DNA Methylation at Birth Predicts Intellectual Functioning and Autism Features in Children with Fragile X Syndrome

 , , , , , , and
, , , , , , and 
Abstract
1. Introduction
2. Results
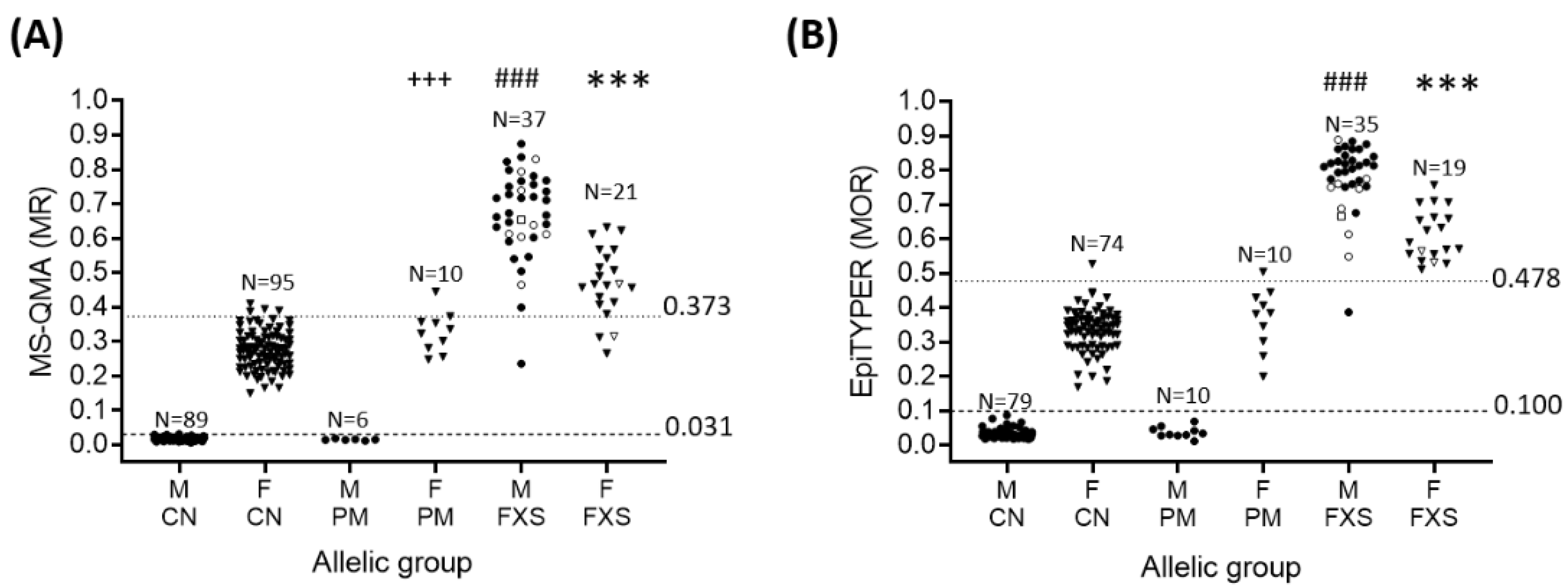
2.1. Sensitivity and Specificity of FREE2m Analysis of NBS (Time-Point 1: Birth)
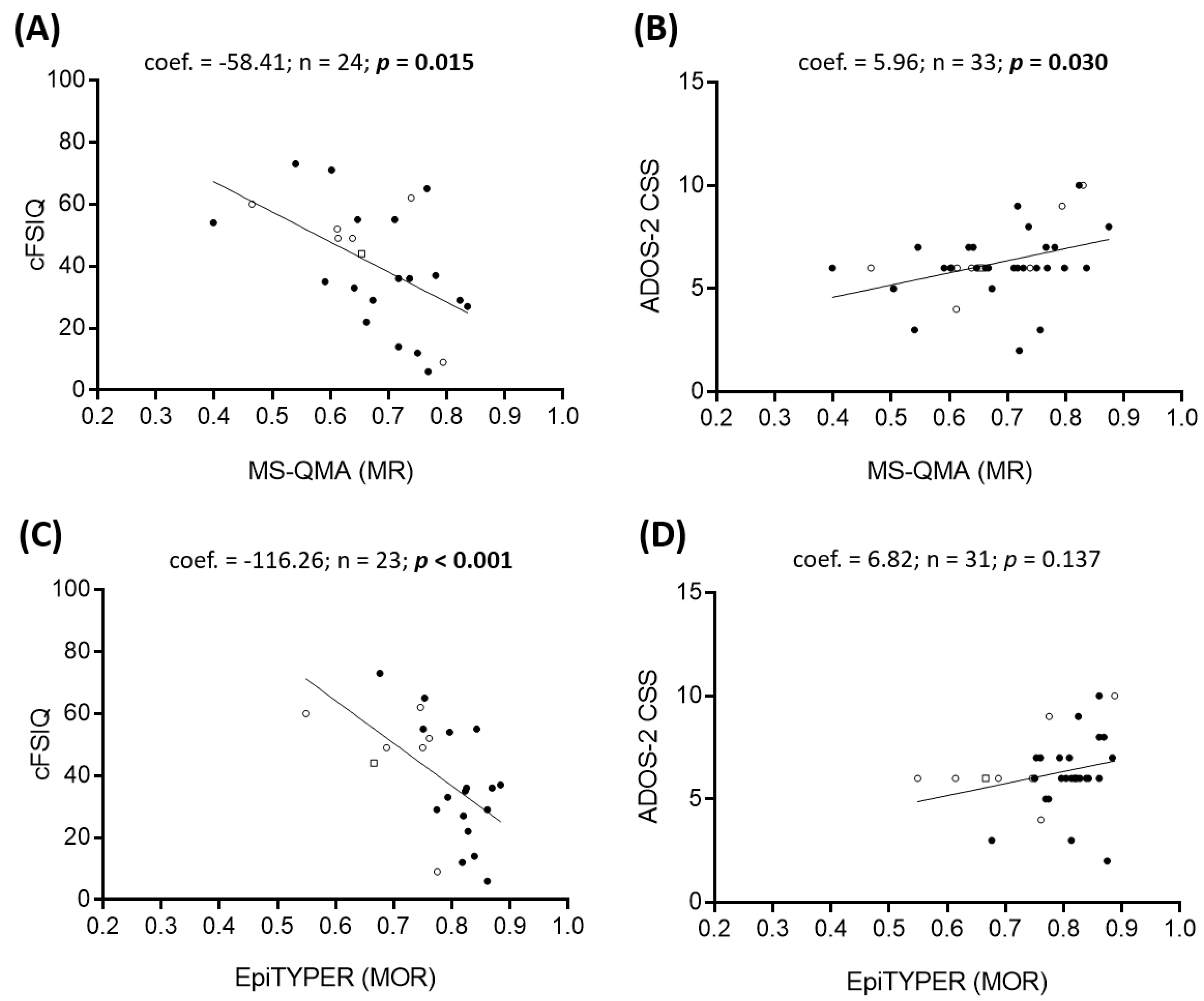
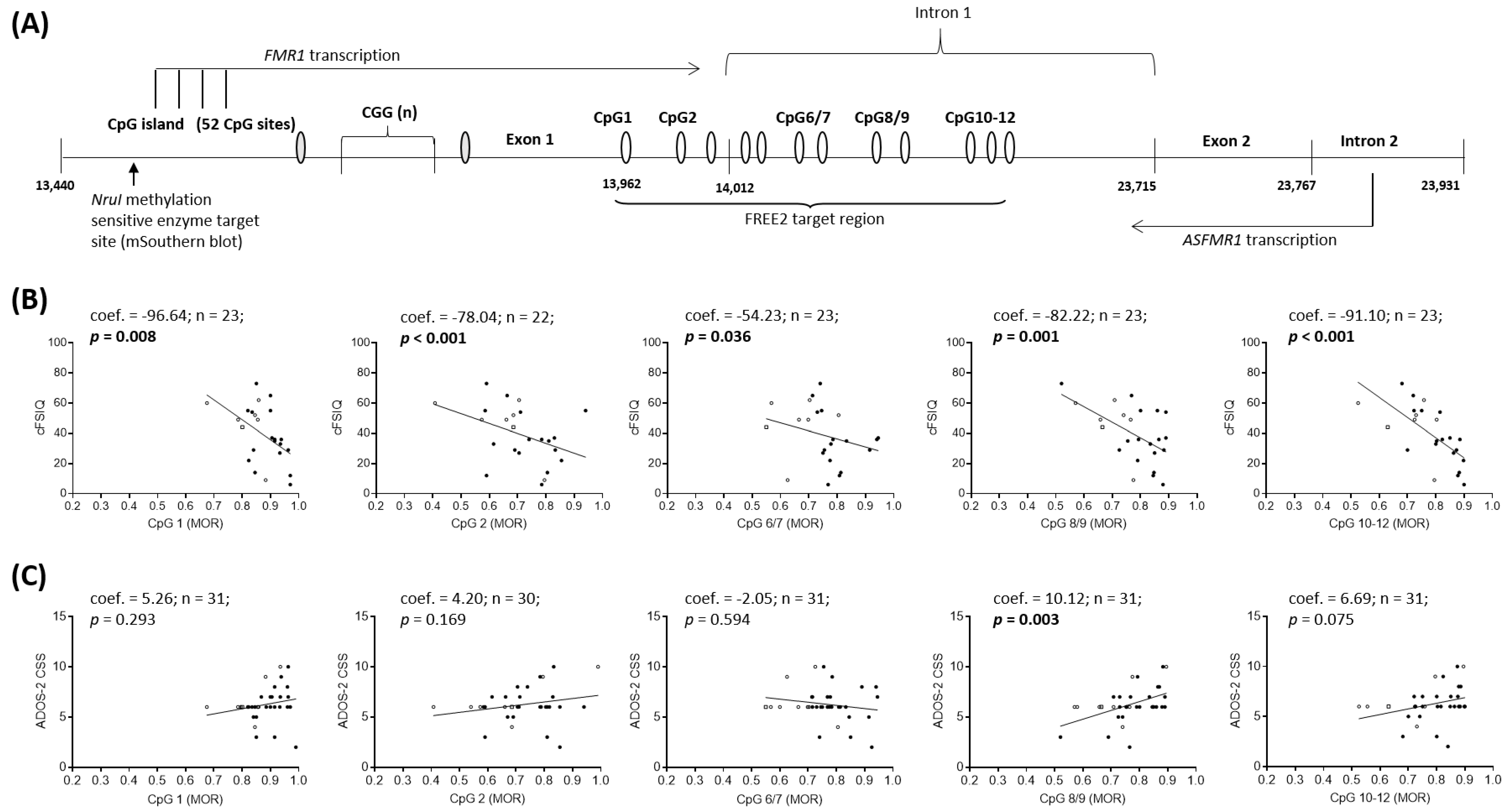
2.2. Epigenotype-Phenotype Correlations using NBS FREE2m (Time-Point 1: Birth)
2.3. Epigenotype-Phenotype Correlations using DBS FREE2m (Time-Point 2: Childhood)
2.4. Correlation between FMR1 mRNA and FREE2m Using NBS (at Birth) and DBS (during Childhood)
2.5. Longitudinal Changes in FREE2m from Birth to Childhood
3. Discussion
3.1. Epigenotype-Phenotype Associations
3.2. Screening and Diagnostic Potential
3.3. Sex Specific Differences in DNAm Longitudinal Trajectories in Blood
3.4. Limitations
4. Materials and Methods
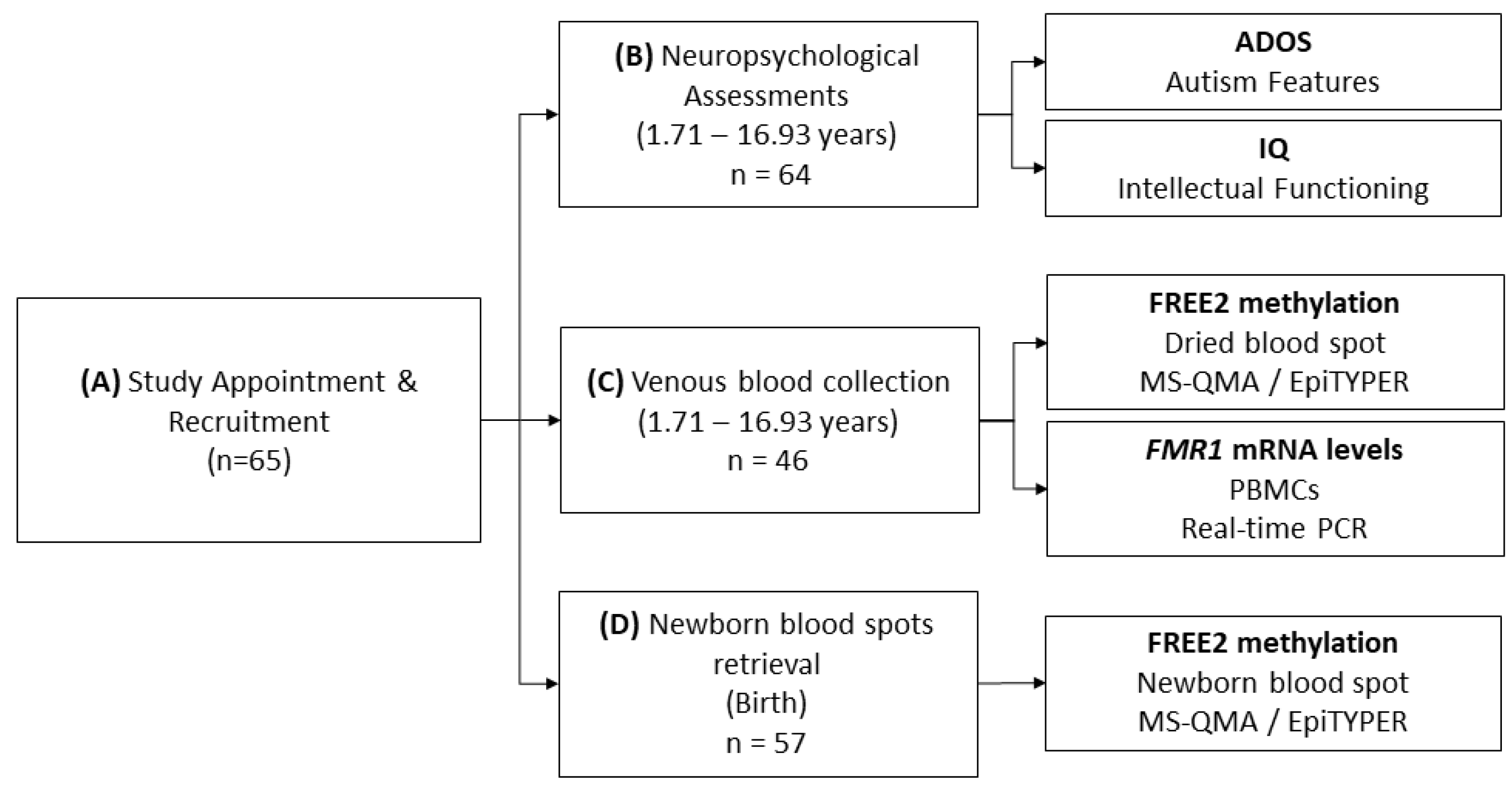
4.1. Participants
4.2. Molecular Testing
4.3. Neuropsychological Testing
4.4. Statistical Analyses
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADOS-2 | Autism Diagnostic Observation Schedule-Second Edition |
| ASD | Autism Spectrum Disorder |
| AUC | Area under the curve |
| cFSIQ | Corrected Full Scale IQ |
| CGG | Cytosine-Guanine-Guanine |
| cVIQ | Corrected Verbal IQ |
| CSS | Calibrated Severity Score |
| cPIQ | Corrected Performance IQ |
| DBS | Dried blood spots |
| DNAm | DNA methylation |
| FDR | False Discovery Rate |
| FM | Full mutation |
| FMRP | Fragile X Mental Retardation Protein |
| FREE2 | Fragile X Related Epigenetic Element 2 |
| FREE2m | Fragile X Related Epigenetic Element 2 methylation |
| FXS | Fragile X syndrome |
| ID | Intellectual Disability |
| IQ | Intelligence Quotient |
| MS-QMA | Methylation Specific-Quantitative Melt Analysis |
| NBS | Newborn blood spots |
| NS | Normal size |
| PBMC | Peripheral Blood Mononuclear Cells |
| PCR | Polyamerase Chain Reaction |
| PM | Premutation |
| PM/FM | Premutation/Full mutation |
| ROC | Receiver operating characteristic curve |
| RRB | Repetitive and Restricted Behaviours |
| SA | Social Affect |
| VCGS | Victorian Clinical Genetic Services |
| WPPSI-III | Wechsler Preschool and Primary Scale of Intelligence—Third edition |
| WISC-IV | Wechsler Intelligence Scale for Children—Fourth edition |
References
- Rahat, B.; Mahajan, A.; Bagga, R.; Hamid, A.; Kaur, J. Epigenetic modifications at DMRs of placental genes are subjected to variations in normal gestation, pathological conditions and folate supplementation. Sci. Rep. 2017, 7, 40774. [Google Scholar] [CrossRef]
- Joubert, B.R.; Felix, J.F.; Yousefi, P.; Bakulski, K.M.; Just, A.C.; Breton, C.; Reese, S.E.; Markunas, C.A.; Richmond, R.C.; Xu, C.-J.; et al. DNA Methylation in Newborns and Maternal Smoking in Pregnancy: Genome-wide Consortium Meta-analysis. Am. J. Hum. Genet. 2016, 98, 680–696. [Google Scholar] [CrossRef] [PubMed]
- Breitling, L.P.; Yang, R.; Korn, B.; Burwinkel, B.; Brenner, H. Tobacco-Smoking-Related Differential DNA Methylation: 27K Discovery and Replication. Am. J. Hum. Genet. 2011, 88, 450–457. [Google Scholar] [CrossRef]
- Aref-Eshghi, E.; Rodenhiser, D.I.; Schenkel, L.C.; Lin, H.; Skinner, C.; Ainsworth, P.; Paré, G.; Hood, R.L.; Bulman, D.E.; Kernohan, K.D.; et al. Genomic DNA Methylation Signatures Enable Concurrent Diagnosis and Clinical Genetic Variant Classification in Neurodevelopmental Syndromes. Am. J. Hum. Genet. 2018, 102, 156–174. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S.V.; Ellis, S.E.; Bakulski, K.M.; Sheppard, B.; Croen, L.A.; Hertz-Picciotto, I.; Newschaffer, C.J.; Feinberg, A.P.; Arking, D.E.; Ladd-Acosta, C.; et al. Cross-tissue integration of genetic and epigenetic data offers insight into autism spectrum disorder. Nat. Commun. 2017, 8, 1011. [Google Scholar] [CrossRef] [PubMed]
- Kraan, C.M.; Godler, D.E.; Amor, D.J. Epigenetics of fragile X syndrome and fragile X-related disorders. Dev. Med. Child Neurol. 2018, 61, 121–127. [Google Scholar] [CrossRef]
- Godler, D.E.; Amor, D.J. DNA methylation analysis for screening and diagnostic testing in neurodevelopmental disorders. Essays Biochem. 2019, 63, 785–795. [Google Scholar] [CrossRef]
- Vangeel, E.B.; Pishva, E.; Hompes, T.; Hove, D.L.A.V.D.; Lambrechts, D.; Allegaert, K.; Freson, K.; Izzi, B.; Claes, S. Newborn genome-wide DNA methylation in association with pregnancy anxiety reveals a potential role for GABBR1. Clin. Epigenetics 2017, 9, 107. [Google Scholar] [CrossRef]
- Sharp, G.C.; Lawlor, D.; Richmond, R.C.; Fraser, A.; Simpkin, A.J.; Suderman, M.; Shihab, H.; Lyttleton, O.; McArdle, W.L.; Ring, S.M.; et al. Maternal pre-pregnancy BMI and gestational weight gain, offspring DNA methylation and later offspring adiposity: Findings from the Avon Longitudinal Study of Parents and Children. Int. J. Epidemiol. 2015, 44, 1288–1304. [Google Scholar] [CrossRef]
- Cecil, C.A.M.; Walton, E.; Smith, R.G.; Viding, E.; McCrory, E.J.; Relton, C.L.; Suderman, M.; Pingault, J.-B.; McArdle, W.; Gaunt, T.R.; et al. DNA methylation and substance-use risk: A prospective, genome-wide study spanning gestation to adolescence. Transl. Psychiatry 2016, 6, e976. [Google Scholar] [CrossRef] [PubMed]
- Richmond, R.C.; Simpkin, A.J.; Woodward, G.; Gaunt, T.R.; Lyttleton, O.; McArdle, W.L.; Ring, S.M.; Smith, A.D.; Timpson, N.J.; Tilling, K.; et al. Prenatal exposure to maternal smoking and offspring DNA methylation across the lifecourse: Findings from the Avon Longitudinal Study of Parents and Children (ALSPAC). Hum. Mol. Genet. 2014, 24, 2201–2217. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, S.J.; Peters, T.J.; Buckley, M.; Zhou, J.; Jones, P.; Gibson, R.; Makrides, M.; Muhlhausler, B.S.; Molloy, P.L. DNA methylation in blood from neonatal screening cards and the association with BMI and insulin sensitivity in early childhood. Int. J. Obes. 2017, 42, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Monaghan, K.G.; Lyon, E.; Spector, E.B. ACMG Standards and Guidelines for fragile X testing: A revision to the disease-specific supplements to the Standards and Guidelines for Clinical Genetics Laboratories of the American College of Medical Genetics and Genomics. Genet. Med. 2013, 15, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Devys, D.; Lutz, Y.; Rouyer, N.; Bellocq, J.-P.; Mandel, J.-L. The FMR–1 protein is cytoplasmic, most abundant in neurons and appears normal in carriers of a fragile X premutation. Nat. Genet. 1993, 4, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Pieretti, M.; Zhang, F.; Fu, Y.-H.; Warren, S.T.; Oostra, B.A.; Caskey, C.; Nelson, D.L. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell 1991, 66, 817–822. [Google Scholar] [CrossRef]
- Aliaga, S.M.; Slater, H.; Francis, D.; Du Sart, D.; Li, X.; Amor, D.J.; Alliende, A.M.; Maria, L.S.; Faundes, V.; Morales, P.; et al. Identification of Males with Cryptic Fragile X Alleles by Methylation-Specific Quantitative Melt Analysis. Clin. Chem. 2016, 62, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Godler, D.E.; Inaba, Y.; Schwartz, C.E.; Bui, M.; Shi, E.Z.; Li, X.; Herlihy, A.S.; Skinner, C.; Hagerman, R.J.; Francis, D.; et al. Detection of skewed X-chromosome inactivation in Fragile X syndrome and X chromosome aneuploidy using quantitative melt analysis. Expert Rev. Mol. Med. 2015, 17, 13. [Google Scholar] [CrossRef]
- Inaba, Y.; Herlihy, A.S.; Schwartz, C.; Skinner, C.; Bui, M.; Cobb, J.; Shi, E.Z.; Francis, D.; Arvaj, A.; Amor, D.J.; et al. Fragile X–related element 2 methylation analysis may provide a suitable option for inclusion of fragile X syndrome and/or sex chromosome aneuploidy into newborn screening: A technical validation study. Genet. Med. 2012, 15, 290–298. [Google Scholar] [CrossRef]
- Inaba, Y.; Schwartz, C.; Bui, M.; Li, X.; Skinner, C.; Field, M.; Wotton, T.; Hagerman, R.J.; Francis, D.; Amor, D.J.; et al. Early Detection of Fragile X Syndrome: Applications of a Novel Approach for Improved Quantitative Methylation Analysis in Venous Blood and Newborn Blood Spots. Clin. Chem. 2014, 60, 963–973. [Google Scholar] [CrossRef]
- Arpone, M.; Baker, E.K.; Bretherton, L.; Bui, M.; Li, X.; Whitaker, S.; Dissanayake, C.; Cohen, J.; Hickerton, C.; Rogers, C.; et al. Intragenic DNA methylation in buccal epithelial cells and intellectual functioning in a paediatric cohort of males with fragile X. Sci. Rep. 2018, 8, 3644. [Google Scholar] [CrossRef]
- Godler, D.E.; Slater, H.; Bui, M.; Storey, E.; Ono, M.Y.; Gehling, F.; Inaba, Y.; Francis, D.; Hopper, J.L.; Kinsella, G.J.; et al. Fragile X Mental Retardation 1 (FMR1) Intron 1 Methylation in Blood Predicts Verbal Cognitive Impairment in Female Carriers of Expanded FMR1 Alleles: Evidence from a Pilot Study. Clin. Chem. 2012, 58, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Godler, D.E.; Tassone, F.; Loesch, D.Z.; Taylor, A.K.; Gehling, F.; Hagerman, R.J.; Burgess, T.; Ganesamoorthy, D.; Hennerich, D.; Gordon, L.; et al. Methylation of novel markers of fragile X alleles is inversely correlated with FMRP expression and FMR1 activation ratio. Hum. Mol. Genet. 2010, 19, 1618–1632. [Google Scholar] [CrossRef] [PubMed]
- Godler, D.E.; Slater, H.R.; Bui, M.; Ono, M.; Gehling, F.; Francis, D.; Amor, D.J.; Hopper, J.L.; Hagerman, R.; Loesch, D.Z. FMR1 Intron 1 Methylation Predicts FMRP Expression in Blood of Female Carriers of Expanded FMR1 Alleles. J. Mol. Diagn. 2011, 13, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Avitzour, M.; Mor-Shaked, H.; Yanovsky-Dagan, S.; Aharoni, S.; Altarescu, G.; Renbaum, P.; Eldar-Geva, T.; Schonberger, O.; Levy-Lahad, E.; Epsztejn-Litman, S.; et al. FMR1 epigenetic silencing commonly occurs in undifferentiated fragile X-affected embryonic stem cells. Stem Cell Rep. 2014, 3, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Esanov, R.; Andrade, N.S.; Bennison, S.; Wahlestedt, C.; Zeier, Z. The FMR1 promoter is selectively hydroxymethylated in primary neurons of fragile X syndrome patients. Hum. Mol. Genet. 2016, 25, 4870–4880. [Google Scholar] [CrossRef]
- Godler, D.E.; Inaba, Y.; Shi, E.Z.; Skinner, C.; Bui, Q.M.; Francis, D.; Amor, D.J.; Hopper, J.L.; Loesch, D.Z.; Hagerman, R.J.; et al. Relationships between age and epi-genotype of the FMR1 exon 1/intron 1 boundary are consistent with non-random X-chromosome inactivation in FM individuals, with the selection for the unmethylated state being most significant between birth and puberty. Hum. Mol. Genet. 2013, 22, 1516–1524. [Google Scholar] [CrossRef] [PubMed]
- Stark, Z.; Francis, D.; Gaffney, L.; Greenberg, J.; Hills, L.; Li, X.; Godler, D.E.; Slater, H.R. Prenatal diagnosis of fragile X syndrome complicated by full mutation retraction. Am. J. Med. Genet. Part A 2015, 167, 2485–2487. [Google Scholar] [CrossRef]
- Colak, D.; Zaninovic, N.; Cohen, M.S.; Rosenwaks, Z.; Yang, W.-Y.; Gerhardt, J.; Disney, M.D.; Jaffrey, S.R. Promoter-Bound Trinucleotide Repeat mRNA Drives Epigenetic Silencing in Fragile X Syndrome. Science 2014, 343, 1002–1005. [Google Scholar] [CrossRef]
- Stöger, R.; Genereux, D.P.; Hagerman, R.J.; Hagerman, P.J.; Tassone, F.; Laird, C.D. Testing the FMR1 Promoter for Mosaicism in DNA Methylation among CpG Sites, Strands, and Cells in FMR1-Expressing Males with Fragile X Syndrome. PLoS ONE 2011, 6, e23648. [Google Scholar] [CrossRef]
- Kraan, C.M.; Cornish, K.M.; Bui, Q.M.; Li, X.; Slater, H.R.; Godler, D.E. β-glucuronidase mRNA levels are correlated with gait and working memory in premutation females: Understanding the role of FMR1 premutation alleles. Sci. Rep. 2016, 6, 29366. [Google Scholar] [CrossRef]
- Will, E.A.; Bishop, S.L.; Roberts, J.E. Developmental divergence: Motor trajectories in children with fragile X syndrome with and without co-occurring autism. J. Neurodev. Disord. 2019, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, W.E.; Kidd, S.A.; Andrews, H.F.; Budimirovic, D.B.; Esler, A.; Haas-Givler, B.; Stackhouse, T.; Riley, C.; Peacock, G.; Sherman, S.L.; et al. Autism Spectrum Disorder in Fragile X Syndrome: Cooccurring Conditions and Current Treatment. Pediatrics 2017, 139, S194–S206. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, R.; Hagerman, P. Advances in clinical and molecular understanding of the FMR1 premutation and fragile X-associated tremor/ataxia syndrome. Lancet Neurol. 2013, 12, 786–798. [Google Scholar] [CrossRef]
- Hensel, C.H.; Vanzo, R.J.; Martin, M.M.; Ling, L.; Aliaga, S.M.; Bui, M.; Francis, D.I.; Twede, H.; Field, M.H.; Morison, J.W.; et al. Abnormally Methylated FMR1 in Absence of a Detectable Full Mutation in a U.S.A Patient Cohort Referred for Fragile X Testing. Sci. Rep. 2019, 9, 15315. [Google Scholar] [CrossRef]
- Alvarez-Mora, M.I.; Guitart, M.; Rodriguez-Revenga, L.; Madrigal, I.; Gabau, E.; Mila, M. Paternal transmission of a FMR1 full mutation allele. Am. J. Med. Genet. Part A 2017, 173, 2795–2797. [Google Scholar] [CrossRef]
- Zeesman, S.; Zwaigenbaum, L.; Whelan, D.T.; Hagerman, R.J.; Tassone, F.; Taylor, S.A. Paternal transmission of fragile X syndrome. Am. J. Med. Genet. 2004, 129, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Alisch, R.S.; Barwick, B.G.; Chopra, P.; Myrick, L.K.; Satten, G.A.; Conneely, K.N.; Warren, S.T. Age-associated DNA methylation in pediatric populations. Genome Res. 2012, 22, 623–632. [Google Scholar] [CrossRef]
- Pérez, R.F.; Santamarina, P.; Tejedor, J.R.; Urdinguio, R.G.; Álvarez-Pitti, J.; Redon, P.; Fernández, A.F.; Fraga, M.F.; Lurbe, E. Longitudinal genome-wide DNA methylation analysis uncovers persistent early-life DNA methylation changes. J. Transl. Med. 2019, 17, 15. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.K.; Safanda, J.F.; Fall, M.Z.; Quince, C.; Lang, K.; Hull, C.; Carpenter, I.; Staley, L.W.; Hagerman, R.J. Molecular predictors of cognitive involvement in female carriers of fragile X syndrome. JAMA 1994, 271, 507–514. [Google Scholar] [CrossRef]
- Rousseau, F.; Heitz, D.; Oberle, I.; Mandel, J.L. Selection in blood cells from female carriers of the fragile X syndrome: Inverse correlation between age and proportion of active X chromosomes carrying the full mutation. J. Med. Genet. 1991, 28, 830–836. [Google Scholar] [CrossRef][Green Version]
- Rousseau, F.; Heitz, D.; Tarleton, J.; MacPherson, J.; Malmgren, H.; Dahl, N.; Barnicoat, A.; Mathew, C.; Mornet, E.; Tejada, I.; et al. A Multicenter Study on Genotype-Phenotype Correlations in the Fragile X Syndrome, Using Direct Diagnosis with Probe StB12.3: The First 2,253 Cases. Am. J. Hum. Genet. 1994, 55, 225–237. [Google Scholar] [PubMed]
- Christie, L.M.; Wotton, T.; Bennetts, B.; Wiley, V.; Wilcken, B.; Rogers, C.; Boyle, J.; Turner, C.; Hansen, J.; Hunter, M.; et al. Maternal attitudes to newborn screening for fragile X syndrome. Am. J. Med. Genet. Part A 2013, 161, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Cornish, K.M.; Kraan, C.M.; Bui, Q.M.; Bellgrove, M.A.; Metcalfe, S.A.; Trollor, J.N.; Hocking, D.R.; Slater, H.R.; Inaba, Y.; Li, X.; et al. Novel methylation markers of the dysexecutive-psychiatric phenotype in FMR1 premutation females. Neurology 2015, 84, 1631–1638. [Google Scholar] [CrossRef] [PubMed]
- Wechsler, D. Wechsler Preschool and Primary Scale of Intelligence—Third Edition Australian Standardised Edition; NCS Pearson Inc.: Sydney, NSW, Australia, 2004. [Google Scholar]
- Wechsler, D. Wechsler Intelligence Scale for Children, 4th ed.; Australian Standardised Edition (WISC-IV Australian); NCS Pearson Inc.: Sydney, NSW, Australia, 2005. [Google Scholar]
- Lord, C.; Rutter, M.; DiLavore, P.C.; Risi, S.; Gotham, K.; Bishop, S.L. Autism Diagnostic Observation Schedule, 2nd ed.; (ADOS-2); Western Psychological Services: Los Angeles, CA, USA, 2012. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FXS (FM + PM/FM) | FM only | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Male (n = 42) | Female (n = 23) | Male (n = 31) | Female (n = 21) | |||||||
| N | Mean ± SD | N | Mean ± SD | p | N | Mean ± SD | N | Mean ± SD | p | |
| Age at assessment a | 42 | 5.09 ± 4.60 | 23 | 5.02 ± 3.63 | 0.8103 | 31 | 5.17 ± 4.80 | 21 | 5.20 ± 4.12 | 0.8447 |
| cFSIQ | 33 | 41.1 ± 18.4 | 18 | 61.3 ± 17.2 | 0.0004+ | 23 | 38.2 ± 19.3 | 16 | 58.6 ± 15.8 | 0.0013+ |
| cPIQ | 33 | 47.3 ± 16.0 | 18 | 61.9 ± 16.0 | 0.0031+ | 23 | 45.6 ± 17.2 | 16 | 60.3 ± 15.9 | 0.0102+ |
| cVIQ | 33 | 51.5 ± 14.8 | 18 | 68.3 ± 16.2 | 0.0005+ | 23 | 49.9 ± 15.4 | 16 | 65.8 ± 15.0 | 0.0028+ |
| ADOS-2 CSS a | 42 | 6.00 ± 1.00 | 22 | 4.00 ± 3.00 | 0.0002+ | 31 | 6.00 ± 1.00 | 20 | 4.00 ± 2.75 | 0.0012+ |
| SA CSS a | 42 | 6.00 ± 3.00 | 22 | 4.00 ± 3.00 | 0.0094+ | 31 | 6.00 ± 3.00 | 20 | 4.00 ± 2.75 | 0.0226+ |
| RRB CSS a | 42 | 8.00 ± 3.00 | 22 | 7.00 ± 2.25 | 0.0012+ | 31 | 8.00 ± 3.00 | 20 | 7.00 ± 2.00 | 0.0207+ |
| Gestational age a | 41 | 39.0 ± 3.00 | 23 | 39.0 ± 2.00 | 0.5256 | 30 | 38.0 ± 3.00 | 21 | 39.0± 2.00 | 0.2914 |
| Medication use b | 42 | 38.1% | 23 | 26.1% | 0.4157 | 31 | 35.5% | 21 | 28.6% | 0.7650 |
| Ethnicity b | 42 | 23 | 0.8537 | 31 | 21 | 0.8305 | ||||
| White | 32 | 76.2% | 18 | 78.3% | 22 | 71.0% | 16 | 76.2% | ||
| Other | 9 | 21.4% | 4 | 17.4% | 8 | 25.8% | 4 | 19.0% | ||
| Not disclosed | 1 | 2.4% | 1 | 4.3% | 1 | 3.2% | 1 | 4.8% | ||
| Male | Female | |||||||
|---|---|---|---|---|---|---|---|---|
| N | Mean | SD | p | N | Mean | SD | p | |
| MS-QMA * | 20 | 0.56 a | 2.43 | 0.067 | 15 | –0.50 | 1.99 | 0.351 |
| EpiTYPER # | 18 | –0.30 a | 1.22 | 0.286 | 15 | –0.76 a | 1.58 | 0.008 |
| CpG 1 | 18 | –0.72 | 3.02 | 0.328 | 15 | –0.38 | 2.67 | 0.585 |
| CpG 2 | 16 | –0.45 a | 2.01 | 0.326 | 13 | –1.06 | 1.13 | 0.006+ |
| CpG 6/7 | 17 | –0.40 a | 1.79 | 0.332 | 12 | –1.06 | 1.64 | 0.047 |
| CpG 8/9 | 17 | –0.12 | 3.91 | 0.897 | 14 | –1.56 | 2.11 | 0.016+ |
| CpG 10-12 | 18 | –0.50 | 1.89 | 0.282 | 14 | –1.13 a | 1.73 | 0.001+ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kraan, C.M.; Baker, E.K.; Arpone, M.; Bui, M.; Ling, L.; Gamage, D.; Bretherton, L.; Rogers, C.; Field, M.J.; Wotton, T.L.; et al. DNA Methylation at Birth Predicts Intellectual Functioning and Autism Features in Children with Fragile X Syndrome. Int. J. Mol. Sci. 2020, 21, 7735. https://doi.org/10.3390/ijms21207735
Kraan CM, Baker EK, Arpone M, Bui M, Ling L, Gamage D, Bretherton L, Rogers C, Field MJ, Wotton TL, et al. DNA Methylation at Birth Predicts Intellectual Functioning and Autism Features in Children with Fragile X Syndrome. International Journal of Molecular Sciences. 2020; 21(20):7735. https://doi.org/10.3390/ijms21207735
Chicago/Turabian StyleKraan, Claudine M, Emma K Baker, Marta Arpone, Minh Bui, Ling Ling, Dinusha Gamage, Lesley Bretherton, Carolyn Rogers, Michael J Field, Tiffany L Wotton, and et al. 2020. "DNA Methylation at Birth Predicts Intellectual Functioning and Autism Features in Children with Fragile X Syndrome" International Journal of Molecular Sciences 21, no. 20: 7735. https://doi.org/10.3390/ijms21207735
APA StyleKraan, C. M., Baker, E. K., Arpone, M., Bui, M., Ling, L., Gamage, D., Bretherton, L., Rogers, C., Field, M. J., Wotton, T. L., Francis, D., Hunter, M. F., Cohen, J., Amor, D. J., & Godler, D. E. (2020). DNA Methylation at Birth Predicts Intellectual Functioning and Autism Features in Children with Fragile X Syndrome. International Journal of Molecular Sciences, 21(20), 7735. https://doi.org/10.3390/ijms21207735