Cerebral Cavernous Malformation Proteins in Barrier Maintenance and Regulation

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
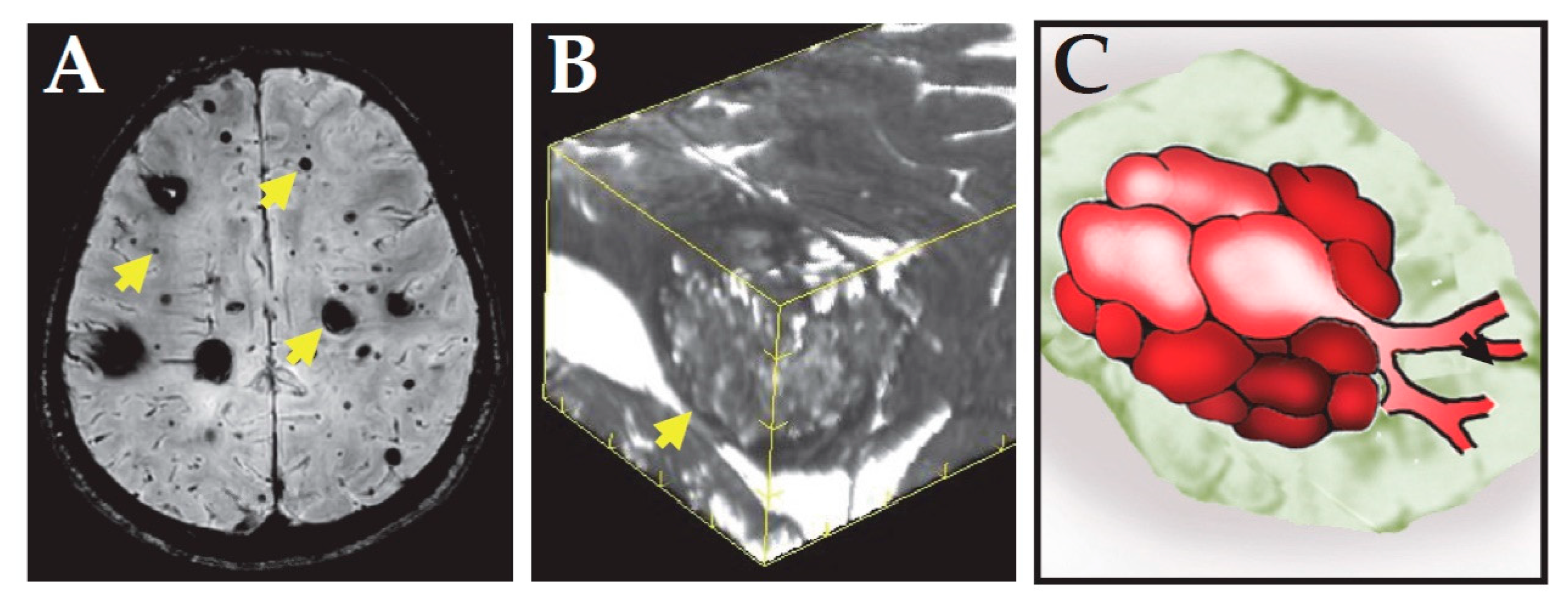
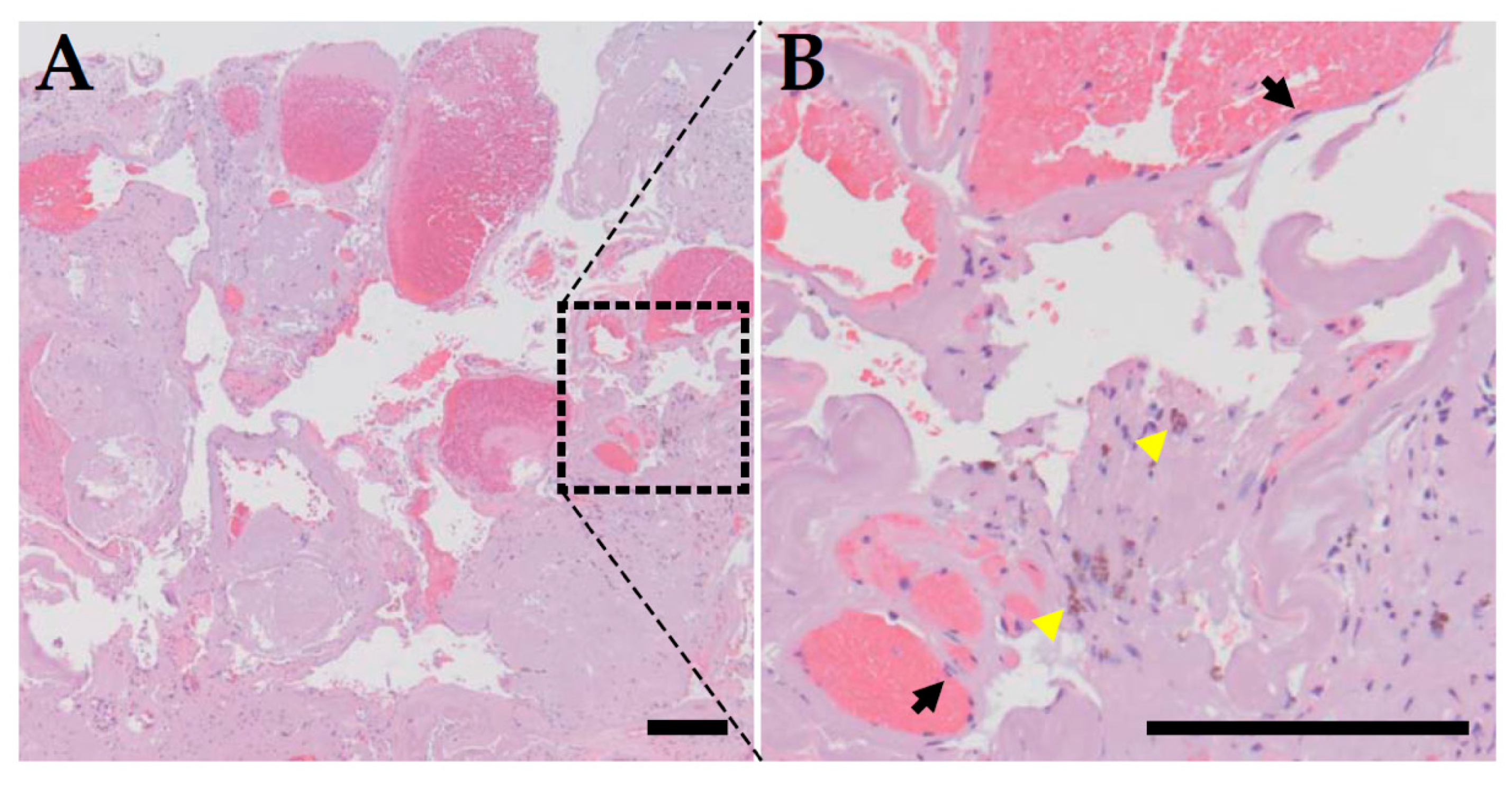
2. Clinical Features of CCM
3. Genetics of CCM
4. CCM Proteins and Their Interactions
5. CCM Proteins and Cellular Signaling
6. CCM Proteins Participate in Endothelial Barrier Maintenance and Regulation
7. Tight Junctions and CCM Disease
8. CCM Proteins Impact Intestinal Homeostasis
9. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CCM | cerebral cavernous malformation |
| EPAC | Rap1 guanine-nucleotide-exchange factor |
| FAT | focal adhesion targeting |
| FERM | band 4.1, ezrin, radixin, moesin |
| GCK | germinal center kinase |
| HHD | harmonin-homology domain |
| JAM | junctional adhesion molecule |
| KRIT1 | Krev interaction trapped protein-1 |
| LPS | lipopolysaccharide |
| MLC | myosin regulatory light chain |
| PDCD10 | programmed cell death 10 |
| PTB | phosphotyrosine-binding domain |
| ROCK | Rho-associated coiled-coil kinase |
| ROS | reactive oxygen species |
| Smurf1 | Smad ubiquitin regulatory factor 1 |
| STRIPAK | striatin interacting phosphatase and kinase |
| TER | transepithelial resistance |
| TNF | tumor necrosis factor |
| VEGF | vascular endothelial growth factor |
| ZO | zonula occludens |
References
- Awad, I.A.; Polster, S.P. Cavernous angiomas: Deconstructing a neurosurgical disease. J. Neurosurg. 2019, 131, 1–13. [Google Scholar] [CrossRef]
- Morris, Z.; Whiteley, W.N.; Longstreth, W.T., Jr.; Weber, F.; Lee, Y.C.; Tsushima, Y.; Alphs, H.; Ladd, S.C.; Warlow, C.; Wardlaw, J.M.; et al. Incidental findings on brain magnetic resonance imaging: Systematic review and meta-analysis. Br. Med. J. 2009, 339, b3016. [Google Scholar] [CrossRef]
- Moore, S.A.; Brown, R.D., Jr.; Christianson, T.J.; Flemming, K.D. Long-term natural history of incidentally discovered cavernous malformations in a single-center cohort. J. Neurosurg. 2014, 120, 1188–1192. [Google Scholar] [CrossRef]
- Flemming, K.D.; Graff-Radford, J.; Aakre, J.; Kantarci, K.; Lanzino, G.; Brown, R.D., Jr.; Mielke, M.M.; Roberts, R.O.; Kremers, W.; Knopman, D.S.; et al. Population-Based Prevalence of Cerebral Cavernous Malformations in Older Adults: Mayo Clinic Study of Aging. JAMA Neurol. 2017, 74, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Otten, P.; Pizzolato, G.P.; Rilliet, B.; Berney, J. [131 cases of cavernous angioma (cavernomas) of the CNS, discovered by retrospective analysis of 24,535 autopsies]. Neurochirurgie 1989, 35, 82–83, 128–131. [Google Scholar]
- Flemming, K.D.; Link, M.J.; Christianson, T.J.; Brown, R.D., Jr. Prospective hemorrhage risk of intracerebral cavernous malformations. Neurology 2012, 78, 632–636. [Google Scholar] [CrossRef] [PubMed]
- Al-Shahi Salman, R.; Berg, M.J.; Morrison, L.; Awad, I.A. Hemorrhage from cavernous malformations of the brain: Definition and reporting standards. Angioma Alliance Scientific Advisory Board. Stroke 2008, 39, 3222–3230. [Google Scholar] [CrossRef] [PubMed]
- Gunel, M.; Awad, I.A.; Anson, J.; Lifton, R.P. Mapping a gene causing cerebral cavernous malformation to 7q11.2-q21. Proc. Natl. Acad. Sci. USA 1995, 92, 6620–6624. [Google Scholar] [CrossRef] [PubMed]
- Marchuk, D.A.; Gallione, C.J.; Morrison, L.A.; Clericuzio, C.L.; Hart, B.L.; Kosofsky, B.E.; Louis, D.N.; Gusella, J.F.; Davis, L.E.; Prenger, V.L. A locus for cerebral cavernous malformations maps to chromosome 7q in two families. Genomics 1995, 28, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Craig, H.D.; Gunel, M.; Cepeda, O.; Johnson, E.W.; Ptacek, L.; Steinberg, G.K.; Ogilvy, C.S.; Berg, M.J.; Crawford, S.C.; Scott, R.M.; et al. Multilocus linkage identifies two new loci for a mendelian form of stroke, cerebral cavernous malformation, at 7p15-13 and 3q25.2-27. Hum. Mol. Genet. 1998, 7, 1851–1858. [Google Scholar] [CrossRef]
- Laberge-le Couteulx, S.; Jung, H.H.; Labauge, P.; Houtteville, J.P.; Lescoat, C.; Cecillon, M.; Marechal, E.; Joutel, A.; Bach, J.F.; Tournier-Lasserve, E. Truncating mutations in CCM1, encoding KRIT1, cause hereditary cavernous angiomas. Nat. Genet. 1999, 23, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, T.; Johnson, E.W.; Thomas, J.W.; Kuehl, P.M.; Jones, T.L.; Dokken, C.G.; Touchman, J.W.; Gallione, C.J.; Lee-Lin, S.Q.; Kosofsky, B.; et al. Mutations in the gene encoding KRIT1, a Krev-1/rap1a binding protein, cause cerebral cavernous malformations (CCM1). Hum. Mol. Genet. 1999, 8, 2325–2333. [Google Scholar] [CrossRef] [PubMed]
- Liquori, C.L.; Berg, M.J.; Siegel, A.M.; Huang, E.; Zawistowski, J.S.; Stoffer, T.; Verlaan, D.; Balogun, F.; Hughes, L.; Leedom, T.P.; et al. Mutations in a gene encoding a novel protein containing a phosphotyrosine-binding domain cause type 2 cerebral cavernous malformations. Am. J. Hum. Genet. 2003, 73, 1459–1464. [Google Scholar] [CrossRef] [PubMed]
- Denier, C.; Goutagny, S.; Labauge, P.; Krivosic, V.; Arnoult, M.; Cousin, A.; Benabid, A.L.; Comoy, J.; Frerebeau, P.; Gilbert, B.; et al. Mutations within the MGC4607 gene cause cerebral cavernous malformations. Am. J. Hum. Genet. 2004, 74, 326–337. [Google Scholar] [CrossRef] [PubMed]
- Guclu, B.; Ozturk, A.K.; Pricola, K.L.; Bilguvar, K.; Shin, D.; O’Roak, B.J.; Gunel, M. Mutations in apoptosis-related gene, PDCD10, cause cerebral cavernous malformation 3. Neurosurgery 2005, 57, 1008–1013. [Google Scholar] [CrossRef] [PubMed]
- Bergametti, F.; Denier, C.; Labauge, P.; Arnoult, M.; Boetto, S.; Clanet, M.; Coubes, P.; Echenne, B.; Ibrahim, R.; Irthum, B.; et al. Mutations within the programmed cell death 10 gene cause cerebral cavernous malformations. Am. J. Hum. Genet. 2005, 76, 42–51. [Google Scholar] [CrossRef]
- Zafar, A.; Quadri, S.A.; Farooqui, M.; Ikram, A.; Robinson, M.; Hart, B.L.; Mabray, M.C.; Vigil, C.; Tang, A.T.; Kahn, M.L.; et al. Familial Cerebral Cavernous Malformations. Stroke 2019, 50, 1294–1301. [Google Scholar] [CrossRef]
- Denier, C.; Labauge, P.; Bergametti, F.; Marchelli, F.; Riant, F.; Arnoult, M.; Maciazek, J.; Vicaut, E.; Brunereau, L.; Tournier-Lasserve, E.; et al. Genotype-phenotype correlations in cerebral cavernous malformations patients. Ann. Neurol. 2006, 60, 550–556. [Google Scholar] [CrossRef]
- Shenkar, R.; Shi, C.; Rebeiz, T.; Stockton, R.A.; McDonald, D.A.; Mikati, A.G.; Zhang, L.; Austin, C.; Akers, A.L.; Gallione, C.J.; et al. Exceptional aggressiveness of cerebral cavernous malformation disease associated with PDCD10 mutations. Genet. Med. Off. J. Am. Coll. Med Genet. 2015, 17, 188–196. [Google Scholar] [CrossRef]
- Spiegler, S.; Rath, M.; Paperlein, C.; Felbor, U. Cerebral Cavernous Malformations: An Update on Prevalence, Molecular Genetic Analyses, and Genetic Counselling. Mol. Syndromol. 2018, 9, 60–69. [Google Scholar] [CrossRef]
- Gault, J.; Shenkar, R.; Recksiek, P.; Awad, I.A. Biallelic somatic and germ line CCM1 truncating mutations in a cerebral cavernous malformation lesion. Stroke 2005, 36, 872–874. [Google Scholar] [CrossRef] [PubMed]
- Akers, A.L.; Johnson, E.; Steinberg, G.K.; Zabramski, J.M.; Marchuk, D.A. Biallelic somatic and germline mutations in cerebral cavernous malformations (CCMs): Evidence for a two-hit mechanism of CCM pathogenesis. Hum. Mol. Genet. 2009, 18, 919–930. [Google Scholar] [CrossRef] [PubMed]
- McDonald, D.A.; Shi, C.; Shenkar, R.; Gallione, C.J.; Akers, A.L.; Li, S.; De Castro, N.; Berg, M.J.; Corcoran, D.L.; Awad, I.A.; et al. Lesions from patients with sporadic cerebral cavernous malformations harbor somatic mutations in the CCM genes: Evidence for a common biochemical pathway for CCM pathogenesis. Hum. Mol. Genet. 2014, 23, 4357–4370. [Google Scholar] [CrossRef]
- Berman, J.R.; Kenyon, C. Germ-cell loss extends C-elegans life span through regulation of DAF-16 by kri-1 and lipophilic-hormone signaling. Cell 2006, 124, 1055–1068. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Greiss, S.; Gartner, A.; Derry, W.B. Cell-Nonautonomous Regulation of C. elegans Germ Cell Death by kri-1. Curr. Biol. 2010, 20, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Chapman, E.M.; Lant, B.; Ohashi, Y.; Yu, B.; Schertzberg, M.; Go, C.; Dogra, D.; Koskimaki, J.; Girard, R.; Li, Y.; et al. A conserved CCM complex promotes apoptosis non-autonomously by regulating zinc homeostasis. Nat. Commun. 2019, 10, 1791. [Google Scholar] [CrossRef]
- Pal, S.; Lant, B.; Yu, B.; Tian, R.L.; Tong, J.F.; Krieger, J.R.; Moran, M.F.; Gingras, A.C.; Derry, W.B. CCM-3 Promotes C. elegans Germline Development by Regulating Vesicle Trafficking Cytokinesis and Polarity. Curr. Biol. 2017, 27, 868–876. [Google Scholar] [CrossRef]
- Lant, B.; Yu, B.; Goudreault, M.; Holmyard, D.; Knight, J.D.R.; Xu, P.; Zhao, L.; Chin, K.; Wallace, E.; Zhen, M.; et al. CCM-3/STRIPAK promotes seamless tube extension through endocytic recycling. Nat. Commun. 2015, 6, 6449. [Google Scholar] [CrossRef]
- Hogan, B.M.; Bussmann, J.; Wolburg, H.; Schulte-Merker, S. Ccm1 cell autonomously regulates endothelial cellular morphogenesis and vascular tubulogenesis in zebrafish. Hum. Mol. Genet. 2008, 17, 2424–2432. [Google Scholar] [CrossRef]
- Zheng, X.J.; Xu, C.; Di Lorenzo, A.; Kleaveland, B.; Zou, Z.Y.; Seiler, C.; Chen, M.; Cheng, L.; Xiao, J.P.; He, J.; et al. CCM3 signaling through sterile 20-like kinases plays an essential role during zebrafish cardiovascular development and cerebral cavernous malformations. J. Clin. Investig. 2010, 120, 2795–2804. [Google Scholar] [CrossRef]
- Plummer, N.W.; Gallione, C.J.; Srinivasan, S.; Zawistowski, J.S.; Louis, D.N.; Marchuk, D.A. Loss of p53 sensitizes mice with a mutation in Ccm1 (KRIT1) to development of cerebral vascular malformations. Am. J. Pathol. 2004, 165, 1509–1518. [Google Scholar] [CrossRef]
- McDonald, D.A.; Shenkar, R.; Shi, C.; Stockton, R.A.; Akers, A.L.; Kucherlapati, M.H.; Kucherlapati, R.; Brainer, J.; Ginsberg, M.H.; Awad, I.A.; et al. A novel mouse model of cerebral cavernous malformations based on the two-hit mutation hypothesis recapitulates the human disease. Hum. Mol. Genet. 2011, 20, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, K.J.; Plummer, N.W.; Adams, J.A.; Marchuk, D.A.; Li, D.Y. Ccm1 is required for arterial morphogenesis: Implications for the etiology of human cavernous malformations. Development 2004, 131, 1437–1448. [Google Scholar] [CrossRef]
- Whitehead, K.J.; Chan, A.C.; Navankasattusas, S.; Koh, W.; London, N.R.; Ling, J.; Mayo, A.H.; Drakos, S.G.; Jones, C.A.; Zhu, W.; et al. The cerebral cavernous malformation signaling pathway promotes vascular integrity via Rho GTPases. Nat. Med. 2009, 15, 177–184. [Google Scholar] [CrossRef]
- He, Y.; Zhang, H.; Yu, L.; Gunel, M.; Boggon, T.J.; Chen, H.; Min, W. Stabilization of VEGFR2 signaling by cerebral cavernous malformation 3 is critical for vascular development. Sci. Signal. 2010, 3, ra26. [Google Scholar] [CrossRef] [PubMed]
- Boulday, G.; Rudini, N.; Maddaluno, L.; Blecon, A.; Arnould, M.; Gaudric, A.; Chapon, F.; Adams, R.H.; Dejana, E.; Tournier-Lasserve, E. Developmental timing of CCM2 loss influences cerebral cavernous malformations in mice. J. Exp. Med. 2011, 208, 1835–1847. [Google Scholar] [CrossRef]
- Mleynek, T.M.; Chan, A.C.; Redd, M.; Gibson, C.C.; Davis, C.T.; Shi, D.S.; Chen, T.; Carter, K.L.; Ling, J.; Blanco, R.; et al. Lack of CCM1 induces hypersprouting and impairs response to flow. Hum. Mol. Genet. 2014, 23, 6223–6234. [Google Scholar] [CrossRef]
- Chan, A.C.; Drakos, S.G.; Ruiz, O.E.; Smith, A.C.; Gibson, C.C.; Ling, J.; Passi, S.F.; Stratman, A.N.; Sacharidou, A.; Revelo, M.P.; et al. Mutations in 2 distinct genetic pathways result in cerebral cavernous malformations in mice. J. Clin. Investig. 2011, 121, 1871–1881. [Google Scholar] [CrossRef]
- Cunningham, K.; Uchida, Y.; O’Donnell, E.; Claudio, E.; Li, W.; Soneji, K.; Wang, H.; Mukouyama, Y.S.; Siebenlist, U. Conditional deletion of Ccm2 causes hemorrhage in the adult brain: A mouse model of human cerebral cavernous malformations. Hum. Mol. Genet. 2011, 20, 3198–3206. [Google Scholar] [CrossRef]
- Detter, M.R.; Snellings, D.A.; Marchuk, D.A. Cerebral Cavernous Malformations Develop Through Clonal Expansion of Mutant Endothelial Cells. Circ. Res. 2018, 123, 1143–1151. [Google Scholar] [CrossRef]
- Serebriiskii, I.; Estojak, J.; Sonoda, G.; Testa, J.R.; Golemis, E.A. Association of Krev-1/rap1a with Krit1, a novel ankyrin repeat-containing protein encoded by a gene mapping to 7q21-22. Oncogene 1997, 15, 1043–1049. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Draheim, K.M.; Zhang, R.; Calderwood, D.A.; Boggon, T.J. Mechanism for KRIT1 release of ICAP1-mediated suppression of integrin activation. Mol. Cell 2013, 49, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Zawistowski, J.S.; Serebriiskii, I.G.; Lee, M.F.; Golemis, E.A.; Marchuk, D.A. KRIT1 association with the integrin-binding protein ICAP-1: A new direction in the elucidation of cerebral cavernous malformations (CCM1) pathogenesis. Hum. Mol. Genet. 2002, 11, 389–396. [Google Scholar] [CrossRef]
- Kleaveland, B.; Zheng, X.; Liu, J.J.; Blum, Y.; Tung, J.J.; Zou, Z.; Sweeney, S.M.; Chen, M.; Guo, L.; Lu, M.M.; et al. Regulation of cardiovascular development and integrity by the heart of glass-cerebral cavernous malformation protein pathway. Nat. Med. 2009, 15, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Gingras, A.R.; Liu, J.J.; Ginsberg, M.H. Structural basis of the junctional anchorage of the cerebral cavernous malformations complex. J. Cell Biol. 2012, 199, 39–48. [Google Scholar] [CrossRef]
- Gingras, A.R.; Puzon-McLaughlin, W.; Ginsberg, M.H. The structure of the ternary complex of Krev interaction trapped 1 (KRIT1) bound to both the Rap1 GTPase and the heart of glass (HEG1) cytoplasmic tail. J. Biol. Chem. 2013, 288, 23639–23649. [Google Scholar] [CrossRef] [PubMed]
- Gunel, M.; Laurans, M.S.; Shin, D.; DiLuna, M.L.; Voorhees, J.; Choate, K.; Nelson-Williams, C.; Lifton, R.P. KRIT1, a gene mutated in cerebral cavernous malformation, encodes a microtubule-associated protein. Proc. Natl. Acad. Sci. USA 2002, 99, 10677–10682. [Google Scholar] [CrossRef]
- Fisher, O.S.; Zhang, R.; Li, X.; Murphy, J.W.; Demeler, B.; Boggon, T.J. Structural studies of cerebral cavernous malformations 2 (CCM2) reveal a folded helical domain at its C-terminus. FEBS Lett. 2013, 587, 272–277. [Google Scholar] [CrossRef]
- Uhlik, M.T.; Abell, A.N.; Johnson, N.L.; Sun, W.; Cuevas, B.D.; Lobel-Rice, K.E.; Horne, E.A.; Dell’Acqua, M.L.; Johnson, G.L. Rac-MEKK3-MKK3 scaffolding for p38 MAPK activation during hyperosmotic shock. Nat. Cell Biol. 2003, 5, 1104–1110. [Google Scholar] [CrossRef]
- Hilder, T.L.; Malone, M.H.; Bencharit, S.; Colicelli, J.; Haystead, T.A.; Johnson, G.L.; Wu, C.C. Proteomic identification of the cerebral cavernous malformation signaling complex. J. Proteome Res. 2007, 6, 4343–4355. [Google Scholar] [CrossRef]
- Zhang, J.; Rigamonti, D.; Dietz, H.C.; Clatterbuck, R.E. Interaction between krit1 and malcavernin: Implications for the pathogenesis of cerebral cavernous malformations. Neurosurgery 2007, 60, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Zawistowski, J.S.; Stalheim, L.; Uhlik, M.T.; Abell, A.N.; Ancrile, B.B.; Johnson, G.L.; Marchuk, D.A. CCM1 and CCM2 protein interactions in cell signaling: Implications for cerebral cavernous malformations pathogenesis. Hum. Mol. Genet. 2005, 14, 2521–2531. [Google Scholar] [CrossRef] [PubMed]
- Draheim, K.M.; Li, X.; Zhang, R.; Fisher, O.S.; Villari, G.; Boggon, T.J.; Calderwood, D.A. CCM2-CCM3 interaction stabilizes their protein expression and permits endothelial network formation. J. Cell Biol. 2015, 208, 987–1001. [Google Scholar] [CrossRef] [PubMed]
- Stockton, R.A.; Shenkar, R.; Awad, I.A.; Ginsberg, M.H. Cerebral cavernous malformations proteins inhibit Rho kinase to stabilize vascular integrity. J. Exp. Med. 2010, 207, 881–896. [Google Scholar] [CrossRef]
- Hilder, T.L.; Malone, M.H.; Johnson, G.L. Hyperosmotic induction of mitogen-activated protein kinase scaffolding. Methods Enzym. 2007, 428, 297–312. [Google Scholar] [CrossRef]
- Zheng, X.; Xu, C.; Smith, A.O.; Stratman, A.N.; Zou, Z.; Kleaveland, B.; Yuan, L.; Didiku, C.; Sen, A.; Liu, X.; et al. Dynamic regulation of the cerebral cavernous malformation pathway controls vascular stability and growth. Dev. Cell 2012, 23, 342–355. [Google Scholar] [CrossRef]
- Cullere, X.; Plovie, E.; Bennett, P.M.; MacRae, C.A.; Mayadas, T.N. The cerebral cavernous malformation proteins CCM2L and CCM2 prevent the activation of the MAP kinase MEKK3. Proc. Natl. Acad. Sci. USA 2015, 112, 14284–14289. [Google Scholar] [CrossRef]
- Rosen, J.N.; Sogah, V.M.; Ye, L.Y.; Mably, J.D. Ccm2-like is required for cardiovascular development as a novel component of the Heg-CCM pathway. Dev. Biol. 2013, 376, 74–85. [Google Scholar] [CrossRef]
- Li, X.; Zhang, R.; Zhang, H.; He, Y.; Ji, W.; Min, W.; Boggon, T.J. Crystal structure of CCM3, a cerebral cavernous malformation protein critical for vascular integrity. J. Biol. Chem. 2010, 285, 24099–24107. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, H.; Zhang, Y.; Ma, D. cDNA cloning and expression of an apoptosis-related gene, humanTFAR15 gene. Sci. China C Life Sci. 1999, 42, 323–329. [Google Scholar] [CrossRef]
- Ma, X.; Zhao, H.; Shan, J.; Long, F.; Chen, Y.; Zhang, Y.; Han, X.; Ma, D. PDCD10 interacts with Ste20-related kinase MST4 to promote cell growth and transformation via modulation of the ERK pathway. Mol. Biol. Cell 2007, 18, 1965–1978. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli, D.F.; Laister, R.C.; Mulligan, V.K.; Kean, M.J.; Goudreault, M.; Scott, I.C.; Derry, W.B.; Chakrabartty, A.; Gingras, A.C.; Sicheri, F. CCM3/PDCD10 heterodimerizes with germinal center kinase III (GCKIII) proteins using a mechanism analogous to CCM3 homodimerization. J. Biol. Chem. 2011, 286, 25056–25064. [Google Scholar] [CrossRef] [PubMed]
- Fidalgo, M.; Fraile, M.; Pires, A.; Force, T.; Pombo, C.; Zalvide, J. CCM3/PDCD10 stabilizes GCKIII proteins to promote Golgi assembly and cell orientation. J. Cell Sci. 2010, 123, 1274–1284. [Google Scholar] [CrossRef]
- Goudreault, M.; D’Ambrosio, L.M.; Kean, M.J.; Mullin, M.J.; Larsen, B.G.; Sanchez, A.; Chaudhry, S.; Chen, G.I.; Sicheri, F.; Nesvizhskii, A.I.; et al. A PP2A phosphatase high density interaction network identifies a novel striatin-interacting phosphatase and kinase complex linked to the cerebral cavernous malformation 3 (CCM3) protein. Mol. Cell Proteom. 2009, 8, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Suryavanshi, N.; Furmston, J.; Ridley, A.J. The STRIPAK complex components FAM40A and FAM40B regulate endothelial cell contractility via ROCKs. BMC Cell Biol. 2018, 19, 26. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.; Hwang, J.; Carrier, K.J.; Jones, C.A.; Kern, Q.L.; Moreno, C.S.; Karas, R.H.; Pallas, D.C. Protein phosphatase 2a (PP2A) binds within the oligomerization domain of striatin and regulates the phosphorylation and activation of the mammalian Ste20-Like kinase Mst3. BMC Biochem. 2011, 12, 54. [Google Scholar] [CrossRef] [PubMed]
- Voss, K.; Stahl, S.; Schleider, E.; Ullrich, S.; Nickel, J.; Mueller, T.D.; Felbor, U. CCM3 interacts with CCM2 indicating common pathogenesis for cerebral cavernous malformations. Neurogenetics 2007, 8, 249–256. [Google Scholar] [CrossRef]
- Li, X.; Ji, W.; Zhang, R.; Folta-Stogniew, E.; Min, W.; Boggon, T.J. Molecular recognition of leucine-aspartate repeat (LD) motifs by the focal adhesion targeting homology domain of cerebral cavernous malformation 3 (CCM3). J. Biol. Chem. 2011, 286, 26138–26147. [Google Scholar] [CrossRef]
- Zhang, Y.; Tang, W.; Zhang, H.; Niu, X.; Xu, Y.; Zhang, J.; Gao, K.; Pan, W.; Boggon, T.J.; Toomre, D.; et al. A network of interactions enables CCM3 and STK24 to coordinate UNC13D-driven vesicle exocytosis in neutrophils. Dev. Cell 2013, 27, 215–226. [Google Scholar] [CrossRef]
- Dibble, C.F.; Horst, J.A.; Malone, M.H.; Park, K.; Temple, B.; Cheeseman, H.; Barbaro, J.R.; Johnson, G.L.; Bencharit, S. Defining the Functional Domain of Programmed Cell Death 10 through Its Interactions with Phosphatidylinositol-3,4,5-Trisphosphate. PLoS ONE 2010, 5, e11740. [Google Scholar] [CrossRef]
- Castro, M.; Lavina, B.; Ando, K.; Alvarez-Aznar, A.; Abu Taha, A.; Brakebusch, C.; Dejana, E.; Betsholtz, C.; Gaengel, K. CDC42 Deletion Elicits Cerebral Vascular Malformations via Increased MEKK3-Dependent KLF4 Expression. Circ. Res. 2019, 124, 1240–1252. [Google Scholar] [CrossRef] [PubMed]
- Mardakheh, F.K.; Self, A.; Marshall, C.J. RHO binding to FAM65A regulates Golgi reorientation during cell migration. J. Cell Sci. 2016, 129, 4466–4479. [Google Scholar] [CrossRef] [PubMed]
- Maddaluno, L.; Rudini, N.; Cuttano, R.; Bravi, L.; Giampietro, C.; Corada, M.; Ferrarini, L.; Orsenigo, F.; Papa, E.; Boulday, G.; et al. EndMT contributes to the onset and progression of cerebral cavernous malformations. Nature 2013, 498, 492–496. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Corricelli, M.; Trapani, E.; Bravi, L.; Pittaro, A.; Delle Monache, S.; Ferroni, L.; Patergnani, S.; Missiroli, S.; Goitre, L.; et al. Defective autophagy is a key feature of cerebral cavernous malformations. EMBO Mol. Med. 2015, 7, 1403–1417. [Google Scholar] [CrossRef]
- Zhou, H.J.; Qin, L.; Zhang, H.; Tang, W.; Ji, W.; He, Y.; Liang, X.; Wang, Z.; Yuan, Q.; Vortmeyer, A.; et al. Endothelial exocytosis of angiopoietin-2 resulting from CCM3 deficiency contributes to cerebral cavernous malformation. Nat. Med. 2016, 22, 1033–1042. [Google Scholar] [CrossRef]
- Crose, L.E.; Hilder, T.L.; Sciaky, N.; Johnson, G.L. Cerebral cavernous malformation 2 protein promotes smad ubiquitin regulatory factor 1-mediated RhoA degradation in endothelial cells. J. Biol. Chem. 2009, 284, 13301–13305. [Google Scholar] [CrossRef]
- Glading, A.; Han, J.; Stockton, R.A.; Ginsberg, M.H. KRIT-1/CCM1 is a Rap1 effector that regulates endothelial cell cell junctions. J. Cell Biol. 2007, 179, 247–254. [Google Scholar] [CrossRef]
- Wang, H.R.; Zhang, Y.; Ozdamar, B.; Ogunjimi, A.A.; Alexandrova, E.; Thomsen, G.H.; Wrana, J.L. Regulation of cell polarity and protrusion formation by targeting RhoA for degradation. Science 2003, 302, 1775–1779. [Google Scholar] [CrossRef]
- Zhou, Z.; Rawnsley, D.R.; Goddard, L.M.; Pan, W.; Cao, X.J.; Jakus, Z.; Zheng, H.; Yang, J.; Arthur, J.S.; Whitehead, K.J.; et al. The cerebral cavernous malformation pathway controls cardiac development via regulation of endocardial MEKK3 signaling and KLF expression. Dev. Cell 2015, 32, 168–180. [Google Scholar] [CrossRef]
- Zhou, Z.; Tang, A.T.; Wong, W.Y.; Bamezai, S.; Goddard, L.M.; Shenkar, R.; Zhou, S.; Yang, J.; Wright, A.C.; Foley, M.; et al. Cerebral cavernous malformations arise from endothelial gain of MEKK3-KLF2/4 signalling. Nature 2016, 532, 122–126. [Google Scholar] [CrossRef]
- Huang, Q.; Yang, J.; Lin, Y.; Walker, C.; Cheng, J.; Liu, Z.G.; Su, B. Differential regulation of interleukin 1 receptor and Toll-like receptor signaling by MEKK3. Nat. Immunol. 2004, 5, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Wang, M.; Hu, Y.; Xu, N.; Yu, Q.; Wang, Q. TAK1 knockdown enhances lipopolysaccharide-induced secretion of proinflammatory cytokines in myeloid cells via unleashing MEKK3 activity. Cell. Immunol. 2016, 310, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Samanta, A.K.; Huang, H.J.; Bast, R.C., Jr.; Liao, W.S. Overexpression of MEKK3 confers resistance to apoptosis through activation of NFkappaB. J. Biol. Chem. 2004, 279, 7576–7583. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lin, Y.; Guo, Z.; Cheng, J.; Huang, J.; Deng, L.; Liao, W.; Chen, Z.; Liu, Z.; Su, B. The essential role of MEKK3 in TNF-induced NF-kappaB activation. Nat. Immunol. 2001, 2, 620–624. [Google Scholar] [CrossRef] [PubMed]
- Tang, A.T.; Choi, J.P.; Kotzin, J.J.; Yang, Y.; Hong, C.C.; Hobson, N.; Girard, R.; Zeineddine, H.A.; Lightle, R.; Moore, T.; et al. Endothelial TLR4 and the microbiome drive cerebral cavernous malformations. Nature 2017, 545, 305–310. [Google Scholar] [CrossRef]
- Fisher, O.S.; Deng, H.; Liu, D.; Zhang, Y.; Wei, R.; Deng, Y.; Zhang, F.; Louvi, A.; Turk, B.E.; Boggon, T.J.; et al. Structure and vascular function of MEKK3-cerebral cavernous malformations 2 complex. Nat. Commun. 2015, 6, 7937. [Google Scholar] [CrossRef]
- Harel, L.; Costa, B.; Tcherpakov, M.; Zapatka, M.; Oberthuer, A.; Hansford, L.M.; Vojvodic, M.; Levy, Z.; Chen, Z.Y.; Lee, F.S.; et al. CCM2 mediates death signaling by the TrkA receptor tyrosine kinase. Neuron 2009, 63, 585–591. [Google Scholar] [CrossRef]
- Costa, B.; Kean, M.J.; Ast, V.; Knight, J.D.; Mett, A.; Levy, Z.; Ceccarelli, D.F.; Badillo, B.G.; Eils, R.; Konig, R.; et al. STK25 protein mediates TrkA and CCM2 protein-dependent death in pediatric tumor cells of neural origin. J. Biol. Chem. 2012, 287, 29285–29289. [Google Scholar] [CrossRef]
- Wu, Z.; Qi, Y.; Guo, Z.; Li, P.; Zhou, D. miR-613 suppresses ischemia-reperfusion-induced cardiomyocyte apoptosis by targeting the programmed cell death 10 gene. Biosci. Trends 2016, 10, 251–257. [Google Scholar] [CrossRef]
- Chen, L.; Tanriover, G.; Yano, H.; Friedlander, R.; Louvi, A.; Gunel, M. Apoptotic functions of PDCD10/CCM3, the gene mutated in cerebral cavernous malformation 3. Stroke 2009, 40, 1474–1481. [Google Scholar] [CrossRef]
- Zhang, H.; Ma, X.; Deng, X.; Chen, Y.; Mo, X.; Zhang, Y.; Zhao, H.; Ma, D. PDCD10 interacts with STK25 to accelerate cell apoptosis under oxidative stress. Front. Biosci. 2012, 17, 2295–2305. [Google Scholar] [CrossRef] [PubMed]
- Fidalgo, M.; Guerrero, A.; Fraile, M.; Iglesias, C.; Pombo, C.M.; Zalvide, J. Adaptor protein cerebral cavernous malformation 3 (CCM3) mediates phosphorylation of the cytoskeletal proteins ezrin/radixin/moesin by mammalian Ste20-4 to protect cells from oxidative stress. J. Biol. Chem. 2012, 287, 11556–11565. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.H.; Awad, I.A.; Kim, J.H. Ultrastructural pathological features of cerebrovascular malformations: A preliminary report. Neurosurgery 2000, 46, 1454–1459. [Google Scholar] [CrossRef] [PubMed]
- Tu, J.; Stoodley, M.A.; Morgan, M.K.; Storer, K.P. Ultrastructural characteristics of hemorrhagic, nonhemorrhagic, and recurrent cavernous malformations. J. Neurosurg. 2005, 103, 903–909. [Google Scholar] [CrossRef]
- Girard, R.; Fam, M.D.; Zeineddine, H.A.; Tan, H.; Mikati, A.G.; Shi, C.; Jesselson, M.; Shenkar, R.; Wu, M.; Cao, Y.; et al. Vascular permeability and iron deposition biomarkers in longitudinal follow-up of cerebral cavernous malformations. J. Neurosurg. 2017, 127, 102–110. [Google Scholar] [CrossRef]
- Mikati, A.G.; Khanna, O.; Zhang, L.; Girard, R.; Shenkar, R.; Guo, X.; Shah, A.; Larsson, H.B.; Tan, H.; Li, L.; et al. Vascular permeability in cerebral cavernous malformations. J. Cereb. Blood Flow Metab. 2015, 35, 1632–1639. [Google Scholar] [CrossRef]
- Murali, A.; Rajalingam, K. Small Rho GTPases in the control of cell shape and mobility. Cell. Mol. Life Sci. 2014, 71, 1703–1721. [Google Scholar] [CrossRef]
- Citalan-Madrid, A.F.; Garcia-Ponce, A.; Vargas-Robles, H.; Betanzos, A.; Schnoor, M. Small GTPases of the Ras superfamily regulate intestinal epithelial homeostasis and barrier function via common and unique mechanisms. Tissue Barriers 2013, 1, e26938. [Google Scholar] [CrossRef]
- van Buul, J.D.; Geerts, D.; Huveneers, S. Rho GAPs and GEFs: Controling switches in endothelial cell adhesion. Cell Adhes. Migr. 2014, 8, 108–124. [Google Scholar] [CrossRef]
- Shen, Q.; Wu, M.H.; Yuan, S.Y. Endothelial contractile cytoskeleton and microvascular permeability. Cell Health Cytoskelet. 2009, 2009, 43–50. [Google Scholar] [CrossRef][Green Version]
- Corr, M.; Lerman, I.; Keubel, J.M.; Ronacher, L.; Misra, R.; Lund, F.; Sarelius, I.H.; Glading, A.J. Decreased Krev interaction-trapped 1 expression leads to increased vascular permeability and modifies inflammatory responses in vivo. Arter. Thromb. Vasc. Biol. 2012, 32, 2702–2710. [Google Scholar] [CrossRef] [PubMed]
- Goitre, L.; DiStefano, P.V.; Moglia, A.; Nobiletti, N.; Baldini, E.; Trabalzini, L.; Keubel, J.; Trapani, E.; Shuvaev, V.V.; Muzykantov, V.R.; et al. Up-regulation of NADPH oxidase-mediated redox signaling contributes to the loss of barrier function in KRIT1 deficient endothelium. Sci. Rep. 2017, 7, 8296. [Google Scholar] [CrossRef] [PubMed]
- Meliton, A.; Meng, F.; Tian, Y.; Shah, A.A.; Birukova, A.A.; Birukov, K.G. Role of Krev Interaction Trapped-1 in Prostacyclin-Induced Protection against Lung Vascular Permeability Induced by Excessive Mechanical Forces and Thrombin Receptor Activating Peptide 6. Am. J. Respir. Cell Mol. Biol. 2015, 53, 834–843. [Google Scholar] [CrossRef] [PubMed]
- McDonald, D.A.; Shi, C.; Shenkar, R.; Stockton, R.A.; Liu, F.; Ginsberg, M.H.; Marchuk, D.A.; Awad, I.A. Fasudil decreases lesion burden in a murine model of cerebral cavernous malformation disease. Stroke 2012, 43, 571–574. [Google Scholar] [CrossRef] [PubMed]
- Shenkar, R.; Shi, C.; Austin, C.; Moore, T.; Lightle, R.; Cao, Y.; Zhang, L.; Wu, M.; Zeineddine, H.A.; Girard, R.; et al. RhoA Kinase Inhibition with Fasudil Versus Simvastatin in Murine Models of Cerebral Cavernous Malformations. Stroke 2017, 48, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Shenkar, R.; Peiper, A.; Pardo, H.; Moore, T.; Lightle, R.; Girard, R.; Hobson, N.; Polster, S.P.; Koskimaki, J.; Zhang, D.; et al. Rho Kinase Inhibition Blunts Lesion Development and Hemorrhage in Murine Models of Aggressive Pdcd10/Ccm3 Disease. Stroke 2019, 50, 738–744. [Google Scholar] [CrossRef] [PubMed]
- Polster, S.P.; Stadnik, A.; Akers, A.L.; Cao, Y.; Christoforidis, G.A.; Fam, M.D.; Flemming, K.D.; Girard, R.; Hobson, N.; Koenig, J.I.; et al. Atorvastatin Treatment of Cavernous Angiomas with Symptomatic Hemorrhage Exploratory Proof of Concept (AT CASH EPOC) Trial. Neurosurgery 2019, 85, 843–853. [Google Scholar] [CrossRef]
- Lange, C.; Storkebaum, E.; de Almodovar, C.R.; Dewerchin, M.; Carmeliet, P. Vascular endothelial growth factor: A neurovascular target in neurological diseases. Nat. Rev. Neurol. 2016, 12, 439–454. [Google Scholar] [CrossRef]
- DiStefano, P.V.; Kuebel, J.M.; Sarelius, I.H.; Glading, A.J. KRIT1 protein depletion modifies endothelial cell behavior via increased vascular endothelial growth factor (VEGF) signaling. J. Biol. Chem. 2014, 289, 33054–33065. [Google Scholar] [CrossRef]
- Goitre, L.; Balzac, F.; Degani, S.; Degan, P.; Marchi, S.; Pinton, P.; Retta, S.F. KRIT1 regulates the homeostasis of intracellular reactive oxygen species. PLoS ONE 2010, 5, e11786. [Google Scholar] [CrossRef]
- Wallez, Y.; Huber, P. Endothelial adherens and tight junctions in vascular homeostasis, inflammation and angiogenesis. Biochim. Biophys. Acta 2008, 1778, 794–809. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [PubMed]
- Gunzel, D.; Yu, A.S. Claudins and the modulation of tight junction permeability. Physiol. Rev. 2013, 93, 525–569. [Google Scholar] [CrossRef] [PubMed]
- Castro Dias, M.; Coisne, C.; Lazarevic, I.; Baden, P.; Hata, M.; Iwamoto, N.; Francisco, D.M.F.; Vanlandewijck, M.; He, L.; Baier, F.A.; et al. Claudin-3-deficient C57BL/6J mice display intact brain barriers. Sci. Rep. 2019, 9, 203. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuki, S.; Yamaguchi, H.; Katsukura, Y.; Asashima, T.; Terasaki, T. mRNA expression levels of tight junction protein genes in mouse brain capillary endothelial cells highly purified by magnetic cell sorting. J. Neurochem. 2008, 104, 147–154. [Google Scholar] [CrossRef]
- Nitta, T.; Hata, M.; Gotoh, S.; Seo, Y.; Sasaki, H.; Hashimoto, N.; Furuse, M.; Tsukita, S. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J. Cell Biol. 2003, 161, 653–660. [Google Scholar] [CrossRef]
- Raleigh, D.R.; Marchiando, A.M.; Zhang, Y.; Shen, L.; Sasaki, H.; Wang, Y.; Long, M.; Turner, J.R. Tight junction-associated MARVEL proteins marveld3, tricellulin, and occludin have distinct but overlapping functions. Mol. Biol. Cell 2010, 21, 1200–1213. [Google Scholar] [CrossRef]
- Buschmann, M.M.; Shen, L.; Rajapakse, H.; Raleigh, D.R.; Wang, Y.; Lingaraju, A.; Zha, J.; Abbott, E.; McAuley, E.M.; Breskin, L.A.; et al. Occludin OCEL-domain interactions are required for maintenance and regulation of the tight junction barrier to macromolecular flux. Mol. Biol. Cell 2013, 24, 3056–3068. [Google Scholar] [CrossRef]
- Krug, S.M.; Amasheh, S.; Richter, J.F.; Milatz, S.; Gunzel, D.; Westphal, J.K.; Huber, O.; Schulzke, J.D.; Fromm, M. Tricellulin Forms a Barrier to Macromolecules in Tricellular Tight Junctions without Affecting Ion Permeability. Mol. Biol. Cell 2009, 20, 3713–3724. [Google Scholar] [CrossRef]
- Saitou, M.; Furuse, M.; Sasaki, H.; Schulzke, J.D.; Fromm, M.; Takano, H.; Noda, T.; Tsukita, S. Complex phenotype of mice lacking occludin, a component of tight junction strands. Mol. Biol. Cell 2000, 11, 4131–4142. [Google Scholar] [CrossRef]
- O’Driscoll, M.C.; Daly, S.B.; Urquhart, J.E.; Black, G.C.; Pilz, D.T.; Brockmann, K.; McEntagart, M.; Abdel-Salam, G.; Zaki, M.; Wolf, N.I.; et al. Recessive mutations in the gene encoding the tight junction protein occludin cause band-like calcification with simplified gyration and polymicrogyria. Am. J. Hum. Genet. 2010, 87, 354–364. [Google Scholar] [CrossRef]
- Laukoetter, M.G.; Nava, P.; Lee, W.Y.; Severson, E.A.; Capaldo, C.T.; Babbin, B.A.; Williams, I.R.; Koval, M.; Peatman, E.; Campbell, J.A.; et al. JAM-A regulates permeability and inflammation in the intestine in vivo. J. Exp. Med. 2007, 204, 3067–3076. [Google Scholar] [CrossRef] [PubMed]
- Otani, T.; Nguyen, T.P.; Tokuda, S.; Sugihara, K.; Sugawara, T.; Furuse, K.; Miura, T.; Ebnet, K.; Furuse, M. Claudins and JAM-A coordinately regulate tight junction formation and epithelial polarity. J. Cell Biol. 2019, 218, 3372–3396. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.M.N.; Megens, R.T.A.; Zernecke, A.; Bidzhekov, K.; van den Akker, N.M.; Rademakers, T.; van Zandvoort, M.A.; Hackeng, T.M.; Koenen, R.R.; Weber, C. Endothelial Junctional Adhesion Molecule-A Guides Monocytes into Flow-Dependent Predilection Sites of Atherosclerosis. Circulation 2014, 129, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Cohen, C.J.; Shieh, J.T.; Pickles, R.J.; Okegawa, T.; Hsieh, J.T.; Bergelson, J.M. The coxsackievirus and adenovirus receptor is a transmembrane component of the tight junction. Proc. Natl. Acad. Sci. USA 2001, 98, 15191–15196. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.; Kim, K.H.; An, S.H.; Lee, S.; Lim, B.K.; Kang, S.W.; Kwon, K. Coxsackievirus and adenovirus receptor mediates the responses of endothelial cells to fluid shear stress. Exp. Mol. Med. 2019, 51, 1–15. [Google Scholar] [CrossRef]
- Fanning, A.S.; Anderson, J.M. Zonula Occludens-1 and-2 Are Cytosolic Scaffolds That Regulate the Assembly of Cellular Junctions. Ann. N. Y. Acad. Sci. 2009, 1165, 113–120. [Google Scholar] [CrossRef]
- Katsuno, T.; Umeda, K.; Matsui, T.; Hata, M.; Tamura, A.; Itoh, M.; Takeuchi, K.; Fujimori, T.; Nabeshima, Y.; Noda, T.; et al. Deficiency of zonula occludens-1 causes embryonic lethal phenotype associated with defected yolk sac angiogenesis and apoptosis of embryonic cells. Mol. Biol. Cell 2008, 19, 2465–2475. [Google Scholar] [CrossRef]
- Tornavaca, O.; Chia, M.; Dufton, N.; Almagro, L.O.; Conway, D.E.; Randi, A.M.; Schwartz, M.A.; Matter, K.; Balda, M.S. ZO-1 controls endothelial adherens junctions, cell-cell tension, angiogenesis, and barrier formation. J. Cell Biol. 2015, 208, 821–838. [Google Scholar] [CrossRef] [PubMed]
- Schossleitner, K.; Rauscher, S.; Groger, M.; Friedl, H.P.; Finsterwalder, R.; Habertheuer, A.; Sibilia, M.; Brostjan, C.; Fodinger, D.; Citi, S.; et al. Evidence That Cingulin Regulates Endothelial Barrier Function In Vitro and In Vivo. Arter. Throm. Vas. 2016, 36, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Chrifi, I.; Hermkens, D.; Brandt, M.M.; van Dijk, C.G.M.; Burgisser, P.E.; Haasdijk, R.; Pei, J.Y.; van de Kamp, E.H.M.; Zhu, C.B.; Blonden, L.; et al. Cgnl1, an endothelial junction complex protein, regulates GTPase mediated angiogenesis. Cardiovasc. Res. 2017, 113, 1776–1788. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, J.A.; Ridley, A.J. Roles of Rho/ROCK and MLCK in TNF-alpha-induced changes in endothelial morphology and permeability. J. Cell. Physiol. 2007, 213, 221–228. [Google Scholar] [CrossRef]
- Persidsky, Y.; Heilman, D.; Haorah, J.; Zelivyanskaya, M.; Persidsky, R.; Weber, G.A.; Shimokawa, H.; Kaibuchi, K.; Ikezu, T. Rho-mediated regulation of tight junctions during monocyte migration across the blood-brain barrier in HIV-1 encephalitis (HIVE). Blood 2006, 107, 4770–4780. [Google Scholar] [CrossRef] [PubMed]
- Wojciak-Stothard, B.; Potempa, S.; Eichholtz, T.; Ridley, A.J. Rho and Rac but not Cdc42 regulate endothelial cell permeability. J. Cell Sci. 2001, 114, 1343–1355. [Google Scholar] [PubMed]
- Terry, S.; Nie, M.; Matter, K.; Balda, M.S. Rho signaling and tight junction functions. Physiology 2010, 25, 16–26. [Google Scholar] [CrossRef]
- Yamamoto, M.; Ramirez, S.H.; Sato, S.; Kiyota, T.; Cerny, R.L.; Kaibuchi, K.; Persidsky, Y.; Ikezu, T. Phosphorylation of claudin-5 and occludin by rho kinase in brain endothelial cells. Am. J. Pathol. 2008, 172, 521–533. [Google Scholar] [CrossRef]
- Izawa, Y.; Gu, Y.H.; Osada, T.; Kanazawa, M.; Hawkins, B.T.; Koziol, J.A.; Papayannopoulou, T.; Spatz, M.; Del Zoppo, G.J. beta1-integrin-matrix interactions modulate cerebral microvessel endothelial cell tight junction expression and permeability. J. Cereb. Blood Flow Metab. 2018, 38, 641–658. [Google Scholar] [CrossRef]
- Faurobert, E.; Rome, C.; Lisowska, J.; Manet-Dupe, S.; Boulday, G.; Malbouyres, M.; Balland, M.; Bouin, A.P.; Keramidas, M.; Bouvard, D.; et al. CCM1-ICAP-1 complex controls beta 1 integrin-dependent endothelial contractility and fibronectin remodeling. J. Cell Biol. 2013, 202, 545–561. [Google Scholar] [CrossRef]
- Millon-Fremillon, A.; Brunner, M.; Abed, N.; Collomb, E.; Ribba, A.S.; Block, M.R.; Albiges-Rizo, C.; Bouvard, D. Calcium and calmodulin-dependent serine/threonine protein kinase type II (CaMKII)-mediated intramolecular opening of integrin cytoplasmic domain-associated protein-1 (ICAP-1alpha) negatively regulates beta1 integrins. J. Biol. Chem. 2013, 288, 20248–20260. [Google Scholar] [CrossRef]
- Stroeken, P.J.M.; Alvarez, B.; Van Rheenen, J.; Wijnands, Y.M.; Geerts, D.; Jalink, K.; Roos, E. Integrin cytoplasmic domain-associated protein-1 (ICAP-1) interacts with the ROCK-I kinase at the plasma membrane. J. Cell. Physiol. 2006, 208, 620–628. [Google Scholar] [CrossRef]
- Wittchen, E.S.; Worthylake, R.A.; Kelly, P.; Casey, P.J.; Quilliam, L.A.; Burridge, K. Rap1 GTPase inhibits leukocyte transmigration by promoting endothelial barrier function. J. Biol. Chem. 2005, 280, 11675–11682. [Google Scholar] [CrossRef] [PubMed]
- Ramos, C.J.; Lin, C.; Liu, X.; Antonetti, D.A. The EPAC-Rap1 pathway prevents and reverses cytokine-induced retinal vascular permeability. J. Biol. Chem. 2018, 293, 717–730. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.W.; Parker, L.H.; Hall, C.J.; Smyczek, T.; Mak, J.; Crow, A.; Posthuma, G.; De Maziere, A.; Sagolla, M.; Chalouni, C.; et al. Rasip1 regulates vertebrate vascular endothelial junction stability through Epac1-Rap1 signaling. Blood 2013, 122, 3678–3690. [Google Scholar] [CrossRef] [PubMed]
- Post, A.; Pannekoek, W.J.; Ross, S.H.; Verlaan, I.; Brouwer, P.M.; Bos, J.L. Rasip1 mediates Rap1 regulation of Rho in endothelial barrier function through ArhGAP29. Proc. Natl. Acad. Sci. USA 2013, 110, 11427–11432. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Sacharidou, A.; Fu, S.; Chong, D.C.; Skaug, B.; Chen, Z.J.; Davis, G.E.; Cleaver, O. Blood vessel tubulogenesis requires Rasip1 regulation of GTPase signaling. Dev. Cell 2011, 20, 526–539. [Google Scholar] [CrossRef]
- de Kreuk, B.J.; Gingras, A.R.; Knight, J.D.R.; Liu, J.J.; Gingras, A.C.; Ginsberg, M.H. Heart of glass anchors Rasip1 at endothelial cell-cell junctions to support vascular integrity. eLife 2016, 5, e11394. [Google Scholar] [CrossRef]
- Severson, E.A.; Parkos, C.A. Structural determinants of Junctional Adhesion Molecule A (JAM-A) function and mechanisms of intracellular signaling. Curr. Opin. Cell Biol. 2009, 21, 701–707. [Google Scholar] [CrossRef]
- Giannotta, M.; Benedetti, S.; Tedesco, F.S.; Corada, M.; Trani, M.; D’Antuono, R.; Millet, Q.; Orsenigo, F.; Galvez, B.G.; Cossu, G.; et al. Targeting endothelial junctional adhesion molecule-A/EPAC/Rap-1 axis as a novel strategy to increase stem cell engraftment in dystrophic muscles. EMBO Mol. Med. 2014, 6, 239–258. [Google Scholar] [CrossRef]
- Schneider, H.; Errede, M.; Ulrich, N.H.; Virgintino, D.; Frei, K.; Bertalanffy, H. Impairment of tight junctions and glucose transport in endothelial cells of human cerebral cavernous malformations. J. Neuropathol. Exp. Neurol. 2011, 70, 417–429. [Google Scholar] [CrossRef]
- Jakimovski, D.; Schneider, H.; Frei, K.; Kennes, L.N.; Bertalanffy, H. Bleeding propensity of cavernous malformations: Impact of tight junction alterations on the occurrence of overt hematoma. J. Neurosurg. 2014, 121, 613–620. [Google Scholar] [CrossRef]
- Lopez-Ramirez, M.A.; Fonseca, G.; Zeineddine, H.A.; Girard, R.; Moore, T.; Pham, A.; Cao, Y.; Shenkar, R.; de Kreuk, B.J.; Lagarrigue, F.; et al. Thrombospondin1 (TSP1) replacement prevents cerebral cavernous malformations. J. Exp. Med. 2017, 214, 3331–3346. [Google Scholar] [CrossRef] [PubMed]
- Stamatovic, S.M.; Sladojevic, N.; Keep, R.F.; Andjelkovic, A.V. PDCD10 (CCM3) regulates brain endothelial barrier integrity in cerebral cavernous malformation type 3: Role of CCM3-ERK1/2-cortactin cross-talk. Acta Neuropathol. 2015, 130, 731–750. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.M.; Roach, J.P.; Hu, A.; Stamatovic, S.M.; Zochowski, M.R.; Keep, R.F.; Andjelkovic, A.V. Connexin 43 gap junctions contribute to brain endothelial barrier hyperpermeability in familial cerebral cavernous malformations type III by modulating tight junction structure. FASEB J. 2018, 32, 2615–2629. [Google Scholar] [CrossRef] [PubMed]
- Glading, A.J.; Ginsberg, M.H. Rap1 and its effector KRIT1/CCM1 regulate beta-catenin signaling. Dis. Models Mech. 2010, 3, 73–83. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Y.; Zou, J.; Polster, S.P.; Lightle, R.; Moore, T.; Dimaano, M.; He, T.C.; Weber, C.R.; Awad, I.A.; et al. The cerebral cavernous malformation disease causing gene KRIT1 participates in intestinal epithelial barrier maintenance and regulation. FASEB J. 2019, 33, 2132–2143. [Google Scholar] [CrossRef]
- McCarthy, K.M.; Francis, S.A.; McCormack, J.M.; Lai, J.; Rogers, R.A.; Skare, I.B.; Lynch, R.D.; Schneeberger, E.E. Inducible expression of claudin-1-myc but not occludin-VSV-G results in aberrant tight junction strand formation in MDCK cells. J. Cell Sci. 2000, 113, 3387–3398. [Google Scholar]
- Tang, A.T.; Sullivan, K.R.; Hong, C.C.; Goddard, L.M.; Mahadevan, A.; Ren, A.; Pardo, H.; Peiper, A.; Griffin, E.; Tanes, C.; et al. Distinct cellular roles for PDCD10 define a gut-brain axis in cerebral cavernous malformation. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, S.; Li, Y.; Polster, S.P.; Weber, C.R.; Awad, I.A.; Shen, L. Cerebral Cavernous Malformation Proteins in Barrier Maintenance and Regulation. Int. J. Mol. Sci. 2020, 21, 675. https://doi.org/10.3390/ijms21020675
Wei S, Li Y, Polster SP, Weber CR, Awad IA, Shen L. Cerebral Cavernous Malformation Proteins in Barrier Maintenance and Regulation. International Journal of Molecular Sciences. 2020; 21(2):675. https://doi.org/10.3390/ijms21020675
Chicago/Turabian StyleWei, Shu, Ye Li, Sean P. Polster, Christopher R. Weber, Issam A. Awad, and Le Shen. 2020. "Cerebral Cavernous Malformation Proteins in Barrier Maintenance and Regulation" International Journal of Molecular Sciences 21, no. 2: 675. https://doi.org/10.3390/ijms21020675
APA StyleWei, S., Li, Y., Polster, S. P., Weber, C. R., Awad, I. A., & Shen, L. (2020). Cerebral Cavernous Malformation Proteins in Barrier Maintenance and Regulation. International Journal of Molecular Sciences, 21(2), 675. https://doi.org/10.3390/ijms21020675