EZH2 as a Potential Target for NAFLD Therapy


Abstract
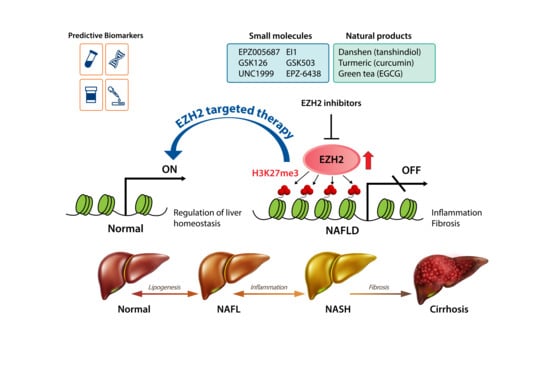
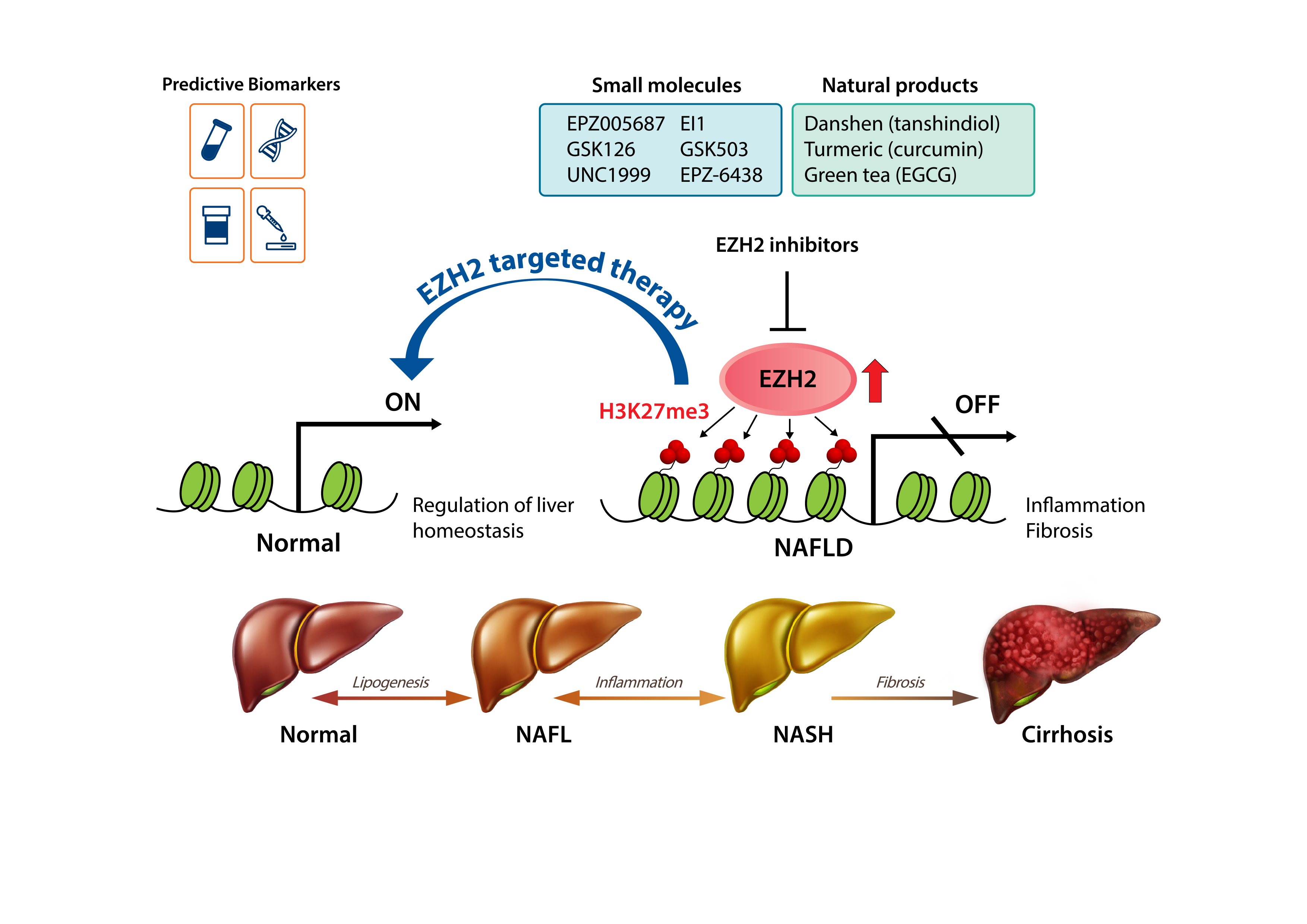
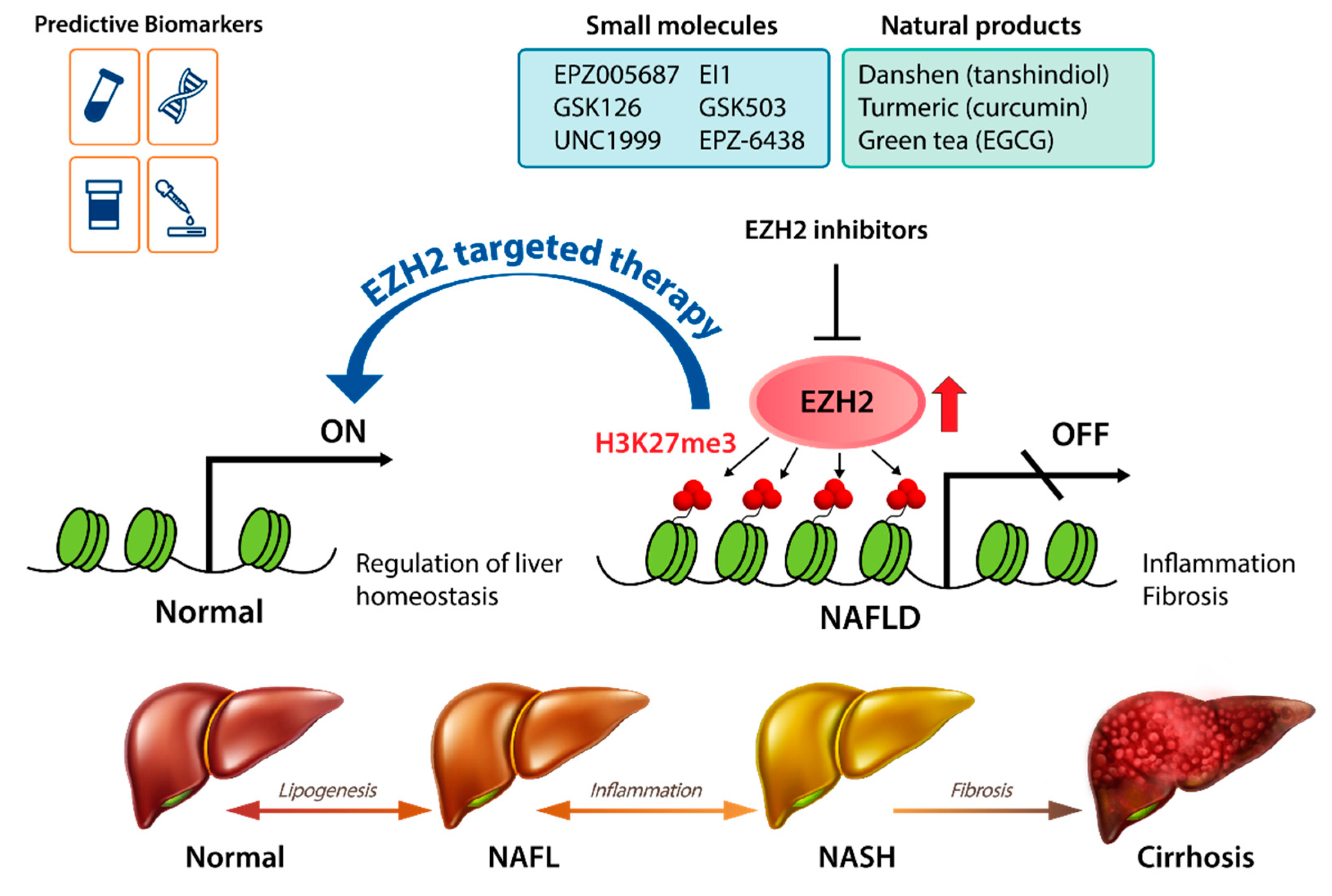
1. Introduction
2. Epigenetic Features of NAFLD
2.1. DNA Methylation in NAFLD
2.2. Histone Modifications in NAFLD
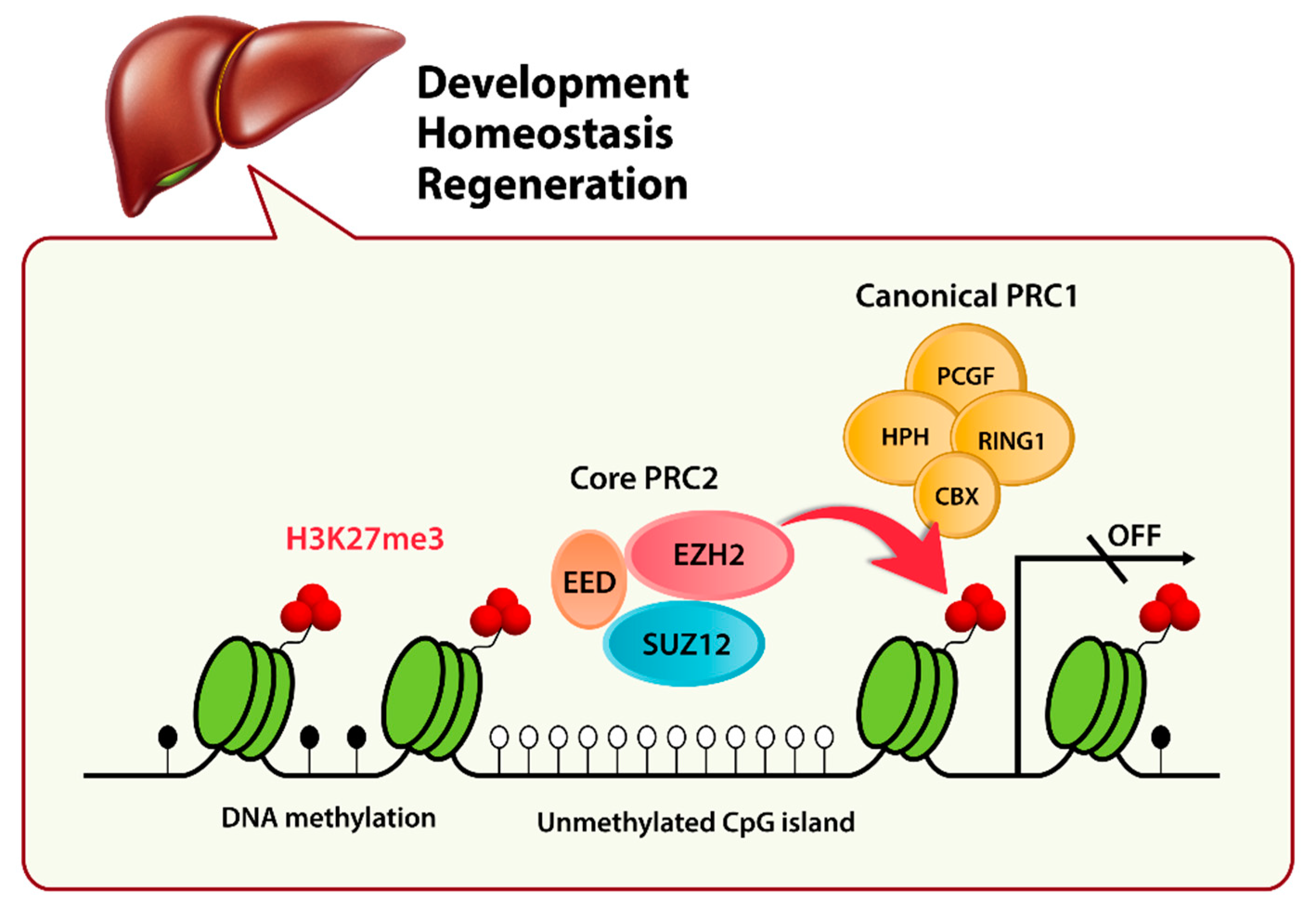
3. Role of EZH2 in the Liver
3.1. Polycomb Group (PcG) Protein EZH2
3.2. EZH2 in Liver Cell Differentiation and Liver Homeostasis
4. EZH2 as a Therapeutic Target in NAFLD
5. EZH2 Inhibitors
5.1. Small-Molecule Inhibitors of EZH2
5.2. Natural Products with EZH2-Inhibiting Activity
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BET | Bromodomain and extra-terminal domain |
| CBX | Chromobox domain |
| ChREBP | Carbohydrate response element binding protein |
| DNMT | DNA methyltransferase |
| DZNep | 3-deazaneplanocin A |
| EED | Embryonic ectoderm development |
| EGCG | Epigallocatechin-3-gallate |
| EZH2 | Enhancer of zeste homolog 2 |
| H3K27me3 | Trimethylation on Lys 27 of histone H3 |
| H3K4me3 | Trimethylation on Lys 27 of histone H3 |
| H3K9me2 | Dimethylation on Lys 9 of H3 |
| HAT | Histone acetyltransferase |
| HCC | Hepatocellular carcinoma |
| HDAC | Histone deacetylase |
| HMT | Histone methyl transferase |
| HPH | Human polyhomeotic homolog |
| HSC | Hepatic stellate cell |
| KDM | Histone lysine demethylase |
| KMT | Histone lysine methyltransferase |
| MeCP2 | Methyl-CpG binding protein 2 |
| NASH | Non-alcoholic steatohepatitis |
| PPARγ | Peroxisome proliferator-activated receptor-gamma |
| PcG | Polycomb group |
| PCGF | Polycomb group ring finger protein |
| Phf2 | Plant homeodomain finger 2 |
| PRC | Polycomb repressive complex |
| SAH | S-adenosyl-l-homocysteine |
| SAM | S-adenosyl-l-methionine |
| SUZ12 | Suppressor of zeste 12 |
References
- Cotter, T.G.; Rinella, M. Nonalcoholic fatty liver disease 2020: The state of the disease. Gastroenterology 2020, 158, 1851–1864. [Google Scholar] [CrossRef]
- Swinburn, B.A.; Sacks, G.; Hall, K.D.; McPherson, K.; Finegood, D.T.; Moodie, M.L.; Gortmaker, S.L. The global obesity pandemic: Shaped by global drivers and local environments. Lancet 2011, 378, 804–814. [Google Scholar] [CrossRef]
- Dowman, J.K.; Tomlinson, J.W.; Newsome, P.N. Pathogenesis of non-alcoholic fatty liver disease. QJM 2010, 103, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Day, C.P. From fat to inflammation. Gastroenterology 2006, 130, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, J.; Viggiano, T.R.; McGill, D.B.; Oh, B.J. Nonalcoholic steatohepatitis: Mayo clinic experiences with a hitherto unnamed disease. Mayo. Clin. Proc. 1980, 55, 434–438. [Google Scholar] [PubMed]
- Eslam, M.; Sanyal, A.J.; George, J.; International Consensus, P. MAFLD: A consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology 2020, 158, 1999–2014.e1991. [Google Scholar] [CrossRef]
- Adams, L.A.; Lymp, J.F.; St Sauver, J.; Sanderson, S.O.; Lindor, K.D.; Feldstein, A.; Angulo, P. The natural history of nonalcoholic fatty liver disease: A population-based cohort study. Gastroenterology 2005, 129, 113–121. [Google Scholar] [CrossRef]
- Fassio, E.; Alvarez, E.; Dominguez, N.; Landeira, G.; Longo, C. Natural history of nonalcoholic steatohepatitis: A longitudinal study of repeat liver biopsies. Hepatology 2004, 40, 820–826. [Google Scholar] [CrossRef]
- Koopman, K.E.; Caan, M.W.; Nederveen, A.J.; Pels, A.; Ackermans, M.T.; Fliers, E.; la Fleur, S.E.; Serlie, M.J. Hypercaloric diets with increased meal frequency, but not meal size, increase intrahepatic triglycerides: A randomized controlled trial. Hepatology 2014, 60, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Vilar-Gomez, E.; Martinez-Perez, Y.; Calzadilla-Bertot, L.; Torres-Gonzalez, A.; Gra-Oramas, B.; Gonzalez-Fabian, L.; Friedman, S.L.; Diago, M.; Romero-Gomez, M. Weight loss through lifestyle modification significantly reduces features of nonalcoholic steatohepatitis. Gastroenterology 2015, 149, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Pydyn, N.; Miekus, K.; Jura, J.; Kotlinowski, J. New therapeutic strategies in nonalcoholic fatty liver disease: A focus on promising drugs for nonalcoholic steatohepatitis. Pharmacol. Rep. 2020, 72, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Munteanu, M.A.; Nagy, G.A.; Mircea, P.A. Current management of NAFLD. Clujul. Med. 2016, 89, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Oseini, A.M.; Sanyal, A.J. Therapies in non-alcoholic steatohepatitis (NASH). Liver Int. 2017, 37 (Suppl. 1), 97–103. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Cassader, M.; Gambino, R. Non-alcoholic steatohepatitis: Emerging molecular targets and therapeutic strategies. Nat. Rev. Drug Discov. 2016, 15, 249–274. [Google Scholar] [CrossRef] [PubMed]
- Bugianesi, E.; Moscatiello, S.; Ciaravella, M.F.; Marchesini, G. Insulin resistance in nonalcoholic fatty liver disease. Curr. Pharm. Des. 2010, 16, 1941–1951. [Google Scholar] [CrossRef]
- Mendez-Sanchez, N.; Cruz-Ramon, V.C.; Ramirez-Perez, O.L.; Hwang, J.P.; Barranco-Fragoso, B.; Cordova-Gallardo, J. New aspects of lipotoxicity in nonalcoholic steatohepatitis. Int. J. Mol. Sci. 2018, 19, 2034. [Google Scholar] [CrossRef]
- Bird, A. Perceptions of epigenetics. Nature 2007, 447, 396–398. [Google Scholar] [CrossRef]
- Egger, G.; Liang, G.; Aparicio, A.; Jones, P.A. Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004, 429, 457–463. [Google Scholar] [CrossRef]
- Lu, Y.; Chan, Y.T.; Tan, H.Y.; Li, S.; Wang, N.; Feng, Y. Epigenetic regulation in human cancer: The potential role of epi-drug in cancer therapy. Mol. Cancer 2020, 19, 79. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Roberts, C.W. Targeting EZH2 in cancer. Nat. Med. 2016, 22, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Valenti, L.; Romeo, S. Genetics and epigenetics of NAFLD and NASH: Clinical impact. J. Hepatol. 2018, 68, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [PubMed]
- Del Campo, J.A.; Gallego-Duran, R.; Gallego, P.; Grande, L. Genetic and epigenetic regulation in nonalcoholic fatty liver disease (NAFLD). Int. J. Mol. Sci. 2018, 19, 911. [Google Scholar] [CrossRef] [PubMed]
- Kim, M. DNA methylation: A cause and consequence of type 2 diabetes. Genom. Inform. 2019, 17, e38. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.K.; Yang, H.; Moylan, C.A.; Pang, H.; Dellinger, A.; Abdelmalek, M.F.; Garrett, M.E.; Ashley-Koch, A.; Suzuki, A.; Tillmann, H.L.; et al. Relationship between methylome and transcriptome in patients with nonalcoholic fatty liver disease. Gastroenterology 2013, 145, 1076–1087. [Google Scholar] [CrossRef]
- Hotta, K.; Kitamoto, T.; Kitamoto, A.; Ogawa, Y.; Honda, Y.; Kessoku, T.; Yoneda, M.; Imajo, K.; Tomeno, W.; Saito, S.; et al. Identification of the genomic region under epigenetic regulation during non-alcoholic fatty liver disease progression. Hepatol. Res. 2018, 48, E320–E334. [Google Scholar] [CrossRef] [PubMed]
- Ahrens, M.; Ammerpohl, O.; von Schonfels, W.; Kolarova, J.; Bens, S.; Itzel, T.; Teufel, A.; Herrmann, A.; Brosch, M.; Hinrichsen, H.; et al. DNA methylation analysis in nonalcoholic fatty liver disease suggests distinct disease-specific and remodeling signatures after bariatric surgery. Cell Metab. 2013, 18, 296–302. [Google Scholar] [CrossRef]
- Kitamoto, T.; Kitamoto, A.; Ogawa, Y.; Honda, Y.; Imajo, K.; Saito, S.; Yoneda, M.; Nakamura, T.; Nakajima, A.; Hotta, K. Targeted-bisulfite sequence analysis of the methylation of CpG islands in genes encoding PNPLA3, SAMM50, and PARVB of patients with non-alcoholic fatty liver disease. J. Hepatol. 2015, 63, 494–502. [Google Scholar] [CrossRef]
- Walle, P.; Mannisto, V.; de Mello, V.D.; Vaittinen, M.; Perfilyev, A.; Hanhineva, K.; Ling, C.; Pihlajamaki, J. Liver DNA methylation of FADS2 associates with FADS2 genotype. Clin. Epigenet. 2019, 11, 10. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G., Jr.; McKnight, S.L. Influence of metabolism on epigenetics and disease. Cell 2013, 153, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Thompson, C.B. Metabolic regulation of epigenetics. Cell Metab. 2012, 16, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [PubMed]
- Niki, T.; Rombouts, K.; De Bleser, P.; De Smet, K.; Rogiers, V.; Schuppan, D.; Yoshida, M.; Gabbiani, G.; Geerts, A. A histone deacetylase inhibitor, trichostatin a, suppresses myofibroblastic differentiation of rat hepatic stellate cells in primary culture. Hepatology 1999, 29, 858–867. [Google Scholar] [CrossRef]
- Park, K.C.; Park, J.H.; Jeon, J.Y.; Kim, S.Y.; Kim, J.M.; Lim, C.Y.; Lee, T.H.; Kim, H.K.; Lee, H.G.; Kim, S.M.; et al. A new histone deacetylase inhibitor improves liver fibrosis in BDL rats through suppression of hepatic stellate cells. Br. J. Pharmacol. 2014, 171, 4820–4830. [Google Scholar] [CrossRef]
- Tian, Y.; Wong, V.W.; Wong, G.L.; Yang, W.; Sun, H.; Shen, J.; Tong, J.H.; Go, M.Y.; Cheung, Y.S.; Lai, P.B.; et al. Histone deacetylase HDAC8 promotes insulin resistance and beta-catenin activation in NAFLD-associated hepatocellular carcinoma. Cancer Res. 2015, 75, 4803–4816. [Google Scholar] [CrossRef]
- Dhalluin, C.; Carlson, J.E.; Zeng, L.; He, C.; Aggarwal, A.K.; Zhou, M.M. Structure and ligand of a histone acetyltransferase bromodomain. Nature 1999, 399, 491–496. [Google Scholar] [CrossRef]
- Belkina, A.C.; Denis, G.V. BET domain co-regulators in obesity, inflammation and cancer. Nat. Rev. Cancer 2012, 12, 465–477. [Google Scholar] [CrossRef]
- Theodoulou, N.H.; Tomkinson, N.C.; Prinjha, R.K.; Humphreys, P.G. Clinical progress and pharmacology of small molecule bromodomain inhibitors. Curr. Opin. Chem. Biol. 2016, 33, 58–66. [Google Scholar] [CrossRef][Green Version]
- Ding, N.; Hah, N.; Yu, R.T.; Sherman, M.H.; Benner, C.; Leblanc, M.; He, M.; Liddle, C.; Downes, M.; Evans, R.M. BRD4 is a novel therapeutic target for liver fibrosis. Proc. Natl. Acad. Sci. USA 2015, 112, 15713–15718. [Google Scholar] [CrossRef]
- Jambhekar, A.; Dhall, A.; Shi, Y. Roles and regulation of histone methylation in animal development. Nat. Rev. Mol. Cell Biol. 2019, 20, 625–641. [Google Scholar] [CrossRef]
- Klose, R.J.; Kallin, E.M.; Zhang, Y. JmjC-domain-containing proteins and histone demethylation. Nat. Rev. Genet. 2006, 7, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Bricambert, J.; Alves-Guerra, M.C.; Esteves, P.; Prip-Buus, C.; Bertrand-Michel, J.; Guillou, H.; Chang, C.J.; Vander Wal, M.N.; Canonne-Hergaux, F.; Mathurin, P.; et al. The histone demethylase Phf2 acts as a molecular checkpoint to prevent NAFLD progression during obesity. Nat. Commun. 2018, 9, 2092. [Google Scholar] [CrossRef] [PubMed]
- Page, A.; Paoli, P.P.; Hill, S.J.; Howarth, R.; Wu, R.; Kweon, S.M.; French, J.; White, S.; Tsukamoto, H.; Mann, D.A.; et al. Alcohol directly stimulates epigenetic modifications in hepatic stellate cells. J. Hepatol. 2015, 62, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Perugorria, M.J.; Wilson, C.L.; Zeybel, M.; Walsh, M.; Amin, S.; Robinson, S.; White, S.A.; Burt, A.D.; Oakley, F.; Tsukamoto, H.; et al. Histone methyltransferase ash1 orchestrates fibrogenic gene transcription during myofibroblast transdifferentiation. Hepatology 2012, 56, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Argemi, J.; Bataller, R. Identifying new epigenetic drivers of liver fibrosis. Cell Mol. Gastroenterol. Hepatol. 2019, 7, 237–238. [Google Scholar] [CrossRef]
- Ringrose, L.; Paro, R. Epigenetic regulation of cellular memory by the polycomb and trithorax group proteins. Ann. Rev. Genet. 2004, 38, 413–443. [Google Scholar] [CrossRef] [PubMed]
- Francis, N.J.; Kingston, R.E.; Woodcock, C.L. Chromatin compaction by a polycomb group protein complex. Science 2004, 306, 1574–1577. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef]
- Di Croce, L.; Helin, K. Transcriptional regulation by polycomb group proteins. Nat. Struct. Mol. Biol. 2013, 20, 1147–1155. [Google Scholar] [CrossRef] [PubMed]
- Margueron, R.; Li, G.; Sarma, K.; Blais, A.; Zavadil, J.; Woodcock, C.L.; Dynlacht, B.D.; Reinberg, D. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol. Cell 2008, 32, 503–518. [Google Scholar] [CrossRef] [PubMed]
- Gunawan, M.; Venkatesan, N.; Loh, J.T.; Wong, J.F.; Berger, H.; Neo, W.H.; Li, L.Y.; La Win, M.K.; Yau, Y.H.; Guo, T.; et al. The methyltransferase Ezh2 controls cell adhesion and migration through direct methylation of the extranuclear regulatory protein talin. Nat. Immunol. 2015, 16, 505–516. [Google Scholar] [CrossRef]
- Kim, E.; Kim, M.; Woo, D.H.; Shin, Y.; Shin, J.; Chang, N.; Oh, Y.T.; Kim, H.; Rheey, J.; Nakano, I.; et al. Phosphorylation of EZH2 activates STAT3 signaling via stat3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell 2013, 23, 839–852. [Google Scholar] [CrossRef] [PubMed]
- Su, I.H.; Dobenecker, M.W.; Dickinson, E.; Oser, M.; Basavaraj, A.; Marqueron, R.; Viale, A.; Reinberg, D.; Wulfing, C.; Tarakhovsky, A. Polycomb group protein Ezh2 controls actin polymerization and cell signaling. Cell 2005, 121, 425–436. [Google Scholar] [CrossRef]
- Vasanthakumar, A.; Xu, D.; Lun, A.T.; Kueh, A.J.; van Gisbergen, K.P.; Iannarella, N.; Li, X.; Yu, L.; Wang, D.; Williams, B.R.; et al. A non-canonical function of Ezh2 preserves immune homeostasis. EMBO Rep. 2017, 18, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Wu, Z.J.; Groner, A.C.; He, H.H.; Cai, C.; Lis, R.T.; Wu, X.; Stack, E.C.; Loda, M.; Liu, T.; et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is polycomb-independent. Science 2012, 338, 1465–1469. [Google Scholar] [CrossRef]
- Batool, A.; Jin, C.; Liu, Y.X. Role of EZH2 in cell lineage determination and relative signaling pathways. Front. Biosci. 2019, 24, 947–960. [Google Scholar] [CrossRef]
- Hu, X.; Eastman, A.E.; Guo, S. Cell cycle dynamics in the reprogramming of cellular identity. FEBS Lett. 2019, 593, 2840–2852. [Google Scholar] [CrossRef]
- Yao, Y.; Hu, H.; Yang, Y.; Zhou, G.; Shang, Z.; Yang, X.; Sun, K.; Zhan, S.; Yu, Z.; Li, P.; et al. Downregulation of enhancer of zeste homolog 2 (EZH2) is essential for the induction of autophagy and apoptosis in colorectal cancer cells. Genes 2016, 7, 83. [Google Scholar] [CrossRef]
- Michalopoulos, G.K.; DeFrances, M.C. Liver regeneration. Science 1997, 276, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Fausto, N. Liver regeneration and repair: Hepatocytes, progenitor cells, and stem cells. Hepatology 2004, 39, 1477–1487. [Google Scholar] [CrossRef] [PubMed]
- Fausto, N.; Campbell, J.S.; Riehle, K.J. Liver regeneration. Hepatology 2006, 43, S45–S53. [Google Scholar] [CrossRef] [PubMed]
- Aoki, R.; Chiba, T.; Miyagi, S.; Negishi, M.; Konuma, T.; Taniguchi, H.; Ogawa, M.; Yokosuka, O.; Iwama, A. The polycomb group gene product Ezh2 regulates proliferation and differentiation of murine hepatic stem/progenitor cells. J. Hepatol. 2010, 52, 854–863. [Google Scholar] [CrossRef] [PubMed]
- Koike, H.; Ouchi, R.; Ueno, Y.; Nakata, S.; Obana, Y.; Sekine, K.; Zheng, Y.W.; Takebe, T.; Isono, K.; Koseki, H.; et al. Polycomb group protein Ezh2 regulates hepatic progenitor cell proliferation and differentiation in murine embryonic liver. PLoS ONE 2014, 9, e104776. [Google Scholar] [CrossRef]
- Bae, W.K.; Kang, K.; Yu, J.H.; Yoo, K.H.; Factor, V.M.; Kaji, K.; Matter, M.; Thorgeirsson, S.; Hennighausen, L. The methyltransferases enhancer of zeste homolog (EZH) 1 and EZH2 control hepatocyte homeostasis and regeneration. FASEB J. 2015, 29, 1653–1662. [Google Scholar] [CrossRef]
- Grindheim, J.M.; Nicetto, D.; Donahue, G.; Zaret, K.S. Polycomb repressive complex 2 proteins EZH1 and EZH2 regulate timing of postnatal hepatocyte maturation and fibrosis by repressing genes with euchromatic promoters in mice. Gastroenterology 2019, 156, 1834–1848. [Google Scholar] [CrossRef]
- Cai, M.Y.; Tong, Z.T.; Zheng, F.; Liao, Y.J.; Wang, Y.; Rao, H.L.; Chen, Y.C.; Wu, Q.L.; Liu, Y.H.; Guan, X.Y.; et al. EZH2 protein: A promising immunomarker for the detection of hepatocellular carcinomas in liver needle biopsies. Gut 2011, 60, 967–976. [Google Scholar] [CrossRef]
- Au, S.L.; Wong, C.C.; Lee, J.M.; Fan, D.N.; Tsang, F.H.; Ng, I.O.; Wong, C.M. Enhancer of zeste homolog 2 epigenetically silences multiple tumor suppressor micrornas to promote liver cancer metastasis. Hepatology 2012, 56, 622–631. [Google Scholar] [CrossRef]
- Chen, S.; Pu, J.; Bai, J.; Yin, Y.; Wu, K.; Wang, J.; Shuai, X.; Gao, J.; Tao, K.; Wang, G.; et al. EZH2 promotes hepatocellular carcinoma progression through modulating miR-22/galectin-9 axis. J. Exp. Clin. Cancer Res. 2018, 37, 3. [Google Scholar] [CrossRef]
- Liu, H.; Liu, Y.; Liu, W.; Zhang, W.; Xu, J. EZH2-mediated loss of miR-622 determines cxcr4 activation in hepatocellular carcinoma. Nat. Commun. 2015, 6, 8494. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Yu, Z.; Tian, Y.; Lee, Y.Y.; Li, M.S.; Go, M.Y.; Cheung, Y.S.; Lai, P.B.; Chan, A.M.; To, K.F.; et al. A CCRK-EZH2 epigenetic circuitry drives hepatocarcinogenesis and associates with tumor recurrence and poor survival of patients. J. Hepatol. 2015, 62, 1100–1111. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.; Chu, D.C.; Maxwell, A.; Oakley, F.; Zhu, N.L.; Tsukamoto, H.; Mann, D.A. MeCP2 controls an epigenetic pathway that promotes myofibroblast transdifferentiation and fibrosis. Gastroenterology 2010, 138, 705–714, 714.e1-4. [Google Scholar] [CrossRef] [PubMed]
- Hazra, S.; Xiong, S.; Wang, J.; Rippe, R.A.; Krishna, V.; Chatterjee, K.; Tsukamoto, H. Peroxisome proliferator-activated receptor gamma induces a phenotypic switch from activated to quiescent hepatic stellate cells. J. Biol. Chem. 2004, 279, 11392–11401. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, X.X.; Li, W.X.; Wu, X.Q.; Huang, C.; Xie, J.; Zhao, Y.X.; Meng, X.M.; Li, J. EZH2-mediated repression of Dkk1 promotes hepatic stellate cell activation and hepatic fibrosis. J. Cell Mol. Med. 2017, 21, 2317–2328. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Wang, Z.; Tang, F.; Zhao, Y.; Feng, D.; Li, Y.; Hu, Y.; Wang, C.; Zhou, J.; Tian, X.; et al. Carnosol-mediated Sirtuin 1 activation inhibits Enhancer of Zeste Homolog 2 to attenuate liver fibrosis. Pharmacol. Res. 2018, 128, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Dooley, S.; ten Dijke, P. Tgf-beta in progression of liver disease. Cell Tissue Res. 2012, 347, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Castilla, A.; Prieto, J.; Fausto, N. Transforming growth factors beta 1 and alpha in chronic liver disease. Effects of interferon alfa therapy. N. Engl. J. Med. 1991, 324, 933–940. [Google Scholar] [CrossRef]
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef]
- Martin-Mateos, R.; De Assuncao, T.M.; Arab, J.P.; Jalan-Sakrikar, N.; Yaqoob, U.; Greuter, T.; Verma, V.K.; Mathison, A.J.; Cao, S.; Lomberk, G.; et al. Enhancer of zeste homologue 2 inhibition attenuates TGF-beta dependent hepatic stellate cell activation and liver fibrosis. Cell Mol. Gastroenterol. Hepatol. 2019, 7, 197–209. [Google Scholar] [CrossRef]
- Vella, S.; Gnani, D.; Crudele, A.; Ceccarelli, S.; De Stefanis, C.; Gaspari, S.; Nobili, V.; Locatelli, F.; Marquez, V.E.; Rota, R.; et al. EZH2 down-regulation exacerbates lipid accumulation and inflammation in in vitro and in vivo NAFLD. Int. J. Mol. Sci. 2013, 14, 24154–24168. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Woo, D.C.; Kang, J.; Ra, M.; Kim, K.H.; Lee, S.R.; Choi, D.K.; Lee, H.; Hong, K.B.; Min, S.H.; et al. The role of the histone methyltransferase EZH2 in liver inflammation and fibrosis in STAM NASH mice. Biology 2020, 9, 93. [Google Scholar] [CrossRef] [PubMed]
- Furman, B.L. Streptozotocin-induced diabetic models in mice and rats. Curr. Protoc. Pharmacol. 2015, 70, 5.47.1–5.47.20. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Uebanso, T.; Maekawa, K.; Ishikawa, M.; Taguchi, R.; Nammo, T.; Nishimaki-Mogami, T.; Udagawa, H.; Fujii, M.; Shibazaki, Y.; et al. Characterization of hepatic lipid profiles in a mouse model with nonalcoholic steatohepatitis and subsequent fibrosis. Sci. Rep. 2015, 5, 12466. [Google Scholar] [CrossRef]
- Tam, E.K.; Nguyen, T.M.; Lim, C.Z.; Lee, P.L.; Li, Z.; Jiang, X.; Santhanakrishnan, S.; Tan, T.W.; Goh, Y.L.; Wong, S.Y.; et al. 3-deazaneplanocin a and neplanocin a analogues and their effects on apoptotic cell death. ChemMedChem 2015, 10, 173–182. [Google Scholar] [CrossRef]
- Fujiwara, T.; Saitoh, H.; Inoue, A.; Kobayashi, M.; Okitsu, Y.; Katsuoka, Y.; Fukuhara, N.; Onishi, Y.; Ishizawa, K.; Ichinohasama, R.; et al. 3-deazaneplanocin a (DZNep), an inhibitor of s-adenosylmethionine-dependent methyltransferase, promotes erythroid differentiation. J. Biol. Chem. 2014, 289, 8121–8134. [Google Scholar] [CrossRef]
- Zeybel, M.; Luli, S.; Sabater, L.; Hardy, T.; Oakley, F.; Leslie, J.; Page, A.; Moran Salvador, E.; Sharkey, V.; Tsukamoto, H.; et al. A proof-of-concept for epigenetic therapy of tissue fibrosis: Inhibition of liver fibrosis progression by 3-deazaneplanocin a. Mol. Ther. 2017, 25, 218–231. [Google Scholar] [CrossRef]
- Knutson, S.K.; Wigle, T.J.; Warholic, N.M.; Sneeringer, C.J.; Allain, C.J.; Klaus, C.R.; Sacks, J.D.; Raimondi, A.; Majer, C.R.; Song, J.; et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat. Chem. Biol. 2012, 8, 890–896. [Google Scholar] [CrossRef]
- Qi, W.; Chan, H.; Teng, L.; Li, L.; Chuai, S.; Zhang, R.; Zeng, J.; Li, M.; Fan, H.; Lin, Y.; et al. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc. Natl. Acad. Sci. USA 2012, 109, 21360–21365. [Google Scholar] [CrossRef]
- McCabe, M.T.; Ott, H.M.; Ganji, G.; Korenchuk, S.; Thompson, C.; Van Aller, G.S.; Liu, Y.; Graves, A.P.; Della Pietra, A., 3rd; Diaz, E.; et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012, 492, 108–112. [Google Scholar] [CrossRef]
- Zhou, T.; Sun, Y.; Li, M.; Ding, Y.; Yin, R.; Li, Z.; Xie, Q.; Bao, S.; Cai, W. Enhancer of zeste homolog 2-catalysed H3K27 trimethylation plays a key role in acute-on-chronic liver failure via TNF-mediated pathway. Cell Death Dis. 2018, 9, 590. [Google Scholar] [CrossRef] [PubMed]
- Konze, K.D.; Ma, A.; Li, F.; Barsyte-Lovejoy, D.; Parton, T.; Macnevin, C.J.; Liu, F.; Gao, C.; Huang, X.P.; Kuznetsova, E.; et al. An orally bioavailable chemical probe of the lysine methyltransferases EZH2 and EZH1. ACS Chem. Biol. 2013, 8, 1324–1334. [Google Scholar] [CrossRef] [PubMed]
- Knutson, S.K.; Warholic, N.M.; Wigle, T.J.; Klaus, C.R.; Allain, C.J.; Raimondi, A.; Porter Scott, M.; Chesworth, R.; Moyer, M.P.; Copeland, R.A.; et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc. Natl. Acad. Sci. USA 2013, 110, 7922–7927. [Google Scholar] [CrossRef] [PubMed]
- Knutson, S.K.; Kawano, S.; Minoshima, Y.; Warholic, N.M.; Huang, K.C.; Xiao, Y.; Kadowaki, T.; Uesugi, M.; Kuznetsov, G.; Kumar, N.; et al. Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in ezh2-mutant non-hodgkin lymphoma. Mol. Cancer Ther. 2014, 13, 842–854. [Google Scholar] [CrossRef]
- Hoy, S.M. Tazemetostat: First approval. Drugs 2020, 80, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Yang, X.; Zhuang, L.; Jiang, X.; Chen, W.; Lee, P.L.; Karuturi, R.K.; Tan, P.B.; Liu, E.T.; Yu, Q. Pharmacologic disruption of polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev. 2007, 21, 1050–1063. [Google Scholar] [CrossRef] [PubMed]
- Beguelin, W.; Popovic, R.; Teater, M.; Jiang, Y.; Bunting, K.L.; Rosen, M.; Shen, H.; Yang, S.N.; Wang, L.; Ezponda, T.; et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell 2013, 23, 677–692. [Google Scholar] [CrossRef]
- Shahabipour, F.; Caraglia, M.; Majeed, M.; Derosa, G.; Maffioli, P.; Sahebkar, A. Naturally occurring anti-cancer agents targeting EZH2. Cancer Lett. 2017, 400, 325–335. [Google Scholar] [CrossRef]
- Zhu, C.; Cao, H.; Zhou, X.; Dong, C.; Luo, J.; Zhang, C.; Liu, J.; Ling, Y. Meta-analysis of the clinical value of danshen injection and huangqi injection in liver cirrhosis. Evid. Based Complement. Alternat. Med. 2013, 2013, 842824. [Google Scholar] [CrossRef]
- Woo, J.; Kim, H.Y.; Byun, B.J.; Chae, C.H.; Lee, J.Y.; Ryu, S.Y.; Park, W.K.; Cho, H.; Choi, G. Biological evaluation of tanshindiols as EZH2 histone methyltransferase inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 2486–2492. [Google Scholar] [CrossRef]
- Shen, L.; Liu, C.C.; An, C.Y.; Ji, H.F. How does curcumin work with poor bioavailability? Clues from experimental and theoretical studies. Sci. Rep. 2016, 6, 20872. [Google Scholar] [CrossRef] [PubMed]
- Maheshwari, R.K.; Singh, A.K.; Gaddipati, J.; Srimal, R.C. Multiple biological activities of curcumin: A short review. Life Sci. 2006, 78, 2081–2087. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, S.; Asgary, S.; Askari, G.; Keshvari, M.; Hatamipour, M.; Feizi, A.; Sahebkar, A. Treatment of non-alcoholic fatty liver disease with curcumin: A randomized placebo-controlled trial. Phytother. Res. 2016, 30, 1540–1548. [Google Scholar] [CrossRef] [PubMed]
- Hua, W.F.; Fu, Y.S.; Liao, Y.J.; Xia, W.J.; Chen, Y.C.; Zeng, Y.X.; Kung, H.F.; Xie, D. Curcumin induces down-regulation of EZH2 expression through the MAPK pathway in MDA-MB-435 human breast cancer cells. Eur. J. Pharmacol. 2010, 637, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.Q.; Chai, K.Q.; Zhu, X.M.; Jiang, H.; Wang, X.; Xue, Q.; Zheng, A.H.; Zhou, H.Y.; Chen, Y.; Chen, X.C.; et al. Anti-cancer effects of curcumin on lung cancer through the inhibition of EZH2 and NOTCH1. Oncotarget 2016, 7, 26535–26550. [Google Scholar] [CrossRef]
- Mansour-Ghanaei, F.; Hadi, A.; Pourmasoumi, M.; Joukar, F.; Golpour, S.; Najafgholizadeh, A. Green tea as a safe alternative approach for nonalcoholic fatty liver treatment: A systematic review and meta-analysis of clinical trials. Phytother. Res. 2018, 32, 1876–1884. [Google Scholar] [CrossRef]
- Shimizu, M.; Fukutomi, Y.; Ninomiya, M.; Nagura, K.; Kato, T.; Araki, H.; Suganuma, M.; Fujiki, H.; Moriwaki, H. Green tea extracts for the prevention of metachronous colorectal adenomas: A pilot study. Cancer Epidemiol. Biomark. Prev. 2008, 17, 3020–3025. [Google Scholar] [CrossRef]
- Borutinskaite, V.; Virksaite, A.; Gudelyte, G.; Navakauskiene, R. Green tea polyphenol egcg causes anti-cancerous epigenetic modulations in acute promyelocytic leukemia cells. Leuk. Lymphoma 2018, 59, 469–478. [Google Scholar] [CrossRef]
- Lau, J.K.; Zhang, X.; Yu, J. Animal models of non-alcoholic fatty liver disease: Current perspectives and recent advances. J. Pathol. 2017, 241, 36–44. [Google Scholar] [CrossRef]
- Ouchi, R.; Togo, S.; Kimura, M.; Shinozawa, T.; Koido, M.; Koike, H.; Thompson, W.; Karns, R.A.; Mayhew, C.N.; McGrath, P.S.; et al. Modeling steatohepatitis in humans with pluripotent stem cell-derived organoids. Cell Metab. 2019, 30, 374–384.e376. [Google Scholar] [CrossRef]
- Mun, S.J.; Ryu, J.S.; Lee, M.O.; Son, Y.S.; Oh, S.J.; Cho, H.S.; Son, M.Y.; Kim, D.S.; Kim, S.J.; Yoo, H.J.; et al. Generation of expandable human pluripotent stem cell-derived hepatocyte-like liver organoids. J. Hepatol. 2019, 71, 970–985. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Bird, G.H.; Neff, T.; Guo, G.; Kerenyi, M.A.; Walensky, L.D.; Orkin, S.H. Targeted disruption of the EZH2-EED complex inhibits EZH2-dependent cancer. Nat. Chem. Biol. 2013, 9, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Tien, J.C.; Vo, J.; Tan, M.; Parolia, A.; Zhang, Y.; Wang, L.; Qiao, Y.; Shukla, S.; Wang, X.; et al. Epigenetic reprogramming with antisense oligonucleotides enhances the effectiveness of androgen receptor inhibition in castration-resistant prostate cancer. Cancer Res. 2018, 78, 5731–5740. [Google Scholar] [CrossRef] [PubMed]
- Duan, R.; Du, W.; Guo, W. EZH2: A novel target for cancer treatment. J. Hematol. Oncol. 2020, 13, 104. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Compound | Mechanism | Selectivity * | Ref(s). |
|---|---|---|---|
| DZNep | SAH hydrolase inhibitor | Unknown | [86,96] |
| EPZ005687 | SAM-competitive inhibitor of PRC2 | >500-fold over other HMTs, ~50-fold over EZH1 | [88] |
| EI1 | SAM-competitive inhibitor of PRC2 | >10,000-fold over other HMTs, ~90-fold over EZH1 | [89] |
| GSK126 | SAM-competitive inhibitor of PRC2 | >1000-fold over other HMTs, 150-fold over EZH1 | [90] |
| UNC1999 | SAM-competitive inhibitor of PRC2 | 10,000-fold over other HMTs, 10-fold over EZH1 | [92] |
| GSK503 | SAM-competitive inhibitor of PRC2 | >4000-fold over other HMTs, 200-fold over EZH1 | [97] |
| EPZ-6438 | SAM-competitive inhibitor of PRC2 | >4500-fold over other HMTs, 35-fold over EZH1 | [93,94] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, H.J.; Kim, M. EZH2 as a Potential Target for NAFLD Therapy. Int. J. Mol. Sci. 2020, 21, 8617. https://doi.org/10.3390/ijms21228617
Lim HJ, Kim M. EZH2 as a Potential Target for NAFLD Therapy. International Journal of Molecular Sciences. 2020; 21(22):8617. https://doi.org/10.3390/ijms21228617
Chicago/Turabian StyleLim, Hyun Jung, and Mirang Kim. 2020. "EZH2 as a Potential Target for NAFLD Therapy" International Journal of Molecular Sciences 21, no. 22: 8617. https://doi.org/10.3390/ijms21228617
APA StyleLim, H. J., & Kim, M. (2020). EZH2 as a Potential Target for NAFLD Therapy. International Journal of Molecular Sciences, 21(22), 8617. https://doi.org/10.3390/ijms21228617