Age-Associated TET2 Mutations: Common Drivers of Myeloid Dysfunction, Cancer and Cardiovascular Disease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Hematopoiesis and Myeloid Malignancies
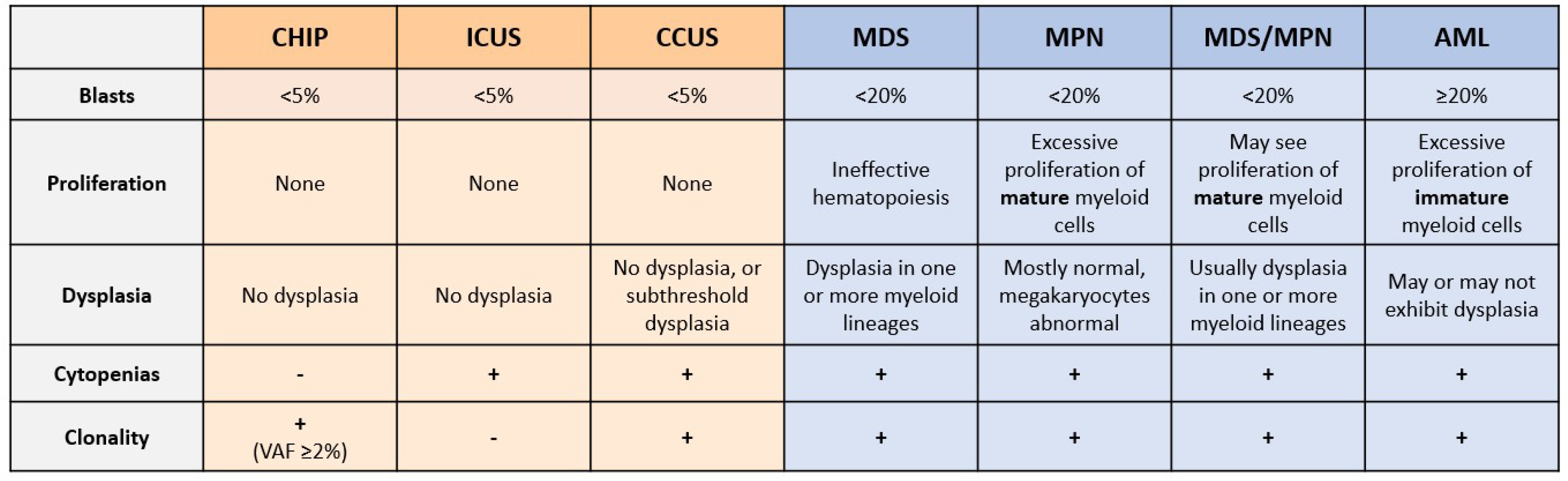
3. Unexplained Cytopenias and Shortcomings of Current Diagnostic Techniques
4. Promise of NGS in Myeloid Malignancy Diagnosis
5. CHIP and Risk of Hematological Malignancy
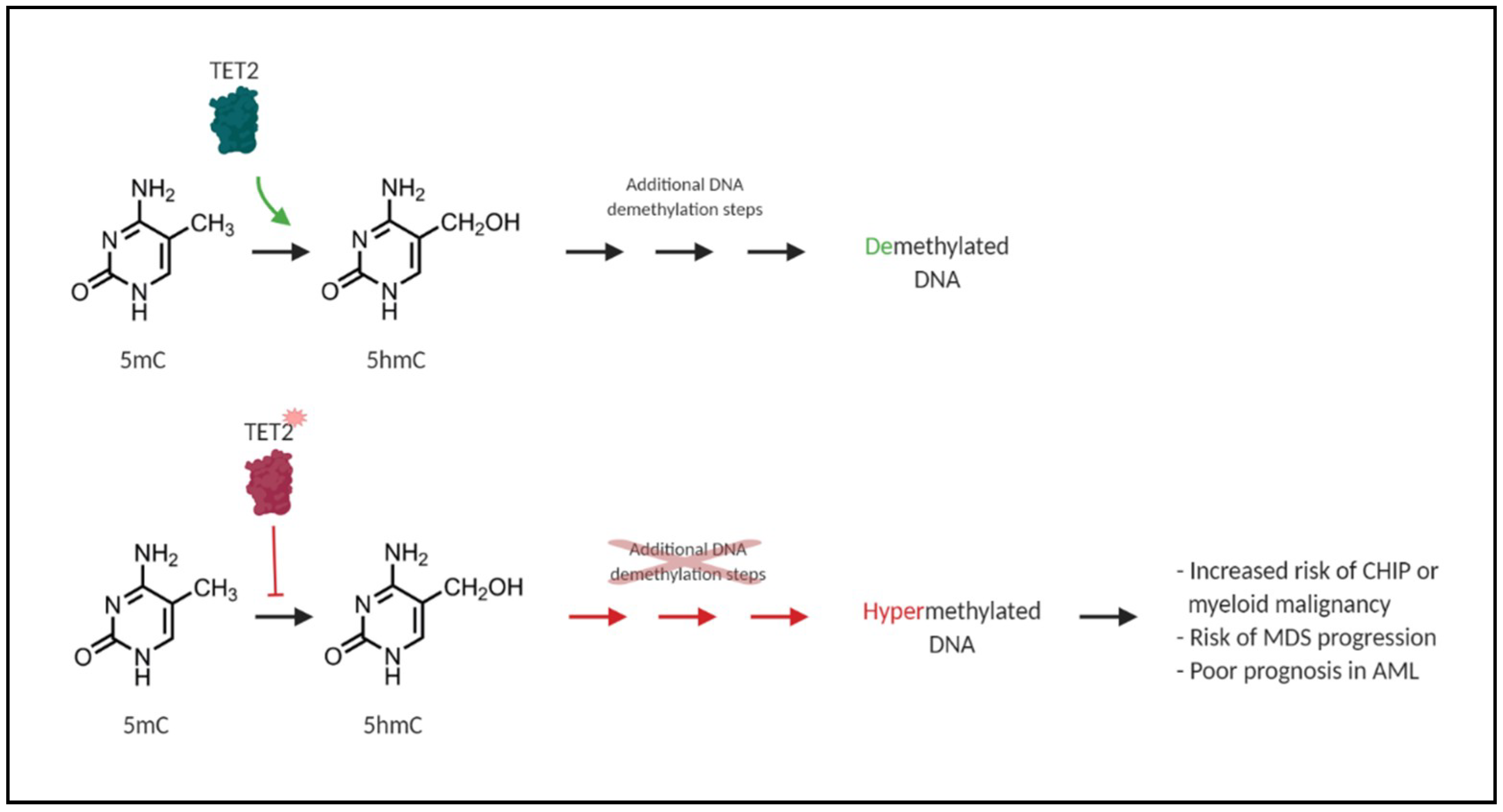
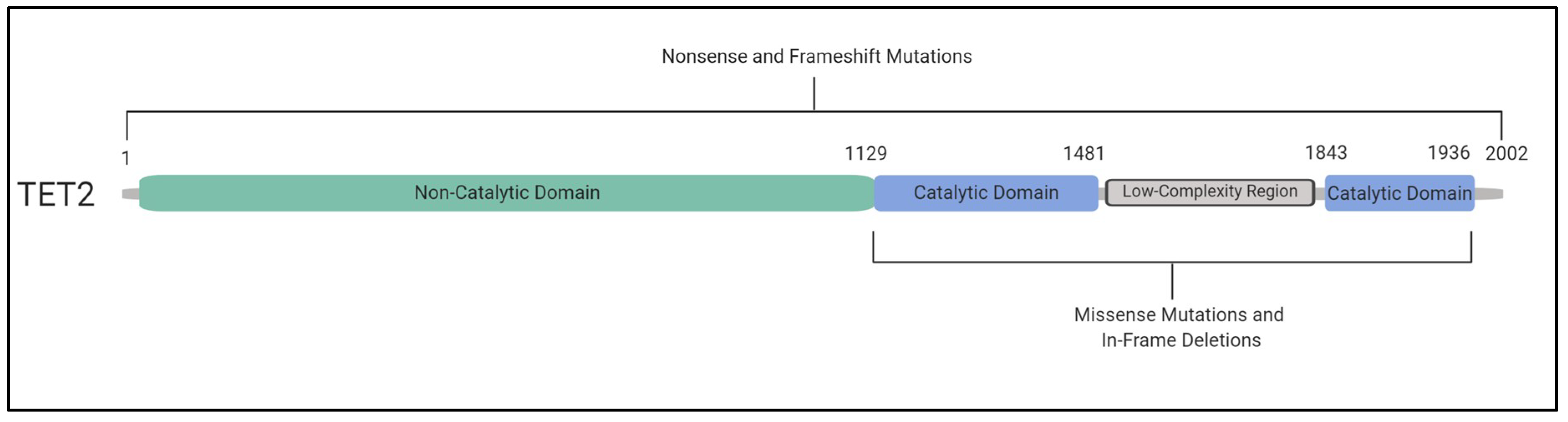
6. TET2 and DNMT3A Mutations and Their Impact
7. CHIP and Comorbid Diseases
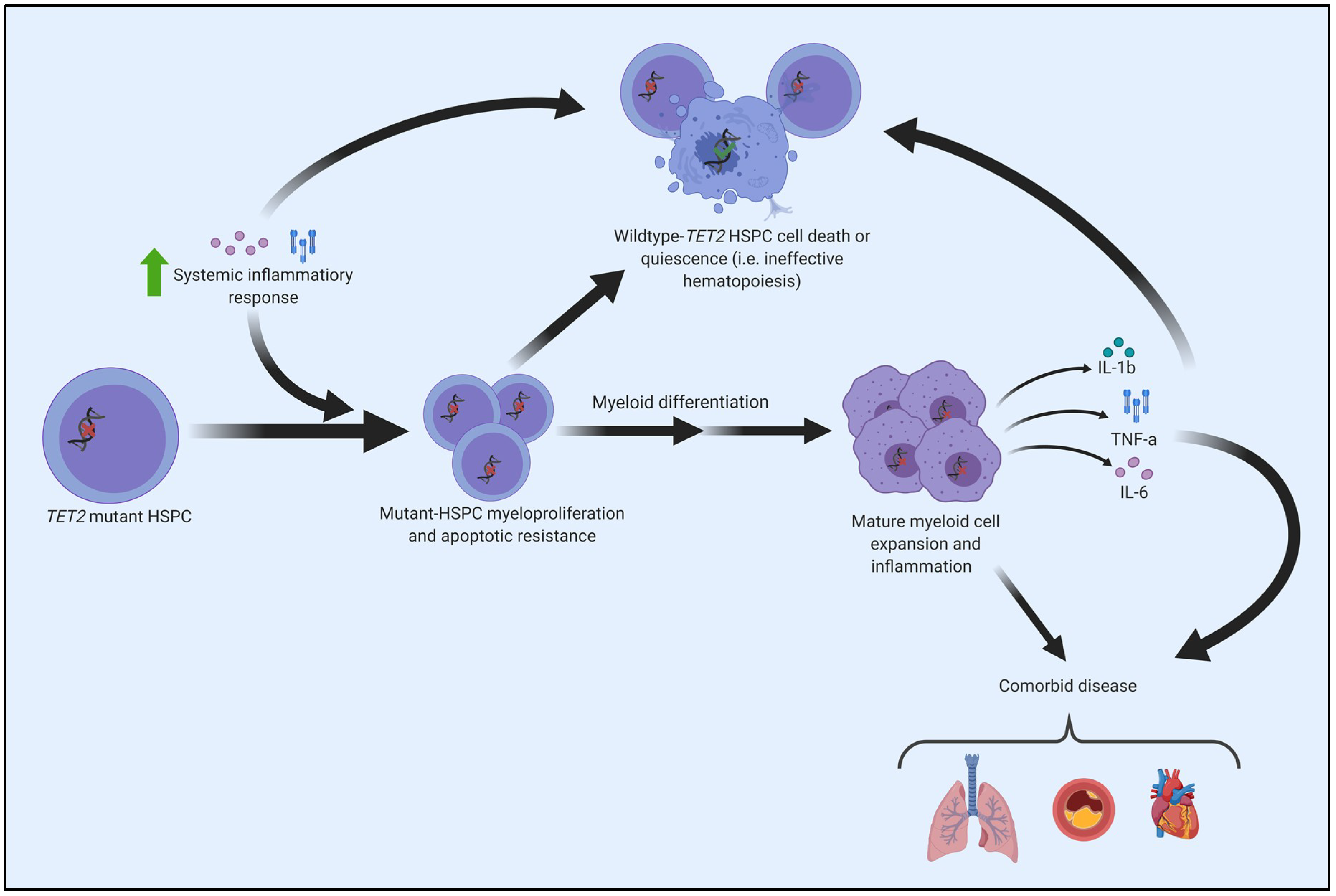
8. CHIP, Inflammation, and the Connection to the Pathogenesis of Myeloid Cancers
9. Promises and Potential Pitfalls of Surveillance for CHIP in Human Aging
10. Promise of Targeting the “Good Copy” of TET2
11. Summary
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| α-KGDDs | Alpha-ketoglutarate- and Fe2+-dependent dioxygenases |
| ARCH | Age-related clonal hematopoiesis |
| Arg1 | Arginase 1 |
| AML | Acute myeloid leukemia |
| CCUS | Clonal cytopenia of undetermined significance |
| CH | Clonal hematopoiesis |
| CHIP | Clonal hematopoiesis of indeterminant potential |
| CMML | Chronic myelomonocytic leukemia |
| COPD | Chronic obstructive pulmonary disease |
| DNMT3A | DNA methyltransferase 3 alpha |
| DOT1L | Disruptor of telomeric silencing 1-like |
| GERD | Gastro-esophageal reflux disease |
| HDAC2 | Histone deacetylase 2 |
| HMA | Hypomethylating agents |
| HSC | Hematopoietic stem cells |
| HSPC | Hematopoietic stem/progenitor cells |
| ICUS | Idiopathic cytopenia of undetermined significance |
| IL-1β | Interleukin 1 beta |
| IL-18 | Interleukin 18 |
| IL-6 | Interleukin 6 |
| IL6R | Interleukin 6 receptor |
| LPS | Lipopolysaccharides |
| MDS | Myelodysplastic syndromes |
| MDS-RS | MDS with ring sideroblasts |
| MDS/MPN | Myelodysplastic syndromes/myeloproliferative neoplasms |
| MPN | Myeloproliferative neoplasm |
| NGS | Next-generation sequencing |
| NLRP3 | Nod-like receptor family pyrin domain containing 3 |
| RARS | Refractory anemia with ring sideroblasts |
| SAM | S-adenosyl-l-methionine |
| TET2 | Tet methylcytosine dioxygenase 2 |
| TNF-α | Tumour necrosis factor alpha |
| VAF | Variant allele frequency |
| 5mC | 5-methylcytosine |
| 5hmC | 5-hydroxymethylcytosine |
References
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2406. [Google Scholar] [CrossRef] [PubMed]
- Barbui, T.; Thiele, J.; Gisslinger, H.; Kvasnicka, H.M.; Vannucchi, A.M.; Guglielmelli, P.; Orazi, A.; Tefferi, A. The 2016 WHO classification and diagnostic criteria for myeloproliferative neoplasms: Document summary and in-depth discussion. Blood Cancer J. 2018, 8, 15. [Google Scholar] [CrossRef] [PubMed]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Mardis, E.R.; Ding, L.; Dooling, D.J.; Larson, D.E.; McLellan, M.D.; Chen, K.; Koboldt, D.C.; Fulton, R.S.; Delehaunty, K.D.; McGrath, S.D.; et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N. Engl. J. Med. 2009, 361, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Loo, P.V.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3627. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network; Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, G.; Hoadley, K.; Triche, T.J.; Laird, P.W.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef]
- Welch, J.S.; Ley, T.J.; Link, D.C.; Miller, C.A.; Larson, D.E.; Koboldt, D.C.; Wartman, L.D.; Lamprecht, T.L.; Liu, F.; Xia, J.; et al. The origin and evolution of mutations in acute myeloid leukemia. Cell 2012, 150, 264–278. [Google Scholar] [CrossRef]
- Delhommeau, F.; Dupont, S.; Della Valle, V.; James, C.; Trannoy, S.; Massé, A.; Kosmider, O.; Le Couedic, J.-P.; Robert, F.; Alberdi, A.; et al. Mutations in TET2 in cancer. N. Engl. J. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef]
- Shlush, L.I.; Zandi, S.; Mitchell, A.; Chen, W.C.; Brandwein, J.M.; Gupta, V.; Kennedy, J.A.; Schimmer, A.D.; Schuh, A.C.; Yee, K.W.; et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014, 506, 328–333. [Google Scholar] [CrossRef]
- Lee-Six, H.; Øbro, N.F.; Shepherd, M.S.; Grossmann, S.; Dawson, K.; Belmonte, M.; Osborne, R.J.; Huntly, B.J.P.; Martincorena, I.; Anderson, E.; et al. Population dynamics of normal human blood inferred from somatic mutations. Nature 2018, 561, 473–478. [Google Scholar] [CrossRef]
- Vardiman, J.W.; Thiele, J.; Arber, D.A.; Brunning, R.D.; Borowitz, M.J.; Porwit, A.; Lee Haris, N.; Le Beau, M.M.; Hellstrom-Lindberg, E.; Tefferi, A.; et al. The 2008 revision of the WHO classification of myeloid neoplasms and acute leukemia: Rationale and important changes. Blood 2009, 114, 937–952. [Google Scholar] [CrossRef] [PubMed]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Bejar, R. CHIP, ICUS, CCUS and other four-letter words. Leukemia 2017, 31, 1869–1871. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Vardiman, J.W. (Eds.) WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed.; International Agency for Research on Cancer: Lyon, France, 2017; Volume 2, Available online: http://publications.iarc.fr/Book-And-Report-Series/Who-Iarc-Classification-Of-Tumours/Who-Classification-Of-Tumours-Of-Haematopoietic-And-Lymphoid-Tissues-2017 (accessed on 3 June 2019).
- Malcovati, L.; Gall, A.; Travaglino, E.; Ambaglio, I.; Rizzo, E.; Molteni, E.; Elena, C.; Ferretti, V.V.; Catrical, S.; Bono, E.; et al. Clinical significance of somatic mutation in unexplained blood cytopenia. Blood 2017, 129, 3371–3379. [Google Scholar] [CrossRef] [PubMed]
- Steensma, D.P. Dysplasia has a differential diagnosis: Distinguishing genuine myelodysplastic syndromes (MDS) from mimics, imitators, copycats and impostors. Curr. Hematol. Malig. Rep. 2012, 7, 310–320. [Google Scholar] [CrossRef]
- Valent, P.; Horny, H.-P.; Bennett, J.M.; Fonatsch, C.; Germing, U.; Greenberg, P.; Haferlach, T.; Haase, D.; Kolb, H.-J.; Krieger, O.; et al. Definitions and standards in the diagnosis and treatment of the myelodysplastic syndromes: Consensus statements and report from a working conference. Leuk. Res. 2007, 31, 727–736. [Google Scholar] [CrossRef]
- Kwok, B.; Hall, J.M.; Witte, J.S.; Xu, Y.; Reddy, P.; Lin, K.; Flamholz, R.; Dabbas, B.; Yung, A.; Al-Hafidh, J.; et al. MDS-associated somatic mutations and clonal hematopoiesis are common in idiopathic cytopenias of undetermined significance. Blood 2015, 126, 2355–2361. [Google Scholar] [CrossRef]
- Cargo, C.A.; Rowbotham, N.; Evans, P.A.; Barrans, S.L.; Bowen, D.T.; Crouch, S.; Jack, A.S. Targeted sequencing identifies patients with preclinical MDS at high risk of disease progression. Blood 2015, 126, 2362–2365. [Google Scholar] [CrossRef]
- Patel, J.; Gönen, M.; Figueroa, M.E.; Fernandez, H.; Sun, Z.; Racevskis, J.; Van Vlierberghe, P.; Dolgalev, I.; Thomas, S.; Aminova, O.; et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N. Engl. J. Med. 2012, 366, 1079–1089. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic classification and prognosis in acute myeloid leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, V.; Kohlmann, A.; Eder, C.; Haferlach, C.; Kern, W.; Cross, N.C.; Haferlach, T.; Schnittger, S. Molecular profiling of chronic myelomonocytic leukemia reveals diverse mutations in >80% of patients with TET2 and EZH2 being of high prognostic relevance. Leukemia 2011, 25, 877–879. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, I.; Elghetany, T.M. Myelodysplastic syndromes and myeloproliferative disorders. In Lanzkowsky’s Manual of Pediatric Hematology and Oncology; Academic Press: Cambridge, MA, USA, 2016; pp. 348–366. Available online: http://linkinghub.elsevier.com/retrieve/pii/B978012801368700017X (accessed on 16 March 2018).
- Chen, T.-C.; Hou, H.-A.; Chou, W.-C.; Tang, J.-L.; Kuo, Y.-Y.; Chen, C.-Y.; Tseng, M.-H.; Huang, C.-F.; Lai, Y.-J.; Chiang, Y.-C.; et al. Dynamics of ASXL1 mutation and other associated genetic alterations during disease progression in patients with primary myelodysplastic syndrome. Blood Cancer J. 2014, 4, e177. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Im, K.; Park, S.N.; Kwon, J.; Kim, J.-A.; Lee, D.S. CALR, JAK2, and MPL mutation profiles in patients with four different subtypes of myeloproliferative neoplasms: Primary myelofibrosis, essential thrombocythemia, polycythemia vera, and myeloproliferative neoplasm, unclassifiable. Am. J. Clin. Pathol. 2015, 143, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Dombret, H. Gene mutation and AML pathogenesis. Blood 2011, 118, 5366–5367. [Google Scholar] [CrossRef] [PubMed]
- Fathi, A.T.; Chen, Y.-B. Treatment of FLT3-ITD acute myeloid leukemia. Am. J. Blood Res. 2011, 1, 175–189. [Google Scholar] [PubMed]
- Quintás-Cardama, A.; Hu, C.; Qutub, A.; Qiu, Y.H.; Zhang, X.; Post, S.M.; Zhang, N.; Coombes, K.; Kornblau, S.M. p53 pathway dysfunction is highly prevalent in acute myeloid leukemia independent of TP53 mutational status. Leukemia 2017, 31, 1296–1305. [Google Scholar] [CrossRef]
- Osorio, F.G.; Rosendahl Huber, A.; Oka, R.; Verheul, M.; Patel, S.H.; Hasaart, K.; de la Fonteijne, L.; Varela, I.; Camargo, F.D.; van Boxtel, R. Somatic mutations reveal lineage relationships and age-related mutagenesis in human hematopoiesis. Cell Rep. 2018, 25, 2308–2316.e4. [Google Scholar] [CrossRef]
- Arends, C.M.; Galan-Sousa, J.; Hoyer, K.; Chan, W.; Jäger, M.; Yoshida, K.; Seemann, R.; Noerenberg, D.; Waldhueter, N.; Fleischer-Notter, H.; et al. Hematopoietic lineage distribution and evolutionary dynamics of clonal hematopoiesis. Leukemia 2018, 32, 1908–1919. [Google Scholar] [CrossRef]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef]
- Zink, F.; Stacey, S.N.; Norddahl, G.L.; Frigge, M.L.; Magnusson, O.T.; Jonsdottir, I.; Thorgeirsson, T.E.; Sigurdsson, A.; Gudjonsson, S.A.; Gudmundsson, J.; et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood 2017, 130, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, O.; Mullally, A.; Hedvat, C.; Garcia-Manero, G.; Patel, J.; Wadleigh, M.; Malinge, S.; Yao, J.; Kilpivaara, O.; Bhat, R.; et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood 2009, 114, 144–147. [Google Scholar] [CrossRef] [PubMed]
- Guillamot, M.; Cimmino, L.; Aifantis, I. The impact of DNA methylation in hematopoietic malignancies. Trends Cancer 2016, 2, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Jan, M.; Snyder, T.M.; Corces-Zimmerman, M.R.; Vyas, P.; Weissman, I.L.; Quake, S.R.; Majeti, R. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci. Transl. Med. 2012, 4, 149ra118. [Google Scholar] [CrossRef] [PubMed]
- Jeong, M.; Goodell, M.A. New answers to old questions from genome-wide maps of DNA methylation in hematopoietic cells. Exp. Hematol. 2014, 42, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Fu, R.; Yu, J.; Wu, X. DNA demethylation: Where genetics meets epigenetics. Curr. Pharm. Des. 2014, 20, 1625–1631. [Google Scholar] [CrossRef]
- Cimmino, L.; Dolgalev, I.; Wang, Y.; Yoshimi, A.; Martin, G.H.; Wang, J.; Ng, V.; Xia, B.; Witkowski, M.T.; Mitchell-Flack, M.; et al. Restoration of TET2 function blocks aberrant self-renewal and leukemia progression. Cell 2017, 170, 1079–1095.e20. [Google Scholar] [CrossRef]
- Hu, L.; Li, Z.; Cheng, J.; Rao, Q.; Gong, W.; Liu, M.; Shi, Y.G.; Zhu, J.; Wang, P.; Xu, Y. Crystal structure of TET2-DNA complex: Insight into TET-mediated 5mC oxidation. Cell 2013, 155, 1545–1555. [Google Scholar] [CrossRef]
- Langemeijer, S.M.C.; Kuiper, R.P.; Berends, M.; Knops, R.; Aslanyan, M.G.; Massop, M.; Stevens-Linders, E.; Van Hoogen, P.; Van Kessel, A.G.; Raymakers, R.A.P.; et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat. Genet. 2009, 41, 838–842. [Google Scholar] [CrossRef]
- Mesa, R.A.; Jamieson, C.; Bhatia, R.; Deininger, M.W.; Fletcher, C.D.; Gerds, A.T.; Gojo, I.; Gotlib, J.; Gundabolu, K.; Hobbs, G.; et al. NCCN guidelines insights: Myeloproliferative neoplasms, version 2.2018. J. Natl. Compr. Cancer Netw. 2017, 15, 1193–1207. [Google Scholar] [CrossRef]
- Bejar, R.; Lord, A.; Stevenson, K.; Bar-Natan, M.; Perez-Ladaga, A.; Zaneveld, J.; Wang, H.; Caughey, B.; Stojanov, P.; Getz, G.; et al. TET2 mutations predict response to hypomethylating agents in myelodysplastic syndrome patients. Blood 2014, 124, 2705–2712. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, M.M.; Lasho, T.L.; Vijayvargiya, P.; Finke, C.M.; Hanson, C.A.; Ketterling, R.P.; Gangat, N.; Tefferi, A. Prognostic interaction between ASXL1 and TET2 mutations in chronic myelomonocytic leukemia. Blood Cancer J. 2016, 6, e385. [Google Scholar] [CrossRef]
- Nazha, A.; Sekeres, M.A.; Bejar, R.; Rauh, M.J.; Othus, M.; Komrokji, R.S.; Barnard, J.; Hilton, C.B.; Kerr, C.M.; Steensma, D.P.; et al. Genomic biomarkers to predict resistance to hypomethylating agents in patients with myelodysplastic syndromes using artificial intelligence. J. Clin. Oncol. Precis. Oncol. 2019, 3, 1–11. [Google Scholar] [CrossRef]
- Sekeres, M.A.; Othus, M.; List, A.F.; Odenike, O.; Stone, R.M.; Gore, S.D.; Litzow, M.R.; Buckstein, R.; Fang, M.; Roulston, D.; et al. Randomized Phase II study of azacitidine alone or in combination with lenalidomide or with vorinostat in higher-risk myelodysplastic syndromes and chronic myelomonocytic leukemia: North American Intergroup Study SWOG S1117. J. Clin. Oncol. 2017, 35, 2745–2753. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Su, J.; Jeong, M.; Ko, M.; Huang, Y.; Park, H.J.; Guzman, A.; Lei, Y.; Huang, Y.-H.; Rao, A.; et al. DNMT3A and TET2 compete and cooperate to repress lineage-specific transcription factors in hematopoietic stem cells. Nat. Genet. 2016, 48, 1014–1023. [Google Scholar] [CrossRef]
- Abelson, S.; Collord, G.; Ng, S.W.K.; Weissbrod, O.; Mendelson Cohen, N.; Niemeyer, E.; Barda, N.; Zuzarte, P.C.; Heisler, L.; Sundaravadanam, Y.; et al. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature 2018, 559, 400–404. [Google Scholar] [CrossRef]
- Heuser, M.; Thol, F.; Ganser, A. Clonal hematopoiesis of indeterminate potential. Dtsch. Arztebl. Int. 2016, 113, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.X.; Riggs, A.D. DNA methylation and demethylation in mammal. J. Biol. Chem. 2011, 286, 18347–18353. [Google Scholar] [CrossRef]
- Yang, L.; Rau, R.; Goodell, M.A. DNMT3A in haematological malignancies. Nat. Rev. Cancer 2015, 15, 152–165. [Google Scholar] [CrossRef]
- Traina, F.; Visconte, V.; Elson, P.; Tabarroki, A.; Jankowska, A.M.; Hasrouni, E.; Sugimoto, Y.; Szpurka, H.; Makishima, H.; O’Keefe, C.L.; et al. Impact of molecular mutations on treatment response to DNMT inhibitors in myelodysplasia and related neoplasms. Leukemia 2014, 28, 78–87. [Google Scholar] [CrossRef]
- Martín, I.; Such, E.; Navarro, B.; Villamón, E.; Vicente, A.; Mora, E.; Pedrola, L.; Ibáñez, M.; López-Pavía, M.; Tormo, M.; et al. Prognostic impact of gene mutations in myelodysplastic syndromes with ring sideroblasts. Blood Cancer J. 2017, 7, 630. [Google Scholar] [CrossRef]
- Martín, I.; Such, E.; Navarro, B.; Vicente, A.; López-Pavía, M.; Ibáñez, M.; Tormo, M.; Villamón, E.; Gómez-Seguí, I.; Luna, I.; et al. Negative impact on clinical outcome of the mutational co-occurrence of SF3B1 and DNMT3A in refractory anemia with ring sideroblasts (RARS). Leuk. Lymphoma 2017, 58, 1686–1693. [Google Scholar] [CrossRef]
- Lin, M.E.; Hou, H.A.; Tsai, C.H.; Wu, S.J.; Kuo, Y.Y.; Tseng, M.H.; Liu, M.C.; Liu, C.W.; Chou, W.C.; Chen, C.Y.; et al. Dynamics of DNMT3A mutation and prognostic relevance in patients with primary myelodysplastic syndrome. Clin. Epigenet. 2018, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Cook, E.K.; Izukawa, T.; Young, S.; Rosen, G.; Jamali, M.; Zhang, L.; Johnson, D.; Bain, E.; Hilland, J.; Ferrone, C.K.; et al. Comorbid and inflammatory characteristics of genetic subtypes of clonal hematopoiesis. Blood Adv. 2019, 3, 2482–2486. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Dorsheimer, L.; Assmus, B.; Rasper, T.; Ortmann, C.A.; Abou-El-Ardat, K.; Kiefer, K.C.; Hoffmann, J.; Seeger, F.; Bonig, H.; Dimmeler, S.; et al. Association of mutations contributing to clonal hematopoiesis with prognosis in chronic ischemic Heart Failure. JAMA Cardiol. 2019, 4, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Mas-Peiro, S.; Hoffmann, J.; Fichtlscherer, S.; Dorsheimer, L.; Rieger, M.A.; Dimmeler, S.; Vasa-Nicotera, M.; Zeiher, A.M. Clonal haematopoiesis in patients with degenerative aortic valve stenosis undergoing transcatheter aortic valve implantation. Eur. Heart J. 2019, 1–7. [Google Scholar] [CrossRef]
- Sano, S.; Oshima, K.; Wang, Y.; MacLauchlan, S.; Katanasaka, Y.; Sano, M.; Zuriaga, M.A.; Yoshiyama, M.; Goukassian, D.; Cooper, M.A.; et al. Tet2-mediated clonal hematopoiesis accelerates heart failure through a mechanism involving the IL-1B/NLRP3 inflammasome. J. Am. Coll. Cardiol. 2018, 71, 875–886. [Google Scholar] [CrossRef]
- Fuster, J.; MacLauchlan, S.; Zuriaga, M.; Polackal, M.N.; Ostriker, A.C.; Chakraborty, R.; Wu, C.L.; Sano, S.; Muralidharan, S.; Rius, C.; et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 2017, 355, 842–847. [Google Scholar] [CrossRef]
- Schaub, F.X.; Looser, R.; Li, S.; Hao-Shen, H.; Lehmann, T.; Tichelli, A.; Skoda, R.C. Clonal analysis of TET2 and JAK2 mutations suggests that TET2 can be a late event in the progression of myeloproliferative neoplasms. Blood 2010, 115, 2003–2007. [Google Scholar] [CrossRef]
- Kaasinen, E.; Kuismin, O.; Rajamäki, K.; Ristolainen, H.; Aavikko, M.; Kondelin, J.; Saarinen, S.; Berta, D.G.; Katainen, R.; Hirvonen, E.A.M.; et al. Impact of constitutional TET2 haploinsufficiency on molecular and clinical phenotype in humans. Nat. Commun. 2019, 10, 1252. [Google Scholar] [CrossRef] [PubMed]
- Duployez, N.; Goursaud, L.; Fenwarth, L.; Bories, C.; Marceau-Renaut, A.; Boyer, T.; Fournier, E.; Nibourel, O.; Roche-Lestienne, C.; Huet, G.; et al. Familial myeloid malignancies with germline TET2 mutation. Leukemia 2019. [Google Scholar] [CrossRef]
- Buscarlet, M.; Provost, S.; Zada, Y.F.; Barhdadi, A.; Bourgoin, V.; Lépine, G.; Mollica, L.; Szuber, N.; Dubé, M.P.; Busque, L. DNMT3A and TET2 dominate clonal hematopoiesis and demonstrate benign phenotypes and different genetic predispositions. Blood 2017, 130, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhao, K.; Shen, Q.; Han, Y.; Gu, Y.; Li, X.; Zhao, D.; Liu, Y.; Wang, C.; Zhang, X.; et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 2015, 525, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Cull, A.H.; Snetsinger, B.; Buckstein, R.; Wells, R.A.; Rauh, M.J. Tet2 restrains inflammatory gene expression in macrophages. Exp. Hematol. 2017, 55, 56–70. [Google Scholar] [CrossRef]
- Ichiyama, K.; Chen, T.; Wang, X.; Yan, X.; Kim, B.S.; Tanaka, S.; Ndiaye-Lobry, D.; Deng, Y.; Zou, Y.; Zheng, P.; et al. The methylcytosine dioxygenase Tet2 promotes DNA demethylation and activation of cytokine gene expression in T cells. Immunity 2015, 42, 613–626. [Google Scholar] [CrossRef]
- Abegunde, S.O.; Buckstein, R.; Wells, R.A.; Rauh, M.J. An inflammatory environment containing TNFα favors Tet2-mutant clonal hematopoiesis. Exp. Hematol. 2018, 59, 60–65. [Google Scholar] [CrossRef]
- Cai, Z.; Kotzin, J.J.; Ramdas, B.; Chen, S.; Nelanuthala, S.; Palam, L.R.; Pandey, R.; Mali, R.S.; Liu, Y.; Kelley, M.R.; et al. Inhibition of inflammatory signaling in Tet2 mutant preleukemic cells mitigates stress-induced abnormalities and clonal hematopoiesis. Cell Stem Cell 2018, 23, 833–849. [Google Scholar] [CrossRef]
- Meisel, M.; Hinterleitner, R.; Pacis, A.; Chen, L.; Earley, Z.M.; Mayassi, T.; Pierre, J.F.; Ernest, J.D.; Galipeau, H.J.; Thuille, N.; et al. Microbial signals drive pre-leukaemic myeloproliferation in a Tet2-deficient host. Nature 2018, 557, 580–584. [Google Scholar] [CrossRef]
- Cull, A.H.; Rauh, M.J. Success in bone marrow failure? Novel therapeutic directions based on the immune environment of myelodysplastic syndromes. J. Leukoc. Biol. 2017, 102, 209–219. [Google Scholar] [CrossRef]
- Heaney, M.L.; Golde, D.W. Myelodysplasia. N. Engl. J. Med. 1999, 340, 1649–1660. [Google Scholar] [CrossRef]
- Flores-Figueroa, E.; Gutiérrez-Espíndola, G.; Montesinos, J.J.; Arana-Trejo, R.M.; Mayani, H. In vitro characterization of hematopoietic microenvironment cells from patients with myelodysplastic syndrome. Leuk. Res. 2002, 26, 677–686. [Google Scholar] [CrossRef]
- Kitagawa, M.; Saito, I.; Kuwata, T.; Yoshida, S.; Yamaguchi, S.; Takahashi, M.; Tanizawa, T.; Kamiyama, R.; Hirokawa, K. Overexpression of tumor necrosis factor (TNF)-α and interferon (IFN)-γ by bone marrow cells from patients with myelodysplastic syndromes. Leukemia 1997, 11, 2049–2054. [Google Scholar] [CrossRef] [PubMed]
- Pang, W.W.; Pluvinage, J.V.; Price, E.A.; Sridhar, K.S.; Arber, D.A.; Greenberg, P.L.; Schrier, S.L.; Park, C.Y. Hematopoietic stem cell and progenitor cell mechanisms in myelodysplastic syndromes. Proc. Natl. Acad. Sci. USA 2013, 110, 3011–3016. [Google Scholar] [CrossRef] [PubMed]
- Basiorka, A.A.; Mcgraw, K.L.; Eksioglu, E.A.; Chen, X.; Johnson, J.; Zhang, L.; Zhang, Q.; Irvine, B.A.; Cluzeau, T.; Sallman, D.A.; et al. The NLRP3 inflammasome functions as a driver of the myelodysplastic syndrome phenotype. Blood 2016, 128, 2960–2975. [Google Scholar] [CrossRef]
- Yang, L.; Qian, Y.; Eksioglu, E.; Epling-Burnette, P.K.; Wei, S. The inflammatory microenvironment in MDS. Cell Mol. Life Sci. 2015, 72, 1959–1966. [Google Scholar] [CrossRef]
- Zambetti, N.A.; Ping, Z.; Chen, S.; Kenswil, K.J.G.; Mylona, M.A.; Sanders, M.A.; Hoogenboezem, R.M.; Bindels, E.M.J.; Adisty, M.N.; Van Strien, P.M.H.; et al. Mesenchymal inflammation drives genotoxic stress in hematopoietic stem cells and predicts disease evolution in human pre-leukemia. Cell Stem Cell 2016, 19, 613–627. [Google Scholar] [CrossRef] [PubMed]
- Gañán-Gómez, I.; Wei, Y.; Starczynowski, D.T.; Colla, S.; Yang, H.; Cabrero-Calvo, M.; Bohannan, Z.S.; Verma, A.; Steidl, U.; Garcia-Manero, G. Deregulation of innate immune and inflammatory signaling in myelodysplastic syndromes. Leukemia 2015, 29, 1458–1469. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; Kastelein, J.J.P.; et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Ridker, P.M.; Macfadyen, J.G.; Thuren, T.; Libby, P. Residual inflammatory risk associated with interleukin-18 and interleukin-6 after successful interleukin-1B inhibition with canakinumab: Further rationale for the development of targeted anti-cytokine therapies for the treatment of atherothrombosis. Eur. Heart J. 2019, 1–11. [Google Scholar] [CrossRef]
- Bick, A.G.; Pirruccello, J.P.; Griffin, G.K.; Gupta, N.; Gabriel, S.; Saleheen, D.; Libby, P.; Kathiresan, S.; Natarajan, P. Genetic IL-6 signaling deficiency attenuates cardiovascular risk in clonal hematopoiesis. Circulation 2019. [Google Scholar] [CrossRef]
- Van den Akker, E.B.; Pitts, S.J.; Deelen, J.; Moed, M.H.; Potluri, S.; van Rooij, J.; Suchiman, H.E.; Lakenberg, N.; de Dijcker, W.J.; Uitterlinden, A.G.; et al. Uncompromised 10-year survival of oldest old carrying somatic mutations in DNMT3A and TET2. Blood 2016, 127, 1512–1515. [Google Scholar] [CrossRef] [PubMed]
- Desai, P.; Mencia-trinchant, N.; Savenkov, O.; Simon, M.S.; Cheang, G.; Lee, S.; Samuel, M.; Ritchie, E.K.; Guzman, M.L.; Ballman, K.V.; et al. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat. Med. 2018, 24, 1015–1023. [Google Scholar] [CrossRef] [PubMed]
- Blaschke, K.; Ebata, K.T.; Karimi, M.M.; Zepeda-Martínez, J.A.; Goyal, P.; Mahapatra, S.; Tam, A.; Laird, D.J.; Hirst, M.; Rao, A.; et al. Vitamin C induces TET-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature 2013, 500, 222–226. [Google Scholar] [CrossRef]
- Fritz, H.; Flower, G.; Weeks, L.; Cooley, K.; Callachan, M.; McGowan, J.; Skidmore, B.; Kirchner, L.; Seely, D. Intravenous vitamin C and cancer. Integr. Cancer Ther. 2014, 13, 280–300. [Google Scholar] [CrossRef]
- Young, J.I.; Züchner, S.; Wang, G. Regulation of the epigenome by vitamin C. Annu. Rev. Nutr. 2015, 35, 545–564. [Google Scholar] [CrossRef]
- Agathocleous, M.; Meacham, C.E.; Burgess, R.J.; Piskounova, E.; Zhao, Z.; Crane, G.M.; Cowin, B.L.; Bruner, E.; Murphy, M.M.; Chen, W.; et al. Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature 2017, 549, 476–481. [Google Scholar] [CrossRef]
- Zhao, H.; Zhu, H.; Huang, J.; Zhu, Y.; Hong, M.; Zhu, H.; Zhang, J.; Li, S.; Yang, L.; Lian, Y.; et al. The synergy of Vitamin C with decitabine activates TET2 in leukemic cells and significantly improves overall survival in elderly patients with acute myeloid leukemia. Leuk. Res. 2018, 66, 1–7. [Google Scholar] [CrossRef]
- Das, A.B.; Kakadia, P.M.; Wojcik, D.; Pemberton, L.; Browett, P.J.; Bohlander, S.K.; Vissers, M.C.M. Clinical remission following ascorbate treatment in a case of acute myeloid leukemia with mutations in TET2 and WT1. Blood Cancer J. 2019, 9, 82. [Google Scholar] [CrossRef]
- Rau, R.E.; Rodriguez, B.A.; Luo, M.; Jeong, M.; Rosen, A.; Rogers, J.H.; Campbell, C.T.; Daigle, S.R.; Deng, L.; Song, Y.; et al. DOT1L as a therapeutic target for the treatment of DNMT3A-mutant acute myeloid leukemia. Blood 2016, 128, 971–981. [Google Scholar] [CrossRef]
- Adema, V.; Balasubramanian, S.K.; Hirsch, C.M.; Przychodzen, B.P.; Phillips, J.G.; Lindner, D.; Radivoyevitch, T.; Mukherjee, S.; Nazha, A.; Carraway, H.E.; et al. Novel therapeutic targets for DNMT3A mutant myeloid neoplasms. Blood 2017, 130, 106. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferrone, C.K.; Blydt-Hansen, M.; Rauh, M.J. Age-Associated TET2 Mutations: Common Drivers of Myeloid Dysfunction, Cancer and Cardiovascular Disease. Int. J. Mol. Sci. 2020, 21, 626. https://doi.org/10.3390/ijms21020626
Ferrone CK, Blydt-Hansen M, Rauh MJ. Age-Associated TET2 Mutations: Common Drivers of Myeloid Dysfunction, Cancer and Cardiovascular Disease. International Journal of Molecular Sciences. 2020; 21(2):626. https://doi.org/10.3390/ijms21020626
Chicago/Turabian StyleFerrone, Christina K., Mackenzie Blydt-Hansen, and Michael J. Rauh. 2020. "Age-Associated TET2 Mutations: Common Drivers of Myeloid Dysfunction, Cancer and Cardiovascular Disease" International Journal of Molecular Sciences 21, no. 2: 626. https://doi.org/10.3390/ijms21020626
APA StyleFerrone, C. K., Blydt-Hansen, M., & Rauh, M. J. (2020). Age-Associated TET2 Mutations: Common Drivers of Myeloid Dysfunction, Cancer and Cardiovascular Disease. International Journal of Molecular Sciences, 21(2), 626. https://doi.org/10.3390/ijms21020626