Hydroxylation of Antitubercular Drug Candidate, SQ109, by Mycobacterial Cytochrome P450

 , , , , and
, , , , and
Abstract
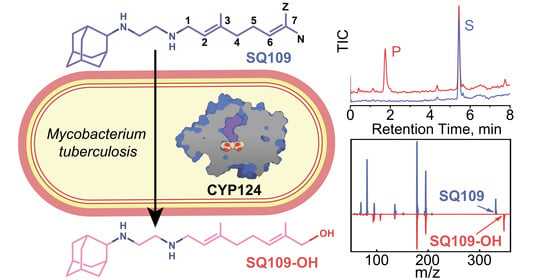
1. Introduction
2. Results
2.1. CYP124, CYP125, and CYP142 Bind SQ109
2.2. CYP124 Catalyses Hydroxylation of SQ109
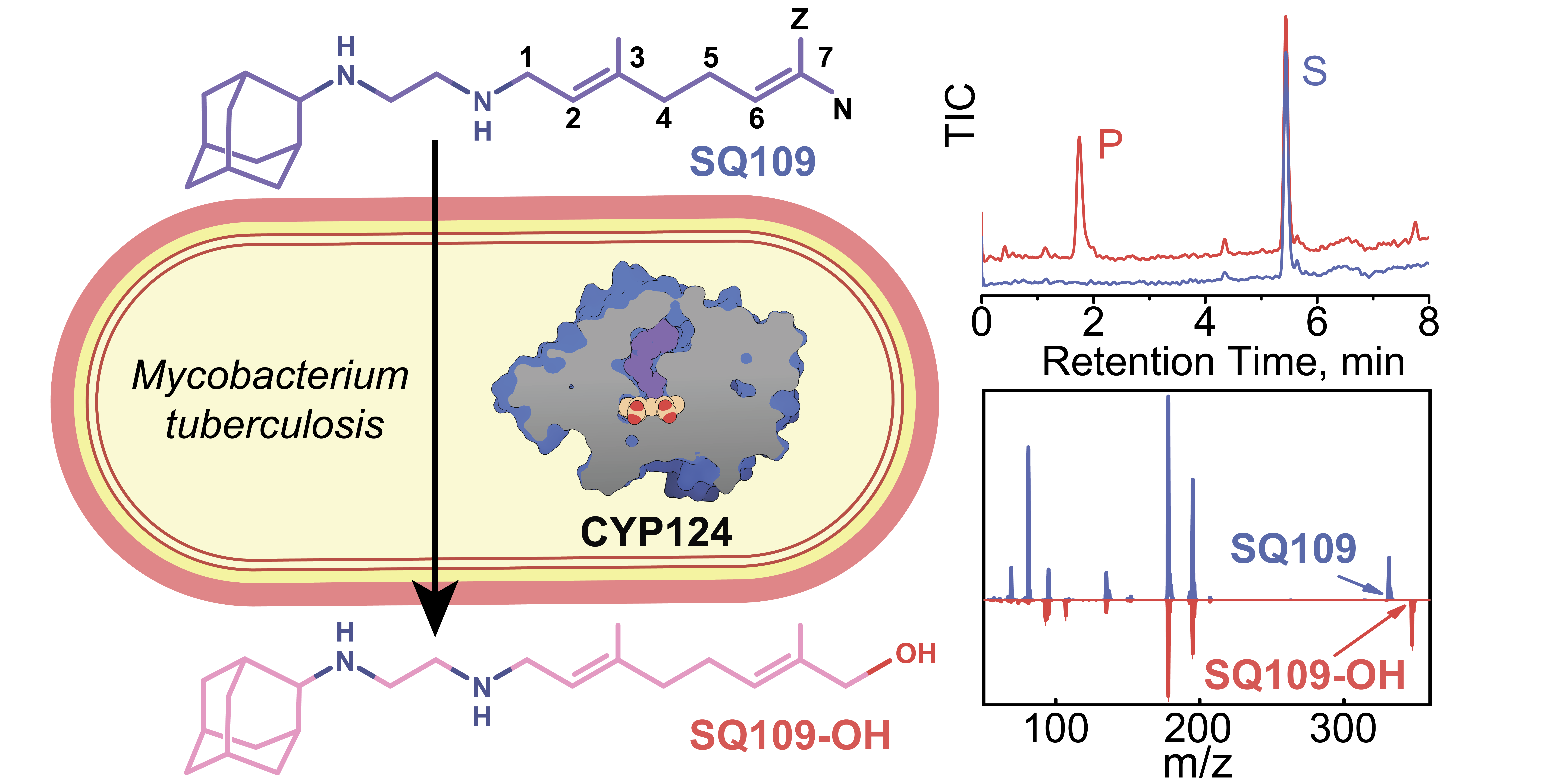
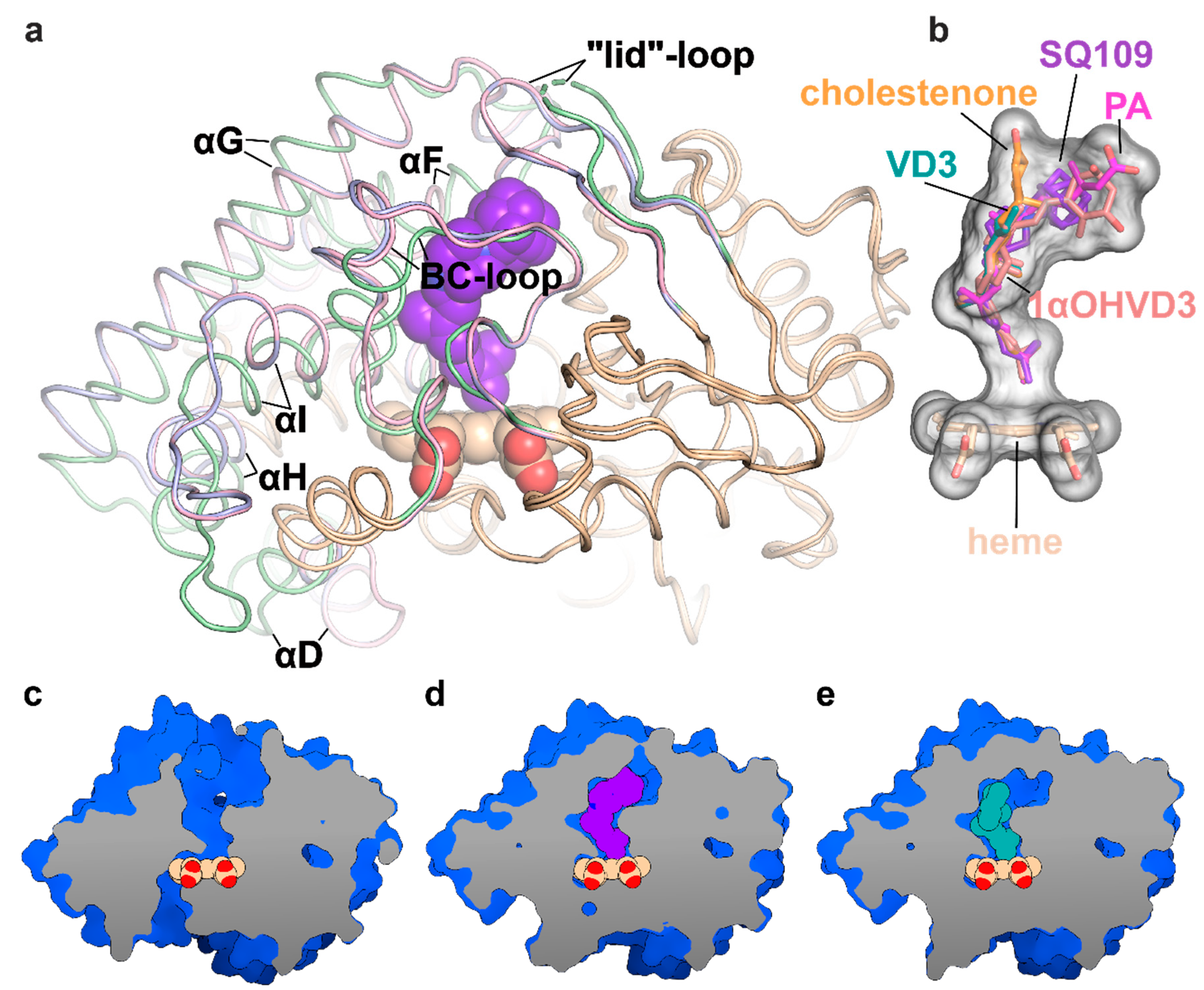
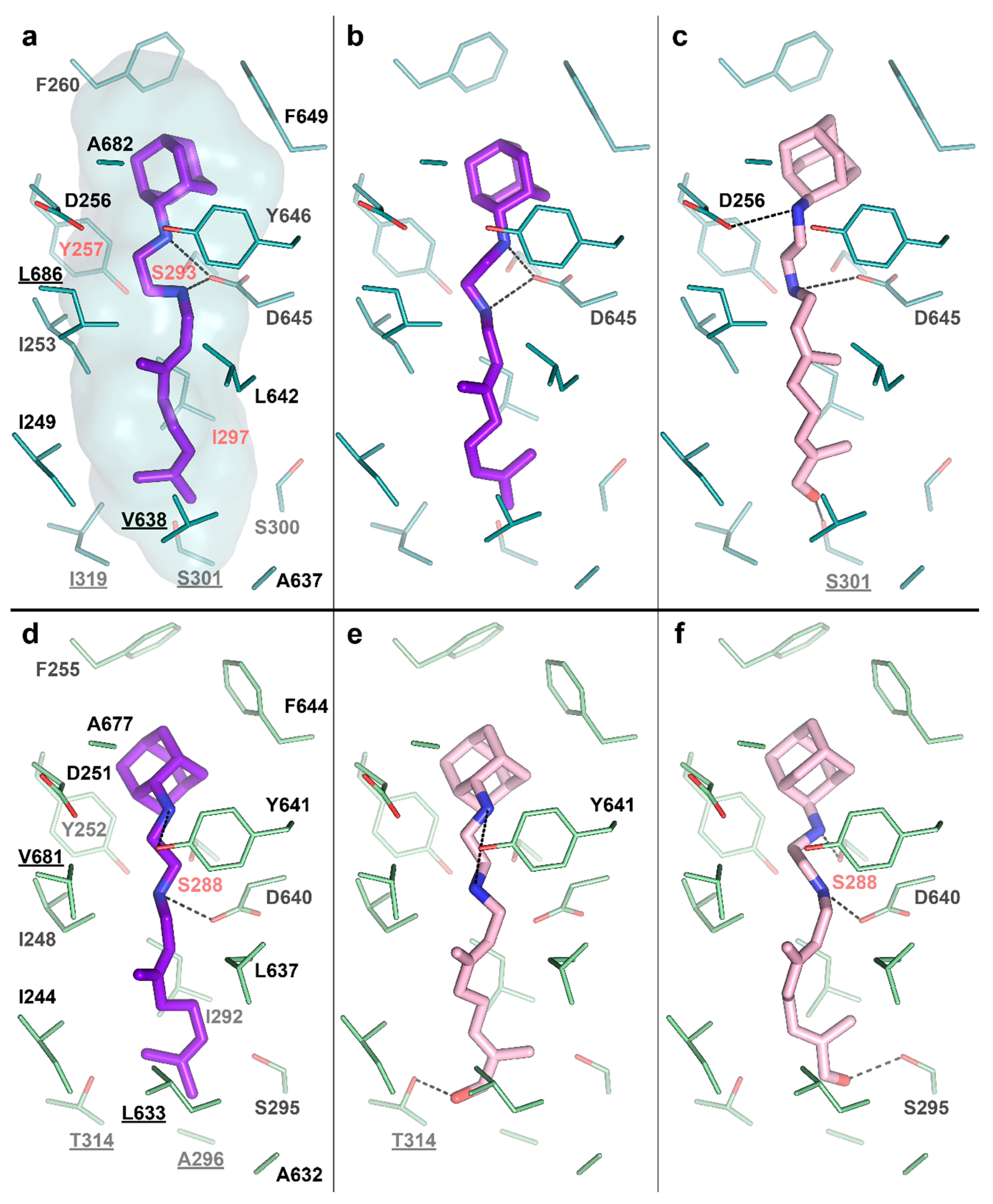
2.3. Crystal Structure of CYP124–SQ109 Complex
3. Discussion
4. Materials and Methods
4.1. Cloning, Expression, and Purification of Recombinant CYP124
4.2. Substrate Binding Studies
4.3. Catalytic Activity Assay
4.4. Identification of SQ109 Product
4.5. Crystallization, Data Collection, and Crystal Structure Determination
4.6. Molecular Docking
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 1αOHVD3 | 1α-hydroxy-vitamin D3 |
| Arh1 | adrenodoxin reductase-like flavoprotein |
| BCG | bacillus Calmette–Guérin |
| cholestenone | cholest-4-en-3-one |
| CYP | Cytochrome P450 |
| ESI | electrospray ionization source |
| ESRF | European Synchrotron Radiation Facility |
| Fdx1 | Ferredoxin-1 |
| Kdapp | ligand-binding constant |
| MDR | multidrug-resistant |
| MIC | minimal inhibitory concentration |
| MmpL3 | Mycobacterial membrane protein Large 3 |
| Msm | Mycobacterium. smegmatis |
| Mtb | Mycobacterium tuberculosis |
| PDB ID | Protein Data Bank accession number |
| PA | phytanic acid |
| RIF | rifampicin |
| VD3 | vitamin D3 |
References
- World Health Organization. Global Tuberculosis Report 2019; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Cohen, K.A.; Manson, A.L.; Abeel, T.; Desjardins, C.A.; Chapman, S.B.; Hoffner, S.; Birren, B.W.; Earl, A.M. Extensive global movement of multidrug-resistant M. tuberculosis strains revealed by whole-genome analysis. Thorax 2019, 74, 882–889. [Google Scholar] [CrossRef]
- Lange, C.; Chesov, D.; Furin, J.; Udwadia, Z.; Dheda, K. Revising the definition of extensively drug-resistant tuberculosis. Lancet Respir. Med. 2018, 6, 893–895. [Google Scholar] [CrossRef]
- Protopopova, M.; Hanrahan, C.; Nikonenko, B.; Samala, R.; Chen, P.; Gearhart, J.; Einck, L.; Nacy, C.A. Identification of a new antitubercular drug candidate, SQ109, from a combinatorial library of 1,2-ethylenediamines. J. Antimicrob. Chemother. 2005, 56, 968–974. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.S.; Paterson, D.L. Antibiotics in the clinical pipeline in October 2019. J. Antibiot. 2020, 73, 329–364. [Google Scholar] [CrossRef] [PubMed]
- Sacksteder, K.A.; Protopopova, M.; Barry, C.E.; Andries, K.; Nacy, C.A. Discovery and development of SQ109: A new antitubercular drug with a novel mechanism of action. Future Microbiol. 2012, 7, 823–837. [Google Scholar] [CrossRef]
- Tahlan, K.; Wilson, R.; Kastrinsky, D.B.; Arora, K.; Nair, V.; Fischer, E.; Whitney Barnes, S.; Walker, J.R.; Alland, D.; Barry, C.E.; et al. SQ109 targets MmpL3, a membrane transporter of trehalose monomycolate involved in mycolic acid donation to the cell wall core of mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2012, 56, 1797–1809. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Schurig-Briccio, L.A.; Feng, X.; Upadhyay, A.; Pujari, V.; Lechartier, B.; Fontes, F.L.; Yang, H.; Rao, G.; Zhu, W.; et al. Multitarget drug discovery for tuberculosis and other infectious diseases. J. Med. Chem. 2014, 57, 3126–3129. [Google Scholar] [CrossRef]
- Chen, P.; Gearhart, J.; Protopopova, M.; Einck, L.; Nacy, C.A. Synergistic interactions of SQ109, a new ethylene diamine, with front-line antitubercular drugs in vitro. J. Antimicrob. Chemother. 2006, 58, 332–337. [Google Scholar] [CrossRef]
- Makobongo, M.O.; Einck, L.; Peek, R.M.; Merrell, D.S. In Vitro Characterization of the Anti-Bacterial Activity of SQ109 against Helicobacter pylori. PLoS ONE 2013, 8, e68917. [Google Scholar] [CrossRef]
- García-García, V.; Oldfield, E.; Benaim, G. Inhibition of Leishmania mexicana Growth by the Tuberculosis Drug SQ109. Antimicrob. Agents Chemother. 2016. [Google Scholar] [CrossRef]
- Veiga-Santos, P.; Li, K.; Lameira, L.; Ulisses De Carvalho, T.M.; Huang, G.; Galizzi, M.; Shang, N.; Li, Q.; Gonzalez-Pacanowska, D.; Hernandez-Rodriguez, V.; et al. SQ109, a new drug lead for chagas disease. Antimicrob. Agents Chemother. 2015. [Google Scholar] [CrossRef] [PubMed]
- Gil, Z.; Martinez-Sotillo, N.; Pinto-Martinez, A.; Mejias, F.; Martinez, J.C.; Galindo, I.; Oldfield, E.; Benaim, G. SQ109 inhibits proliferation of Leishmania donovani by disruption of intracellular Ca2+ homeostasis, collapsing the mitochondrial electrochemical potential (ΔΨm) and affecting acidocalcisomes. Parasitol. Res. 2020, 119, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.M.; Einck, L.; Andries, K.; Nacy, C.A. In vitro interactions between new antitubercular drug candidates SQ109 and TMC207. Antimicrob. Agents Chemother. 2010, 54, 2840–2846. [Google Scholar] [CrossRef] [PubMed]
- Sotgiu, G.; Centis, R.; D’Ambrosio, L.; Battista Migliori, G. Tuberculosis treatment and drug regimens. Cold Spring Harb. Perspect. Med. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Noker, P.E.; Coward, L.; Gorman, G.S.; Protopopova, M.; Tomaszewski, J.E. Interspecies pharmacokinetics and in vitro metabolism of SQ109. Br. J. Pharmacol. 2006, 147, 476–485. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.B.; Kells, P.M.; Podust, L.M.; Ortiz de Montellano, P.R. Biochemical and structural characterization of CYP124: A methyl-branched lipid omega-hydroxylase from Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2009, 106, 20687–20692. [Google Scholar] [CrossRef]
- Vasilevskaya, A.V.; Yantsevich, A.V.; Sergeev, G.V.; Lemish, A.P.; Usanov, S.A.; Gilep, A.A. Identification of Mycobacterium tuberculosis enzyme involved in vitamin D and 7-dehydrocholesterol metabolism. J. Steroid Biochem. Mol. Biol. 2017, 169, 202–209. [Google Scholar] [CrossRef]
- Johnston, J.B.; Ouellet, H.; Ortiz De Montellano, P.R. Functional redundancy of steroid C26-monooxygenase activity in Mycobacterium tuberculosis revealed by biochemical and genetic analyses. J. Biol. Chem. 2010, 285, 36352–36360. [Google Scholar] [CrossRef]
- Varaksa, T.; Bukhdruker, S.; Grabovec, I.; Marin, E.; Kavaleuski, A.; Gusach, A.; Kovalev, K.; Maslov, I.; Luginina, A.; Zabelskiy, D.; et al. Metabolic fate of human immunoactive sterols in Mycobacterium tuberculosis. bioRxiv 2020, arXiv:2020.07.07.192294. [Google Scholar] [CrossRef]
- Makarov, V.; Salina, E.; Reynolds, R.C.; Kyaw Zin, P.P.; Ekins, S. Molecule Property Analyses of Active Compounds for Mycobacterium tuberculosis. J. Med. Chem. 2020. [Google Scholar] [CrossRef]
- Weinert, L.A.; Welch, J.J. Why Might Bacterial Pathogens Have Small Genomes? Trends Ecol. Evol. 2017, 32, 936–947. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Stevens, C.M.; Pandya, A.N.; Darzynkiewicz, Z.; Bhattarai, P.; Tong, W.; Gonzalez-Juarrero, M.; North, E.J.; Zgurskaya, H.I.; Jackson, M. Direct Inhibition of MmpL3 by Novel Antitubercular Compounds. ACS Infect. Dis. 2019, 5, 1001–1012. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Li, J.; Yang, X.; Wu, L.; Zhang, J.; Yang, Y.; Zhao, Y.; Zhang, L.; Yang, X.; Yang, X.; et al. Crystal Structures of Membrane Transporter MmpL3, an Anti-TB Drug Target. Cell 2019, 176, 636–648.e13. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Meshcheryakov, V.A.; Poce, G.; Chng, S.S. MmpL3 is the flippase for mycolic acids in mycobacteria. Proc. Natl. Acad. Sci. USA 2017, 114, 7993–7998. [Google Scholar] [CrossRef]
- Ramachandran, G.; Gurumurthy, P. Effect of Rifampicin & Isoniazid on Cytochrome P-450 in Mycobacteria. Indian J. Med. Res. 2002, 116, 140–144. [Google Scholar]
- Grzegorzewicz, A.E.; Pham, H.; Gundi, V.A.K.B.; Scherman, M.S.; North, E.J.; Hess, T.; Jones, V.; Gruppo, V.; Born, S.E.M.; Korduláková, J.; et al. Inhibition of mycolic acid transport across the Mycobacterium tuberculosis plasma membrane. Nat. Chem. Biol. 2012, 8, 334–341. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, V.; Poce, G.; Canseco, J.O.; Buroni, S.; Pasca, M.R.; Biava, M.; Raju, R.M.; Porretta, G.C.; Alfonso, S.; Battilocchio, C.; et al. MmpL3 is the cellular target of the antitubercular pyrrole derivative BM212. Antimicrob. Agents Chemother. 2012, 56, 324–331. [Google Scholar] [CrossRef]
- Awasthi, D.; Freundlich, J.S. Antimycobacterial metabolism: Illuminating mycobacterium tuberculosis biology and drug discovery. Trends Microbiol. 2017, 25, 756–767. [Google Scholar] [CrossRef]
- Zhang, Y.; Heym, B.; Allen, B.; Young, D.; Cole, S. The catalase—Peroxidase gene and isoniazid resistance of Mycobacterium tuberculosis. Nature 1992, 358, 591–593. [Google Scholar] [CrossRef]
- Konno, K.; Feldmann, F.M.; McDermott, W. Pyrazinamide susceptibility and amidase activity of tubercle bacilli. Am. Rev. Respir. Dis. 1967, 95, 461–469. [Google Scholar] [CrossRef]
- Fraaije, M.W.; Kamerbeek, N.M.; Heidekamp, A.J.; Fortin, R.; Janssen, D.B. The Prodrug Activator EtaA from Mycobacterium tuberculosis Is a Baeyer-Villiger Monooxygenase. J. Biol. Chem. 2004, 279, 3354–3360. [Google Scholar] [CrossRef]
- Aldridge, B.B.; Rhee, K.Y. Microbial metabolomics: Innovation, application, insight. Curr. Opin. Microbiol. 2014, 19, 90–96. [Google Scholar] [CrossRef]
- Hartman, T.E.; Rhee, K.Y. Microbial Metabolomics: Fifty Shades of Metabolism. ACS Infect. Dis. 2015, 1, 73–75. [Google Scholar] [CrossRef]
- Bourenkov, G.P.; Popov, A.N. Optimization of data collection taking radiation damage into account. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 409–419. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef]
- McCoy, A.J. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, 63, 32–41. [Google Scholar] [CrossRef]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef]
- Terwilliger, T.C.; Grosse-Kunstleve, R.W.; Afonine, P.V.; Moriarty, N.W.; Zwart, P.H.; Hung, L.W.; Read, R.J.; Adams, P.D. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. Sect. D Biol. Crystallogr. 2007, 64, 61–69. [Google Scholar] [CrossRef]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012, 68, 352–367. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Diederichs, K.; Karplus, P.A. Better models by discarding data? Acta Crystallogr. Sect. D Biol. Crystallogr. 2013, 69, 1215–1222. [Google Scholar] [CrossRef]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef]
- Grabowski, M.; Langner, K.M.; Cymborowski, M.; Porebski, P.J.; Sroka, P.; Zheng, H.; Cooper, D.R.; Zimmerman, M.D.; Elsliger, M.A.; Burley, S.K.; et al. A public database of macromolecular diffraction experiments. Acta Crystallogr. Sect. D Struct. Biol. 2016, 72, 1181–1193. [Google Scholar] [CrossRef]
- Morris, G.M.; Ruth, H.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31. [Google Scholar] [CrossRef]
- Kadukova, M.; Grudinin, S. Convex-PL: A novel knowledge-based potential for protein-ligand interactions deduced from structural databases using convex optimization. J. Comput. Aided. Mol. Des. 2017, 31, 943–958. [Google Scholar] [CrossRef]
- Kadukova, M.; Chupin, V.; Grudinin, S. Docking rigid macrocycles using Convex-PL, AutoDock Vina, and RDKit in the D3R Grand Challenge 4. J. Comput. Aided. Mol. Des. 2020, 34, 191–200. [Google Scholar] [CrossRef]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein-ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Processing | |
| PDB ID code | 6T0J |
| Source | ESRF ID23-1 |
| Wavelength (Å) | 0.972 |
| Space group | P1211 |
| Cell dimensions | |
| a, b, c (Å) | 51.54, 75.10, 56.59 |
| α, β, γ (°) | 90, 106.840, 90 |
| No. of observations | 1,056,761 (63,680) |
| No. of unique reflections | 162,381 (11,133) |
| Resolution (Å) | 30–1.10 (1.13–1.10) |
| Rmeas | 0.193 (2.751) |
| Rpim | 0.075 (1.126) |
| I/σI | 5.54 (0.40) |
| CC1/2 | 99.7 (16.8) |
| Completeness (%) | 97.4 (90.9) |
| Redundancy | 6.5 (5.7) |
| Refinement Statistics | |
| Resolution (Å) | 30–1.25 (1.28–1.25) |
| No. of reflections (total/unique) | 743,229/111,159 |
| Rwork/Rfree | 0.1479/0.1829 |
| CC* in highest shell | 0.816 (7601) |
| CCwork/CCfree in highest shell | 0.787/0.729 |
| No. of atoms | |
| Protein | 3861 |
| Heme | 86 |
| SQ109 | 48 |
| Solvent | 830 |
| B-factors (Å2) | |
| Protein | 14.0 |
| Heme | 9.7 |
| SQ109 | 16.6 |
| Solvent | 34.3 |
| R.m.s.d | |
| Bond lengths (Å) | 0.008 |
| Bond angles (°) | 1.09 |
| Ramachandran statistics | |
| Favoured (%) | 97.89 |
| Allowed (%) | 2.11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bukhdruker, S.; Varaksa, T.; Grabovec, I.; Marin, E.; Shabunya, P.; Kadukova, M.; Grudinin, S.; Kavaleuski, A.; Gusach, A.; Gilep, A.; et al. Hydroxylation of Antitubercular Drug Candidate, SQ109, by Mycobacterial Cytochrome P450. Int. J. Mol. Sci. 2020, 21, 7683. https://doi.org/10.3390/ijms21207683
Bukhdruker S, Varaksa T, Grabovec I, Marin E, Shabunya P, Kadukova M, Grudinin S, Kavaleuski A, Gusach A, Gilep A, et al. Hydroxylation of Antitubercular Drug Candidate, SQ109, by Mycobacterial Cytochrome P450. International Journal of Molecular Sciences. 2020; 21(20):7683. https://doi.org/10.3390/ijms21207683
Chicago/Turabian StyleBukhdruker, Sergey, Tatsiana Varaksa, Irina Grabovec, Egor Marin, Polina Shabunya, Maria Kadukova, Sergei Grudinin, Anton Kavaleuski, Anastasiia Gusach, Andrei Gilep, and et al. 2020. "Hydroxylation of Antitubercular Drug Candidate, SQ109, by Mycobacterial Cytochrome P450" International Journal of Molecular Sciences 21, no. 20: 7683. https://doi.org/10.3390/ijms21207683
APA StyleBukhdruker, S., Varaksa, T., Grabovec, I., Marin, E., Shabunya, P., Kadukova, M., Grudinin, S., Kavaleuski, A., Gusach, A., Gilep, A., Borshchevskiy, V., & Strushkevich, N. (2020). Hydroxylation of Antitubercular Drug Candidate, SQ109, by Mycobacterial Cytochrome P450. International Journal of Molecular Sciences, 21(20), 7683. https://doi.org/10.3390/ijms21207683