TGF-β/Smad3 Signalling Modulates GABA Neurotransmission: Implications in Parkinson’s Disease


Abstract
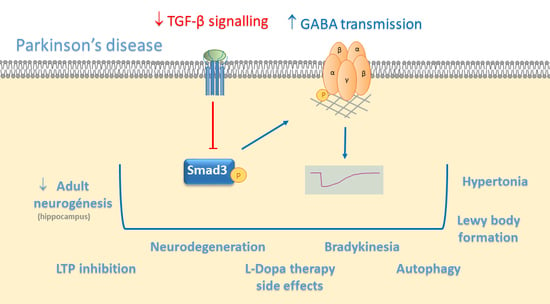
{kind=link}
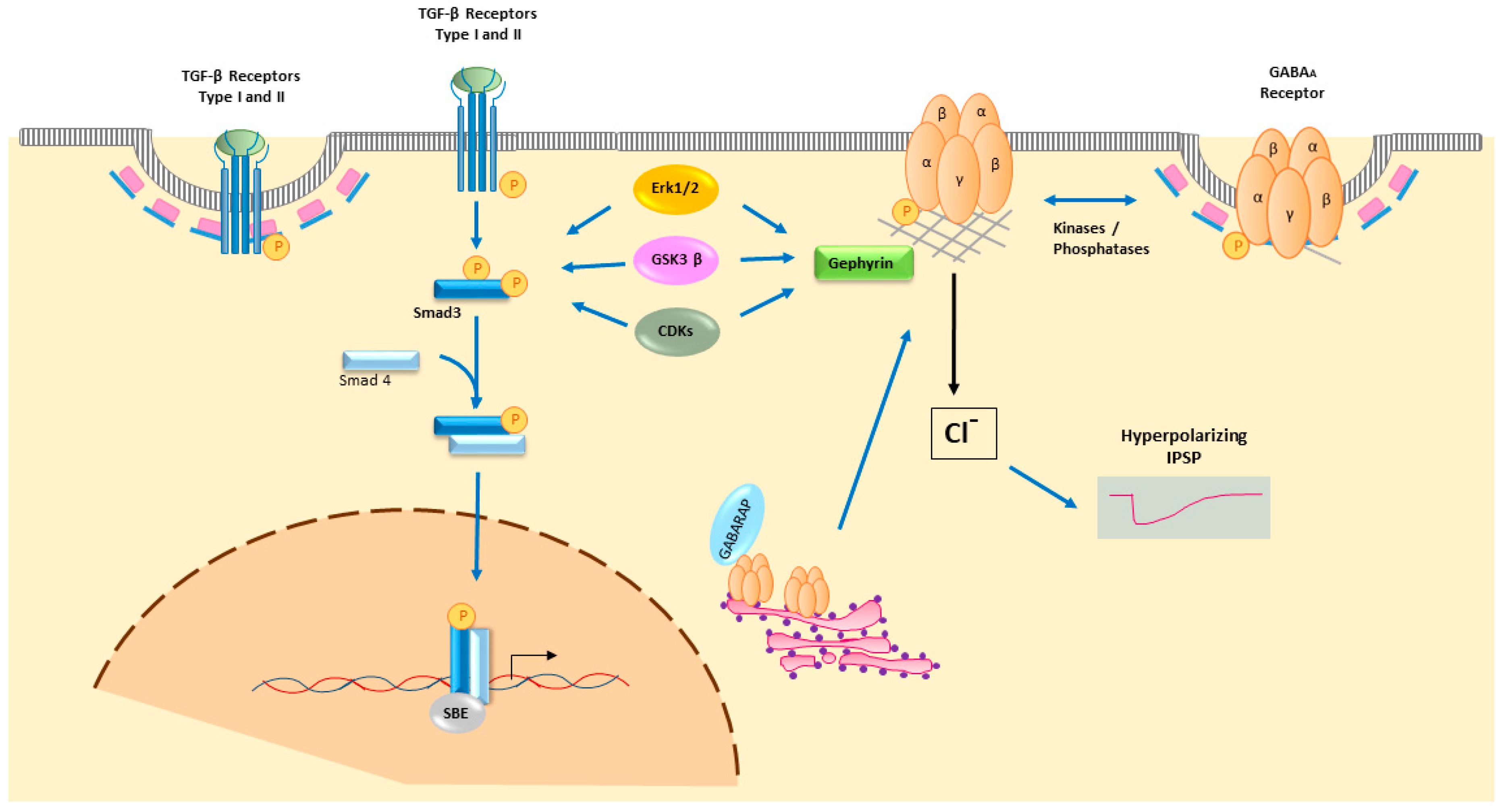{kind=link}
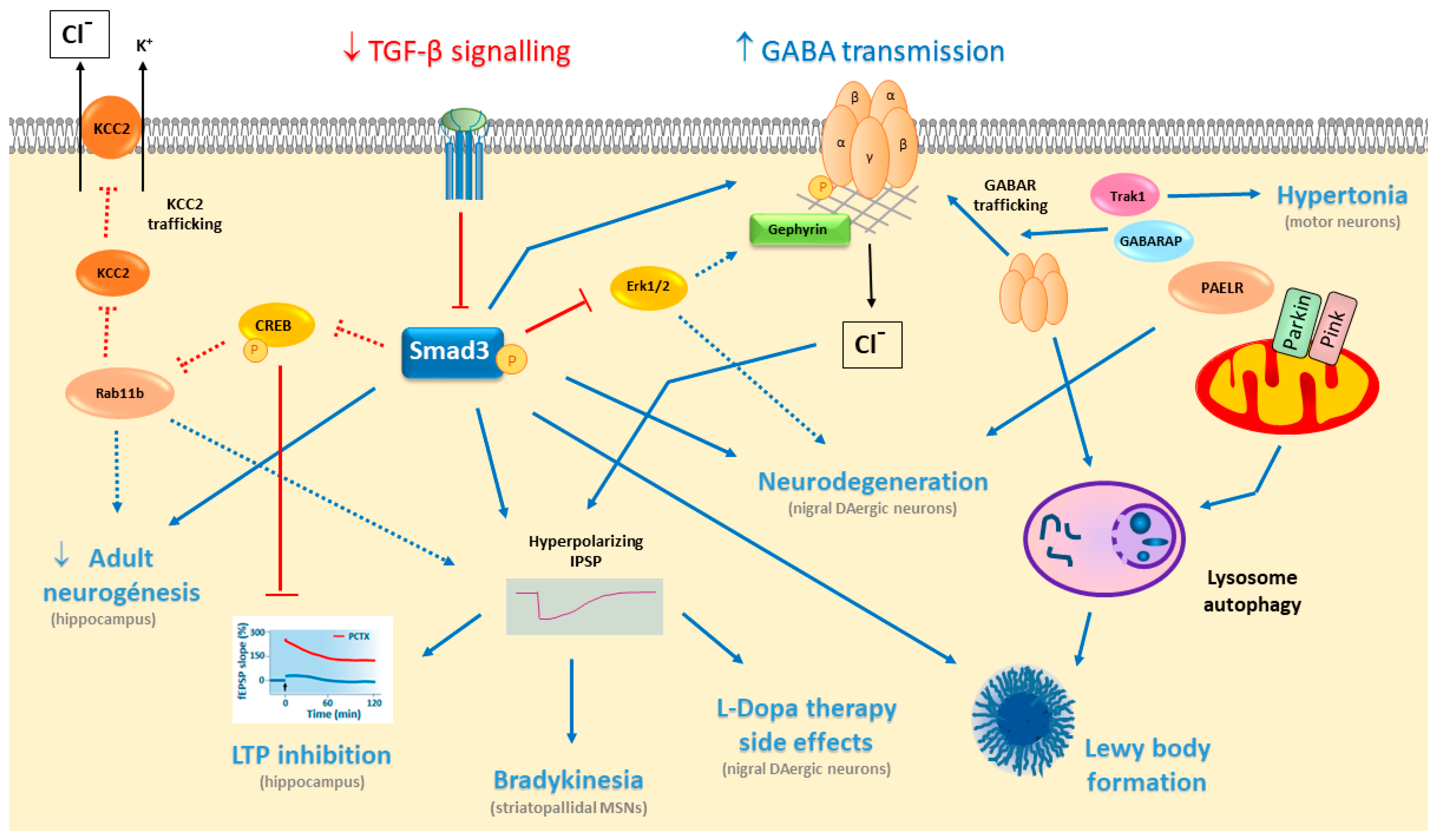{kind=link}
1. Introduction
2. GABA Neurotransmission
2.1. GABA Biosynthesis and Turnover
2.2. Synaptic and Non-Synaptic GABA Neurotransmission
2.3. Postsynaptic Activation by GABARs
3. GABA in PD
3.1. Striatal GABA Neurotransmission
3.2. Striatal GABA Neurotransmission in PD
3.3. Nigral GABA Neurotransmission in PD
3.4. Dysfunctional GABAAR Trafficking in PD
4. TGF-β Signalling
4.1. Secreted TGF-β Ligands
4.2. TGF-β Receptor Activation
4.3. Smad2/3 Intracellular TGF-β Signalling
5. TGF-β/Smad3 in PD
5.1. Deficient TGF-β/Smad3 Signalling in Parkinsonism
5.2. TGF-β/Smad3 Signalling in Cognition
6. TGF-β Signalling Modulates GABA Neurotransmission
7. Treatment of PD With GABA Modulators
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| ATG | Autophagy-related gene |
| ATP | Adenosine triphosphate |
| BGT1 | Betaine-GABA transporter |
| CNS | Central Nervous System |
| COMT | Catechol-O-methyltransferase |
| CREB | cAMP response element-binding protein |
| DA | Dopamine |
| DG | Dentate gyrus |
| DLB | Dementia with Lewy bodies |
| ECM | Extracellular matrix |
| GABA | γ-Amino-butyric acid |
| GABARs | GABA receptors |
| GABAAR | GABAA receptor |
| GABABR | GABAB receptor |
| GABARAP | GABA receptor-associated protein |
| GAD | Glutamic acid decarboxylase |
| GAT | GABA transporter |
| GP | Globus pallidus |
| GPe | external globus pallidus |
| GPi | internal globus pallidus |
| GODZ | Golgi-specific DHHC (Asp-His-His-Cys) zinc finger protein |
| IPSP | Inhibitory postsynaptic potential |
| LAP | Latent associated protein |
| LTP | Long term potentiation |
| MAO | Monoamine oxidase |
| miRs | microRNAs |
| MSNs | Medium-sized spiny neurons |
| NAcc | Nucleus accumbens |
| NO | Nitric Oxide |
| NOS | Nitric oxide synthase |
| PAELR | Parkin-associated endothelin-like receptor |
| PD | Parkinson’s disease |
| PSD | Postsynaptic density |
| SARA | Smad anchor for receptor activation |
| SN | Substantia nigra |
| SNpr | Substantia nigra pars reticulata |
| SNpc | Substantia nigra pars compacta |
| ST | Striatum |
| STN | Subthalamic nucleus |
| TCA | Tricarboxylic acid |
| TGF-β | Transforming Growth Factor β |
| Trak1 | Trafficking kinesin binding 1 protein |
| VGAT | vesicular GABA transporter |
| VTA | ventral tegmental area |
References
- Siucinska, E. Gamma-Aminobutyric Acid in Adult Brain: An Updated. Behav. Brain Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Chagnac-Amitai, Y.; Connors, B.W. Horizontal spread of synchronized activity in neocortex and its control by GABA-mediated inhibition. J. Neurophysiol. 1989, 61, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Swanson, O.K.; Maffei, A. From Hiring to Firing: Activation of Inhibitory Neurons and Their Recruitment in Behavior. Front. Mol. Neurosci. 2019, 12, 168. [Google Scholar] [CrossRef] [PubMed]
- Giraldez-Perez, R.; Antolin-Vallespin, M.; Muñoz, M.; Sanchez-Capelo, A. Models of alpha-synuclein aggregation in Parkinson’s disease. Acta Neuropathol. Commun. 2014, 2, 176. [Google Scholar] [CrossRef] [PubMed]
- Schousboe, A.; Bak, L.K.; Waagepetersen, H.S. Astrocytic Control of Biosynthesis and Turnover of the Neurotransmitters Glutamate and GABA. Front. Endocrinol. 2013, 4, 102. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Wu, J.Y. Post-translational regulation of L-glutamic acid decarboxylase in the brain. Neurochem. Res. 2008, 33, 1459–1465. [Google Scholar] [CrossRef]
- Madsen, K.K.; White, H.S.; Clausen, R.P.; Frolund, B.; Larsson, O.M.; Krogsgaard-Larsen, P.; Schousboe, A. Functional and pharmacological aspects of GABA transporters. In Beural Membranes and Transport; Reith, M.E.A., Ed.; Springer: New York, NY, USA, 2007; pp. 285–304. [Google Scholar]
- Rowley, N.M.; Madsen, K.K.; Schousboe, A.; Steve White, H. Glutamate and GABA synthesis, release, transport and metabolism as targets for seizure control. Neurochem. Int. 2012, 61, 546–558. [Google Scholar] [CrossRef]
- Farrant, M.; Nusser, Z. Variations on an inhibitory theme: Phasic and tonic activation of GABA(A) receptors. Nat. Rev. Neurosci. 2005, 6, 215–229. [Google Scholar] [CrossRef]
- Choquet, D.; Triller, A. The dynamic synapse. Neuron 2013, 80, 691–703. [Google Scholar] [CrossRef]
- Hannan, S.; Minere, M.; Harris, J.; Izquierdo, P.; Thomas, P.; Tench, B.; Smart, T.G. GABAAR isoform and subunit structural motifs determine synaptic and extrasynaptic receptor localisation. Neuropharmacol 2019. [Google Scholar] [CrossRef]
- Rudolph, U.; Knoflach, F. Beyond classical benzodiazepines: Novel therapeutic potential of GABAA receptor subtypes. Nat. Rev. Drug Discov. 2011, 10, 685–697. [Google Scholar] [CrossRef] [PubMed]
- Waldvogel, H.J.; Faull, R.L. The diversity of GABA(A) receptor subunit distribution in the normal and Huntington’s disease human brain. Adv. Pharmacol. 2015, 73, 223–264. [Google Scholar] [PubMed]
- Padgett, C.L.; Slesinger, P.A. GABAB receptor coupling to G-proteins and ion channels. Adv. Pharmacol. 2010, 58, 123–147. [Google Scholar] [PubMed]
- Sodickson, D.L.; Bean, B.P. GABAB receptor-activated inwardly rectifying potassium current in dissociated hippocampal CA3 neurons. J. Neurosci. 1996, 16, 6374–6385. [Google Scholar] [CrossRef]
- Chiu, C.Q.; Barberis, A.; Higley, M.J. Preserving the balance: Diverse forms of long-term GABAergic synaptic plasticity. Nat. Rev. Neurosci. 2019, 20, 272–281. [Google Scholar] [CrossRef]
- Mele, M.; Costa, R.O.; Duarte, C.B. Alterations in GABAA-Receptor Trafficking and Synaptic Dysfunction in Brain Disorders. Front. Cell. Neurosci. 2019, 13, 77. [Google Scholar] [CrossRef]
- Nakamura, Y.; Darnieder, L.M.; Deeb, T.Z.; Moss, S.J. Regulation of GABAARs by phosphorylation. Adv. Pharmacol. 2015, 72, 97–146. [Google Scholar]
- Chen, Z.W.; Olsen, R.W. GABAA receptor associated proteins: A key factor regulating GABAA receptor function. J. Neurochem. 2007, 100, 279–294. [Google Scholar] [CrossRef]
- Kanematsu, T.; Mizokami, A.; Watanabe, K.; Hirata, M. Regulation of GABA(A)-receptor surface expression with special reference to the involvement of GABARAP (GABA(A) receptor-associated protein) and PRIP (phospholipase C-related, but catalytically inactive protein). J. Pharmacol. Sci. 2007, 104, 285–292. [Google Scholar] [CrossRef]
- Kittler, J.T.; Moss, S.J. Modulation of GABAA receptor activity by phosphorylation and receptor trafficking: Implications for the efficacy of synaptic inhibition. Curr. Opin. Neurobiol. 2003, 13, 341–347. [Google Scholar] [CrossRef]
- Kittler, J.T.; Thomas, P.; Tretter, V.; Bogdanov, Y.D.; Haucke, V.; Smart, T.G.; Moss, S.J. Huntingtin-associated protein 1 regulates inhibitory synaptic transmission by modulating gamma-aminobutyric acid type A receptor membrane trafficking. Proc. Natl. Acad. Sci. USA 2004, 101, 12736–12741. [Google Scholar] [CrossRef] [PubMed]
- Khayenko, V.; Maric, H.M. Targeting GABAAR-Associated Proteins: New Modulators, Labels and Concepts. Front. Mol. Neurosci. 2019, 12, 162. [Google Scholar] [CrossRef] [PubMed]
- Pizzarelli, R.; Griguoli, M.; Zacchi, P.; Petrini, E.M.; Barberis, A.; Cattaneo, A.; Cherubini, E. Tuning GABAergic Inhibition: Gephyrin Molecular Organization and Functions. Neuroscience 2019. [Google Scholar] [CrossRef] [PubMed]
- Tyagarajan, S.K.; Fritschy, J.M. Gephyrin: A master regulator of neuronal function? Nat. Rev. Neurosci. 2014, 15, 141–156. [Google Scholar] [CrossRef]
- Loebrich, S.; Bahring, R.; Katsuno, T.; Tsukita, S.; Kneussel, M. Activated radixin is essential for GABAA receptor alpha5 subunit anchoring at the actin cytoskeleton. EMBO J. 2006, 25, 987–999. [Google Scholar] [CrossRef]
- Hausrat, T.J.; Muhia, M.; Gerrow, K.; Thomas, P.; Hirdes, W.; Tsukita, S.; Heisler, F.F.; Herich, L.; Dubroqua, S.; Breiden, P.; et al. Radixin regulates synaptic GABAA receptor density and is essential for reversal learning and short-term memory. Nat. Commun. 2015, 6, 6872. [Google Scholar] [CrossRef]
- Lanciego, J.L.; Luquin, N.; Obeso, J.A. Functional neuroanatomy of the basal ganglia. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef]
- Burke, D.A.; Rotstein, H.G.; Alvarez, V.A. Striatal Local Circuitry: A New Framework for Lateral Inhibition. Neuron 2017, 96, 267–284. [Google Scholar] [CrossRef]
- Gerfen, C.R.; Bolam, J.P. The neuroanatomical organization of the basal ganglia. In Handbook of Basal Ganglia Structure and Function, 2nd ed.; Heinz, S., Kuei, T., Eds.; Academic: London, UK, 2016; pp. 3–30. [Google Scholar]
- Gagnon, D.; Petryszyn, S.; Sanchez, M.G.; Bories, C.; Beaulieu, J.M.; De Koninck, Y.; Parent, A.; Parent, M. Striatal Neurons Expressing D1 and D2 Receptors are Morphologically Distinct and Differently Affected by Dopamine Denervation in Mice. Sci. Rep. 2017, 7, 41432. [Google Scholar] [CrossRef]
- Tecuapetla, F.; Jin, X.; Lima, S.Q.; Costa, R.M. Complementary Contributions of Striatal Projection Pathways to Action Initiation and Execution. Cell 2016, 166, 703–715. [Google Scholar] [CrossRef]
- Ding, J.; Peterson, J.D.; Surmeier, D.J. Corticostriatal and thalamostriatal synapses have distinctive properties. J. Neurosci. 2008, 28, 6483–6492. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.B.; Guzman, J.N.; Peterson, J.D.; Goldberg, J.A.; Surmeier, D.J. Thalamic gating of corticostriatal signaling by cholinergic interneurons. Neuron 2010, 67, 294–307. [Google Scholar] [CrossRef] [PubMed]
- Melzer, S.; Gil, M.; Koser, D.E.; Michael, M.; Huang, K.W.; Monyer, H. Distinct Corticostriatal GABAergic Neurons Modulate Striatal Output Neurons and Motor Activity. Cell Rep. 2017, 19, 1045–1055. [Google Scholar] [PubMed]
- Kim, J.I.; Ganesan, S.; Luo, S.X.; Wu, Y.W.; Park, E.; Huang, E.J.; Chen, L.; Ding, J.B. Aldehyde dehydrogenase 1a1 mediates a GABA synthesis pathway in midbrain dopaminergic neurons. Science 2015, 350, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Tritsch, N.X.; Oh, W.J.; Gu, C.; Sabatini, B.L. Midbrain dopamine neurons sustain inhibitory transmission using plasma membrane uptake of GABA, not synthesis. eLife 2014, 3. [Google Scholar] [CrossRef]
- Lopes, E.F.; Roberts, B.M.; Siddorn, R.E.; Clements, M.A.; Cragg, S.J. Inhibition of Nigrostriatal Dopamine Release by Striatal GABAA and GABAB Receptors. J. Neurosci. 2019, 39, 1058–1065. [Google Scholar] [CrossRef] [PubMed]
- Tomioka, R.; Sakimura, K.; Yanagawa, Y. Corticofugal GABAergic projection neurons in the mouse frontal cortex. Front. Neuroanat. 2015, 9, 133. [Google Scholar] [CrossRef]
- Mallet, N.; Micklem, B.R.; Henny, P.; Brown, M.T.; Williams, C.; Bolam, J.P.; Nakamura, K.C.; Magill, P.J. Dichotomous organization of the external globus pallidus. Neuron 2012, 74, 1075–1086. [Google Scholar] [CrossRef]
- Smith, J.B.; Klug, J.R.; Ross, D.L.; Howard, C.D.; Hollon, N.G.; Ko, V.I.; Hoffman, H.; Callaway, E.M.; Gerfen, C.R.; Jin, X. Genetic-Based Dissection Unveils the Inputs and Outputs of Striatal Patch and Matrix Compartments. Neuron 2016, 91, 1069–1084. [Google Scholar] [CrossRef]
- Wilson, C.J. GABAergic inhibition in the neostriatum. Prog. Brain Res. 2007, 160, 91–110. [Google Scholar]
- Yung, K.K.; Smith, A.D.; Levey, A.I.; Bolam, J.P. Synaptic connections between spiny neurons of the direct and indirect pathways in the neostriatum of the rat: Evidence from dopamine receptor and neuropeptide immunostaining. Eur. J. Neurosci. 1996, 8, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Sagi, Y.; Heiman, M.; Peterson, J.D.; Musatov, S.; Scarduzio, M.; Logan, S.M.; Kaplitt, M.G.; Surmeier, D.J.; Heintz, N.; Greengard, P. Nitric oxide regulates synaptic transmission between spiny projection neurons. Proc. Natl. Acad. Sci. USA 2014, 111, 17636–17641. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Ding, S.; Zhou, F.M. Dopaminergic treatment weakens medium spiny neuron collateral inhibition in the parkinsonian striatum. J. Neurophysiol. 2017, 117, 987–999. [Google Scholar] [CrossRef] [PubMed]
- Xenias, H.S.; Ibanez-Sandoval, O.; Koos, T.; Tepper, J.M. Are striatal tyrosine hydroxylase interneurons dopaminergic? J. Neurosci. 2015, 35, 6584–6599. [Google Scholar] [CrossRef]
- Vernier, P.; Julien, J.F.; Rataboul, P.; Fourrier, O.; Feuerstein, C.; Mallet, J. Similar time course changes in striatal levels of glutamic acid decarboxylase and proenkephalin mRNA following dopaminergic deafferentation in the rat. J. Neurochem. 1988, 51, 1375–1380. [Google Scholar] [CrossRef] [PubMed]
- Soghomonian, J.J.; Laprade, N. Glutamate decarboxylase (GAD67 and GAD65) gene expression is increased in a subpopulation of neurons in the putamen of Parkinsonian monkeys. Synapse 1997, 27, 122–132. [Google Scholar] [CrossRef]
- Lemos, J.C.; Friend, D.M.; Kaplan, A.R.; Shin, J.H.; Rubinstein, M.; Kravitz, A.V.; Alvarez, V.A. Enhanced GABA Transmission Drives Bradykinesia Following Loss of Dopamine D2 Receptor Signaling. Neuron 2016, 90, 824–838. [Google Scholar] [CrossRef]
- Emir, U.E.; Tuite, P.J.; Oz, G. Elevated pontine and putamenal GABA levels in mild-moderate Parkinson disease detected by 7 tesla proton MRS. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Kish, S.J.; Rajput, A.; Gilbert, J.; Rozdilsky, B.; Chang, L.J.; Shannak, K.; Hornykiewicz, O. Elevated gamma-aminobutyric acid level in striatal but not extrastriatal brain regions in Parkinson’s disease: Correlation with striatal dopamine loss. Ann. Neurol. 1986, 20, 26–31. [Google Scholar] [CrossRef]
- Dydak, U.; Jiang, Y.M.; Long, L.L.; Zhu, H.; Chen, J.; Li, W.M.; Edden, R.A.; Hu, S.; Fu, X.; Long, Z.; et al. In vivo measurement of brain GABA concentrations by magnetic resonance spectroscopy in smelters occupationally exposed to manganese. Environ. Health Perspect. 2011, 119, 219–224. [Google Scholar] [CrossRef]
- Tanaka, Y.; Niijima, K.; Mizuno, Y.; Yoshida, M. Changes in gamma-aminobutyrate, glutamate, aspartate, glycine, and taurine contents in the striatum after unilateral nigrostriatal lesions in rats. Exp. Neurol. 1986, 91, 259–268. [Google Scholar] [CrossRef]
- Griffiths, P.D.; Sambrook, M.A.; Perry, R.; Crossman, A.R. Changes in benzodiazepine and acetylcholine receptors in the globus pallidus in Parkinson’s disease. J. Neurol. Sci. 1990, 100, 131–136. [Google Scholar] [CrossRef]
- Robertson, R.G.; Clarke, C.A.; Boyce, S.; Sambrook, M.A.; Crossman, A.R. The role of striatopallidal neurones utilizing gamma-aminobutyric acid in the pathophysiology of MPTP-induced parkinsonism in the primate: Evidence from [3H] flunitrazepam autoradiography. Brain Res. 1990, 531, 95–104. [Google Scholar] [CrossRef]
- Fieblinger, T.; Graves, S.M.; Sebel, L.E.; Alcacer, C.; Plotkin, J.L.; Gertler, T.S.; Chan, C.S.; Heiman, M.; Greengard, P.; Cenci, M.A.; et al. Cell type-specific plasticity of striatal projection neurons in parkinsonism and L-DOPA-induced dyskinesia. Nature Commun. 2014, 5, 5316. [Google Scholar] [CrossRef] [PubMed]
- Taverna, S.; Ilijic, E.; Surmeier, D.J. Recurrent collateral connections of striatal medium spiny neurons are disrupted in models of Parkinson’s disease. J. Neurosci. 2008, 28, 5504–5512. [Google Scholar] [CrossRef] [PubMed]
- Tepper, J.M.; Lee, C.R. GABAergic control of substantia nigra dopaminergic neurons. Prog. Brain Res. 2007, 160, 189–208. [Google Scholar]
- Lobb, C.J.; Wilson, C.J.; Paladini, C.A. A dynamic role for GABA receptors on the firing pattern of midbrain dopaminergic neurons. J. Neurophysiol 2010, 104, 403–413. [Google Scholar] [CrossRef]
- Friend, D.M.; Kravitz, A.V. Working together: Basal ganglia pathways in action selection. Trends Neurosci. 2014, 37, 301–303. [Google Scholar] [CrossRef]
- Paladini, C.A.; Tepper, J.M. Neurophysiology of Substantia Nigra Dopamine Neurons: Modulation by GABA and Glutamate. In Handbook of Basal Ganglia Structure and Function, 2nd ed.; Heinz, S., Tseng, K.Y., Eds.; Elsevier Cambridge: Cambridge, MA, USA, 2016; Volume 24, pp. 335–360. [Google Scholar]
- Ding, S.; Li, L.; Zhou, F.M. Nigral dopamine loss induces a global upregulation of presynaptic dopamine D1 receptor facilitation of the striatonigral GABAergic output. J. Neurophysiol. 2015, 113, 1697–1711. [Google Scholar] [CrossRef]
- Brazhnik, E.; Shah, F.; Tepper, J.M. GABAergic afferents activate both GABAA and GABAB receptors in mouse substantia nigra dopaminergic neurons in vivo. J. Neurosci. 2008, 28, 10386–10398. [Google Scholar] [CrossRef]
- Gulacsi, A.; Lee, C.R.; Sik, A.; Viitanen, T.; Kaila, K.; Tepper, J.M.; Freund, T.F. Cell type-specific differences in chloride-regulatory mechanisms and GABA(A) receptor-mediated inhibition in rat substantia nigra. J. Neurosci. 2003, 23, 8237–8246. [Google Scholar] [CrossRef] [PubMed]
- Borgkvist, A.; Avegno, E.M.; Wong, M.Y.; Kheirbek, M.A.; Sonders, M.S.; Hen, R.; Sulzer, D. Loss of Striatonigral GABAergic Presynaptic Inhibition Enables Motor Sensitization in Parkinsonian Mice. Neuron 2015, 87, 976–988. [Google Scholar] [CrossRef] [PubMed]
- Bar-On, L.; Molenaers, G.; Aertbelien, E.; Van Campenhout, A.; Feys, H.; Nuttin, B.; Desloovere, K. Spasticity and its contribution to hypertonia in cerebral palsy. BioMed Res. Int. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, S.L.; Zhang, L.; Forster, M.L.; Anderson, J.R.; Iwase, T.; Soliven, B.; Donahue, L.R.; Sweet, H.O.; Bronson, R.T.; Davisson, M.T.; et al. Trak1 mutation disrupts GABA(A) receptor homeostasis in hypertonic mice. Nat. Genet. 2006, 38, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Rowland, A.M.; Richmond, J.E.; Olsen, J.G.; Hall, D.H.; Bamber, B.A. Presynaptic terminals independently regulate synaptic clustering and autophagy of GABAA receptors in Caenorhabditis elegans. J. Neurosci. 2006, 26, 1711–1720. [Google Scholar] [CrossRef] [PubMed]
- Tanji, K.; Mori, F.; Kakita, A.; Takahashi, H.; Wakabayashi, K. Alteration of autophagosomal proteins (LC3, GABARAP and GATE-16) in Lewy body disease. Neurobiol. Dis. 2011, 43, 690–697. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef]
- Dutta, P.; Dargahi, L.; O’Connell, K.E.; Bolia, A.; Ozkan, B.; Sailer, A.W.; Dev, K.K. A novel modelling mechanism of PAEL receptor and GABARAPL2 interaction involved in Parkinson’s disease. Neurosci. Lett. 2018, 673, 12–18. [Google Scholar] [CrossRef]
- Murakami, T.; Shoji, M.; Imai, Y.; Inoue, H.; Kawarabayashi, T.; Matsubara, E.; Harigaya, Y.; Sasaki, A.; Takahashi, R.; Abe, K. Pael-R is accumulated in Lewy bodies of Parkinson’s disease. Ann. Neurol. 2004, 55, 439–442. [Google Scholar] [CrossRef]
- Derynck, R.; Miyazono, K. The TGFβ Family; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2008. [Google Scholar]
- Van Caam, A.; Madej, W.; Garcia de Vinuesa, A.; Goumans, M.J.; Ten Dijke, P.; Blaney Davidson, E.; van der Kraan, P. TGFbeta1-induced SMAD2/3 and SMAD1/5 phosphorylation are both ALK5-kinase-dependent in primary chondrocytes and mediated by TAK1 kinase activity. Arthritis Res. Ther. 2017, 19, 112. [Google Scholar] [CrossRef]
- Macias, M.J.; Martin-Malpartida, P.; Massague, J. Structural determinants of Smad function in TGF-beta signaling. Trends Biochem. Sci. 2015, 40, 296–308. [Google Scholar] [CrossRef] [PubMed]
- Horiguchi, M.; Ota, M.; Rifkin, D.B. Matrix control of transforming growth factor-beta function. J. Biochem. 2012, 152, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Budi, E.H. Specificity, versatility, and control of TGF-beta family signaling. Sci. Signal. 2019, 12, eaav5183. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Kornberg, T.B. Paracrine signaling mediated at cell-cell contacts. Bioessays 2015, 37, 25–33. [Google Scholar] [CrossRef]
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-beta and the TGF-beta Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb. Perspect. Biol. 2016, 8, a021873. [Google Scholar] [CrossRef]
- Murray-Rust, J.; McDonald, N.Q.; Blundell, T.L.; Hosang, M.; Oefner, C.; Winkler, F.; Bradshaw, R.A. Topological similarities in TGF-beta 2, PDGF-BB and NGF define a superfamily of polypeptide growth factors. Structure 1993, 1, 153–159. [Google Scholar] [CrossRef]
- Yakymovych, I.; Yakymovych, M.; Heldin, C.H. Intracellular trafficking of transforming growth factor beta receptors. Acta Biochim. Biophys. Sin. 2018, 50, 3–11. [Google Scholar] [CrossRef]
- Chen, Y.G. Endocytic regulation of TGF-beta signaling. Cell Res. 2009, 19, 58–70. [Google Scholar] [CrossRef]
- Miyazawa, K.; Miyazono, K. Regulation of TGF-beta Family Signaling by Inhibitory Smads. Cold Spring Harb. Perspect. Biol. 2017, 9, a022095. [Google Scholar] [CrossRef]
- Budi, E.H.; Muthusamy, B.P.; Derynck, R. The insulin response integrates increased TGF-beta signaling through Akt-induced enhancement of cell surface delivery of TGF-beta receptors. Sci. Signal. 2015, 8, ra96. [Google Scholar] [CrossRef]
- Kim, S.I.; Choi, M.E. TGF-beta-activated kinase-1: New insights into the mechanism of TGF-beta signaling and kidney disease. Kidney Res. Clin. Pract. 2012, 31, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Zi, Z.; Chapnick, D.A.; Liu, X. Dynamics of TGF-beta/Smad signaling. FEBS Lett. 2012, 586, 1921–1928. [Google Scholar] [CrossRef] [PubMed]
- Yagi, K.; Goto, D.; Hamamoto, T.; Takenoshita, S.; Kato, M.; Miyazono, K. Alternatively spliced variant of Smad2 lacking exon 3. Comparison with wild-type Smad2 and Smad3. J. Biol. Chem. 1999, 274, 703–709. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Liu, X.; Ren, X.; Tian, Y.; Chen, Z.; Xu, X.; Du, Y.; Jiang, C.; Fang, Y.; Liu, Z.; et al. Smad2 and Smad3 have differential sensitivity in relaying TGFbeta signaling and inversely regulate early lineage specification. Sci. Rep. 2016, 6, 21602. [Google Scholar] [CrossRef]
- Hill, C.S. Transcriptional Control by the SMADs. Cold Spring Harb. Perspect. Biol. 2016, 8, a022079. [Google Scholar] [CrossRef]
- Luo, K. Signaling Cross Talk between TGF-beta/Smad and Other Signaling Pathways. Cold Spring Harb. Perspect. Biol. 2017, 9, a022137. [Google Scholar] [CrossRef]
- Koinuma, D.; Tsutsumi, S.; Kamimura, N.; Taniguchi, H.; Miyazawa, K.; Sunamura, M.; Imamura, T.; Miyazono, K.; Aburatani, H. Chromatin immunoprecipitation on microarray analysis of Smad2/3 binding sites reveals roles of ETS1 and TFAP2A in transforming growth factor beta signaling. Mol. Cell. Biol. 2009, 29, 172–186. [Google Scholar] [CrossRef]
- Hata, A.; Lieberman, J. Dysregulation of microRNA biogenesis and gene silencing in cancer. Sci. Signal. 2015, 8, re3. [Google Scholar] [CrossRef]
- Horiguchi, K.; Sakamoto, K.; Koinuma, D.; Semba, K.; Inoue, A.; Inoue, S.; Fujii, H.; Yamaguchi, A.; Miyazawa, K.; Miyazono, K.; et al. TGF-beta drives epithelial-mesenchymal transition through deltaEF1-mediated downregulation of ESRP. Oncogene 2012, 31, 3190–3201. [Google Scholar] [CrossRef]
- Tripathi, V.; Sixt, K.M.; Gao, S.; Xu, X.; Huang, J.; Weigert, R.; Zhou, M.; Zhang, Y.E. Direct Regulation of Alternative Splicing by SMAD3 through PCBP1 Is Essential to the Tumor-Promoting Role of TGF-beta. Mol. Cell 2016, 64, 549–564. [Google Scholar] [CrossRef]
- Bertero, A.; Brown, S.; Madrigal, P.; Osnato, A.; Ortmann, D.; Yiangou, L.; Kadiwala, J.; Hubner, N.C.; de Los Mozos, I.R.; Sadee, C.; et al. The SMAD2/3 interactome reveals that TGFbeta controls m(6)A mRNA methylation in pluripotency. Nature 2018, 555, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Capelo, A. Dual role for TGF-beta1 in apoptosis. Cytokine Growth Factor Rev. 2005, 16, 15–34. [Google Scholar] [CrossRef] [PubMed]
- Vawter, M.P.; Dillon-Carter, O.; Tourtellotte, W.W.; Carvey, P.; Freed, W.J. TGFbeta1 and TGFbeta2 concentrations are elevated in Parkinson’s disease in ventricular cerebrospinal fluid. Exp. Neurol. 1996, 142, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Mogi, M.; Harada, M.; Kondo, T.; Narabayashi, H.; Riederer, P.; Nagatsu, T. Transforming growth factor-beta 1 levels are elevated in the striatum and in ventricular cerebrospinal fluid in Parkinson’s disease. Neurosci. Lett. 1995, 193, 129–132. [Google Scholar] [CrossRef]
- Van der Wal, E.A.; Gómez-Pinilla, F.; Cotman, C.W. Transforming growth factor-beta 1 is in plaques in Alzheimer and Down pathologies. Neuroreport 1993, 4, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Peress, N.S.; Perillo, E. Differential expression of TGF-beta 1, 2 and 3 isotypes in Alzheimer’s disease: A comparative immunohistochemical study with cerebral infarction, aged human and mouse control brains. J. Neuropathol. Exp. Neurol. 1995, 54, 802–811. [Google Scholar] [CrossRef][Green Version]
- Tarkowski, E.; Issa, R.; Sjögren, M.; Wallin, A.; Blennow, K.; Tarkowski, A.; Kumar, P. Increased intrathecal levels of the angiogenic factors VEGF and TGF-beta in Alzheimer’s disease and vascular dementia. Neurobiol. Aging 2002, 23, 237–243. [Google Scholar] [CrossRef]
- Zetterberg, H.; Andreasen, N.; Blennow, K. Increased cerebrospinal fluid levels of transforming growth factor-beta1 in Alzheimer’s disease. Neurosci. Lett. 2004, 367, 194–196. [Google Scholar] [CrossRef]
- Ilzecka, J.; Stelmasiak, Z.; Dobosz, B. Transforming growth factor-Beta 1 (tgf-Beta 1) in patients with amyotrophic lateral sclerosis. Cytokine 2002, 20, 239–243. [Google Scholar] [CrossRef]
- Krupinski, J.; Kumar, P.; Kumar, S.; Kaluza, J. Increased expression of TGF-beta 1 in brain tissue after ischemic stroke in humans. Stroke 1996, 27, 852–857. [Google Scholar] [CrossRef]
- Buss, A.; Pech, K.; Kakulas, B.A.; Martin, D.; Schoenen, J.; Noth, J.; Brook, G.A. TGF-beta1 and TGF-beta2 expression after traumatic human spinal cord injury. Spinal Cord 2008, 46, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Capelo, A.; Corti, O.; Mallet, J. Adenovirus-mediated over-expression of TGFbeta1 in the striatum decreases dopaminergic cell survival in embryonic nigral grafts. Neuroreport 1999, 10, 2169–2173. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Canabal, A.; Wheeler, A.L.; Sarkis, D.; Lerch, J.P.; Lu, W.Y.; Buckwalter, M.S.; Wyss-Coray, T.; Josselyn, S.A.; Frankland, P.W. Chronic over-expression of TGFβ1 alters hippocampal structure and causes learning deficits. Hippocampus 2013, 23, 1198–1211. [Google Scholar] [CrossRef] [PubMed]
- Ueberham, U.; Ueberham, E.; Brückner, M.K.; Seeger, G.; Gärtner, U.; Gruschka, H.; Gebhardt, R.; Arendt, T. Inducible neuronal expression of transgenic TGF-beta1 in vivo: Dissection of short-term and long-term effects. Eur. J. Neurosci. 2005, 22, 50–64. [Google Scholar] [CrossRef]
- Sanchez-Capelo, A.; Colin, P.; Guibert, B.; Biguet, N.F.; Mallet, J. Transforming growth factor beta1 overexpression in the nigrostriatal system increases the dopaminergic deficit of MPTP mice. Mol. Cell. Neurosci. 2003, 23, 614–625. [Google Scholar] [CrossRef]
- Tapia-Gonzalez, S.; Giraldez-Perez, R.M.; Cuartero, M.I.; Casarejos, M.J.; Mena, M.A.; Wang, X.F.; Sanchez-Capelo, A. Dopamine and alpha-synuclein dysfunction in Smad3 null mice. Mol. Neurodegener. 2011, 6, 72. [Google Scholar] [CrossRef]
- Andrews, Z.B.; Zhao, H.; Frugier, T.; Meguro, R.; Grattan, D.R.; Koishi, K.; McLennan, I.S. Transforming growth factor beta2 haploinsufficient mice develop age-related nigrostriatal dopamine deficits. Neurobiol. Dis. 2006, 21, 568–575. [Google Scholar] [CrossRef]
- Ueberham, U.; Ueberham, E.; Gruschka, H.; Arendt, T. Altered subcellular location of phosphorylated Smads in Alzheimer’s disease. Eur. J. Neurosci. 2006, 24, 2327–2334. [Google Scholar] [CrossRef]
- Baig, S.; van Helmond, Z.; Love, S. Tau hyperphosphorylation affects Smad 2/3 translocation. Neuroscience 2009, 163, 561–570. [Google Scholar] [CrossRef]
- Villapol, S.; Wang, Y.; Adams, M.; Symes, A.J. Smad3 deficiency increases cortical and hippocampal neuronal loss following traumatic brain injury. Exp. Neurol. 2013, 250, 353–365. [Google Scholar] [CrossRef]
- Tesseur, I.; Nguyen, A.; Chang, B.; Li, L.; Woodling, N.S.; Wyss-Coray, T.; Luo, J. Deficiency in Neuronal TGF-beta Signaling Leads to Nigrostriatal Degeneration and Activation of TGF-beta Signaling Protects against MPTP Neurotoxicity in Mice. J. Neurosci. 2017, 37, 4584–4592. [Google Scholar] [CrossRef] [PubMed]
- Goris, A.; Williams-Gray, C.H.; Foltynie, T.; Brown, J.; Maranian, M.; Walton, A.; Compston, D.A.; Barker, R.A.; Sawcer, S.J. Investigation of TGFB2 as a candidate gene in multiple sclerosis and Parkinson’s disease. J. Neurol. 2007, 254, 846–848. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Pho, V.; Bonasera, S.J.; Holtzman, J.; Tang, A.T.; Hellmuth, J.; Tang, S.; Janak, P.H.; Tecott, L.H.; Huang, E.J. Essential function of HIPK2 in TGFbeta-dependent survival of midbrain dopamine neurons. Nat. Neurosci. 2007, 10, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Diniz, L.P.; Matias, I.; Araujo, A.P.B.; Garcia, M.N.; Barros-Aragao, F.G.Q.; Alves-Leon, S.V.; de Souza, J.M.; Foguel, D.; Figueiredo, C.P.; Braga, C.; et al. alpha-synuclein oligomers enhance astrocyte-induced synapse formation through TGF-beta1 signaling in a Parkinson’s disease model. J. Neurochem. 2019, 150, 138–157. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.X.; Timbang, L.; Kim, J.I.; Shang, Y.; Sandoval, K.; Tang, A.A.; Whistler, J.L.; Ding, J.B.; Huang, E.J. TGF-beta Signaling in Dopaminergic Neurons Regulates Dendritic Growth, Excitatory-Inhibitory Synaptic Balance, and Reversal Learning. Cell Rep. 2016, 17, 3233–3245. [Google Scholar] [CrossRef] [PubMed]
- Tapia-Gonzalez, S.; Munoz, M.D.; Cuartero, M.I.; Sanchez-Capelo, A. Smad3 is required for the survival of proliferative intermediate progenitor cells in the dentate gyrus of adult mice. Cell Commun. Signal. 2013, 11, 93. [Google Scholar] [CrossRef]
- Leggio, L.; Vivarelli, S.; L’Episcopo, F.; Tirolo, C.; Caniglia, S.; Testa, N.; Marchetti, B.; Iraci, N. microRNAs in Parkinson’s Disease: From Pathogenesis to Novel Diagnostic and Therapeutic Approaches. Int. J. Mol. Sci. 2017, 18, 2698. [Google Scholar] [CrossRef]
- Cho, H.J.; Liu, G.; Jin, S.M.; Parisiadou, L.; Xie, C.; Yu, J.; Sun, L.; Ma, B.; Ding, J.; Vancraenenbroeck, R.; et al. MicroRNA-205 regulates the expression of Parkinson’s disease-related leucine-rich repeat kinase 2 protein. Hum. Mol. Genet. 2013, 22, 608–620. [Google Scholar] [CrossRef]
- Yarnall, A.J.; Breen, D.P.; Duncan, G.W.; Khoo, T.K.; Coleman, S.Y.; Firbank, M.J.; Nombela, C.; Winder-Rhodes, S.; Evans, J.R.; Rowe, J.B.; et al. Characterizing mild cognitive impairment in incident Parkinson disease: The ICICLE-PD study. Neurology 2014, 82, 308–316. [Google Scholar] [CrossRef]
- Elgh, E.; Domellöf, M.; Linder, J.; Edström, M.; Stenlund, H.; Forsgren, L. Cognitive function in early Parkinson’s disease: A population-based study. Eur. J. Neurol. 2009, 16, 1278–1284. [Google Scholar] [CrossRef]
- Aarsland, D.; Kurz, M.W. The epidemiology of dementia associated with Parkinson’s disease. Brain Pathol. 2010, 20, 633–639. [Google Scholar] [CrossRef]
- Muñoz, M.; Antolín-Vallespín, M.; Tapia-González, S.; Sánchez-Capelo, A. Smad3 deficiency inhibits dentate gyrus LTP by enhancing GABAA neurotransmission. J. Neurochem. 2016, 137, 190–199. [Google Scholar] [CrossRef]
- Catavero, C.; Bao, H.; Song, J. Neural mechanisms underlying GABAergic regulation of adult hippocampal neurogenesis. Cell Tissue Res. 2018, 371, 33–46. [Google Scholar] [CrossRef]
- Lim, J.; Bang, Y.; Choi, H.J. Abnormal hippocampal neurogenesis in Parkinson’s disease: Relevance to a new therapeutic target for depression with Parkinson’s disease. Arch. Pharm. Res. 2018, 41, 943–954. [Google Scholar] [CrossRef]
- Hoglinger, G.U.; Rizk, P.; Muriel, M.P.; Duyckaerts, C.; Oertel, W.H.; Caille, I.; Hirsch, E.C. Dopamine depletion impairs precursor cell proliferation in Parkinson disease. Nat. Neurosci. 2004, 7, 726–735. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Endo, S.; Cleary, L.J.; Eskin, A.; Byrne, J.H. Role of transforming growth factor-beta in long-term synaptic facilitation in Aplysia. Science 1997, 275, 1318–1320. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, S.; Kim, S.M.; Ramaswami, M. Retrograde regulation in the CNS; neuron-specific interpretations of TGF-beta signaling. Neuron 2004, 41, 845–848. [Google Scholar] [CrossRef]
- Chin, J.; Angers, A.; Cleary, L.J.; Eskin, A.; Byrne, J.H. TGF-beta1 in Aplysia: Role in long-term changes in the excitability of sensory neurons and distribution of TbetaR-II-like immunoreactivity. Learn. Mem. 1999, 6, 317–330. [Google Scholar] [PubMed]
- Chin, J.; Angers, A.; Cleary, L.J.; Eskin, A.; Byrne, J.H. Transforming growth factor beta1 alters synapsin distribution and modulates synaptic depression in Aplysia. J. Neurosci. 2002, 22, RC220. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, T.; Liu, R.Y.; Byrne, J.H. Transforming growth factor-beta2 modulates synaptic efficacy and plasticity and induces phosphorylation of CREB in hippocampal neurons. Hippocampus 2007, 17, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Caraci, F.; Gulisano, W.; Guida, C.A.; Impellizzeri, A.A.; Drago, F.; Puzzo, D.; Palmeri, A. A key role for TGF-beta1 in hippocampal synaptic plasticity and memory. Sci. Rep. 2015, 5, 11252. [Google Scholar] [CrossRef] [PubMed]
- Kida, S. A Functional Role for CREB as a Positive Regulator of Memory Formation and LTP. Exp. Neurobiol. 2012, 21, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Nenov, M.N.; Malkov, A.E.; Konakov, M.V.; Levin, S.G. Interleukin-10 and transforming growth factor-beta1 facilitate long-term potentiation in CA1 region of hippocampus. Biochem. Biophys. Res. Commun. 2019, 518, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Ageta, H.; Ikegami, S.; Miura, M.; Masuda, M.; Migishima, R.; Hino, T.; Takashima, N.; Murayama, A.; Sugino, H.; Setou, M.; et al. Activin plays a key role in the maintenance of long-term memory and late-LTP. Learn. Mem. 2010, 17, 176–185. [Google Scholar] [CrossRef]
- Hasegawa, Y.; Mukai, H.; Asashima, M.; Hojo, Y.; Ikeda, M.; Komatsuzaki, Y.; Ooishi, Y.; Kawato, S. Acute modulation of synaptic plasticity of pyramidal neurons by activin in adult hippocampus. Front. Neural Circuits 2014, 8, 56. [Google Scholar] [CrossRef]
- Arkhipov, V.I.; Pershina, E.V.; Levin, S.G. Deficiency of transforming growth factor-beta signaling disrupts memory processes in rats. Neuroreport 2018, 29, 353–355. [Google Scholar] [CrossRef]
- Athauda, D.; Foltynie, T. Insulin resistance and Parkinson’s disease: A new target for disease modification? Prog. Neurobiol. 2016, 145–146, 98–120. [Google Scholar] [CrossRef]
- Yang, L.; Chen, Z.; Li, B.; Wang, M.; Yu, L.; Wan, Y.; Gan, J.; Zhang, Y.; Liu, Z.; Wang, X. Multiple Evidences for Association between Cognitive Impairment and Dysglycemia in Parkinson’s Disease: Implications for Clinical Practice. Front. Aging Neurosci. 2017, 9, 355. [Google Scholar] [CrossRef]
- Peters, A. The selfish brain: Competition for energy resources. Am. J. Hum. Biol. 2011, 23, 29–34. [Google Scholar] [CrossRef]
- Yang, L.; Wang, H.; Liu, L.; Xie, A. The Role of Insulin/IGF-1/PI3K/Akt/GSK3beta Signaling in Parkinson’s Disease Dementia. Front. Neurosci. 2018, 12, 73. [Google Scholar] [CrossRef]
- Ge, S.; Goh, E.L.; Sailor, K.A.; Kitabatake, Y.; Ming, G.L.; Song, H. GABA regulates synaptic integration of newly generated neurons in the adult brain. Nature 2006, 439, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Jagasia, R.; Steib, K.; Englberger, E.; Herold, S.; Faus-Kessler, T.; Saxe, M.; Gage, F.H.; Song, H.; Lie, D.C. GABA-cAMP response element-binding protein signaling regulates maturation and survival of newly generated neurons in the adult hippocampus. J. Neurosci. 2009, 29, 7966–7977. [Google Scholar] [CrossRef] [PubMed]
- Roussa, E.; Speer, J.M.; Chudotvorova, I.; Khakipoor, S.; Smirnov, S.; Rivera, C.; Krieglstein, K. The membrane trafficking and functionality of the K+-Cl- co-transporter KCC2 is regulated by TGF-beta2. J. Cell Sci. 2016, 129, 3485–3498. [Google Scholar] [CrossRef] [PubMed]
- Frahm, C.; Draguhn, A. GAD and GABA transporter (GAT-1) mRNA expression in the developing rat hippocampus. Brain Res. Dev. Brain Res. 2001, 132, 1–13. [Google Scholar] [CrossRef]
- Arima-Yoshida, F.; Watabe, A.M.; Manabe, T. The mechanisms of the strong inhibitory modulation of long-term potentiation in the rat dentate gyrus. Eur. J. Neurosci. 2011, 33, 1637–1646. [Google Scholar] [CrossRef]
- Zhuang, J.L.; Wang, C.Y.; Zhou, M.H.; Duan, K.Z.; Mei, Y.A. TGF-beta1 enhances Kv2.1 potassium channel protein expression and promotes maturation of cerebellar granule neurons. J. Cell. Physiol. 2012, 227, 297–307. [Google Scholar] [CrossRef]
- Martinez-Cue, C.; Martinez, P.; Rueda, N.; Vidal, R.; Garcia, S.; Vidal, V.; Corrales, A.; Montero, J.A.; Pazos, A.; Florez, J.; et al. Reducing GABAA alpha5 receptor-mediated inhibition rescues functional and neuromorphological deficits in a mouse model of down syndrome. J. Neurosci. 2013, 33, 3953–3966. [Google Scholar] [CrossRef]
- Yoshiike, Y.; Kimura, T.; Yamashita, S.; Furudate, H.; Mizoroki, T.; Murayama, M.; Takashima, A. GABA(A) receptor-mediated acceleration of aging-associated memory decline in APP/PS1 mice and its pharmacological treatment by picrotoxin. PLoS ONE 2008, 3, e3029. [Google Scholar] [CrossRef]
- Na, E.S.; Morris, M.J.; Nelson, E.D.; Monteggia, L.M. GABAA receptor antagonism ameliorates behavioral and synaptic impairments associated with MeCP2 overexpression. Neuropsychopharmacology 2014, 39, 1946–1954. [Google Scholar] [CrossRef][Green Version]
- Cui, Y.; Costa, R.M.; Murphy, G.G.; Elgersma, Y.; Zhu, Y.; Gutmann, D.H.; Parada, L.F.; Mody, I.; Silva, A.J. Neurofibromin regulation of ERK signaling modulates GABA release and learning. Cell 2008, 135, 549–560. [Google Scholar] [CrossRef]
- Bruce, D.L.; Sapkota, G.P. Phosphatases in SMAD regulation. FEBS Lett. 2012, 586, 1897–1905. [Google Scholar] [CrossRef]
- Horbelt, D.; Denkis, A.; Knaus, P. A portrait of Transforming Growth Factor beta superfamily signalling: Background matters. Int. J. Biochem. Cell Biol. 2012, 44, 469–474. [Google Scholar] [CrossRef]
- Hales, C.M.; Rees, H.; Seyfried, N.T.; Dammer, E.B.; Duong, D.M.; Gearing, M.; Montine, T.J.; Troncoso, J.C.; Thambisetty, M.; Levey, A.I.; et al. Abnormal gephyrin immunoreactivity associated with Alzheimer disease pathologic changes. J. Neuropathol. Exp. Neurol. 2013, 72, 1009–1015. [Google Scholar] [CrossRef]
- Irwin, D.J.; Lee, V.M.; Trojanowski, J.Q. Parkinson’s disease dementia: Convergence of alpha-synuclein, tau and amyloid-beta pathologies. Nat. Rev. Neurosci. 2013, 14, 626–636. [Google Scholar] [CrossRef]
- Ongali, B.; Nicolakakis, N.; Lecrux, C.; Aboulkassim, T.; Rosa-Neto, P.; Papadopoulos, P.; Tong, X.K.; Hamel, E. Transgenic mice overexpressing APP and transforming growth factor-beta1 feature cognitive and vascular hallmarks of Alzheimer’s disease. Am. J. Pathol. 2010, 177, 3071–3080. [Google Scholar] [CrossRef]
- Estrada, L.D.; Oliveira-Cruz, L.; Cabrera, D. Transforming Growth Factor Beta Type I Role in Neurodegeneration: Implications for Alzheimer’s Disease. Curr. Protein Pept. Sci. 2018, 19, 1180–1188. [Google Scholar] [CrossRef] [PubMed]
- Guridi, J.; Rodriguez-Rojas, R.; Carmona-Abellan, M.; Parras, O.; Becerra, V.; Lanciego, J.L. History and future challenges of the subthalamic nucleus as surgical target: Review article. Mov. Disord. 2018, 33, 1540–1550. [Google Scholar] [CrossRef] [PubMed]
- Baron, M.S.; Wichmann, T.; Ma, D.; DeLong, M.R. Effects of transient focal inactivation of the basal ganglia in parkinsonian primates. J. Neurosci. 2002, 22, 592–599. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Kaplitt, M.G.; Fitzsimons, H.L.; Zuzga, D.S.; Liu, Y.; Oshinsky, M.L.; During, M.J. Subthalamic GAD gene therapy in a Parkinson’s disease rat model. Science 2002, 298, 425–429. [Google Scholar] [CrossRef] [PubMed]
- Niethammer, M.; Tang, C.C.; LeWitt, P.A.; Rezai, A.R.; Leehey, M.A.; Ojemann, S.G.; Flaherty, A.W.; Eskandar, E.N.; Kostyk, S.K.; Sarkar, A.; et al. Long-term follow-up of a randomized AAV2-GAD gene therapy trial for Parkinson’s disease. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Ondo, W.G.; Hunter, C. Flumazenil, a GABA antagonist, may improve features of Parkinson’s disease. Mov. Disord. 2003, 18, 683–685. [Google Scholar] [CrossRef] [PubMed]
- Hulse, G.; Kelty, E.; Hood, S.; Norman, A.; Basso, M.R.; Reece, A.S. Novel Indications for Benzodiazepine Antagonist Flumazenil in GABA Mediated Pathological Conditions of the Central Nervous System. Curr. Pharm. Des. 2015, 21, 3325–3342. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muñoz, M.D.; de la Fuente, N.; Sánchez-Capelo, A. TGF-β/Smad3 Signalling Modulates GABA Neurotransmission: Implications in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 590. https://doi.org/10.3390/ijms21020590
Muñoz MD, de la Fuente N, Sánchez-Capelo A. TGF-β/Smad3 Signalling Modulates GABA Neurotransmission: Implications in Parkinson’s Disease. International Journal of Molecular Sciences. 2020; 21(2):590. https://doi.org/10.3390/ijms21020590
Chicago/Turabian StyleMuñoz, Mª Dolores, Nerea de la Fuente, and Amelia Sánchez-Capelo. 2020. "TGF-β/Smad3 Signalling Modulates GABA Neurotransmission: Implications in Parkinson’s Disease" International Journal of Molecular Sciences 21, no. 2: 590. https://doi.org/10.3390/ijms21020590
APA StyleMuñoz, M. D., de la Fuente, N., & Sánchez-Capelo, A. (2020). TGF-β/Smad3 Signalling Modulates GABA Neurotransmission: Implications in Parkinson’s Disease. International Journal of Molecular Sciences, 21(2), 590. https://doi.org/10.3390/ijms21020590