Structural Insights into the Active Site Formation of DUSP22 in N-loop-containing Protein Tyrosine Phosphatases


Abstract
1. Introduction
2. Results
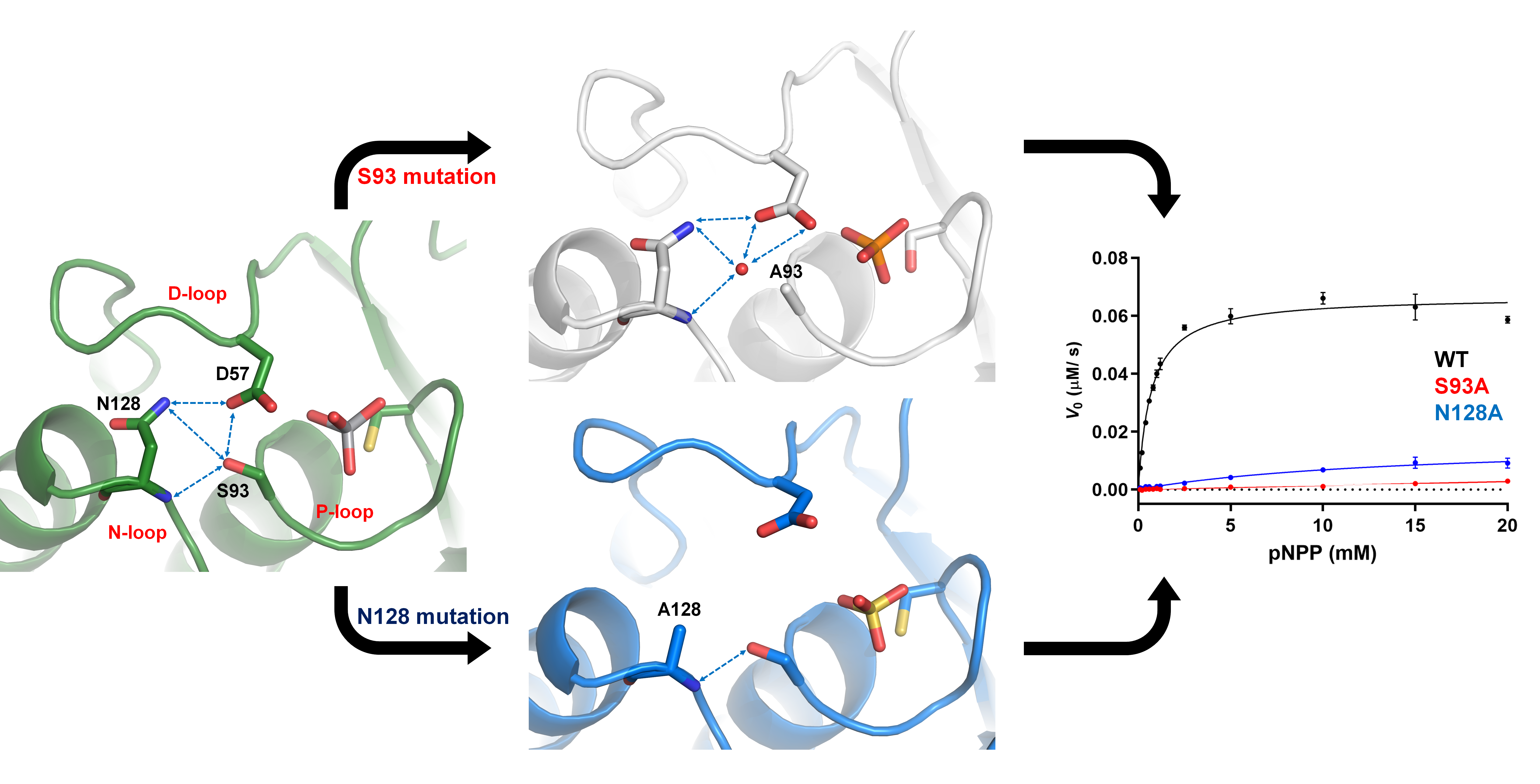
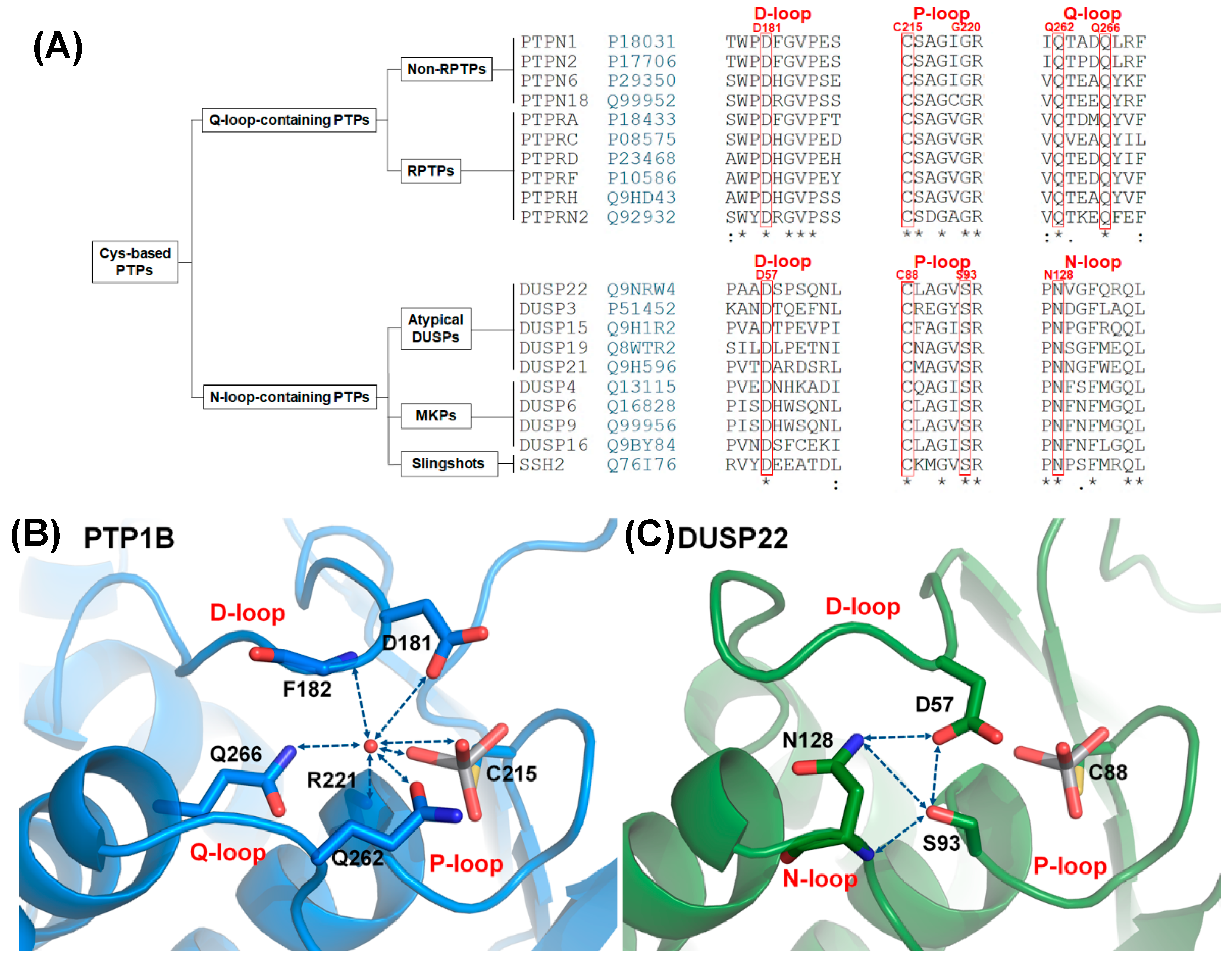
2.1. Characteristics of the DPN–triloop Interaction
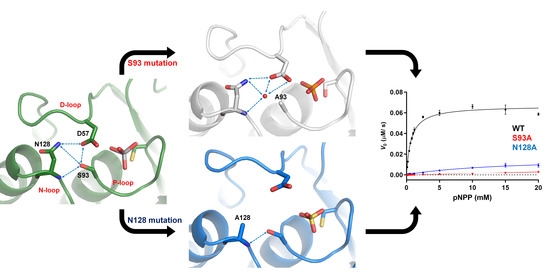
2.2. The Role of DPN–triloop Interaction in Phosphatase Activity
2.3. D57, S93, and N128 Mutants Perturbed the Active Site Conformation in the Solution Structure
2.4. The Structural Role of the DPN–triloop Interaction in Crystal Structures
3. Discussion
4. Materials and Methods
4.1. Protein Expression and Purification
4.2. Kinetic Assay
4.3. NMR Spectroscopy
4.4. Protein Crystallization
4.5. X-Ray Data Collection and Structure Determination
4.6. Data Availability
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Cys-based PTPs | Cysteine-based protein tyrosine phosphatases |
| DUSPs | Dual-specificity phosphatases |
| NMR | Nuclear magnetic resonance |
| P-loop | Phosphate binding loop |
References
- Hobiger, K.; Friedrich, T. Voltage sensitive phosphatases: Emerging kinship to protein tyrosine phosphatases from structure-function research. Front. Pharmacol. 2015, 6, 20. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Alonso, A.; Pulido, R. The extended human PTPome: A growing tyrosine phosphatase family. FEBS J. 2016, 283, 1404–1429. [Google Scholar] [CrossRef] [PubMed]
- Patterson, K.I.; Brummer, T.; O’Brien, P.M.; Daly, R.J. Dual-specificity phosphatases: Critical regulators with diverse cellular targets. Biochem. J. 2009, 418, 475–489. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Shen, D.; Parsons, D.W.; Bardelli, A.; Sager, J.; Szabo, S.; Ptak, J.; Silliman, N.; Peters, B.A.; Van der Heijden, M.S.; et al. Mutational analysis of the tyrosine phosphatome in colorectal cancers. Science 2004, 304, 1164–1166. [Google Scholar] [CrossRef]
- Pramanik, K.; Chun, C.Z.; Garnaas, M.K.; Samant, G.V.; Li, K.; Horswill, M.A.; North, P.E.; Ramchandran, R. Dusp-5 and Snrk-1 coordinately function during vascular development and disease. Blood 2009, 113, 1184–1191. [Google Scholar] [CrossRef]
- Zhao, S.; Sedwick, D.; Wang, Z. Genetic alterations of protein tyrosine phosphatases in human cancers. Oncogene 2015, 34, 3885–3894. [Google Scholar] [CrossRef]
- Brandao, T.A.; Hengge, A.C.; Johnson, S.J. Insights into the reaction of protein-tyrosine phosphatase 1B: Crystal structures for transition state analogs of both catalytic steps. J. Biol. Chem. 2010, 285, 15874–15883. [Google Scholar] [CrossRef]
- Pannifer, A.D.; Flint, A.J.; Tonks, N.K.; Barford, D. Visualization of the cysteinyl-phosphate intermediate of a protein-tyrosine phosphatase by x-ray crystallography. J. Biol. Chem. 1998, 273, 10454–10462. [Google Scholar] [CrossRef]
- Zhao, Y.; Wu, L.; Noh, S.J.; Guan, K.L.; Zhang, Z.Y. Altering the nucleophile specificity of a protein-tyrosine phosphatase-catalyzed reaction. Probing the function of the invariant glutamine residues. J. Biol. Chem. 1998, 273, 5484–5492. [Google Scholar] [CrossRef]
- Kuznetsov, V.I.; Hengge, A.C.; Johnson, S.J. New aspects of the phosphatase VHZ revealed by a high-resolution structure with vanadate and substrate screening. Biochemistry 2012, 51, 9869–9879. [Google Scholar] [CrossRef]
- Yuvaniyama, J.; Denu, J.M.; Dixon, J.E.; Saper, M.A. Crystal structure of the dual specificity protein phosphatase VHR. Science 1996, 272, 1328–1331. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.N.; Mortensen, O.H.; Peters, G.H.; Drake, P.G.; Iversen, L.F.; Olsen, O.H.; Jansen, P.G.; Andersen, H.S.; Tonks, N.K.; Moller, N.P. Structural and evolutionary relationships among protein tyrosine phosphatase domains. Mol. Cell. Biol. 2001, 21, 7117–7136. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.J.; Dixon, J.E.; Manning, G. Genomics and evolution of protein phosphatases. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Barford, D.; Flint, A.J.; Tonks, N.K. Structural basis for phosphotyrosine peptide recognition by protein tyrosine phosphatase 1B. Science 1995, 268, 1754–1758. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef]
- Zhang, J.; Bajari, R.; Andric, D.; Gerthoffert, F.; Lepsa, A.; Nahal-Bose, H.; Stein, L.D.; Ferretti, V. The International Cancer Genome Consortium Data Portal. Nat. Biotechnol. 2019, 37, 367–369. [Google Scholar] [CrossRef]
- Yokota, T.; Nara, Y.; Kashima, A.; Matsubara, K.; Misawa, S.; Kato, R.; Sugio, S. Crystal structure of human dual specificity phosphatase, JNK stimulatory phosphatase-1, at 1.5 A resolution. Proteins 2007, 66, 272–278. [Google Scholar] [CrossRef]
- Schwertassek, U.; Buckley, D.A.; Xu, C.F.; Lindsay, A.J.; McCaffrey, M.W.; Neubert, T.A.; Tonks, N.K. Myristoylation of the dual-specificity phosphatase c-JUN N-terminal kinase (JNK) stimulatory phosphatase 1 is necessary for its activation of JNK signaling and apoptosis. FEBS J. 2010, 277, 2463–2473. [Google Scholar] [CrossRef]
- Li, J.P.; Yang, C.Y.; Chuang, H.C.; Lan, J.L.; Chen, D.Y.; Chen, Y.M.; Wang, X.; Chen, A.J.; Belmont, J.W.; Tan, T.H. The phosphatase JKAP/DUSP22 inhibits T-cell receptor signalling and autoimmunity by inactivating Lck. Nat. Commun. 2014, 5, 3618. [Google Scholar] [CrossRef]
- Shen, Y.; Luche, R.; Wei, B.; Gordon, M.L.; Diltz, C.D.; Tonks, N.K. Activation of the Jnk signaling pathway by a dual-specificity phosphatase, JSP-1. Proc. Natl. Acad. Sci. USA 2001, 98, 13613–13618. [Google Scholar] [CrossRef]
- Chen, A.J.; Zhou, G.; Juan, T.; Colicos, S.M.; Cannon, J.P.; Cabriera-Hansen, M.; Meyer, C.F.; Jurecic, R.; Copeland, N.G.; Gilbert, D.J.; et al. The dual specificity JKAP specifically activates the c-Jun N-terminal kinase pathway. J. Biol. Chem. 2002, 277, 36592–36601. [Google Scholar] [CrossRef] [PubMed]
- Li, J.P.; Fu, Y.N.; Chen, Y.R.; Tan, T.H. JNK pathway-associated phosphatase dephosphorylates focal adhesion kinase and suppresses cell migration. J. Biol. Chem. 2010, 285, 5472–5478. [Google Scholar] [CrossRef] [PubMed]
- Chuang, H.C.; Chen, Y.M.; Hung, W.T.; Li, J.P.; Chen, D.Y.; Lan, J.L.; Tan, T.H. Downregulation of the phosphatase JKAP/DUSP22 in T cells as a potential new biomarker of systemic lupus erythematosus nephritis. Oncotarget 2016, 7, 57593–57605. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Chang, Y.; Liu, J.; Chen, M.; Wang, H.; Huang, M.; Liu, S.; Wang, X.; Zhao, Q. JNK Pathway-Associated Phosphatase/DUSP22 Suppresses CD4(+) T-Cell Activation and Th1/Th17-Cell Differentiation and Negatively Correlates with Clinical Activity in Inflammatory Bowel Disease. Front. Immunol. 2017, 8, 781. [Google Scholar] [CrossRef]
- Xie, L.; Zhang, Y.L.; Zhang, Z.Y. Design and characterization of an improved protein tyrosine phosphatase substrate-trapping mutant. Biochemistry 2002, 41, 4032–4039. [Google Scholar] [CrossRef]
- Denu, J.M.; Lohse, D.L.; Vijayalakshmi, J.; Saper, M.A.; Dixon, J.E. Visualization of intermediate and transition-state structures in protein-tyrosine phosphatase catalysis. Proc. Natl. Acad. Sci. USA 1996, 93, 2493–2498. [Google Scholar] [CrossRef]
- Alonso, A.; Burkhalter, S.; Sasin, J.; Tautz, L.; Bogetz, J.; Huynh, H.; Bremer, M.C.; Holsinger, L.J.; Godzik, A.; Mustelin, T. The minimal essential core of a cysteine-based protein-tyrosine phosphatase revealed by a novel 16-kDa VH1-like phosphatase, VHZ. J. Biol. Chem. 2004, 279, 35768–35774. [Google Scholar] [CrossRef]
- Lountos, G.T.; Cherry, S.; Tropea, J.E.; Waugh, D.S. Structural analysis of human dual-specificity phosphatase 22 complexed with a phosphotyrosine-like substrate. Acta Crystallogr Sect. F 2015, 71, 199–205. [Google Scholar] [CrossRef]
- Flint, A.J.; Tiganis, T.; Barford, D.; Tonks, N.K. Development of “substrate-trapping” mutants to identify physiological substrates of protein tyrosine phosphatases. Proc. Natl. Acad. Sci. USA 1997, 94, 1680–1685. [Google Scholar] [CrossRef]
- Cui, D.S.; Beaumont, V.; Ginther, P.S.; Lipchock, J.M.; Loria, J.P. Leveraging Reciprocity to Identify and Characterize Unknown Allosteric Sites in Protein Tyrosine Phosphatases. J. Mol. Biol. 2017, 429, 2360–2372. [Google Scholar] [CrossRef]
- Gannam, Z.T.K.; Min, K.; Shillingford, S.R.; Zhang, L.; Herrington, J.; Abriola, L.; Gareiss, P.C.; Pantouris, G.; Tzouvelekis, A.; Kaminski, N.; et al. An allosteric site on MKP5 reveals a strategy for small-molecule inhibition. Sci. Signal. 2020, 13. [Google Scholar] [CrossRef] [PubMed]
- Meeusen, B.; Janssens, V. Tumor suppressive protein phosphatases in human cancer: Emerging targets for therapeutic intervention and tumor stratification. Int. J. Biochem. Cell Biol. 2018, 96, 98–134. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.F.; Chuang, H.C.; Tan, T.H. Regulation of Dual-Specificity Phosphatase (DUSP) Ubiquitination and Protein Stability. Int. J. Mol. Sci. 2019, 20, 2668. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Ma, Z.; Fang, C.; Ding, J.; Yang, W.; Chen, P.; Huang, L.; Wang, C.; Yu, Y.; Yang, L.; et al. Pseudophosphatase STYX promotes tumor growth and metastasis by inhibiting FBXW7 function in colorectal cancer. Cancer Lett. 2019, 454, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kuramitsu, Y.; Kitagawa, T.; Baron, B.; Yoshino, S.; Maehara, S.; Maehara, Y.; Oka, M.; Nakamura, K. Cofilin-phosphatase slingshot-1L (SSH1L) is over-expressed in pancreatic cancer (PC) and contributes to tumor cell migration. Cancer Lett. 2015, 360, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.H.; Lu, Y.X.; Zhang, Z.Y.; Zhang, J.M.; Zhang, W.J.; Zheng, L.; Lin, W.H.; Zhang, W.; Li, X.N. SSH3 facilitates colorectal cancer cell invasion and metastasis by affecting signaling cascades involving LIMK1/Rac1. Am. J. Cancer Res. 2019, 9, 1061–1073. [Google Scholar]
- Lorenz, U. Protein tyrosine phosphatase assays. Curr. Protoc. Immunol. 2011, 93. [Google Scholar] [CrossRef]
- Alonso, A.; Narisawa, S.; Bogetz, J.; Tautz, L.; Hadzic, R.; Huynh, H.; Williams, S.; Gjorloff-Wingren, A.; Bremer, M.C.; Holsinger, L.J.; et al. VHY, a novel myristoylated testis-restricted dual specificity protein phosphatase related to VHX. J. Biol. Chem. 2004, 279, 32586–32591. [Google Scholar] [CrossRef]
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar] [CrossRef]
- Lee, W.; Tonelli, M.; Markley, J.L. NMRFAM-SPARKY: Enhanced software for biomolecular NMR spectroscopy. Bioinformatics 2015, 31, 1325–1327. [Google Scholar] [CrossRef]
- Van Ingen, H.; Bonvin, A.M. Information-driven modeling of large macromolecular assemblies using NMR data. J. Magn. Reson. 2014, 241, 103–114. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. [Google Scholar] [PubMed]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr Sect. D 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- The PyMOL Molecular Graphics System; Version 1.3; Schrödinger LLC: New York, NY, USA, 2011.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KM (mM) | kcat (s−1) | kcat/KM (s−1·M−1) | |
|---|---|---|---|
| WT | 0.64 ± 0.03 | 0.65 ± 0.01 | 1.0 × 103 |
| D57A | 5.6 ± 0.7 | 0.025 ± 0.001 | 4.5 |
| D57N | 0.83 ± 0.07 | 0.021 ± 0.0005 | 25 |
| S93A | 12 ± 2 | 0.045 ± 0.003 | 3.8 |
| S93N | 8.8 ± 0.5 | 0.057 ± 0.001 | 6.5 |
| N128A | 26 ± 3 | 0.26 ± 0.01 | 10 |
| N128D | 21 ± 2 | 0.041 ± 0.002 | 2.0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lai, C.-H.; Chang, C.-C.; Chuang, H.-C.; Tan, T.-H.; Lyu, P.-C. Structural Insights into the Active Site Formation of DUSP22 in N-loop-containing Protein Tyrosine Phosphatases. Int. J. Mol. Sci. 2020, 21, 7515. https://doi.org/10.3390/ijms21207515
Lai C-H, Chang C-C, Chuang H-C, Tan T-H, Lyu P-C. Structural Insights into the Active Site Formation of DUSP22 in N-loop-containing Protein Tyrosine Phosphatases. International Journal of Molecular Sciences. 2020; 21(20):7515. https://doi.org/10.3390/ijms21207515
Chicago/Turabian StyleLai, Chih-Hsuan, Co-Chih Chang, Huai-Chia Chuang, Tse-Hua Tan, and Ping-Chiang Lyu. 2020. "Structural Insights into the Active Site Formation of DUSP22 in N-loop-containing Protein Tyrosine Phosphatases" International Journal of Molecular Sciences 21, no. 20: 7515. https://doi.org/10.3390/ijms21207515
APA StyleLai, C.-H., Chang, C.-C., Chuang, H.-C., Tan, T.-H., & Lyu, P.-C. (2020). Structural Insights into the Active Site Formation of DUSP22 in N-loop-containing Protein Tyrosine Phosphatases. International Journal of Molecular Sciences, 21(20), 7515. https://doi.org/10.3390/ijms21207515