Fatty Acid Synthase Is a Key Enabler for Endocrine Resistance in Heregulin-Overexpressing Luminal B-Like Breast Cancer

 ,
,  , ,
, ,
Abstract
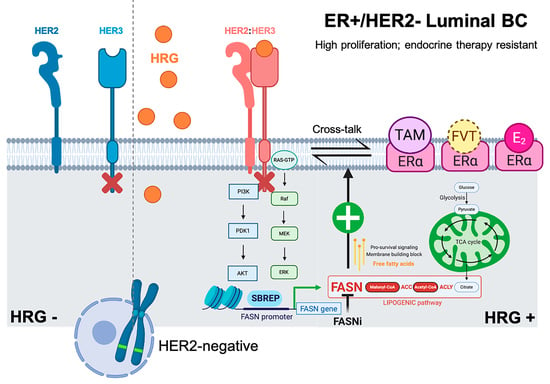
{kind=link}
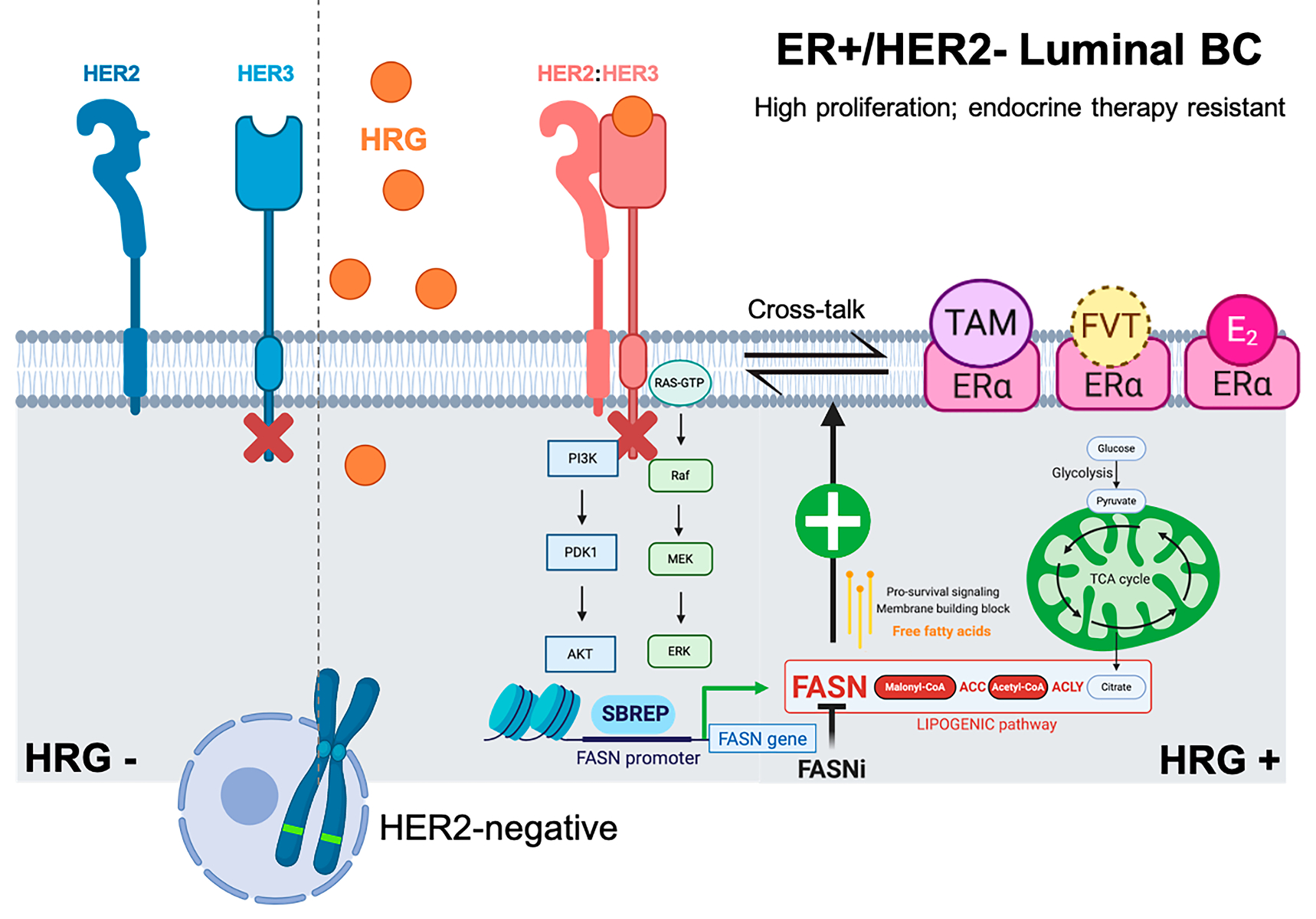
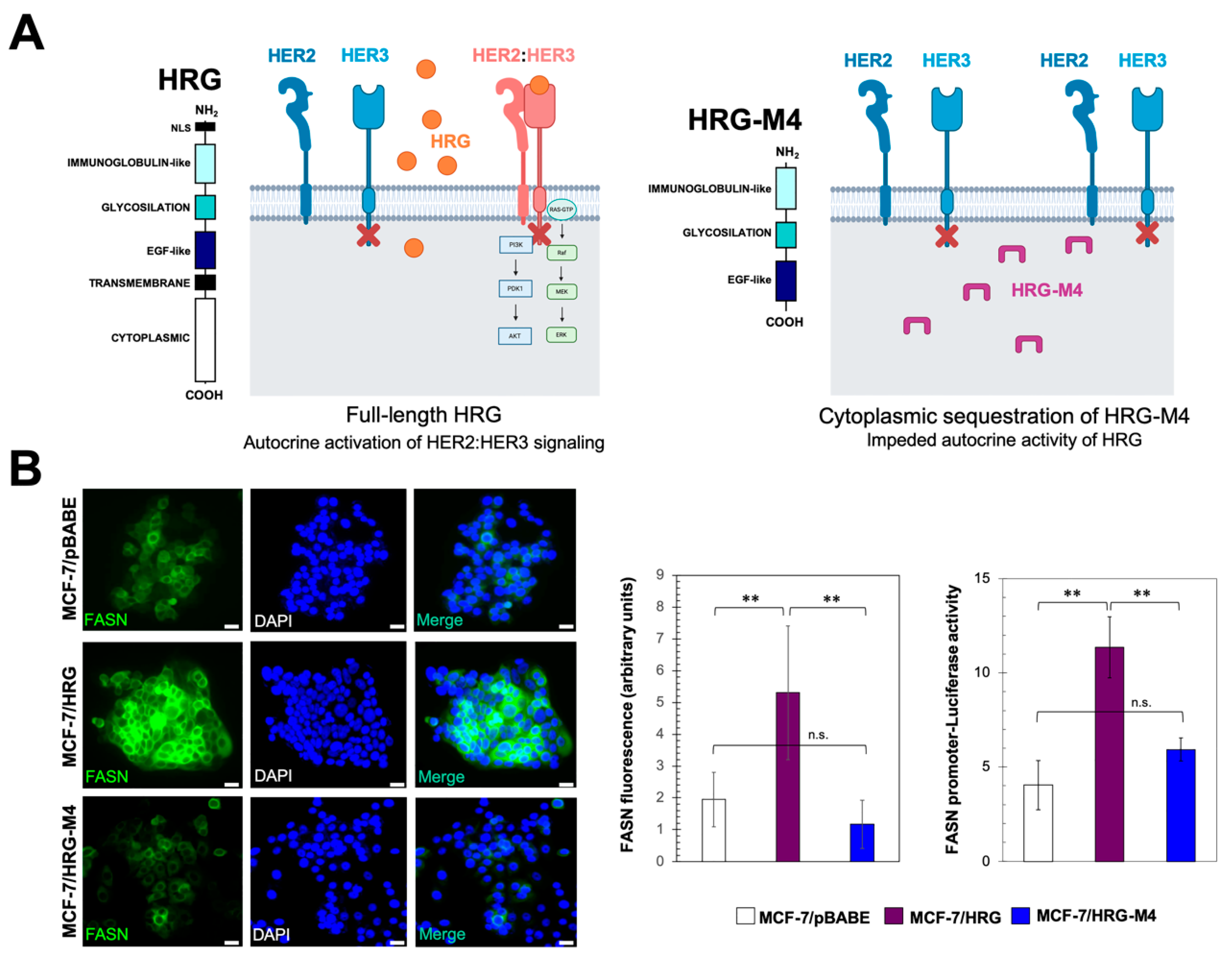{kind=link}
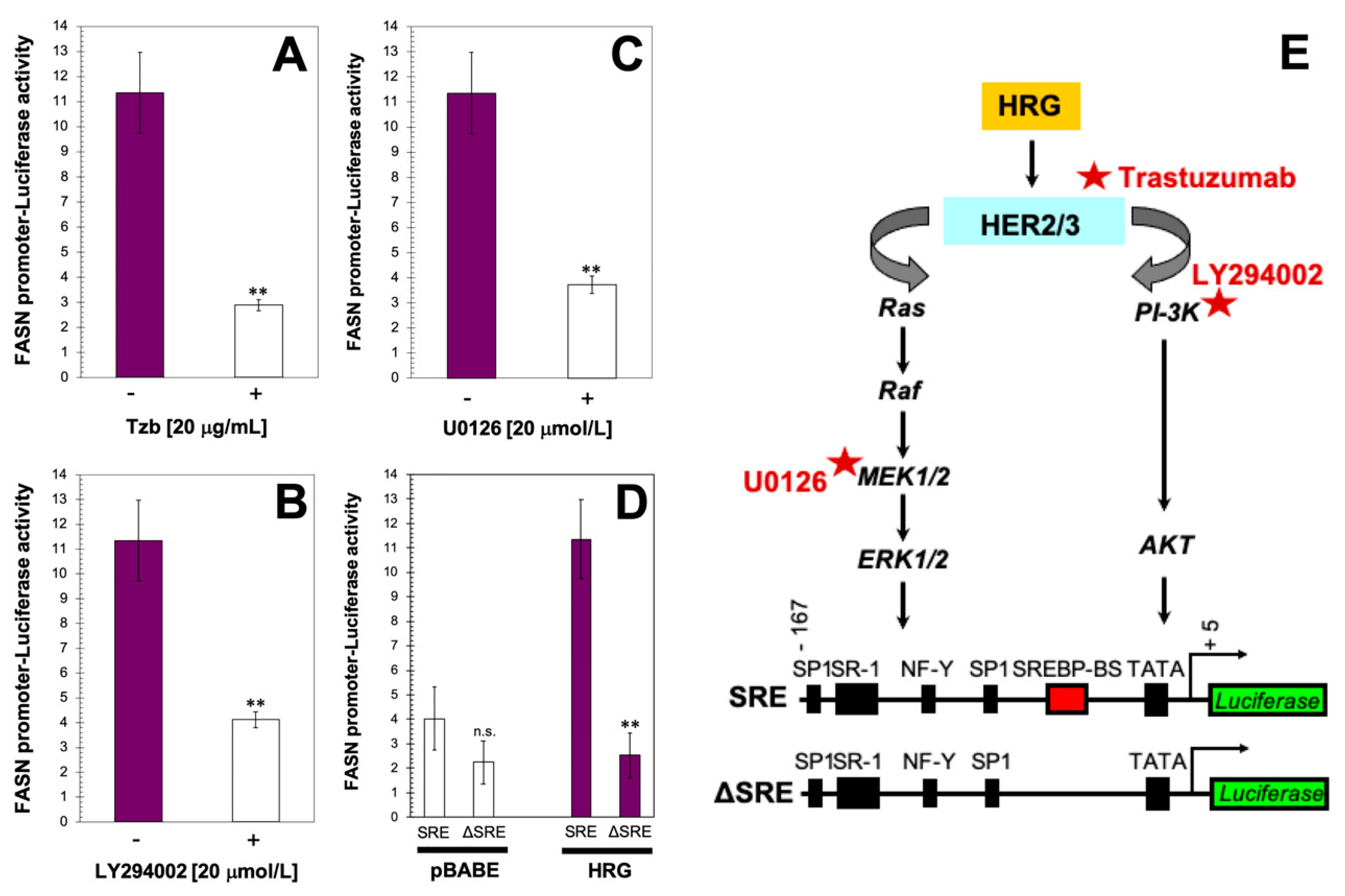{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
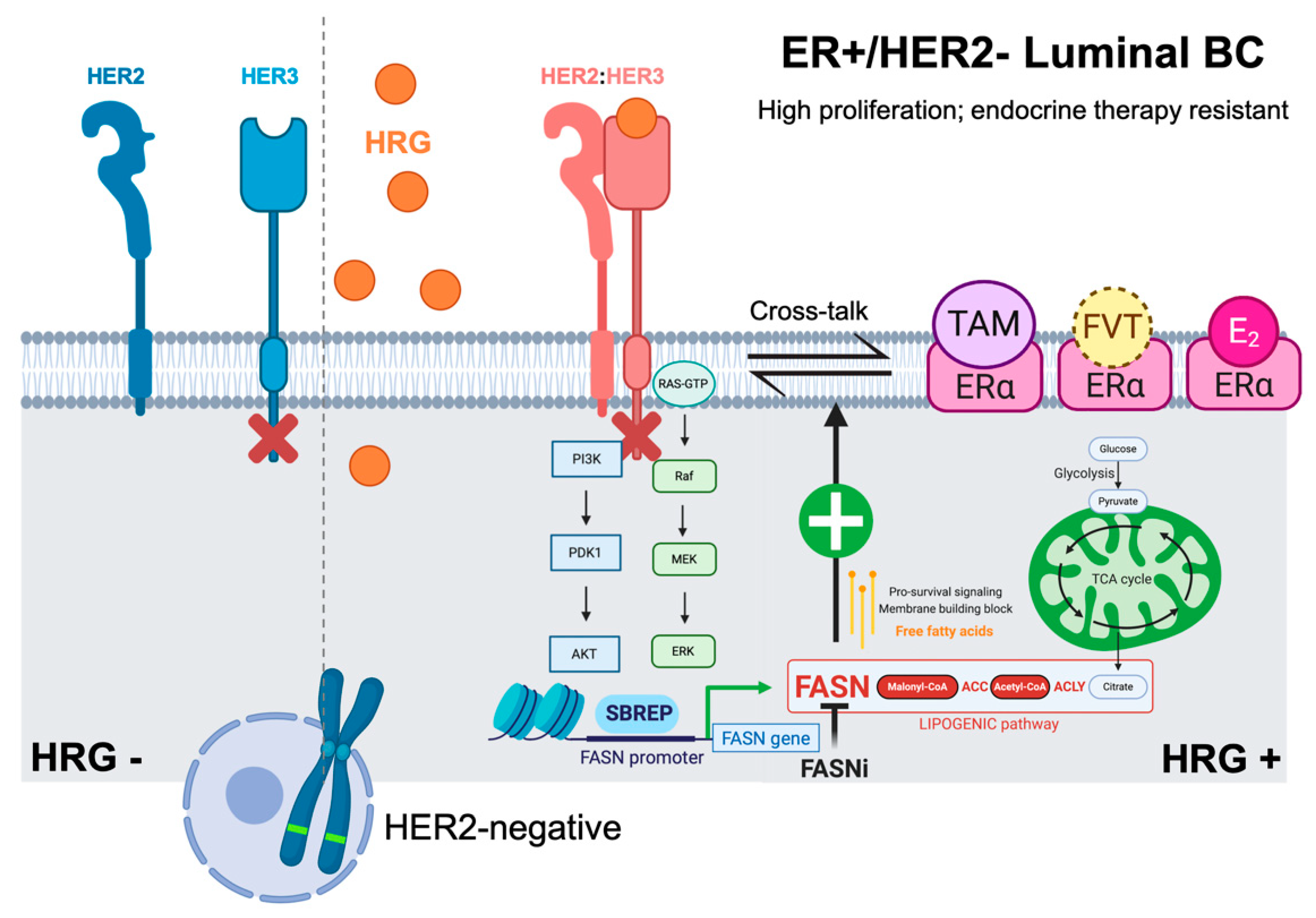
2.1. Heregulin Up-Regulates FASN Gene Expression by Autocrine Transactivation of HER2 Signaling
2.2. PI-3K/AKT and MAPK ERK1/2 Signaling Pathways Drive Heregulin-Stimulated, SBREP-Dependent FASN Gene Expression in Breast Cancer Cells
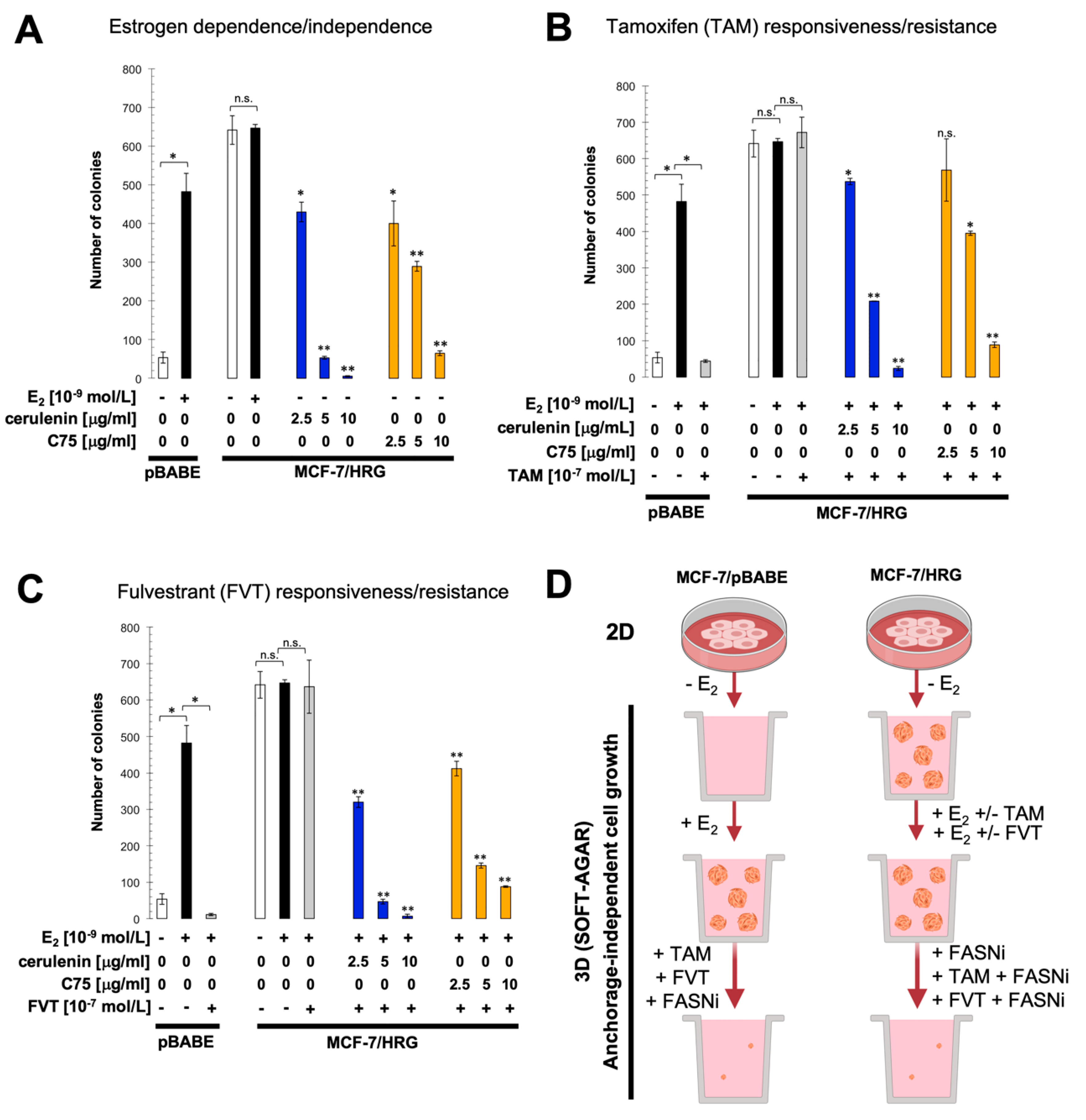
2.3. Pharmacological Blockade of FASN Activity Reverses HRG-Promoted Estrogen Independence and Endocrine Therapy-Resistance In Vitro
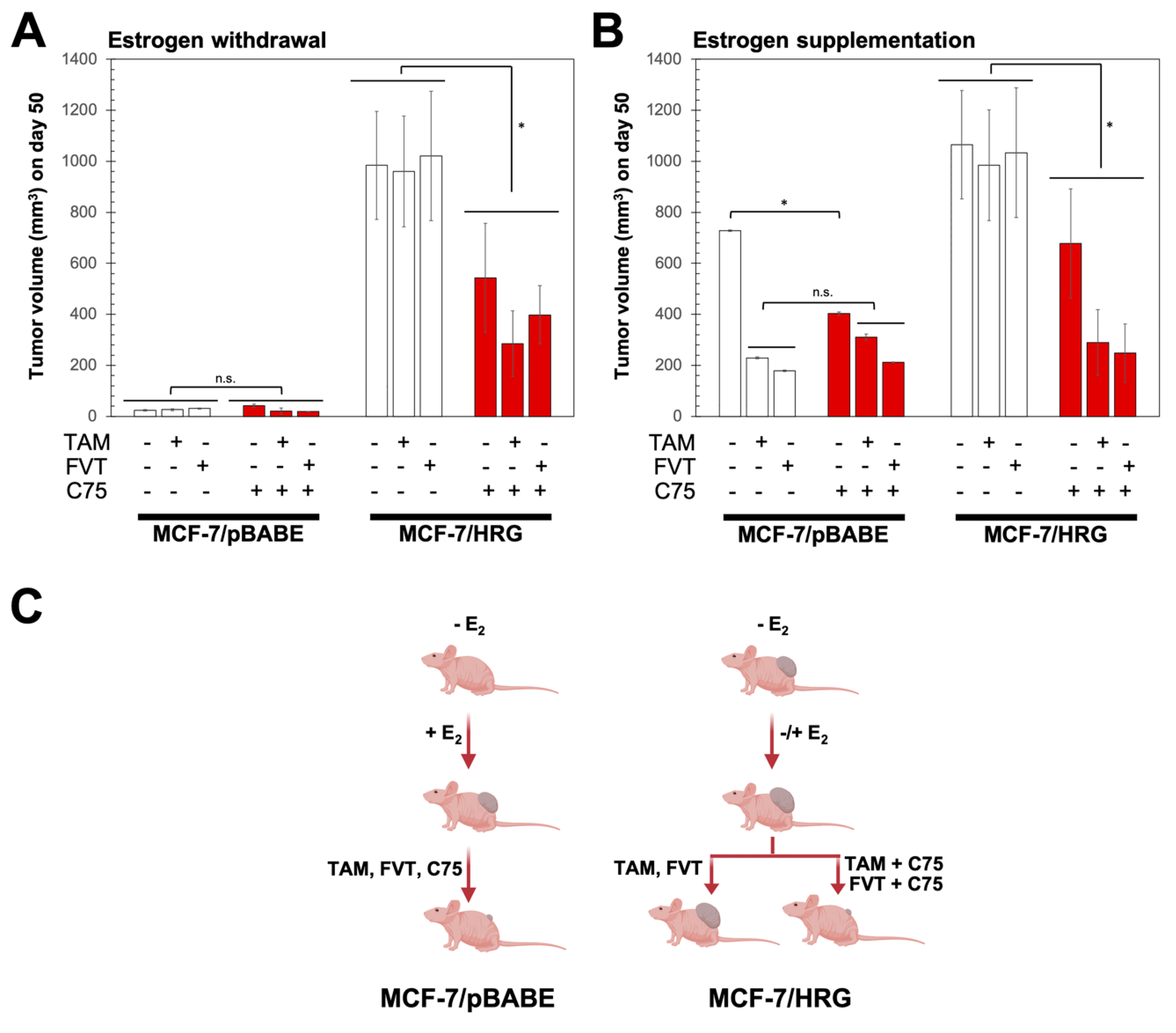
2.4. FASN Inhibition Reverses HRG-Mediated Resistance to Tamoxifen and Fulvestrant In Vivo
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Heregulin Constructs and Retroviral Infection of Cell Lines
4.3. Immunofluorescence Staining
4.4. FASN Promoter Activity
4.5. Xenograft Studies
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| FASN | Fatty acid synthase |
| ER | Estrogen Receptor |
| HRG | Heregulin |
| SERMs | Selective Estrogen Receptor Modulators |
| SERDs | Selective Estrogen Receptor Down-regulators |
| SBREP-1 | Sterol Regulatory Element-Binding Protein 1 |
| FASNi | FASN inhibitor |
References
- Santen, R.J.; Fan, P.; Zhang, Z.; Bao, Y.; Song, R.X.-D.; Yue, W. Estrogen signals via an extra-nuclear pathway involving IGF-1R and EGFR in tamoxifen-sensitive and -resistant breast cancer cells. Steroids 2009, 74, 586–594. [Google Scholar] [CrossRef] [PubMed]
- Arpino, G.; De Angelis, C.; Giuliano, M.; Giordano, A.; Falato, C.; De Laurentiis, M.; De Placido, S. Molecular Mechanism and Clinical Implications of Endocrine Therapy Resistance in Breast Cancer. Oncology 2009, 77, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Alluri, P.G.; Speers, C.; Chinnaiyan, A.M. Estrogen receptor mutations and their role in breast cancer progression. Breast Cancer Res. 2014, 16, 494. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Chang, J.; Fu, P. Endocrine therapy resistance in breast cancer: Current status, possible mechanisms and overcoming strategies. Future Med. Chem. 2015, 7, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Haque, M.; Desai, K.V. Pathways to Endocrine Therapy Resistance in Breast Cancer. Front. Endocrinol. 2019, 10, 573. [Google Scholar] [CrossRef]
- Loi, S.; Haibe-Kains, B.; Desmedt, C.; Lallemand, F.; Tutt, A.; Gillet, C.; Ellis, P.A.; Harris, A.; Bergh, J.; Foekens, J.A.; et al. Definition of Clinically Distinct Molecular Subtypes in Estrogen Receptor—Positive Breast Carcinomas Through Genomic Grade. J. Clin. Oncol. 2007, 25, 1239–1246. [Google Scholar] [CrossRef] [PubMed]
- Cheang, M.C.U.; Chia, S.K.; Voduc, D.; Gao, D.; Leung, S.; Snider, J.; Watson, M.; Davies, S.; Bernard, P.S.; Parker, J.S.; et al. Ki67 Index, HER2 Status, and Prognosis of Patients With Luminal B Breast Cancer. J. Natl. Cancer Inst. 2009, 101, 736–750. [Google Scholar] [CrossRef]
- Lange, C.A.; Yee, D. Killing the second messenger: Targeting loss of cell cycle control in endocrine-resistant breast cancer. Endocrine Relat. Cancer 2011, 18, C19–C24. [Google Scholar] [CrossRef]
- Munzone, E.; Curigliano, G.; Colleoni, M. Tailoring Adjuvant Treatments for the Individual Patient with Luminal Breast Cancer. Hematol. Clin. N. Am. 2013, 27, 703–714. [Google Scholar] [CrossRef]
- Araki, K.; Miyoshi, Y. Mechanism of resistance to endocrine therapy in breast cancer: The important role of PI3K/Akt/mTOR in estrogen receptor-positive, HER2-negative breast cancer. Breast Cancer 2017, 25, 392–401. [Google Scholar] [CrossRef]
- Loi, S.M.; Sotiriou, C.; Haibe-Kains, B.; Lallemand, F.; Conus, N.M.; Piccart, M.; Speed, T.P.; McArthur, G.A. Gene expression profiling identifies activated growth factor signaling in poor prognosis (Luminal-B) estrogen receptor positive breast cancer. BMC Med. Genom. 2009, 2, 37. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.K.; Perez, C.; Grunt, T.; Waibel, C.; Cho, C.; Lupu, R. Involvement of heregulin-beta2 in the acquisition of the hormone-independent phenotype of breast cancer cells. Cancer Res. 1996, 56, 3350–3358. [Google Scholar] [PubMed]
- Saceda, M.; Grunt, T.W.; Colomer, R.; Lippman, M.E.; Lupu, R.; Martin, M.B. Regulation of estrogen receptor concentration and activity by an erbB/HER ligand in breast carcinoma cell lines. Endocrinology 1996, 137, 4322–4330. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lupu, R.; Cardillo, M.; Cho, C.; Harris, L.; Hijazi, M.; Perez, C.; Rosenberg, K.; Yang, D.; Tang, C. The significance ofheregulin in breast cancer tumor progression and drug resistance. Breast Cancer Res. Treat. 1996, 38, 57–66. [Google Scholar] [CrossRef]
- Du, T.; Sikora, M.J.; Levine, K.; Oesterreich, S.; Riggins, R.B.; Wendell, S.G.; Van Houten, B.; Oesterreich, S. Key regulators of lipid metabolism drive endocrine resistance in invasive lobular breast cancer. Breast Cancer Res. 2018, 20, 106. [Google Scholar] [CrossRef]
- Gökmen-Polar, Y.; Neelamraju, Y.; Goswami, C.P.; Gu, Y.; Gu, X.; Nallamothu, G.; Vieth, E.; Janga, S.C.; Ryan, M.; Badve, S. Splicing factor ESRP 1 controls ER -positive breast cancer by altering metabolic pathways. EMBO Rep. 2019, 20, e46078. [Google Scholar] [CrossRef]
- Menendez, J.A.; Lupu, R. Fatty acid synthase regulates estrogen receptor-α signaling in breast cancer cells. Oncogenesis 2017, 6, e299. [Google Scholar] [CrossRef]
- Menendez, J.A.; Mehmi, I.; Lupu, R. Trastuzumab in Combination With Heregulin-Activated Her-2 (erbB-2) Triggers a Receptor-Enhanced Chemosensitivity Effect in the Absence of Her-2 Overexpression. J. Clin. Oncol. 2006, 24, 3735–3746. [Google Scholar] [CrossRef]
- Atlas, E.; Bojanowski, K.; Mehmi, I.; Lupu, R. A deletion mutant of heregulin increases the sensitivity of breast cancer cells to chemotherapy without promoting tumorigenicity. Oncogene 2003, 22, 3441–3451. [Google Scholar] [CrossRef]
- Corominas-Faja, B.; Vellon, L.; Cuyàs, E.; Buxó, M.; Martin-Castillo, B.; Serra, L.; García, J.; Lupu, R.; Menendez, J.A. Clinical and therapeutic relevance of the metabolic oncogene fatty acid synthase in HER2+ breast cancer. Histol. Histopathol. 2016, 32, 687–698. [Google Scholar] [CrossRef]
- Menendez, J.A.; Vellon, L.; Espinoza, I.; Lupu, R. The metastasis inducer CCN1 (CYR61) activates the fatty acid synthase (FASN)-driven lipogenic phenotype in breast cancer cells. Oncoscience 2016, 3, 242–257. [Google Scholar] [CrossRef] [PubMed]
- Swinnen, J.V.; Ulrix, W.; Heyns, W.; Verhoeven, G. Coordinate regulation of lipogenic gene expression by androgens: Evidence for a cascade mechanism involving sterol regulatory element binding proteins. Proc. Natl. Acad. Sci. USA 1997, 94, 12975–12980. [Google Scholar] [CrossRef] [PubMed]
- Swinnen, J.V.; Heemers, H.; Deboel, L.; Foufelle, F.; Heyns, W.; Verhoeven, G. Stimulation of tumor-associated fatty acid synthase expression by growth factor activation of the sterol regulatory element-binding protein pathway. Oncogene 2000, 19, 5173–5181. [Google Scholar] [CrossRef] [PubMed]
- Heemers, H.; Maes, B.; Foufelle, F.; Heyns, W.; Verhoeven, G.; Swinnen, J.V. Androgens stimulate lipogenic gene expression in prostate cancer cells by activation of the sterol regulatory element-binding protein cleavage activating protein/sterol regulatory element-binding protein pathway. Mol. Endocrinol. 2001, 15, 1817–1828. [Google Scholar] [CrossRef] [PubMed]
- Kumar-Sinha, C.; Ignatoski, K.W.; Lippman, M.E.; Ethier, S.P.; Chinnaiyan, A.M. Transcriptome analysis of HER2 reveals a molecular connection to fatty acid synthesis. Cancer Res. 2003, 63, 132–139. [Google Scholar]
- Menendez, J.A.; Mehmi, I.; Verma, V.A.; Teng, P.K.; Lupu, R. Pharmacological inhibition of fatty acid synthase (FAS): A novel therapeutic approach for breast cancer chemoprevention through its ability to suppress Her-2/neu (erbB-2) oncogene-induced malignant transformation. Mol. Carcinog. 2004, 41, 164–178. [Google Scholar] [CrossRef]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef]
- Menendez, J.A.; Lupu, R. Fatty acid synthase (FASN) as a therapeutic target in breast cancer. Expert Opin. Ther. Targets 2017, 21, 1001–1016. [Google Scholar] [CrossRef]
- Pan, M.H.; Lin, C.C.; Lin, J.K.; Chen, W.J. Tea polyphenol (-)-epigallocatechin 3-gallate suppresses heregulin-beta1-induced fatty acid synthase expression in human breast cancer cells by inhibiting phosphatidylinositol 3-kinase/Akt and mitogen-activated protein kinase cascade signaling. J. Agric. Food Chem. 2007, 55, 5030–5037. [Google Scholar] [CrossRef]
- Veigel, D.; Wagner, R.; Stübiger, G.; Wuczkowski, M.; Filipits, M.; Horvat, R.; Benhamú, B.; López-Rodríguez, M.L.; Leisser, A.; Valent, P.; et al. Fatty acid synthase is a metabolic marker of cell proliferation rather than malignancy in ovarian cancer and its precursor cells. Int. J. Cancer 2014, 136, 2078–2090. [Google Scholar] [CrossRef]
- Montesdeoca, N.; López, M.; Ariza, X.; Herrero, L.; Makowski, K. Inhibitors of lipogenic enzymes as a potential therapy against cancer. FASEB J. 2020, 34, 11355–11381. [Google Scholar] [CrossRef] [PubMed]
- Pandey, P.R.; Liu, W.; Xing, F.; Fukuda, K.; Watabe, K. Anti-cancer drugs targeting fatty acid synthase (FAS). Recent Pat. Anti Cancer Drug Discov. 2012, 7, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.F.; Infante, J.R. Molecular Pathways: Fatty Acid Synthase. Clin. Cancer Res. 2015, 21, 5434–5438. [Google Scholar] [CrossRef]
- Jiang, L.; Wang, H.; Li, J.; Fang, X.; Pan, H.; Yuan, X.; Zhang, P. Up-Regulated FASN Expression Promotes Transcoelomic Metastasis of Ovarian Cancer Cell through Epithelial-Mesenchymal Transition. Int. J. Mol. Sci. 2014, 15, 11539–11554. [Google Scholar] [CrossRef]
- Ricklefs, F.L.; Maire, C.L.; Matschke, J.; Dührsen, L.; Sauvigny, T.; Holz, M.; Kolbe, K.; Peine, S.; Herold-Mende, C.; Carter, B.S.; et al. FASN is a Biomarker Enriched in Malignant Glioma-Derived Extracellular Vesicles. Int. J. Mol. Sci. 2020, 21, 1931. [Google Scholar] [CrossRef]
- Gonzalez-Guerrico, A.M.; Espinoza, I.; Schroeder, B.; Park, C.H.; Kvp, C.M.; Khurana, A.; Corominas-Faja, B.; Cuyàs, E.; Alarcón, T.; Kleer, C.; et al. Suppression of endogenous lipogenesis induces reversion of the malignant phenotype and normalized differentiation in breast cancer. Oncotarget 2016, 7, 71151–71168. [Google Scholar] [CrossRef]
- Su, Y.-W.; Wu, P.-S.; Lin, S.-H.; Huang, W.-Y.; Kuo, Y.-S.; Lin, H.-P. Prognostic Value of the Overexpression of Fatty Acid Metabolism-Related Enzymes in Squamous Cell Carcinoma of the Head and Neck. Int. J. Mol. Sci. 2020, 21, 6851. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Menendez, J.A.; Mehmi, I.; Papadimitropoulou, A.; Vander Steen, T.; Cuyàs, E.; Verdura, S.; Espinoza, I.; Vellon, L.; Atlas, E.; Lupu, R. Fatty Acid Synthase Is a Key Enabler for Endocrine Resistance in Heregulin-Overexpressing Luminal B-Like Breast Cancer. Int. J. Mol. Sci. 2020, 21, 7661. https://doi.org/10.3390/ijms21207661
Menendez JA, Mehmi I, Papadimitropoulou A, Vander Steen T, Cuyàs E, Verdura S, Espinoza I, Vellon L, Atlas E, Lupu R. Fatty Acid Synthase Is a Key Enabler for Endocrine Resistance in Heregulin-Overexpressing Luminal B-Like Breast Cancer. International Journal of Molecular Sciences. 2020; 21(20):7661. https://doi.org/10.3390/ijms21207661
Chicago/Turabian StyleMenendez, Javier A., Inderjit Mehmi, Adriana Papadimitropoulou, Travis Vander Steen, Elisabet Cuyàs, Sara Verdura, Ingrid Espinoza, Luciano Vellon, Ella Atlas, and Ruth Lupu. 2020. "Fatty Acid Synthase Is a Key Enabler for Endocrine Resistance in Heregulin-Overexpressing Luminal B-Like Breast Cancer" International Journal of Molecular Sciences 21, no. 20: 7661. https://doi.org/10.3390/ijms21207661
APA StyleMenendez, J. A., Mehmi, I., Papadimitropoulou, A., Vander Steen, T., Cuyàs, E., Verdura, S., Espinoza, I., Vellon, L., Atlas, E., & Lupu, R. (2020). Fatty Acid Synthase Is a Key Enabler for Endocrine Resistance in Heregulin-Overexpressing Luminal B-Like Breast Cancer. International Journal of Molecular Sciences, 21(20), 7661. https://doi.org/10.3390/ijms21207661