriction and Retards Aging Parameters in Mice. PLoS ONE 2008, 3, e2264. [Google Scholar] [CrossRef]
- Wang, N.; Luo, Z.; Jin, M.; Sheng, W.; Wang, H.-T.; Long, X.; Wu, Y.; Hu, P.; Xu, H.; Zhang, X. Exploration of Age-Related Mitochondrial Dysfunction and the Anti-Aging Effects of Resveratrol in Zebrafish Retina. Aging 2019, 11, 3117–3137. [Google Scholar] [CrossRef]
- Pallauf, K.; Chin, D.; Gunther, I.; Birringer, M.; Luersen, K.; Schultheiss, G.; Vieten, S.; Krauss, J.; Bracher, F.; Danylec, N.; et al. Resveratrol, Lunularin and Dihydroresveratrol do not Act as Caloric Restriction Mimetics when Administered Intraperitoneally in Mice. Sci. Rep. 2019, 9, 4445. [Google Scholar] [CrossRef]
- Pallauf, K.; Rimbach, G. Resveratrol and Lifespan in Model Organisms. Curr. Med. Chem. 2016, 23, 4639–4680. [Google Scholar] [CrossRef] [PubMed]
- Van der Made, S.M.; Plat, J.; Mensink, R.P. Resveratrol does not Influence Metabolic Risk Markers Related to Cardiovascular Health in Overweight and Slightly Obese Subjects: A Randomized, Placebo-Controlled Crossover Trial. PLoS ONE 2015, 10, e0118393. [Google Scholar] [CrossRef]
- Timmers, S.; Konings, E.; Bilet, L.; Houtkooper, R.H.; van de Weijer, T.; Goossens, G.H.; Hoeks, J.; van der Krieken, S.; Ryu, D.; Kersten, S.; et al. Calorie Restriction-Like Effects of 30 Days of Resveratrol Supplementation on Energy Metabolism and Metabolic Profile in Obese Humans. Cell Metab. 2011, 14, 612–622. [Google Scholar] [CrossRef]
- Kirkland, J.L.; Tchkonia, T. Cellular Senescence: A Translational Perspective. EBioMedicine 2017, 21, 21–28. [Google Scholar] [CrossRef]
- Kirkland, J.L.; Tchkonia, T.; Zhu, Y.; Niedernhofer, L.J.; Robbins, P.D. The Clinical Potential of Senolytic Drugs. J. Am. Geriatr. Soc. 2017, 65, 2297–2301. [Google Scholar] [CrossRef]
- Xiong, S.; Patrushev, N.; Forouzandeh, F.; Hilenski, L.; Alexander, W.R. PGC-1α Modulates Telomere Function and DNA Damage in Protecting against Aging-Related Chronic Diseases. Cell Rep. 2015, 12, 1391–1399. [Google Scholar] [CrossRef]
- Hofmann, J.W.; Zhao, X.; De Cecco, M.; Peterson, A.L.; Pagliaroli, L.; Manivannan, J.; Hubbard, G.B.; Ikeno, Y.; Zhang, Y.; Feng, B.; et al. Reduced Expression of MYC Increases Longevity and Enhances Healthspan. Cell 2015, 160, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Laberge, R.-M.; Zhou, L.; Sarantos, M.R.; Rodier, F.; Freund, A.; De Keizer, P.L.J.; Liu, S.; DeMaria, M.; Cong, Y.-S.; Kapahi, P.; et al. Glucocorticoids suppress selected components of the senescence-associated secretory phenotype. Aging Cell 2012, 11, 569–578. [Google Scholar] [CrossRef]
- Liu, S.; Uppal, H.; DeMaria, M.; Desprez, P.-Y.; Campisi, J.; Kapahi, P. Simvastatin Suppresses Breast Cancer Cell Proliferation Induced by Senescent Cells. Sci. Rep. 2015, 5, 17895. [Google Scholar] [CrossRef]
- Correia-Melo, C.; Marques, F.D.M.; Anderson, R.; Hewitt, G.; Hewitt, R.; Cole, J.; Carroll, B.M.; Miwa, S.; Birch, J.; Merz, A.; et al. Mitochondria are Required for Pro-Ageing Features of the Senescent Phenotype. EMBO J. 2016, 35, 724–742. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.E.; Strong, R.; Allison, D.B.; Ames, B.N.; Astle, C.M.; Atamna, H.; Fernandez, E.; Flurkey, K.; Javors, M.A.; Nadon, N.L.; et al. Acarbose, 17-α-estradiol, and Nordihydroguaiaretic Acid Extend Mouse Lifespan Preferentially in Males. Aging Cell 2013, 13, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Shen, W.J.; Cortez, Y.; Kraemer, F.B.; Azhar, S. Nordihydroguaiaretic Acid Improves Metabolic Dysregulation and Aberrant Hepatic Lipid Metabolism in Mice by both PPARalpha-Dependent and -Independent Pathways. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, G72–G86. [Google Scholar] [CrossRef][Green Version]
- Perez, E.; Liu, R.; Yang, S.-H.; Cai, Z.Y.; Covey, D.F.; Simpkins, J.W. Neuroprotective effects of an estratriene analog are estrogen receptor independent in vitro and in vivo. Brain Res. 2005, 1038, 216–222. [Google Scholar] [CrossRef]
- Miller, R.A.; Harrison, D.E.; Astle, C.M.; Fernandez, E.; Flurkey, K.; Han, M.; Javors, M.A.; Li, X.; Nadon, N.L.; Nelson, J.F.; et al. Rapamycin-mediated lifespan increase in mice is dose and sex dependent and metabolically distinct from dietary restriction. Aging Cell 2014, 13, 468–477. [Google Scholar] [CrossRef]
- Weir, H.J.; Yao, P.; Huynh, F.K.; Escoubas, C.C.; Goncalves, R.L.; Burkewitz, K.; Laboy, R.; Hirschey, M.D.; Mair, W.B. Dietary restriction and AMPK increase lifespan via mitochondrial network and peroxisome remodeling. Cell Metab. 2017, 26, 884–896.e5. [Google Scholar] [CrossRef]
- Morselli, E.; Maiuri, M.C.; Markaki, M.; Megalou, E.; Pasparaki, A.; Palikaras, K.; Criollo, A.; Galluzzi, L.; A Malik, S.; Vitale, I.; et al. Caloric Restriction and Resveratrol Promote Longevity Through the Sirtuin-1-Dependent Induction of Autophagy. Cell Death Dis. 2010, 1, e10. [Google Scholar] [CrossRef]
- Morselli, E.; Galluzzi, L.; Kepp, O.; Criollo, A.; Maiuri, M.C.; Tavernarakis, N.; Madeo, F.; Kroemer, G. Autophagy Mediates Pharmacological Lifespan Extension by Spermidineand Resveratrol. Aging 2009, 1, 961–970. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, T.; Knauer, H.; Schauer, A.; Büttner, S.; Ruckenstuhl, C.; Carmona-Gutierrez, D.; Ring, J.; Schroeder, S.; Magnes, C.; Antonacci, L.; et al. Induction of Autophagy by Spermidine Promotes Longevity. Nat. Cell Biol. 2009, 11, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Janić, M.; Lunder, M.; Novakovic, S.; Skerl, P.; Šabovič, M. Expression of Longevity Genes Induced by a Low-Dose Fluvastatin and Valsartan Combination with the Potential to Prevent/Treat “Aging-Related Disorders”. Int. J. Mol. Sci. 2019, 20, 1844. [Google Scholar] [CrossRef] [PubMed]
- Buler, M.; Aatsinki, S.; Izzi, V.; Uusimaa, J.; Hakkola, J. SIRT5 is under the control of PGC-1α and AMPK and is involved in regulation of mitochondrial energy metabolism. FASEB J. 2014, 28, 3225–3237. [Google Scholar] [CrossRef]
- Buendia, P.; Ramirez, R.; Aljama, P.; Carracedo, J. Klotho prevents translocation of NFkappaB. Vitam. Horm. 2016, 101, 119–150. [Google Scholar]
- Kuk, M.U.; Kim, J.W.; Lee, Y.-S.; A Cho, K.; Park, J.T.; Park, S.C. Alleviation of senescence via ATM inhibition in accelerated aging models. Mol. Cells 2019, 42, 210–217. [Google Scholar]
- McClintock, D.; Gordon, L.B.; Djabali, K. Hutchinson–gilford progeria mutant lamin a primarily targets human vascular cells as detected by an anti-lamin a G608G antibody. Proc. Natl. Acad. Sci. USA 2006, 103, 2154–2159. [Google Scholar] [CrossRef]
- Gray, M.D.; Shen, J.-C.; Kamath-Loeb, A.S.; Blank, A.; Sopher, B.L.; Martin, G.M.; Oshima, J.; Loeb, L.A. The Werner syndrome protein is a DNA helicase. Nat. Genet. 1997, 17, 100–103. [Google Scholar] [CrossRef]
- Chang, L.; Liu, X.; Wang, D.; Ma, J.; Zhou, T.; Chen, Y.; Sheng, R.; Hu, Y.; Du, Y.; He, Q.; et al. Hypoxia-Targeted Drug Q6 Induces G2-M Arrest and Apoptosis via Poisoning Topoisomerase II under Hypoxia. PLoS ONE 2015, 10, e0144506. [Google Scholar] [CrossRef]
- Savitsky, K.; Bar-Shira, A.; Gilad, S.; Rotman, G.; Ziv, Y.; Vanagaite, L.; Tagle, D.A.; Smith, S.; Uziel, T.; Sfez, S.; et al. A Single Ataxia Telangiectasia Gene with a Product Similar to PI-3 Kinase. Science 1995, 268, 1749–1753. [Google Scholar] [CrossRef]
- Shemiakova, T.; Ivanova, E.; Grechko, A.V.; Gerasimova, E.V.; Sobenin, I.A.; Orekhov, A.N. Mitochondrial Dysfunction and DNA Damage in the Context of Pathogenesis of Atherosclerosis. Biomedicines 2020, 8, 166. [Google Scholar] [CrossRef]
- Stelmashook, E.V.; Isaev, N.K.; Genrikhs, E.E.; Novikova, S.V. Mitochondria-Targeted Antioxidants as Potential Therapy for the Treatment of Traumatic Brain Injury. Antioxidants 2019, 8, 124. [Google Scholar] [CrossRef] [PubMed]
- Agapova, L.S.; Chernyak, B.V.; Domnina, L.V.; Dugina, V.B.; Efimenko, A.Y.; Fetisova, E.K.; Ivanova, O.Y.; Kalinina, N.I.; Khromova, N.V.; Kopnin, B.P.; et al. Mitochondria-Targeted Plastoquinone Derivatives as Tools to Interrupt Execution of the Aging Program. 3. Inhibitory Effect of SkQ1 on Tumor Development from p53-Deficient Cells. Biochemistry (Moscow) 2008, 73, 1300–1316. [Google Scholar] [CrossRef]
- Zhao, K.; Luo, G.; Giannelli, S.; Szeto, H.H. Mitochondria-Targeted Peptide Prevents Mitochondrial Depolarization and Apoptosis Induced by Tert-Butyl Hydroperoxide in Neuronal Cell Lines. Biochem. Pharmacol. 2005, 70, 1796–1806. [Google Scholar] [CrossRef] [PubMed]
- Navarro, A.; Lopez-Cepero, J.M.; Bandez, M.J.; Sanchez-Pino, M.J.; Gomez, C.; Cadenas, E.; Boveris, A. Hippocampal Mitochondrial Dysfunction in Rat Aging. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 294, R501–R509. [Google Scholar] [CrossRef]
- Navarro, A.; Sanchez Del Pino, M.J.; Gomez, C.; Peralta, J.L.; Boveris, A. Behavioral Dysfunction, Brain Oxidative Stress, and Impaired Mitochondrial Electron Transfer in Aging Mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002, 282, R985–R992. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Killilea, D.W.; Ames, B.N. Age-Associated Mitochondrial Oxidative Decay: Improvement of Carnitine Acetyltransferase Substrate-Binding Affinity and Activity in Brain by Feeding Old Rats Acetyl-L-Carnitine and/or R-Alpha-Lipoic Acid. Proc. Natl. Acad. Sci. USA 2002, 99, 1876–1881. [Google Scholar] [CrossRef] [PubMed]
- Hagen, T.M.; Ingersoll, R.T.; Wehr, C.M.; Lykkesfeldt, J.; Vinarsky, V.; Bartholomew, J.C.; Song, M.H.; Ames, B.N. Acetyl-L-Carnitine Fed to Old Rats Partially Restores Mitochondrial Function and Ambulatory Activity. Proc. Natl. Acad. Sci. USA 1998, 95, 9562–9566. [Google Scholar] [CrossRef]
- Sastre, J.; Millan, A.; Garcia de la Asuncion, J.; Pla, R.; Juan, G.; Pallardo; O’Connor, E.; Martin, J.A.; Droy-Lefaix, M.T.; Vina, J. A Ginkgo Biloba Extract (EGb 761) Prevents Mitochondrial Aging by Protecting Against Oxidative Stress. Free Radic. Biol. Med. 1998, 24, 298–304. [Google Scholar] [CrossRef]
- Dikalova, A.E.; Kirilyuk, I.A.; Dikalov, S.I. Antihypertensive Effect of Mitochondria-Targeted Proxyl Nitroxides. Redox Biol. 2015, 4, 355–362. [Google Scholar] [CrossRef]
- Cheng, Y.; Liu, D.Z.; Zhang, C.X.; Cui, H.; Liu, M.; Zhang, B.L.; Mei, Q.B.; Lu, Z.F.; Zhou, S.Y. Mitochondria-Targeted Antioxidant Delivery for Precise Treatment of Myocardial Ischemia-Reperfusion Injury through a Multistage Continuous Targeted Strategy. Nanomed. Nanotechnol. Biol. Med. 2019, 16, 236–249. [Google Scholar] [CrossRef]
- Goldring, S.R.; Goldring, M.B. The Role of Cytokines in Cartilage Matrix Degeneration in Osteoarthritis. Clin. Orthop. Relat. Res. 2004, 427, S27–S36. [Google Scholar] [CrossRef]
- Kim, J.; Xu, M.; Xo, R.; Mates, A.; Wilson, G.L.; Pearsall, A.W.T.; Grishko, V. Mitochondrial DNA Damage is Involved in Apoptosis Caused by Pro-Inflammatory Cytokines in Human OA Chondrocytes. Osteoarthr. Cartil. 2010, 18, 424–432. [Google Scholar] [CrossRef]
- Thoppil, H.; Riabowol, K. Senolytics: A Translational Bridge Between Cellular Senescence and Organismal Aging. Front. Cell Dev. Biol. 2019, 7, 367. [Google Scholar] [CrossRef]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics Improve Physical Function and Increase Lifespan in Old Age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef]
- Cavalcante, M.B.; Saccon, T.D.; Nunes, A.D.; Kirkland, J.L.; Tchkonia, T.; Schneider, A.; Masternak, M.M. Dasatinib Plus Quercetin Prevents Uterine Age-Related Dysfunction and Fibrosis in Mice. Aging 2020, 12, 2711–2722. [Google Scholar] [CrossRef]
- Hickson, L.J.; Prata, L.G.P.L.; Bobart, S.A.; Evans, T.K.; Giorgadze, N.; Hashmi, S.K.; Herrmann, S.M.; Jensen, M.D.; Jia, Q.; Jordan, K.L.; et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of dasatinib plus quercetin in individuals with diabetic kidney disease. EBioMedicine 2019, 47, 446–456. [Google Scholar] [CrossRef]
- Lewinska, A.; Adamczyk-Grochala, J.; Bloniarz, D.; Olszowka, J.; Kulpa-Greszta, M.; Litwinienko, G.; Tomaszewska, A.; Wnuk, M.; Pązik, R. AMPK-Mediated Senolytic and Senostatic Activity of Quercetin Surface Functionalized Fe3O4 Nanoparticles during Oxidant-Induced Senescence in Human Fibroblasts. Redox Biol. 2020, 28, 101337. [Google Scholar] [CrossRef]
- Shen, L.-R.; Parnell, L.D.; Ordovas, J.M.; Lai, C.-Q. Curcumin and Aging. BioFactors 2013, 39, 133–140. [Google Scholar] [CrossRef]
- He, Y.; Li, W.; Hu, G.; Sun, H.; Kong, Q. Bioactivities of EF24, a novel curcumin analog: A review. Front. Oncol. 2018, 8, 614. [Google Scholar] [CrossRef]
- Li, W.; He, Y.; Zhang, R.; Zheng, G.; Zhou, D. The curcumin analog EF24 is a novel senolytic agent. Aging 2019, 11, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Tin, L.; Zhang, Y.; Wu, Y.; Jin, Y.; Jin, X.; Zhang, F.; Li, X. EF24 Suppresses Invasion and Migration of Hepatocellular Carcinoma Cells In Vitro via Inhibiting the Phosphorylation of Src. BioMed Res. Int. 2016, 2016, 1–10. [Google Scholar] [CrossRef]
- Capra, J.; Eskelinen, S. Correlation between E-Cadherin Interactions, Survivin Expression, and Apoptosis in MDCK and ts-Src MDCK Cell Culture Models. Lab. Investig. 2017, 97, 1453–1470. [Google Scholar] [CrossRef]
- Kumar, S.; Agnihotri, N. Piperlongumine, a Piper Alkaloid Targets Ras/PI3K/Akt/mTOR Signaling Axis to Inhibit Tumor Cell Growth and Proliferation in DMH/DSS Induced Experimental Colon Cancer. Biomed. Pharmacother. 2019, 109, 1462–1477. [Google Scholar] [CrossRef]
- Fuhrmann-Stroissnigg, H.; Ling, Y.Y.; Zhao, J.; McGowan, S.J.; Zhu, Y.; Brooks, R.W.; Grassi, D.; Gregg, S.Q.; Stripay, J.L.; Dorronsoro, A.; et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat. Commun. 2017, 8, 422. [Google Scholar] [CrossRef]
- Taipale, M.; Jarosz, D.F.; Lindquist, S. HSP90 at the Hub of Protein Homeostasis: Emerging Mechanistic Insights. Nat. Rev. Mol. Cell Biol. 2010, 11, 515–528. [Google Scholar] [CrossRef]
- Pacey, S.; Wilson, R.H.; Walton, M.; Eatock, M.M.; Hardcastle, A.; Zetterlund, A.; Arkenau, H.-T.; Moreno-Farre, J.; Banerji, U.; Roels, B.; et al. A Phase I Study of the Heat Shock protein 90 Inhibitor Alvespimycin (17-DMAG) Given Intravenously to Patients with Advanced Solid Tumors. Clin. Cancer Res. 2011, 17, 1561–1570. [Google Scholar] [CrossRef]
- Xu, G.; Ma, X.; Chen, F.; Wu, D.; Miao, J.; Fan, Y. 17-DMAG disrupted the autophagy flux leading to the apoptosis of acute lymphoblastic leukemia cells by inducing heat shock cognate protein. Life Sci. 2020, 249, 117532. [Google Scholar] [CrossRef]
- Zhu, Y.; Doornebal, E.J.; Pirtskhalava, T.; Giorgadze, N.; Wentworth, M.; Fuhrmann-Stroissnigg, H.; Niedernhofer, L.J.; Robbins, P.D.; Tchkonia, T.; Kirkland, J.L. New agents that target senescent cells: The flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging 2017, 9, 955–963. [Google Scholar] [CrossRef]
- Moujalled, D.M.; Hanna, D.T.; Hediyeh-Zadeh, S.; Pomilio, G.; Brown, L.; Litalien, V.; Bartolo, R.; Fleming, S.; Chanrion, M.; Banquet, S.; et al. Cotargeting BCL-2 and MCL-1 in high-risk B-ALL. Blood Adv. 2020, 4, 2762–2767. [Google Scholar] [CrossRef] [PubMed]
- Bierbrauer, A.; Jacob, M.; Vogler, M.; Fulda, S. A direct comparison of selective BH3-mimetics reveals BCL-XL, BCL-2 and MCL-1 as promising therapeutic targets in neuroblastoma. Br. J. Cancer 2020, 122, 1544–1551. [Google Scholar] [CrossRef]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.-M. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef]
- Ozsvari, B.; Nuttall, J.R.; Sotgia, F.; Lisanti, M.P. Azithromycin and Roxithromycin define a new family of “senolytic” drugs that target senescent human fibroblasts. Aging 2018, 10, 3294–3307. [Google Scholar] [CrossRef]
- Status of the Health-Related SDGs. Available online: https://www.who.int/gho/publications/world_health_statistics/2018/EN_WHS2018_Part2.pdf?ua=1 (accessed on 28 March 2018).
- Global Action Plan on the Public Health Response to Dementia. Available online: https://apps.who.int/iris/bitstream/handle/10665/259615/9789241513487-eng.pdf;jsessionid=CAD21A0B1FB6D45E2742A834F2856D85?sequence=1 (accessed on 28 March 2018).
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.-M.; et al. Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nat. Cell Biol. 2016, 530, 184–189. [Google Scholar] [CrossRef]
- Brungs, A.; Vollmar, A.; Reese, S.; Poulsen Nautrup, C. Echocardiographic indices of age- and gender-dependent cardiac remodeling over the adult lifespan in Irish Wolfhounds. J. Vet. Cardiol. 2018, 20, 307–318. [Google Scholar] [CrossRef]
- Cotugno, G.; Formisano, P.; Giacco, F.; Colella, P.; Beguinot, F.; Auricchio, A. AP20187-mediated activation of a chimeric insulin receptor results in insulin-like actions in skeletal muscle and liver of diabetic mice. Hum. Gene Ther. 2007, 18, 106–117. [Google Scholar] [CrossRef]
- Roos, C.M.; Zhang, B.; Palmer, A.K.; Ogrodnik, M.B.; Pirtskhalava, T.; Thalji, N.M.; Hagler, M.; Jurk, D.; Smith, L.A.; Casaclang-Verzosa, G.; et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell 2016, 15, 973–977. [Google Scholar] [CrossRef]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; E Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef]
- Walaszczyk, A.; Dookun, E.; Redgrave, R.; Tual-Chalot, S.; Victorelli, S.; Spyridopoulos, I.; Owens, A.; Arthur, H.M.; Passos, J.F.; Richardson, G.D. Pharmacological clearance of senescent cells improves survival and recovery in aged mice following acute myocardial infarction. Aging Cell 2019, 18, e12945. [Google Scholar] [CrossRef]
- Triana-Martínez, F.; Picallos-Rabina, P.; Da Silva-Álvarez, S.; Pietrocola, F.; Llanos, S.; Rodilla, V.; Soprano, E.; Pedrosa, P.; Ferreirós, A.; Barradas, M.; et al. Identification and characterization of Cardiac Glycosides as senolytic compounds. Nat. Commun. 2019, 10, 4731. [Google Scholar] [CrossRef]
- Aoyama, M.; Kawase, H.; Bando, Y.K.; Monji, A.; Murohara, T. Dipeptidyl Peptidase 4 Inhibition Alleviates Shortage of Circulating Glucagon-Like Peptide-1 in Heart Failure and Mitigates Myocardial Remodeling and Apoptosis via the Exchange Protein Directly Activated by Cyclic AMP 1/Ras-Related Protein 1 Axis. Circ. Heart Fail. 2016, 9, e002081. [Google Scholar] [CrossRef]
- Drucker, D.J. The biology of incretin hormones. Cell Metab. 2006, 3, 153–165. [Google Scholar] [CrossRef]
- Wang, J.; Yao, Y.; Zhang, J.; Tang, X.; Meng, X.; Wang, M.; Song, L.; Yuan, J. Platelet microRNA-15b protects against high platelet reactivity in patients undergoing percutaneous coronary intervention through Bcl-2-mediated platelet apoptosis. Ann. Transl. Med. 2020, 8, 364. [Google Scholar] [CrossRef]
- Justice, J.N.; Nambiar, A.M.; Tchkonia, T.; Lebrasseur, N.K.; Pascual, R.; Hashmi, S.K.; Prata, L.; Masternak, M.M.; Kritchevsky, S.B.; Musi, N.; et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine 2019, 40, 554–563. [Google Scholar] [CrossRef]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 14532. [Google Scholar] [CrossRef]
- Zhai, W.; Cardell, M.; De Meester, I.; Augustyns, K.; Hillinger, S.; Inci, I.; Arni, S.; Jungraithmayr, W.; Scharpe, S.; Weder, W.; et al. Intragraft DPP IV Inhibition Attenuates Post-transplant Pulmonary Ischemia/Reperfusion Injury After Extended Ischemia. J. Heart Lung Transplant. 2007, 26, 174–180. [Google Scholar] [CrossRef]
- Shiobara, T.; Chibana, K.; Watanabe, T.; Arai, R.; Horigane, Y.; Nakamura, Y.; Hayashi, Y.; Shimizu, Y.; Takemasa, A.; Ishii, Y. Dipeptidyl peptidase-4 is highly expressed in bronchial epithelial cells of untreated asthma and it increases cell proliferation along with fibronectin production in airway constitutive cells. Respir. Res. 2016, 17, 28. [Google Scholar] [CrossRef]
- Jang, J.H.; Janker, F.; De Meester, I.; Arni, S.; Borgeaud, N.; Yamada, Y.; Gil Bazo, I.; Weder, W.; Jungraithmayr, W. The CD26/DPP4-inhibitor vildagliptin suppresses lung cancer growth via macrophage-mediated NK cell activity. Carcinogenesis 2019, 40, 324–334. [Google Scholar] [CrossRef]
- Nakamaru, Y.; Vuppusetty, C.; Wada, H.; Milne, J.C.; Ito, M.; Rossios, C.; Elliot, M.; Hogg, J.; Kharitonov, S.; Goto, H.; et al. A protein deacetylase SIRT1 is a negative regulator of metalloproteinase-9. FASEB J. 2009, 23, 2810–2819. [Google Scholar] [CrossRef]
- Mikawa, R.; Suzuki, Y.; Baskoro, H.; Kanayama, K.; Sugimoto, K.; Sato, T.; Sugimoto, M. Elimination of p19ARF-expressing cells protects against pulmonary emphysema in mice. Aging Cell 2018, 17, e12827. [Google Scholar] [CrossRef]
- Ritz, S.A.; Wan, J.; Diaz-Sanchez, D. Sulforaphane-stimulated phase II enzyme induction inhibits cytokine production by airway epithelial cells stimulated with diesel extract. Am. J. Physiol. Cell. Mol. Physiol. 2007, 292, L33–L39. [Google Scholar] [CrossRef]
- Cho, H.-Y.; Miller-DeGraff, L.; Blankenship-Paris, T.; Wang, X.; Bell, D.A.; Lih, F.; Deterding, L.; Panduri, V.; Morgan, D.L.; Yamamoto, M.; et al. Sulforaphane enriched transcriptome of lung mitochondrial energy metabolism and provided pulmonary injury protection via Nrf2 in mice. Toxicol. Appl. Pharmacol. 2018, 364, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Bijangi-Vishehsaraei, K.; Saadatzadeh, M.R.; Wang, H.; Nguyen, A.; Kamocka, M.M.; Cai, W.; Cohen-Gadol, A.A.; Halum, S.L.; Sarkaria, J.N.; Pollok, K.E.; et al. Sulforaphane suppresses the growth of glioblastoma cells, glioblastoma stem cell–like spheroids, and tumor xenografts through multiple cell signaling pathways. J. Neurosurg. 2017, 127, 1219–1230. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-W.; Yang, L.-H.; Ren, Z.-C.; Mu, D.-G.; Li, Y.-Q.; Yan, J.-P.; Wang, L.-X.; Chen, C. Resveratrol downregulates TNF-α-induced monocyte chemoattractant protein-1 in primary rat pulmonary artery endothelial cells by P38 mitogen-activated protein kinase signaling. Drug Des. Dev. Ther. 2019, 13, 1843–1853. [Google Scholar] [CrossRef] [PubMed]
- Musi, N.; Valentine, J.M.; Sickora, K.R.; Baeuerle, E.; Thompson, C.S.; Shen, Q.; Orr, M.E. Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell 2018, 17, e12840. [Google Scholar] [CrossRef]
- KoSIK, K.S.; Joachim, C.J.; Selkoe, D.J. Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1986, 83, 4044–4048. [Google Scholar] [CrossRef]
- Guo, J.; Chang, L.; Li, C.; Li, M.; Yan, P.; Guo, Z.; Wang, C.; Zha, Q.; Wang, Q. SB203580 reverses memory deficits and depression-like behavior induced by microinjection of Abeta1-42 into hippocampus of mice. Metab. Brain Dis. 2017, 32, 57–68. [Google Scholar] [CrossRef]
- Zhang, P.; Kishimoto, Y.; Grammatikakis, I.; Gottimukkala, K.; Cutler, R.G.; Zhang, S.; Abdelmohsen, K.; Bohr, V.A.; Misra Sen, J.; Gorospe, M.; et al. Senolytic therapy alleviates Abeta-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat. Neurosci. 2019, 22, 719–728. [Google Scholar] [CrossRef]
- Chalichem, N.S.S.; Kiran, P.S.S.S.; Basavan, D. Possible role of DPP4 inhibitors to promote hippocampal neurogenesis in Alzheimer’s disease. J. Drug Target. 2018, 26, 670–675. [Google Scholar] [CrossRef]
- Riessland, M.; Kolisnyk, B.; Kim, T.W.; Cheng, J.; Ni, J.; Pearson, J.A.; Park, E.J.; Dam, K.; Acehan, D.; Ramos-Espiritu, L.S.; et al. Loss of SATB1 Induces p21-Dependent Cellular Senescence in Post-mitotic Dopaminergic Neurons. Cell Stem Cell 2019, 25, 514–530.e8. [Google Scholar] [CrossRef]
- Singh, A.; Naidu, P.S.; Kulkarni, S.K. Quercetin potentiates L-Dopa reversal of drug-induced catalepsy in rats: Possible COMT/MAO inhibition. Pharmacology 2003, 68, 81–88. [Google Scholar] [CrossRef]
- Yabluchanskiy, A.; Tarantini, S.; Balasubramanian, P.; Kiss, T.; Csipo, T.; Fulop, G.A.; Lipecz, A.; Ahire, C.; DelFavero, J.; Nyul-Toth, A.; et al. Pharmacological or genetic depletion of senescent astrocytes prevents whole brain irradiation-induced impairment of neurovascular coupling responses protecting cognitive function in mice. GeroScience 2020, 42, 409–428. [Google Scholar] [CrossRef] [PubMed]
- WHO. Health Topics: Diabetes. 2020. Available online: https://www.who.int/health-topics/diabetes#tab=tab_1 (accessed on 18 August 2020).
- Palmer, A.K.; Xu, M.; Zhu, Y.; Pirtskhalava, T.; Weivoda, M.M.; Hachfeld, C.M.; Prata, L.G.; Van Dijk, T.H.; Verkade, E.; Casaclang-Verzosa, G.; et al. Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell 2019, 18, e12950. [Google Scholar] [CrossRef] [PubMed]
- Aguayo-Mazzucato, C.; Andle, J.; Lee, T.B.; Midha, A.; Talemal, L.; Chipashvili, V.; Hollister-Lock, J.; Van Deursen, J.; Weir, G.; Bonner-Weir, S. Acceleration of β Cell Aging Determines Diabetes and Senolysis Improves Disease Outcomes. Cell Metab. 2019, 30, 129–142.e4. [Google Scholar] [CrossRef]
- Thompson, P.J.; Shah, A.; Ntranos, V.; Van Gool, F.; Atkinson, M.; Bhushan, A. Targeted Elimination of Senescent Beta Cells Prevents Type 1 Diabetes. Cell Metab. 2019, 29, 1045–1060.e10. [Google Scholar] [CrossRef]
- Velmurugan, G.V.; White, C. Calcium homeostasis in vascular smooth muscle cells is altered in type 2 diabetes by Bcl-2 protein modulation of InsP3R calcium release channels. Am. J. Physiol. Circ. Physiol. 2012, 302, H124–H134. [Google Scholar] [CrossRef] [PubMed]
- Al-Rasheed, N.M.; Al-Rasheed, N.M.; Hasan, I.H.; Al-Amin, M.A.; Al-Ajmi, H.N.; Mohamad, R.A.; Mahmoud, A.M. Simvastatin Ameliorates Diabetic Cardiomyopathy by Attenuating Oxidative Stress and Inflammation in Rats. Oxidative Med. Cell. Longev. 2017, 2017, 1092015. [Google Scholar] [CrossRef]


{kind=link}
| Senomorphic | Target Pathway | Effects | Reference |
|---|---|---|---|
| NDGA | Upregulation of PPARγ | Regulation of dyslipidemia | [45,46] |
| Acarbose | Upregulation of PPARγ | Increase of lifespan | [45] |
| Estradiol | Upregulation of PPARγ | Increase of lifespan | [45] |
| Rapamycin | mTOR is inhibited | Increase of lifespan | [48] |
| Sirt1 | Upregulation of AMPK | Increase of fatty acid oxidation and improvement of mitochondrial functions | [49] |
| RSV | Sirt1 | Amelioration of oxidative stress | [50] |
| Spermidine | Histone deacetylase | Increase of lifespan | [51,52] |
| Fluvastatin and Valsartan | Upregulation of Sirt1, PRKAA, telomerase, and KLOTHO | Amelioration of glucose and fatty acid oxidation | [53,54] |
| KU-60019 | Inhibition of ATM | Improvement of mitochondrial function | [56] |
| Senolytic Molecule | Target Pathway | Effects | Reference |
|---|---|---|---|
| D + Q | Upregulation of AMPK | Reduction of senescent adipocyte and senescent skin cells | [77] |
| Quercetin | Upregulation of AMPK | Reduction of inflammation and senescent cell death | [75] |
| EF24 | Proteasome degradation of BLC-2 family members | Apoptosis in senescent cells | [79,81] |
| Hsp90 inhibitors | Alteration of the PI3K/AKT pathway | Activation of a pro-apoptotic pathway in senescent cells | [85] |
| 17-DMAG | Upregulation of HSP70 | Increase of autophagic flux | [88] |
| A-1155463 and A-1331852 | Inhibition of BCL-XL pathway | Lysis of senescent cells in specific cell lines | [89] |
| Azithromycin and Roxithromycin | Enhancement of aerobic glycolysis | Induction of senescent cell death | [93] |
| DRUG | DISEASE | EFFECT | REFERENCE |
|---|---|---|---|
| ATB-737 | HPR | Might reduce the risk of ischemic and bleeding events | [105] |
| T2D | Amelioration of Ca2+ signaling in vessel cells | [129] | |
| AP20187 | cardiovascular | Cardiac fibrosis and myocardial hypertrophy are reduced | [96] |
| T2D | Improvement of glucose tolerance. Increase of hepatic glucagon and muscular glucose uptake | [98] | |
| D + Q | atherosclerosis | DNA damage is reduced, and improvement of vasoconstriction | [99] |
| IPF | Amelioration of walk speed and resistance | [106] | |
| AD | Improvement of learning and memory | [120] | |
| T2D | Increase of adipogenesis | [125] | |
| Skeletal health | ONGOING CLINICAL TRIAL | (ClinicalTrial identifier: NCT04313634) | |
| AD | ONGOING CLINICAL TRIAL (D + Q cerebrospinal diffusion) | ClinicalTrial identifier NCT04063124 | |
| Diabetic chronic kidney disease | ONGOING CLINICAL TRIAL | ClinicalTrial identifier NCT02848131 | |
| Digoxin | Na+/K+ATPase pump disbalance | Regulation of cellular pH | [102] |
| DPP4 inhibitors | Heart failure | Amelioration of heart functions | [104] |
| Pulmonary disease | Amelioration of oxygenation and reduction of edema | [108] | |
| Lung adenocarcinoma | Block of lung cancer growth | [110] | |
| Fisetin | Knee osteoarthritis | ONGOING CLINICAL TRIAL | ClinicalTrial identifier NCT03675724 |
| Frail Elderly syndrome | ONGOING CLINICAL TRIAL | ClinicalTrial identifier NCT04210986 | |
| Quercetin | PD | Amelioration of catalepsy | [123] |
| Rapamycin | Reducing clinical aging measures | ONGOING CLINICAL TRIAL | ClinicalTrial identifier NCT04488601 |
| RSV | PTE | Reduction of inflammation | [116] |
| Navitoclax | Pulmonary emphysema | Improvement of pressure-volume loop and reduction of inflammation | [124] |
| Brain metastasis | Cognitive performance is increased | [124] | |
| SB203580 | AD | Improvement of memory deficit | [119] |
| SRT2172 | COPD | Sirt1 activity is increased, and the oxygenation is improved | [111] |
| Sulforaphane | Lung injury | The inflammation and SASPs are reduced | [114] |
| UBX0101 | Knee osteoarthritis | FAILED CLINICAL TRIAL | ClinicalTrial identifier NCT04349956 |
| Knee osteoarthritis | ONGOING CLINICAL TRIAL | ClinicalTrial identifier NCT04229225 | |
| Venetoclax | T1D | The production of pro-inflammation cytokines is decreased | [128] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mongelli, A.; Atlante, S.; Barbi, V.; Bachetti, T.; Martelli, F.; Farsetti, A.; Gaetano, C. Treating Senescence like Cancer: Novel Perspectives in Senotherapy of Chronic Diseases. Int. J. Mol. Sci. 2020, 21, 7984. https://doi.org/10.3390/ijms21217984
Mongelli A, Atlante S, Barbi V, Bachetti T, Martelli F, Farsetti A, Gaetano C. Treating Senescence like Cancer: Novel Perspectives in Senotherapy of Chronic Diseases. International Journal of Molecular Sciences. 2020; 21(21):7984. https://doi.org/10.3390/ijms21217984
Chicago/Turabian StyleMongelli, Alessia, Sandra Atlante, Veronica Barbi, Tiziana Bachetti, Fabio Martelli, Antonella Farsetti, and Carlo Gaetano. 2020. "Treating Senescence like Cancer: Novel Perspectives in Senotherapy of Chronic Diseases" International Journal of Molecular Sciences 21, no. 21: 7984. https://doi.org/10.3390/ijms21217984
APA StyleMongelli, A., Atlante, S., Barbi, V., Bachetti, T., Martelli, F., Farsetti, A., & Gaetano, C. (2020). Treating Senescence like Cancer: Novel Perspectives in Senotherapy of Chronic Diseases. International Journal of Molecular Sciences, 21(21), 7984. https://doi.org/10.3390/ijms21217984