Slow Dissolution Kinetics of Model Peptide Fibrils



Abstract
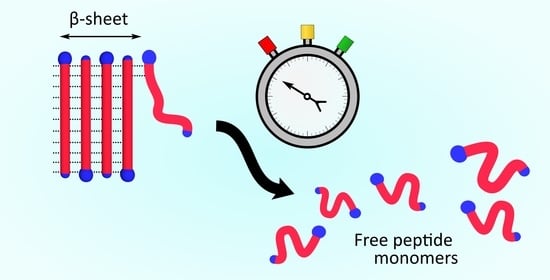
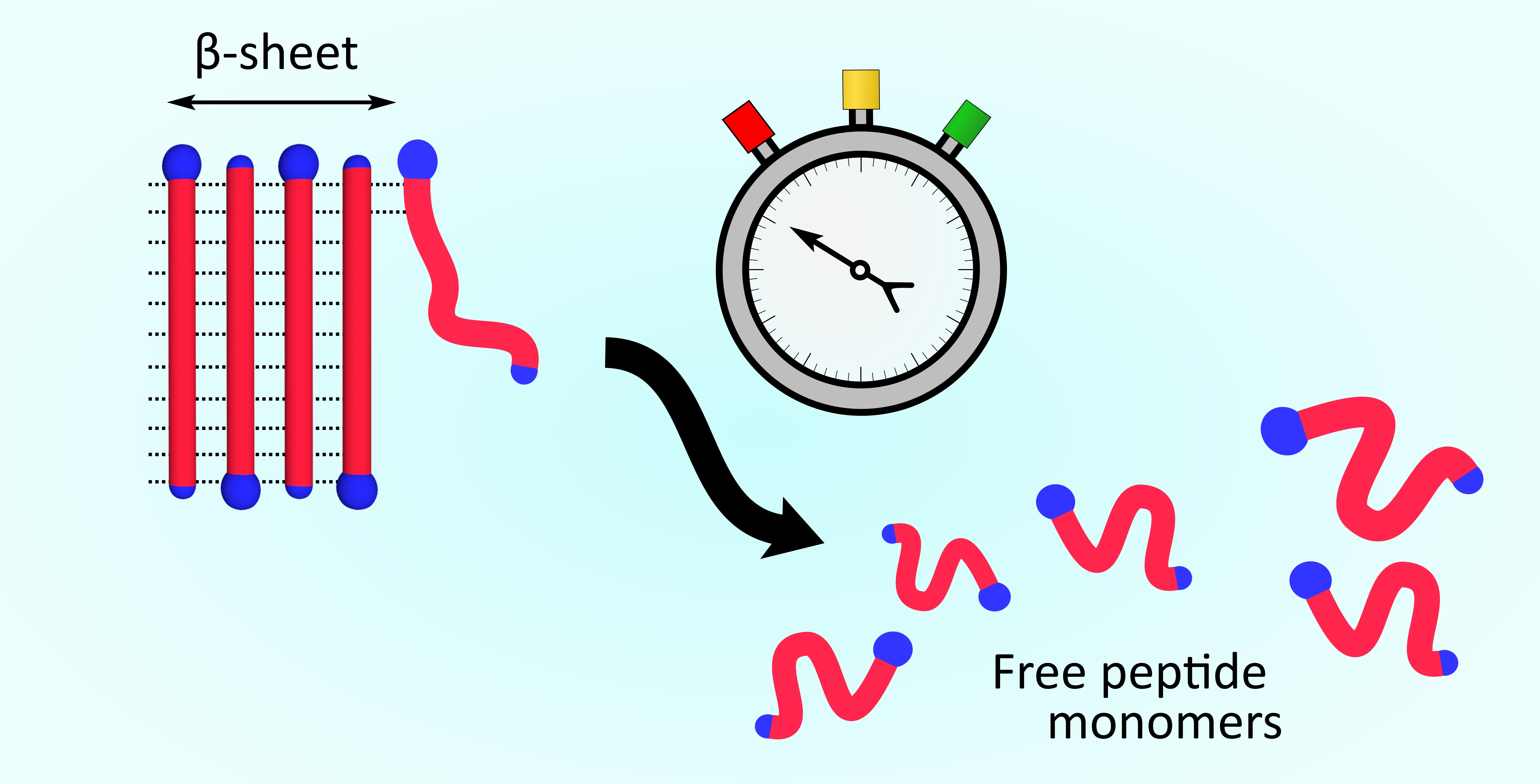{kind=link}
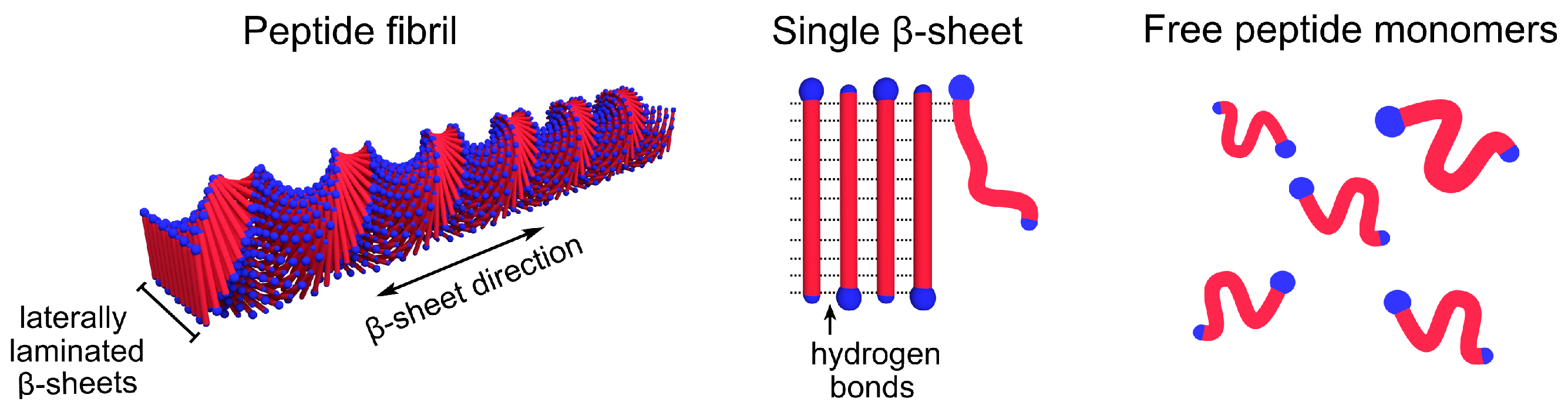{kind=link}
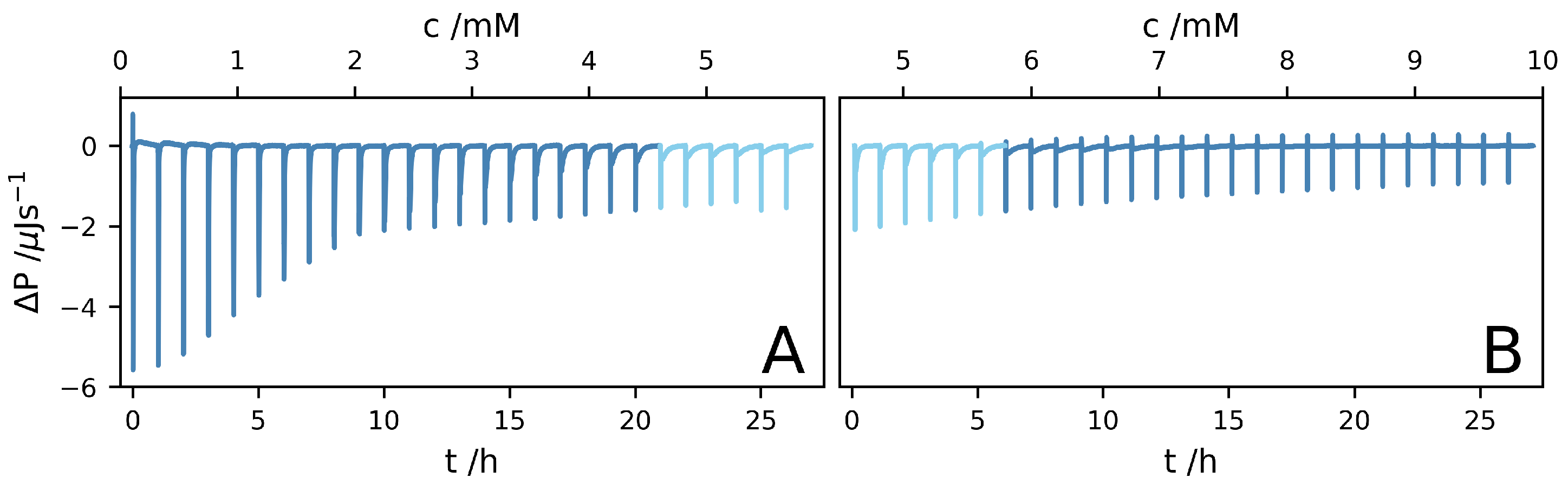{kind=link}
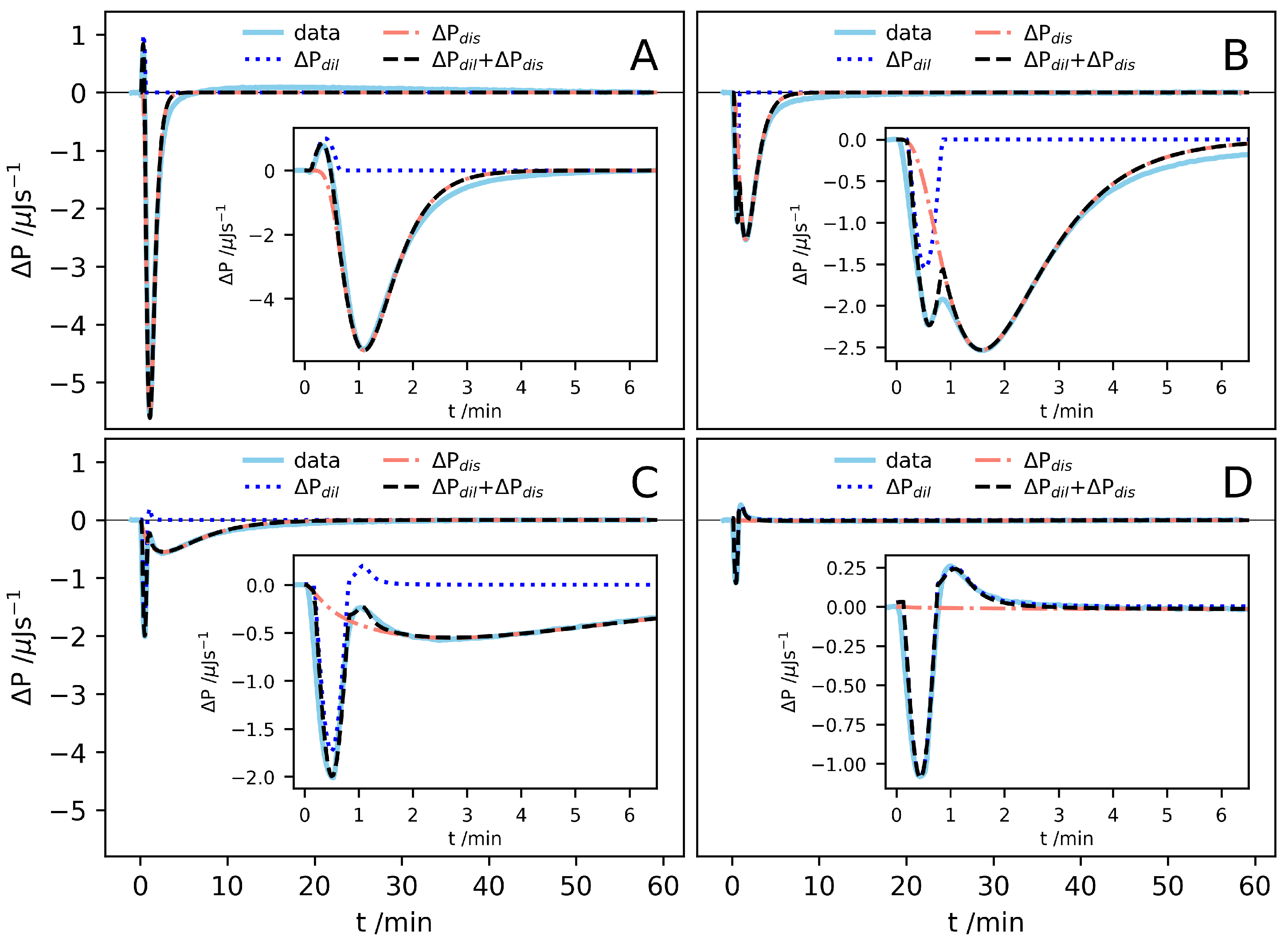{kind=link}
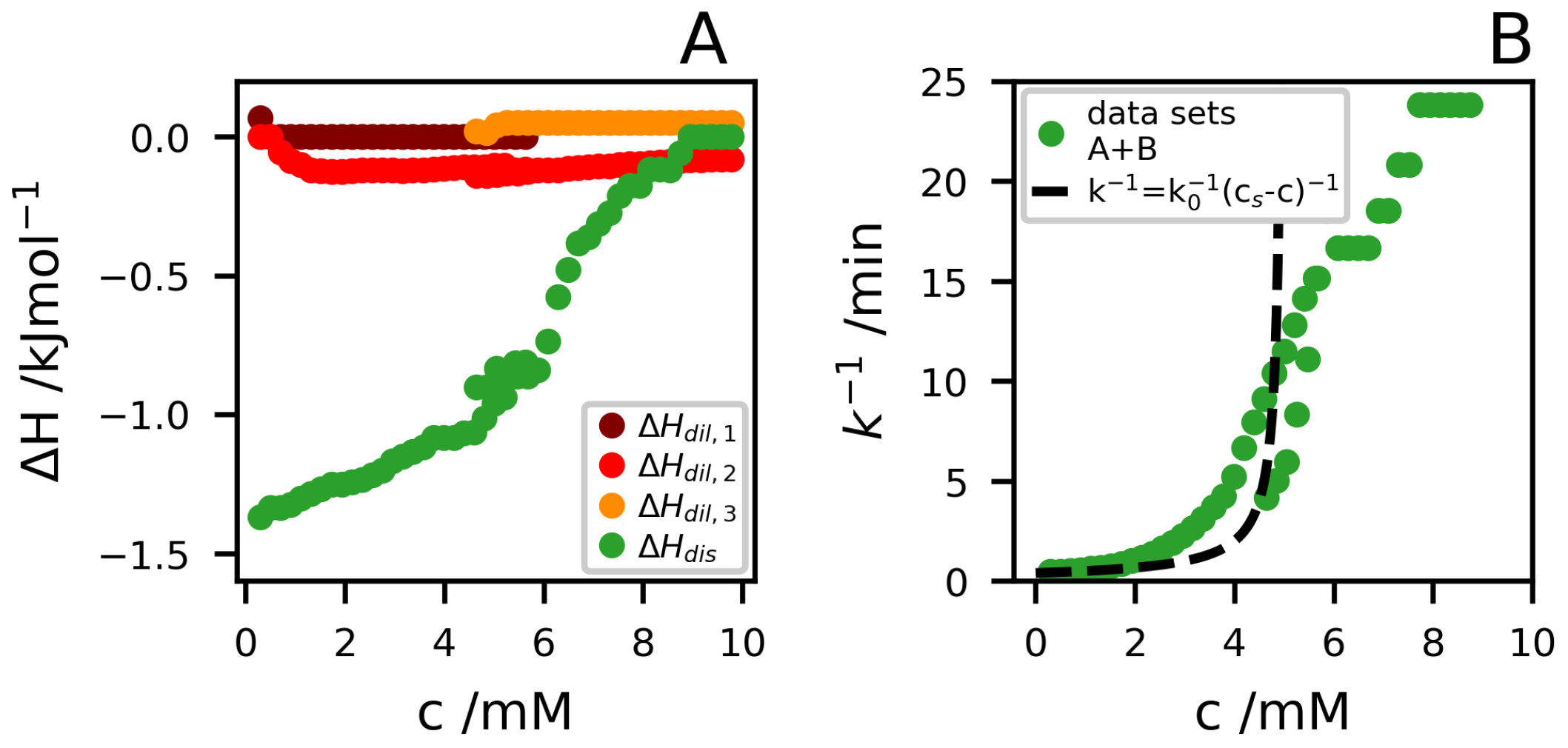{kind=link}
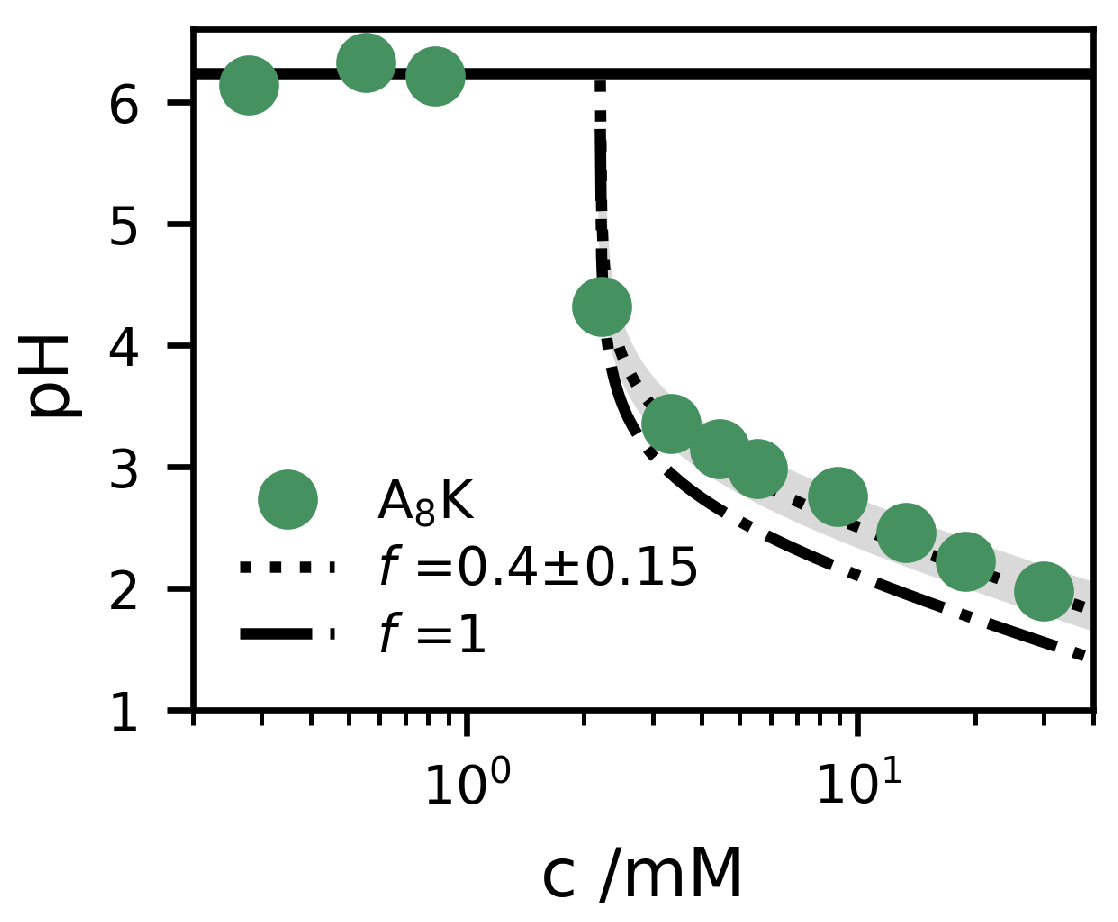{kind=link}
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. Materials
3.2. Isothermal Titration Calorimetry
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dobson, C.M. Protein Misfolding, Evolution and Disease. Trends Biochem. Sci. 1999, 24, 329–332. [Google Scholar] [CrossRef]
- Sipe, J.D.; Cohen, A.S. Review: History of the Amyloid Fibril. J. Struct. Biol. 2000, 130, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Poirier, M.A. Protein Aggregation and Neurodegenerative Disease. Nat. Med. 2004, 10, S10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S. Fabrication of Novel Biomaterials through Molecular Self-Assembly. Nat. Biotechnol. 2003, 21, 1171–1178. [Google Scholar] [CrossRef]
- Zhao, X.; Pan, F.; Xu, H.; Yaseen, M.; Shan, H.; Hauser, C.A.E.; Zhang, S.; Lu, J.R. Molecular Self-Assembly and Applications of Designer Peptide Amphiphiles. Chem. Soc. Rev. 2010, 39, 3480–3498. [Google Scholar] [CrossRef]
- Santis, E.D.; Ryadnov, M.G. Peptide Self-Assembly for Nanomaterials: The Old New Kid on the Block. Chem. Soc. Rev. 2015, 44, 8288–8300. [Google Scholar] [CrossRef]
- Fosgerau, K.; Hoffmann, T. Peptide Therapeutics: Current Status and Future Directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef]
- Araste, F.; Abnous, K.; Hashemi, M.; Taghdisi, S.M.; Ramezani, M.; Alibolandi, M. Peptide-Based Targeted Therapeutics: Focus on Cancer Treatment. J. Control. Release 2018, 292, 141–162. [Google Scholar] [CrossRef]
- Cui, H.; Webber, M.J.; Stupp, S.I. Self-Assembly of Peptide Amphiphiles: From Molecules to Nanostructures to Biomaterials. Biopolymers 2010, 94, 1–18. [Google Scholar] [CrossRef]
- Ulijn, R.V.; Smith, A.M. Designing Peptide Based Nanomaterials. Chem. Soc. Rev. 2008, 37, 664–675. [Google Scholar] [CrossRef]
- Hellstrand, E.; Boland, B.; Walsh, D.M.; Linse, S. Amyloid β-Protein Aggregation Produces Highly Reproducible Kinetic Data and Occurs by a Two-Phase Process. ACS Chem. Neurosci. 2009, 1, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Galvagnion, C. The Role of Lipids Interacting with α-Synuclein in the Pathogenesis of Parkinson’s Disease. J. Park. Dis. 2017, 7, 433–450. [Google Scholar] [CrossRef]
- Bellomo, G.; Bologna, S.; Gonnelli, L.; Ravera, E.; Fragai, M.; Lelli, M.; Luchinat, C. Aggregation Kinetics of the Aβ1–40 Peptide Monitored by NMR. Chem. Commun. 2018, 54, 7601–7604. [Google Scholar] [CrossRef] [PubMed]
- Cenker, Ç.Ç.; Bomans, P.H.H.; Friedrich, H.; Dedeoğlu, B.; Aviyente, V.; Olsson, U.; Sommerdijk, N.A.J.M.; Bucak, S. Peptide Nanotube Formation: A Crystal Growth Process. Soft Matter 2012, 8, 7463–7470. [Google Scholar] [CrossRef]
- Cohen, S.I.A.; Cukalevski, R.; Michaels, T.C.T.; Šarić, A.; Törnquist, M.; Vendruscolo, M.; Dobson, C.M.; Buell, A.K.; Knowles, T.P.J.; Linse, S. Distinct Thermodynamic Signatures of Oligomer Generation in the Aggregation of the Amyloid-β Peptide. Nat. Chem. 2018, 10, 523–531. [Google Scholar] [CrossRef]
- Lee, C.T.; Terentjev, E.M. Mechanisms and Rates of Nucleation of Amyloid Fibrils. J. Chem. Phys. 2017, 147, 105103. [Google Scholar] [CrossRef]
- Šarić, A.; Michaels, T.C.T.; Zaccone, A.; Knowles, T.P.J.; Frenkel, D. Kinetics of Spontaneous Filament Nucleation via Oligomers: Insights from Theory and Simulation. J. Chem. Phys. 2016, 145, 211926. [Google Scholar] [CrossRef]
- Dear, A.J.; Meisl, G.; Michaels, T.C.T.; Zimmermann, M.R.; Linse, S.; Knowles, T.P.J. The Catalytic Nature of Protein Aggregation. J. Chem. Phys. 2020, 152, 045101. [Google Scholar] [CrossRef]
- Cenker, Ç.Ç.; Bucak, S.; Olsson, U. Aqueous Self-Assembly within the Homologous Peptide Series AnK. Langmuir 2014, 30, 10072–10079. [Google Scholar] [CrossRef]
- Rüter, A.; Kuczera, S.; Pochan, D.J.; Olsson, U. Twisted Ribbon Aggregates in a Model Peptide System. Langmuir 2019, 35, 5802–5808. [Google Scholar] [CrossRef]
- Kuczera, S.; Rüter, A.; Roger, K.; Olsson, U. Two Dimensional Oblique Molecular Packing within a Model Peptide Ribbon Aggregate. ChemPhysChem 2020, 21, 1519–1523. [Google Scholar] [CrossRef]
- Olofsson, G.; Loh, W. On the Use of Titration Calorimetry to Study the Association of Surfactants in Aqueous Solutions. J. Braz. Chem. Soc. 2009, 20, 577–593. [Google Scholar] [CrossRef]
- Loh, W.; Brinatti, C.; Tam, K.C. Use of Isothermal Titration Calorimetry to Study Surfactant Aggregation in Colloidal Systems. Biochim. Biophys. Acta (BBA) Gen. Subj. 2016, 1860, 999–1016. [Google Scholar] [CrossRef] [PubMed]
- Kabiri, M.; Unsworth, L.D. Application of Isothermal Titration Calorimetry for Characterizing Thermodynamic Parameters of Biomolecular Interactions: Peptide Self-Assembly and Protein Adsorption Case Studies. Biomacromolecules 2014, 15, 3463–3473. [Google Scholar] [CrossRef]
- Kabiri, M.; Bushnak, I.; McDermot, M.T.; Unsworth, L.D. Toward a Mechanistic Understanding of Ionic Self-Complementary Peptide Self-Assembly: Role of Water Molecules and Ions. Biomacromolecules 2013, 14, 3943–3950. [Google Scholar] [CrossRef]
- Swanekamp, R.J.; DiMaio, J.T.M.; Bowerman, C.J.; Nilsson, B.L. Coassembly of Enantiomeric Amphipathic Peptides into Amyloid-Inspired Rippled β-Sheet Fibrils. J. Am. Chem. Soc. 2012, 134, 5556–5559. [Google Scholar] [CrossRef]
- Evans, D.F.; Wennerström, H. The Colloidal Domain: Where Physics, Chemistry, Biology and Technology Meet, 2nd ed.; Wiley-VCH: New York, NY, USA, 1999. [Google Scholar]
- Southall, N.T.; Dill, K.A.; Haymet, A.D.J. A View of the Hydrophobic Effect. J. Phys. Chem. 2002, 106, 521–533. [Google Scholar] [CrossRef]
- Rodante, F. Thermodynamics of the “Standard” α-Amino Acids in Water at 25° C. Thermochim. Acta 1989, 149, 157–171. [Google Scholar] [CrossRef]
- Lindfors, L.; Skantze, P.; Skantze, U.; Westergren, J.; Olsson, U. Amorphous Drug Nanosuspensions. 3. Particle Dissolution and Crystal Growth. Langmuir 2007, 23, 9866–9874. [Google Scholar] [CrossRef]
- Kabalnov, A.S.; Shchukin, E.D. Ostwald Ripening Theory: Applications to Fluorocarbon Emulsion Stability. Adv. Colloid Interface Sci. 1992, 38, 69–97. [Google Scholar] [CrossRef]
- Cenker, Ç.Ç.; Bucak, S.; Olsson, U. Nanotubes and Bilayers in a Model Peptide System. Soft Matter 2011, 7, 4868–4875. [Google Scholar] [CrossRef]
- Konak, A.R. Surface Reaction-Controlled Dissolution of Crystals in a Solvent or Solution. Chem. Eng. Sci. 1974, 29, 1785–1788. [Google Scholar] [CrossRef]
- Irbäck, A.; Mitternacht, S.; Mohanty, S. An Effective All-Atom Potential for Proteins. PMC Biophys. 2009, 2, 2. [Google Scholar] [CrossRef]
- Israelachvili, J.N. Intermolecular and Surface Forces, 3rd ed.; Academic Press: Burlington, MA, USA, 2011. [Google Scholar]
- Kauzmann, W. Some Factors in the Interpretation of Protein Denaturation. In Advances in Protein Chemistry; Anfinsen, C.B., Anson, M.L., Bailey, K., Edsall, J.T., Eds.; Academic Press: New York, NY, USA, 1959; pp. 1–63. [Google Scholar]
- Baldwin, R.L. In Search of the Energetic Role of Peptide Hydrogen Bonds. J. Biol. Chem. 2003, 278, 17581–17588. [Google Scholar] [CrossRef] [PubMed]
- Milne, J.B.; Parker, T.J. Dissociation Constant of Aqueous Trifluoroacetic Acid by Cryoscopy and Conductivity. J. Solut. Chem. 1981, 10, 479–487. [Google Scholar] [CrossRef]
- Lynch, M.L. Acid-Soaps. Curr. Opin. Colloid Interface Sci. 1997, 2, 495–500. [Google Scholar] [CrossRef]
- Rüter, A.; Olsson, U. Slow Kinetics in a Model Peptide System. To be submitted.
- Tesauro, D.; Accardo, A.; Diaferia, C.; Milano, V.; Guillon, J.; Ronga, L.; Rossi, F. Peptide-Based Drug-Delivery Systems in Biotechnological Applications: Recent Advances and Perspectives. Molecules 2019, 24, 351. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koder Hamid, M.; Rüter, A.; Kuczera, S.; Olsson, U. Slow Dissolution Kinetics of Model Peptide Fibrils. Int. J. Mol. Sci. 2020, 21, 7671. https://doi.org/10.3390/ijms21207671
Koder Hamid M, Rüter A, Kuczera S, Olsson U. Slow Dissolution Kinetics of Model Peptide Fibrils. International Journal of Molecular Sciences. 2020; 21(20):7671. https://doi.org/10.3390/ijms21207671
Chicago/Turabian StyleKoder Hamid, Mona, Axel Rüter, Stefan Kuczera, and Ulf Olsson. 2020. "Slow Dissolution Kinetics of Model Peptide Fibrils" International Journal of Molecular Sciences 21, no. 20: 7671. https://doi.org/10.3390/ijms21207671
APA StyleKoder Hamid, M., Rüter, A., Kuczera, S., & Olsson, U. (2020). Slow Dissolution Kinetics of Model Peptide Fibrils. International Journal of Molecular Sciences, 21(20), 7671. https://doi.org/10.3390/ijms21207671