Relative Centrifugal Force (RCF; G-Force) Affects the Distribution of TGF-β in PRF Membranes Produced Using Horizontal Centrifugation


 , and
, and
Abstract
1. Introduction
2. Results
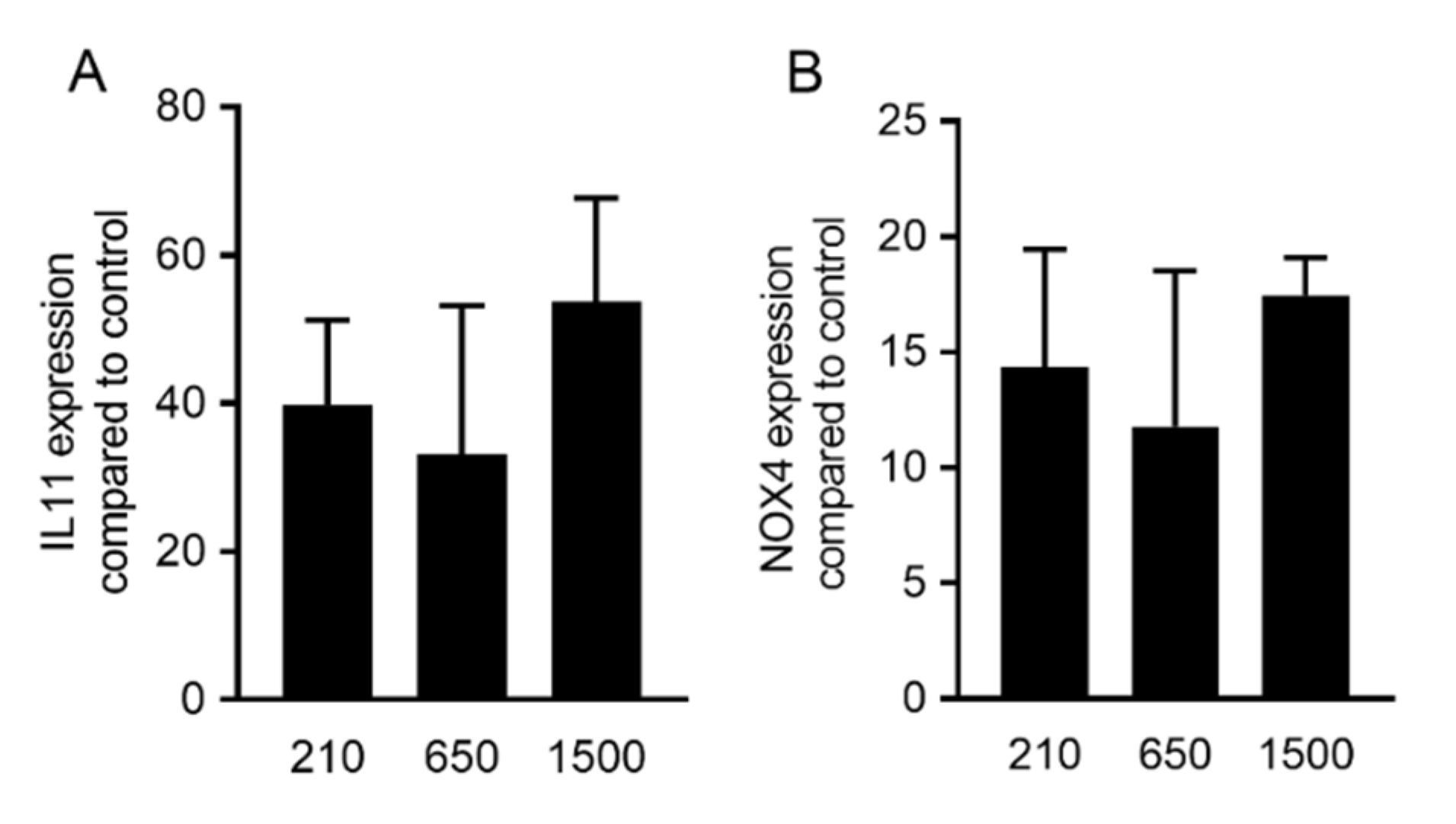
2.1. PRF Lysates Prepared at 210 g, 650 g and 1500 g Provoked an Equal Increase of TGF-β Target Genes
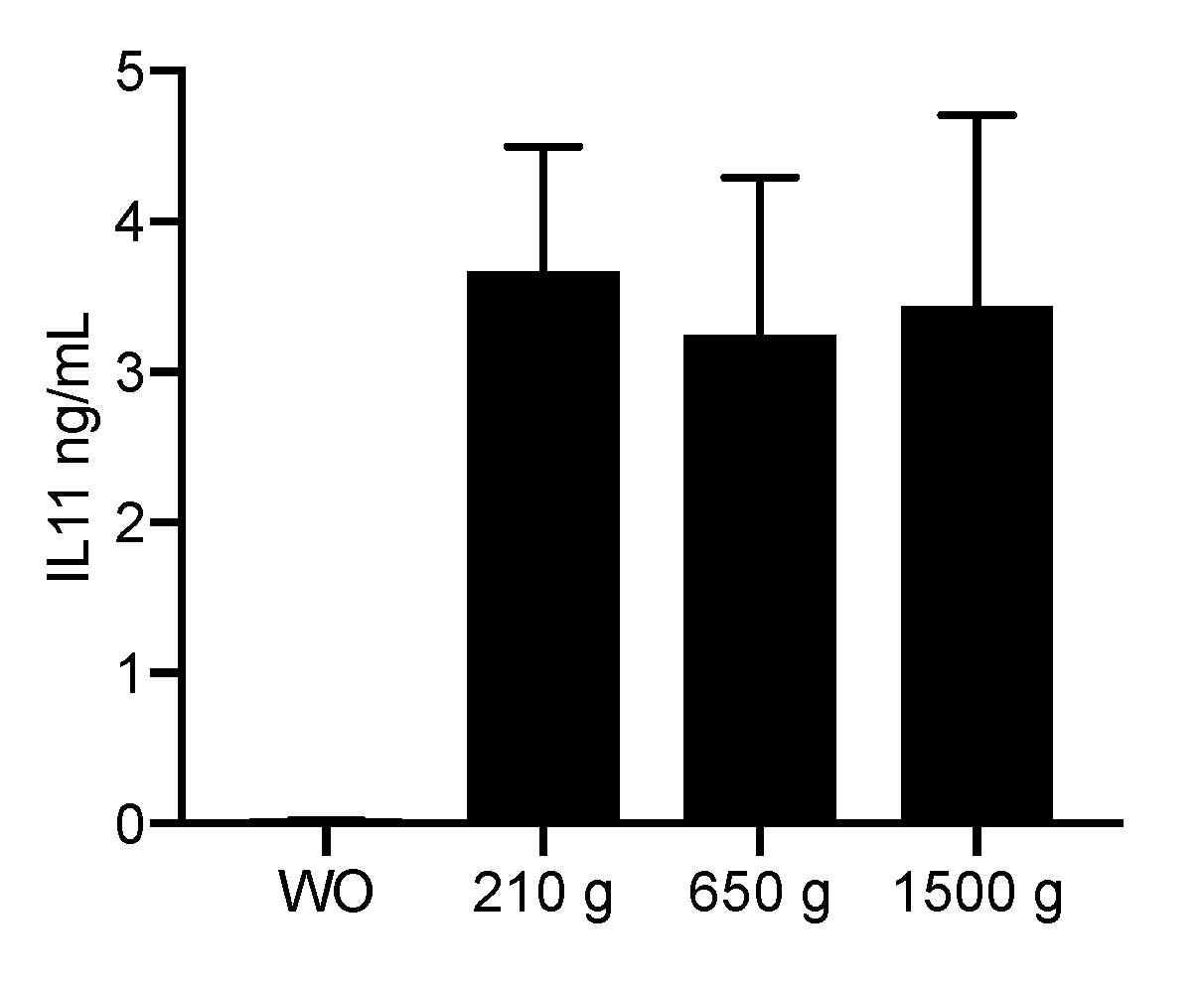
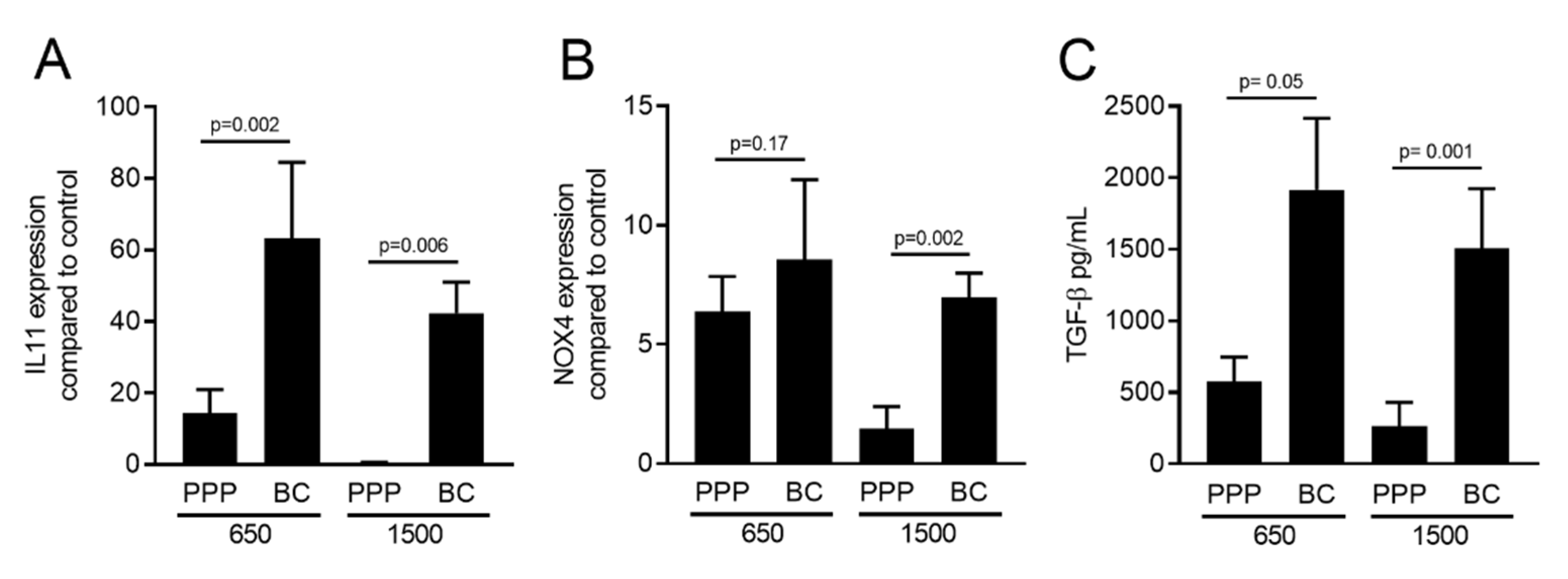
2.2. PRF Lysates Prepared at 210 g, 650 g and 1500 g Caused an Equal Production of IL11 Protein
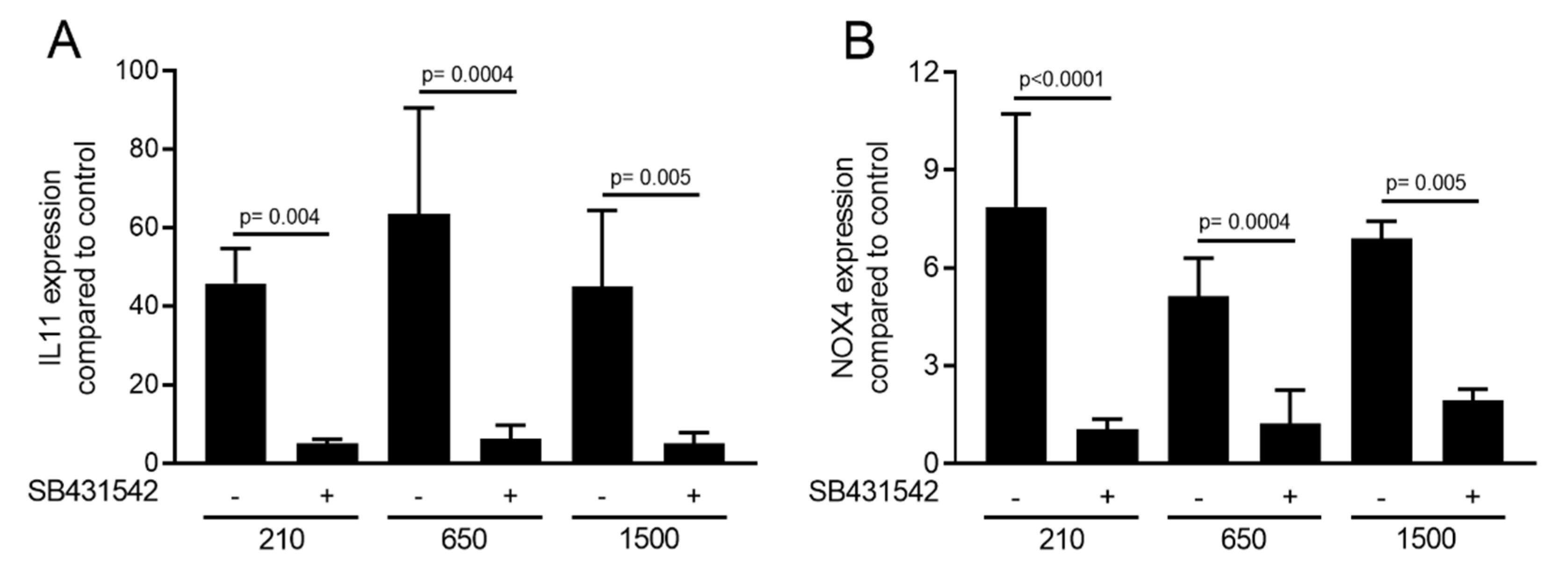
2.3. Expression Changes Were Blocked in the Presence of the TGF-β Receptor Type I Kinase Inhibitor
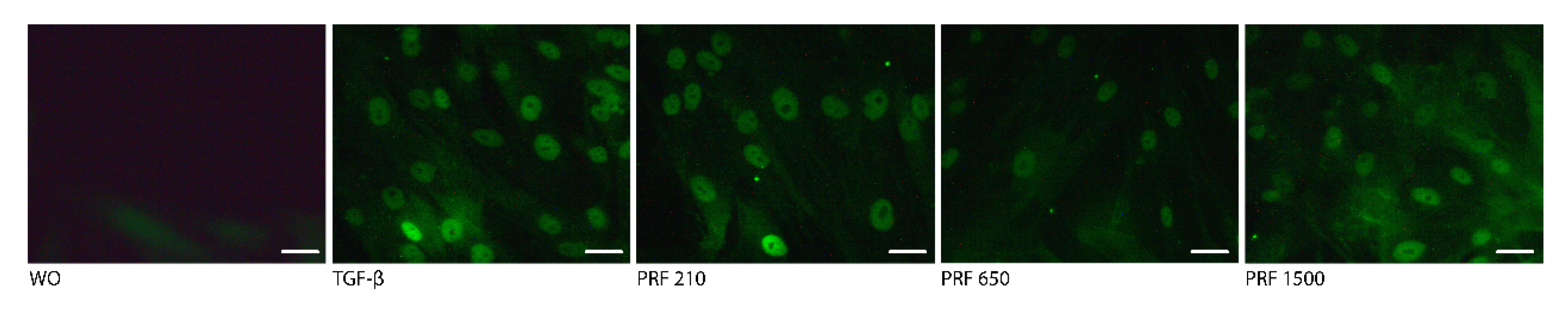
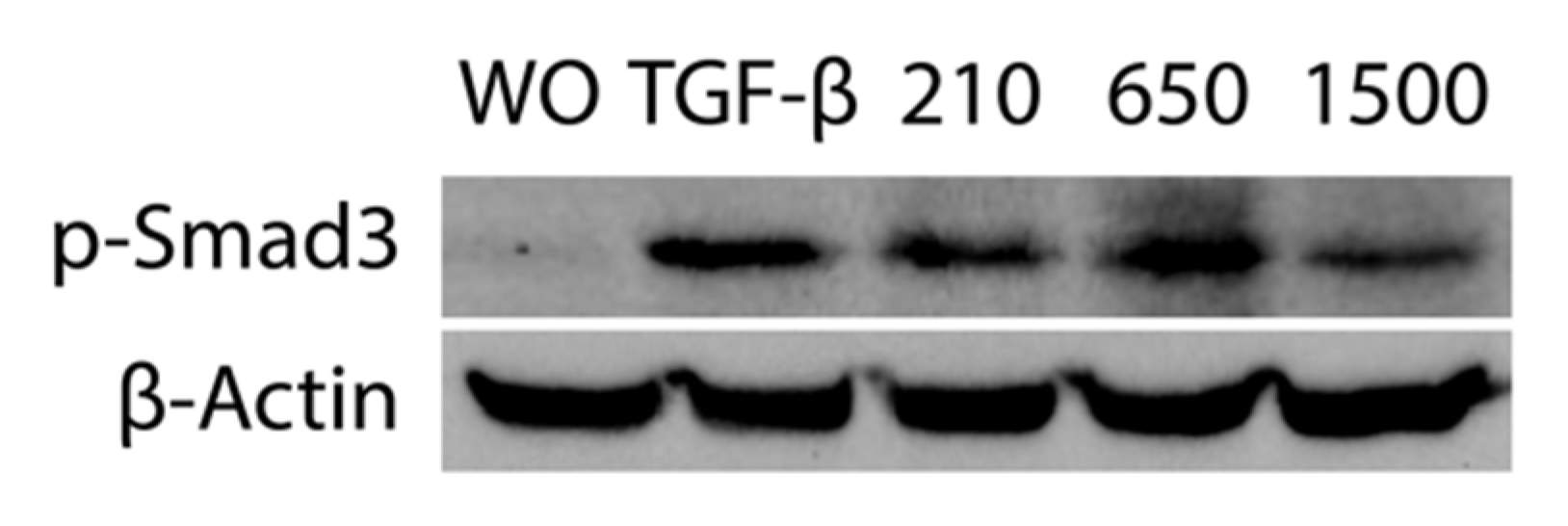
2.4. PRF Lysates Prepared at 210 g, 650 g and 1500 g Provoked an Equal Activation of Smad2/3 Signaling
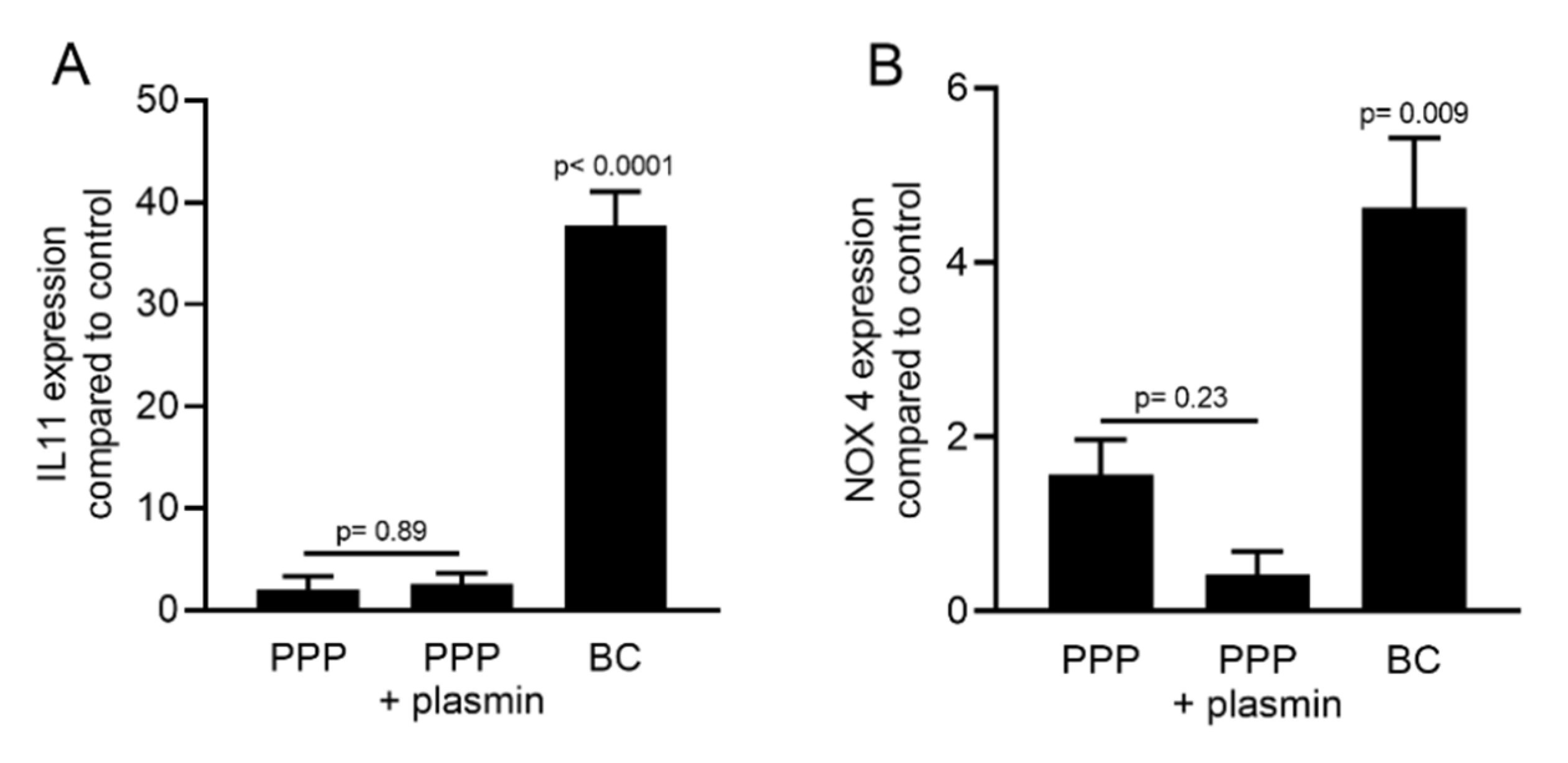
2.5. PPP Lysates Prepared at 1500 g Failed to Increase the Expression of TGF-β Target Genes
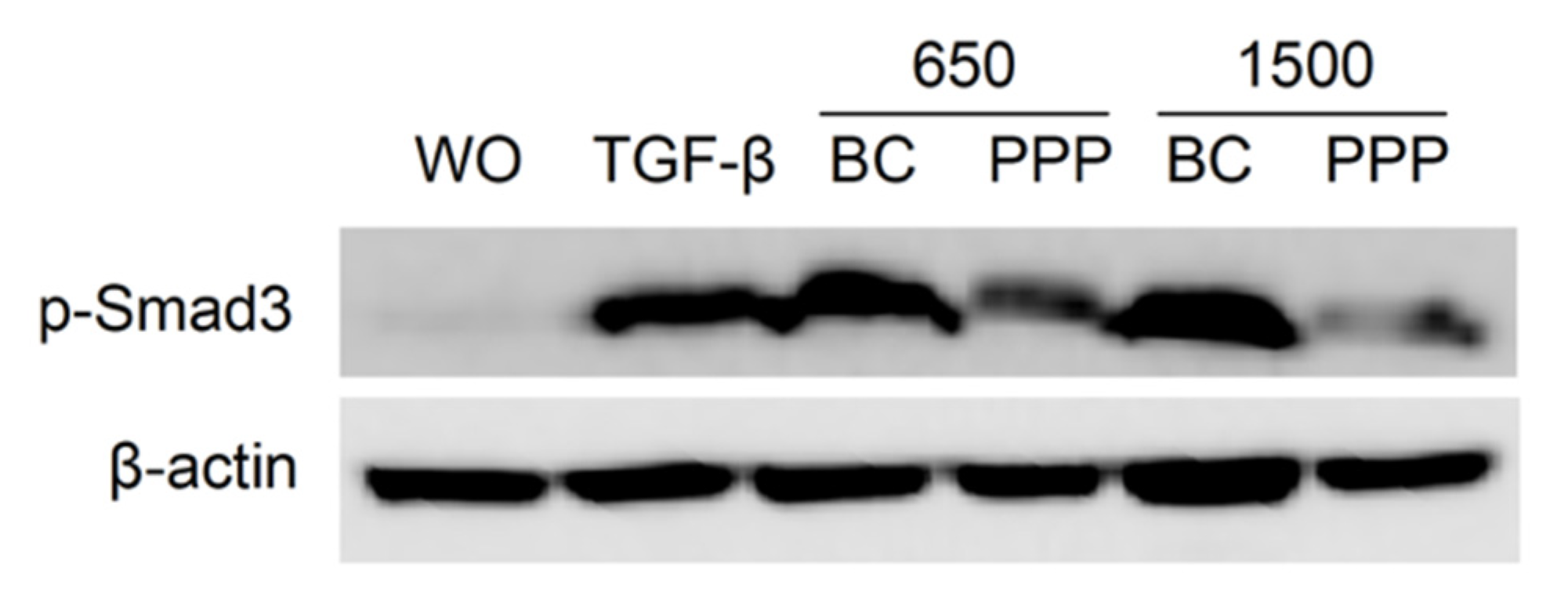
2.6. PPP Lysates Prepared at 1500 g Failed to Considerably Activate Phosphorylation of Smad3
3. Discussion
4. Materials and Methods
4.1. Cell Culture
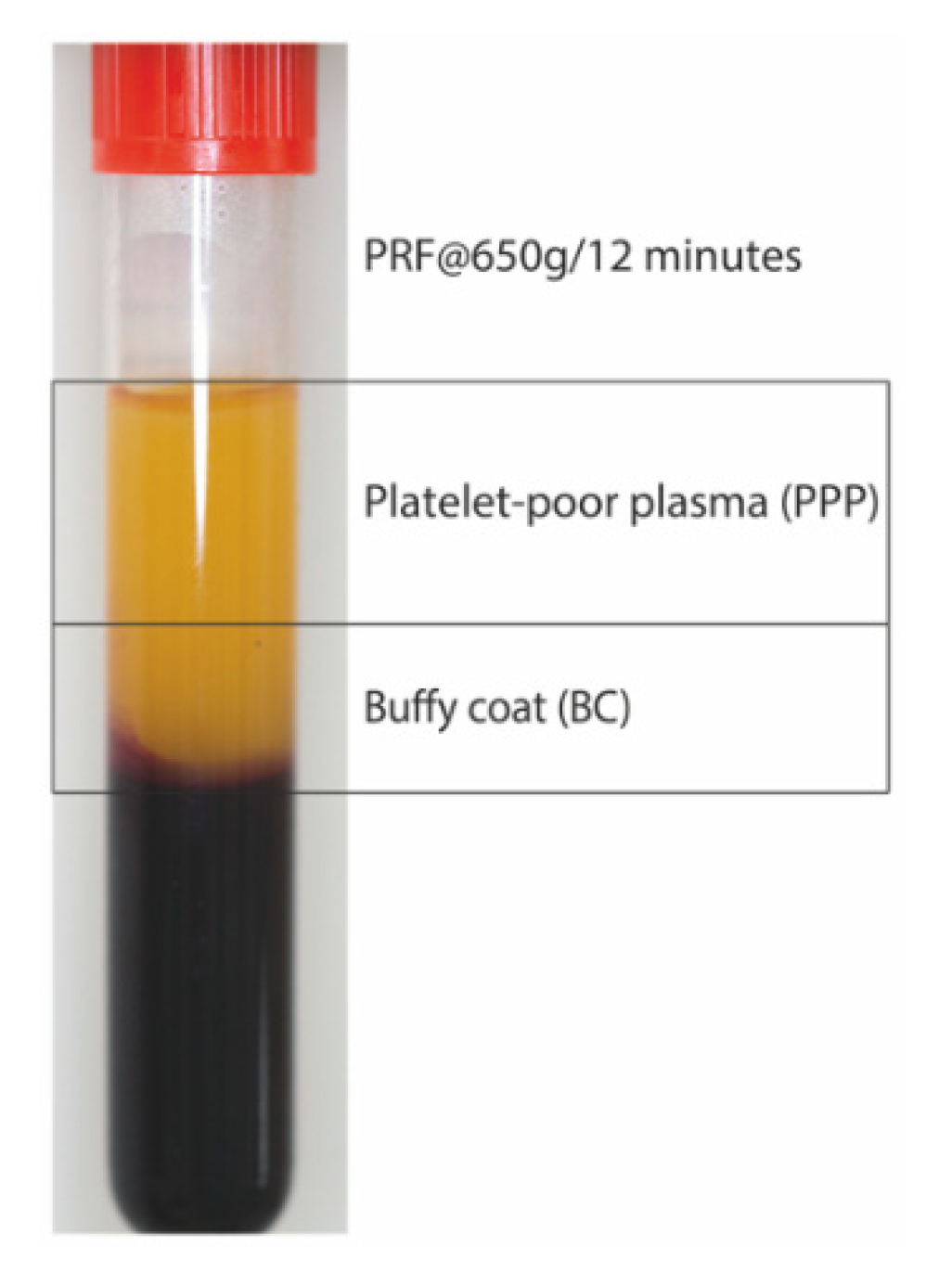
4.2. Preparation of PRF Lysates
4.3. RT-qPCR Analysis and Immunoassay
4.4. Immunofluorescence
4.5. Western Blot
4.6. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dohan, D.M.; Choukroun, J.; Diss, A.; Dohan, S.L.; Dohan, A.J.; Mouhyi, J.; Gogly, B. Platelet-rich fibrin (PRF): A second-generation platelet concentrate. Part I: Technological concepts and evolution. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endodontol. 2006, 101, e37–e44. [Google Scholar] [CrossRef] [PubMed]
- Temmerman, A.; Vandessel, J.; Castro, A.; Jacobs, R.; Teughels, W.; Pinto, N.; Quirynen, M. The use of leucocyte and platelet-rich fibrin in socket management and ridge preservation: A split-mouth, randomized, controlled clinical trial. J. Clin. Periodontol. 2016, 43, 990–999. [Google Scholar] [CrossRef] [PubMed]
- Temmerman, A.; Cleeren, G.J.; Castro, A.B.; Teughels, W.; Quirynen, M. L-PRF for increasing the width of keratinized mucosa around implants: A split-mouth, randomized, controlled pilot clinical trial. J. Periodontal Res. 2018, 53, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Cortellini, S.; Castro, A.B.; Temmerman, A.; Van Dessel, J.; Pinto, N.; Jacobs, R.; Quirynen, M. Leucocyte- and platelet-rich fibrin block for bone augmentation procedure: A proof-of-concept study. J. Clin. Periodontol. 2018, 45, 624–634. [Google Scholar] [CrossRef] [PubMed]
- Strauss, F.J.; Stähli, A.; Gruber, R. The use of platelet-rich fibrin to enhance the outcomes of implant therapy: A systematic review. Clin. Oral Implants Res. 2018, 29, 6–19. [Google Scholar] [CrossRef] [PubMed]
- Pinto, N.R.; Ubilla, M.; Zamora, Y.; Del Rio, V.; Ehrenfest, D.M.D.; Quirynen, M. Leucocyte- and platelet-rich fibrin (L-PRF) as a regenerative medicine strategy for the treatment of refractory leg ulcers: A prospective cohort study. Platelets 2017, 29, 468–475. [Google Scholar] [CrossRef]
- Tsujino, T.; Takahashi, A.; Yamaguchi, S.; Watanabe, T.; Isobe, K.; Kitamura, Y.; Tanaka, T.; Nakata, K.; Kawase, T. Evidence for Contamination of Silica Microparticles in Advanced Platelet-Rich Fibrin Matrices Prepared Using Silica-Coated Plastic Tubes. Biomedicines 2019, 7, 45. [Google Scholar] [CrossRef]
- Aizawa, H.; Tsujino, T.; Watanabe, T.; Isobe, K.; Kitamura, Y.; Sato, A.; Yamaguchi, S.; Okudera, H.; Okuda, K.; Kawase, T. Quantitative Near-Infrared Imaging of Platelets in Platelet-Rich Fibrin (PRF) Matrices: Comparative Analysis of Bio-PRF, Leukocyte-Rich PRF, Advanced-PRF and Concentrated Growth Factors. Int. J. Mol. Sci. 2020, 21, 4426. [Google Scholar] [CrossRef]
- Fujioka-Kobayashi, M.; Miron, R.J.; Hernandez, M.; Kandalam, U.; Zhang, Y.; Choukroun, J. Optimized Platelet-Rich Fibrin with the Low-Speed Concept: Growth Factor Release, Biocompatibility, and Cellular Response. J. Periodontol. 2017, 88, 112–121. [Google Scholar] [CrossRef]
- Ehrenfest, D.M.D.; Pinto, N.R.; Pereda, A.; Jiménez, P.; Del Corso, M.; Kang, B.-S.; Nally, M.; Lanata, N.; Wang, H.-L.; Quirynen, M. The impact of the centrifuge characteristics and centrifugation protocols on the cells, growth factors, and fibrin architecture of a leukocyte- and platelet-rich fibrin (L-PRF) clot and membrane. Platelets 2017, 29, 171–184. [Google Scholar] [CrossRef]
- Miron, R.J.; Xu, H.; Chai, J.; Wang, J.; Zheng, S.; Feng, M.; Zhang, X.; Wei, Y.; Chen, Y.; Mourão, C.F.d.A.B.; et al. Comparison of platelet-rich fibrin (PRF) produced using 3 commercially available centrifuges at both high (~ 700 g) and low (~ 200 g) relative centrifugation forces. Clin. Oral Investig. 2019, 24, 1171–1182. [Google Scholar] [CrossRef] [PubMed]
- Strauss, F.J.; Nasirzade, J.; Kargarpoor, Z.; Stähli, A.; Gruber, R. Effect of platelet-rich fibrin on cell proliferation, migration, differentiation, inflammation, and osteoclastogenesis: A systematic review of in vitro studies. Clin. Oral Investig. 2019, 24, 569–584. [Google Scholar] [CrossRef] [PubMed]
- Fujioka-Kobayashi, M.; Kono, M.; Katagiri, H.; Schaller, B.; Zhang, Y.; Sculean, A.; Miron, R.J. Histological comparison of Platelet rich fibrin clots prepared by fixed-angle versus horizontal centrifugation. Platelets 2020, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Miron, R.J.; Chai, J.; Zheng, S.; Feng, M.; Sculean, A.; Zhang, Y. A novel method for evaluating and quantifying cell types in platelet rich fibrin and an introduction to horizontal centrifugation. J. Biomed. Mater. Res. Part A 2019, 107, 2257–2271. [Google Scholar] [CrossRef]
- Choukroun, J.; Ghanaati, S. Reduction of relative centrifugation force within injectable platelet-rich-fibrin (PRF) concentrates advances patients’ own inflammatory cells, platelets and growth factors: The first introduction to the low speed centrifugation concept. Eur. J. Trauma Emerg. Surg. 2017, 44, 87–95. [Google Scholar] [CrossRef]
- Söderström, A.; Nybo, M.; Nielsen, C.; Vinholt, P.J. The effect of centrifugation speed and time on pre-analytical platelet activation. Clin. Chem. Lab. Med. 2016, 54, 1913–1920. [Google Scholar] [CrossRef]
- Scaravilli, V.; Di Girolamo, L.; Scotti, E.; Busana, M.; Biancolilli, O.; Leonardi, P.; Carlin, A.; Lonati, C.; Panigada, M.; Pesenti, A.; et al. Effects of sodium citrate, citric acid and lactic acid on human blood coagulation. Perfus 2018, 33, 577–583. [Google Scholar] [CrossRef]
- Miron, R.J.; Dham, A.; Dham, U.; Zhang, Y.; Pikos, M.A.; Sculean, A. The effect of age, gender, and time between blood draw and start of centrifugation on the size outcomes of platelet-rich fibrin (PRF) membranes. Clin. Oral Investig. 2018, 23, 2179–2185. [Google Scholar] [CrossRef]
- Jonnalagadda, D.; Izu, L.T.; Whiteheart, S.W. Platelet secretion is kinetically heterogeneous in an agonist-responsive manner. Blood 2012, 120, 5209–5216. [Google Scholar] [CrossRef]
- Martino, M.M.; Briquez, P.S.; Ranga, A.; Lutolf, M.P.; Hubbell, J.A. Heparin-binding domain of fibrin(ogen) binds growth factors and promotes tissue repair when incorporated within a synthetic matrix. Proc. Natl. Acad. Sci. USA. 2013, 110, 4563–4568. [Google Scholar] [CrossRef]
- Martino, M.M.; Hubbell, J.A. The 12th–14th type III repeats of fibronectin function as a highly promiscuous growth factor-binding domain. FASEB J. 2010, 24, 4711–4721. [Google Scholar] [CrossRef] [PubMed]
- Schoppet, M.; Chavakis, T.; Al-Fakhri, N.; Kanse, S.M.; Preissner, K.T. Molecular Interactions and Functional Interference between Vitronectin and Transforming Growth Factor-β. Lab. Investig. 2002, 82, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gallant, R.C.; Ni, H. Extracellular matrix proteins in the regulation of thrombus formation. Curr. Opin. Hematol. 2016, 23, 280–287. [Google Scholar] [CrossRef]
- Takahashi, A.; Tsujino, T.; Yamaguchi, S.; Isobe, K.; Watanabe, T.; Kitamura, Y.; Okuda, K.; Nakata, K.; Kawase, T. Distribution of platelets, transforming growth factor-β1, platelet-derived growth factor-BB, vascular endothelial growth factor and matrix metalloprotease-9 in advanced platelet-rich fibrin and concentrated growth factor matrices. J. Investig. Clin. Dent. 2019, 10, 12458. [Google Scholar] [CrossRef] [PubMed]
- Kargarpour, Z.; Nasirzade, J.; Panahipour, L.; Miron, R.J.; Gruber, R. Liquid Platelet-Rich Fibrin and Heat-Coagulated Albumin Gel: Bioassays for TGF-β Activity. Materials 2020, 13, 3466. [Google Scholar] [CrossRef] [PubMed]
- Varjú, I.; Tenekedjiev, K.; Keresztes, Z.; Pap, A.E.; Szabó, L.; Thelwell, C.; Longstaff, C.; Machovich, R.; Kolev, K.N. Fractal Kinetic Behavior of Plasmin on the Surface of Fibrin Meshwork. Biochemistry 2014, 53, 6348–6356. [Google Scholar] [CrossRef] [PubMed]
- Miyazono, K. TGF-beta signaling by Smad proteins. Cytokine Growth Factor Rev. 2000, 11, 115–157. [Google Scholar] [CrossRef]
- Masuki, H.; Isobe, K.; Kawabata, H.; Tsujino, T.; Yamaguchi, S.; Watanabe, T.; Sato, A.; Aizawa, H.; Mourão, C.F.; Kawase, T. Acute cytotoxic effects of silica microparticles used for coating of plastic blood-collection tubes on human periosteal cells. Odontology 2020, 108, 545–552. [Google Scholar] [CrossRef]
- Fava, R.A.; Casey, T.T.; Wilcox, J.; Pelton, R.W.; Moses, H.L.; Nanney, L.B. Synthesis of transforming growth factor-beta 1 by megakaryocytes and its localization to megakaryocyte and platelet alpha-granules. Blood 1990, 76, 1946–1955. [Google Scholar] [CrossRef] [PubMed]
- Grotendorst, G.R.; Smale, G.; Pencev, D. Production of transforming growth factor beta by human peripheral blood monocytes and neutrophils. J. Cell. Physiol. 1989, 140, 396–402. [Google Scholar] [CrossRef]
- Fulde, M.; Steinert, M.; Bergmann, S. Interaction of streptococcal plasminogen binding proteins with the host fibrinolytic system. Front. Cell. Infect. Microbiol. 2013, 3, 85. [Google Scholar] [CrossRef] [PubMed]
- Jong, A.Y.; Chen, S.H.M.; Stins, M.F.; Kim, K.S.; Tuan, T.-L.; Huang, S.-H. Binding of Candida albicans enolase to plasmin(ogen) results in enhanced invasion of human brain microvascular endothelial cells. J. Med. Microbiol. 2003, 52, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Grenier, D. Degradation of host protease inhibitors and activation of plasminogen by proteolytic enzymes from Porphyromonas gingivalis and Treponema denticola. Microbiology 1996, 142, 955–961. [Google Scholar] [CrossRef][Green Version]
- Castro, A.B.; Herrero, E.R.; Slomka, V.; Pinto, N.; Teughels, W.; Quirynen, M. Antimicrobial capacity of Leucocyte-and Platelet Rich Fibrin against periodontal pathogens. Sci. Rep. 2019, 9, 1–8. [Google Scholar] [CrossRef]
- Strauss, F.J.; Stahli, A.; Beer, L.; Mitulovic, G.; Gilmozzi, V.; Haspel, N.; Schwab, G.; Gruber, R. Acid bone lysate activates TGFbeta signalling in human oral fibroblasts. Sci. Rep. 2018, 8, 16065. [Google Scholar] [CrossRef]
- Di Summa, F.; Kargarpour, Z.; Nasirzade, J.; Stahli, A.; Mitulovic, G.; Panic-Jankovic, T.; Koller, V.; Kaltenbach, C.; Muller, H.; Panahipour, L.; et al. TGFbeta activity released from platelet-rich fibrin adsorbs to titanium surface and collagen membranes. Sci. Rep. 2020, 10, 10203. [Google Scholar] [CrossRef]
- Nasirzade, J.; Kargarpour, Z.; Hasannia, S.; Strauss, F.J.; Gruber, R. Platelet-rich fibrin elicits an anti-inflammatory response in macrophages in vitro. J. Periodontol. 2019, 91, 244–252. [Google Scholar] [CrossRef]
- Kargarpour, Z.; Nasirzade, J.; Strauss, F.J.; Di Summa, F.; Hasannia, S.; Müller, H.; Gruber, R. Platelet-rich fibrin suppresses in vitro osteoclastogenesis. J. Periodontol. 2020, 91, 413–421. [Google Scholar] [CrossRef]
- Kim, P.Y.; Stewart, R.J.; Lipson, S.M.; Nesheim, M.E. The relative kinetics of clotting and lysis provide a biochemical rationale for the correlation between elevated fibrinogen and cardiovascular disease. J. Thromb. Haemost. 2007, 5, 1250–1256. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 210 g (cm) | 650 g (cm) | 1500 g (cm) | |
|---|---|---|---|
| Donor 1 | 1.5 | 3.0 | 4.0 |
| Donor 2 | 2.0 | 2.5 | 3.5 |
| Donor 3 | 2.0 | 3.0 | 4.0 |
| Donor 4 | 2.0 | 3.0 | 3.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kargarpour, Z.; Nasirzade, J.; Panahipour, L.; Miron, R.J.; Gruber, R. Relative Centrifugal Force (RCF; G-Force) Affects the Distribution of TGF-β in PRF Membranes Produced Using Horizontal Centrifugation. Int. J. Mol. Sci. 2020, 21, 7629. https://doi.org/10.3390/ijms21207629
Kargarpour Z, Nasirzade J, Panahipour L, Miron RJ, Gruber R. Relative Centrifugal Force (RCF; G-Force) Affects the Distribution of TGF-β in PRF Membranes Produced Using Horizontal Centrifugation. International Journal of Molecular Sciences. 2020; 21(20):7629. https://doi.org/10.3390/ijms21207629
Chicago/Turabian StyleKargarpour, Zahra, Jila Nasirzade, Layla Panahipour, Richard J. Miron, and Reinhard Gruber. 2020. "Relative Centrifugal Force (RCF; G-Force) Affects the Distribution of TGF-β in PRF Membranes Produced Using Horizontal Centrifugation" International Journal of Molecular Sciences 21, no. 20: 7629. https://doi.org/10.3390/ijms21207629
APA StyleKargarpour, Z., Nasirzade, J., Panahipour, L., Miron, R. J., & Gruber, R. (2020). Relative Centrifugal Force (RCF; G-Force) Affects the Distribution of TGF-β in PRF Membranes Produced Using Horizontal Centrifugation. International Journal of Molecular Sciences, 21(20), 7629. https://doi.org/10.3390/ijms21207629