Potential Therapeutic Strategies for Lung and Breast Cancers through Understanding the Anti-Angiogenesis Resistance Mechanisms

 , ,
, , _Talaat.jpg) and
and 
Abstract
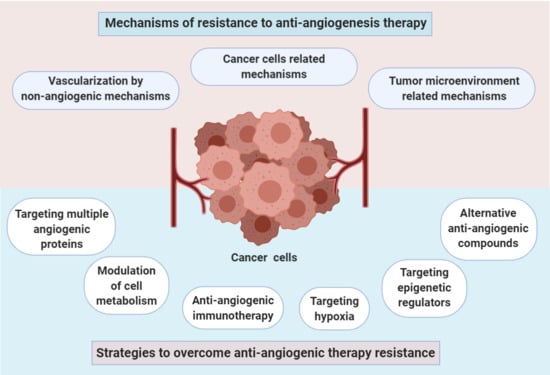
1. Introduction
2. Principles of Anti-Angiogenic Therapy
3. Mechanisms of Resistance to Anti-Angiogenesis Therapy
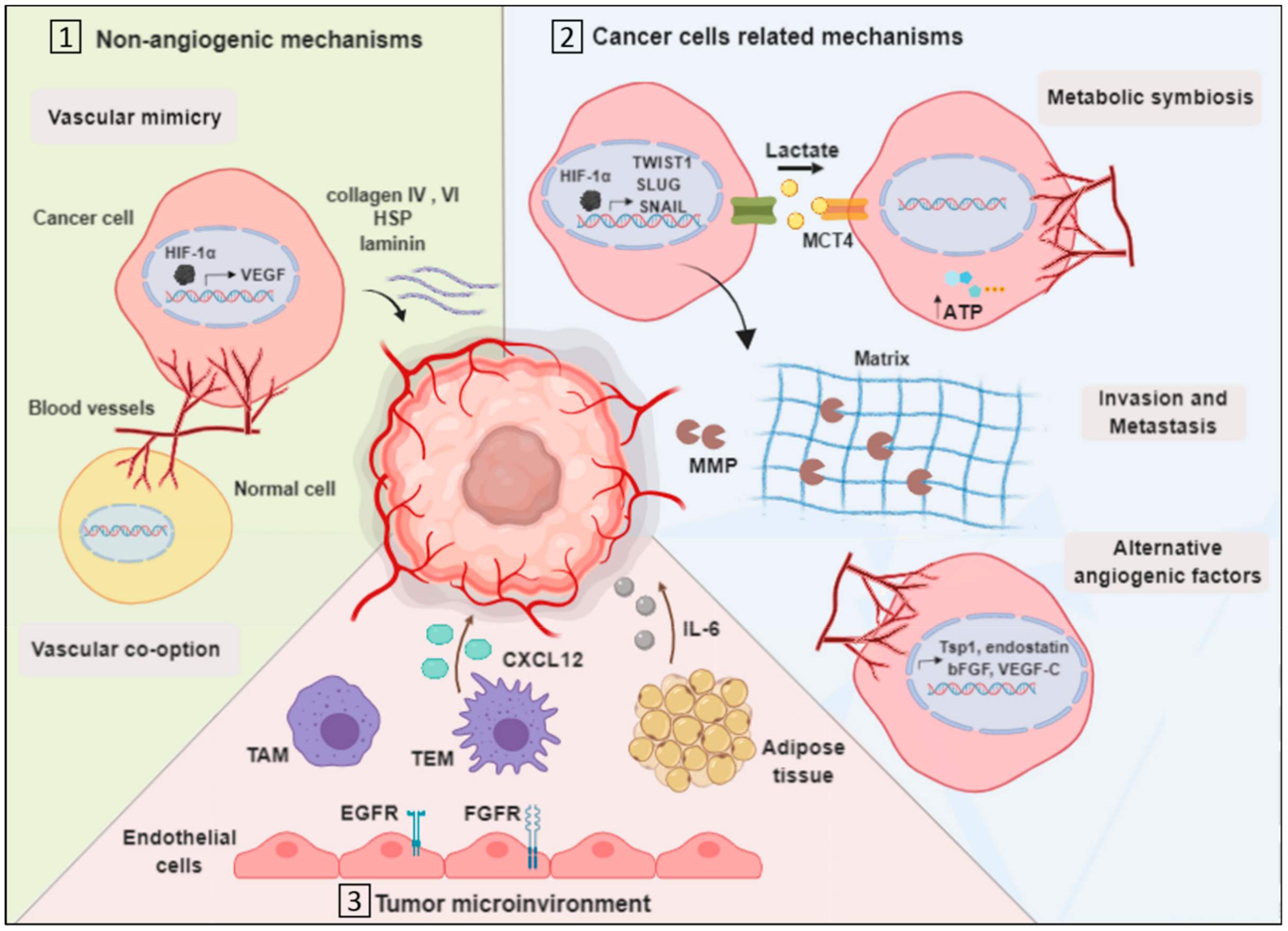
3.1. Vascularization by Non-Angiogenic Mechanisms
3.1.1. Vascular Mimicry
3.1.2. Vascular Co-Option
3.2. Cancer Cell-Related Mechanisms
3.2.1. Metabolic Symbiosis
3.2.2. Invasion
3.2.3. Upregulation of Alternative Angiogenic Factors
3.3. Tumor Microenvironment-Related Mechanisms
3.3.1. Endothelial Cells Mediated Resistance
3.3.2. Tumor-Associated Macrophages
3.3.3. TIE2 Expressing Macrophages
3.3.4. Myeloid and Adipose Cells
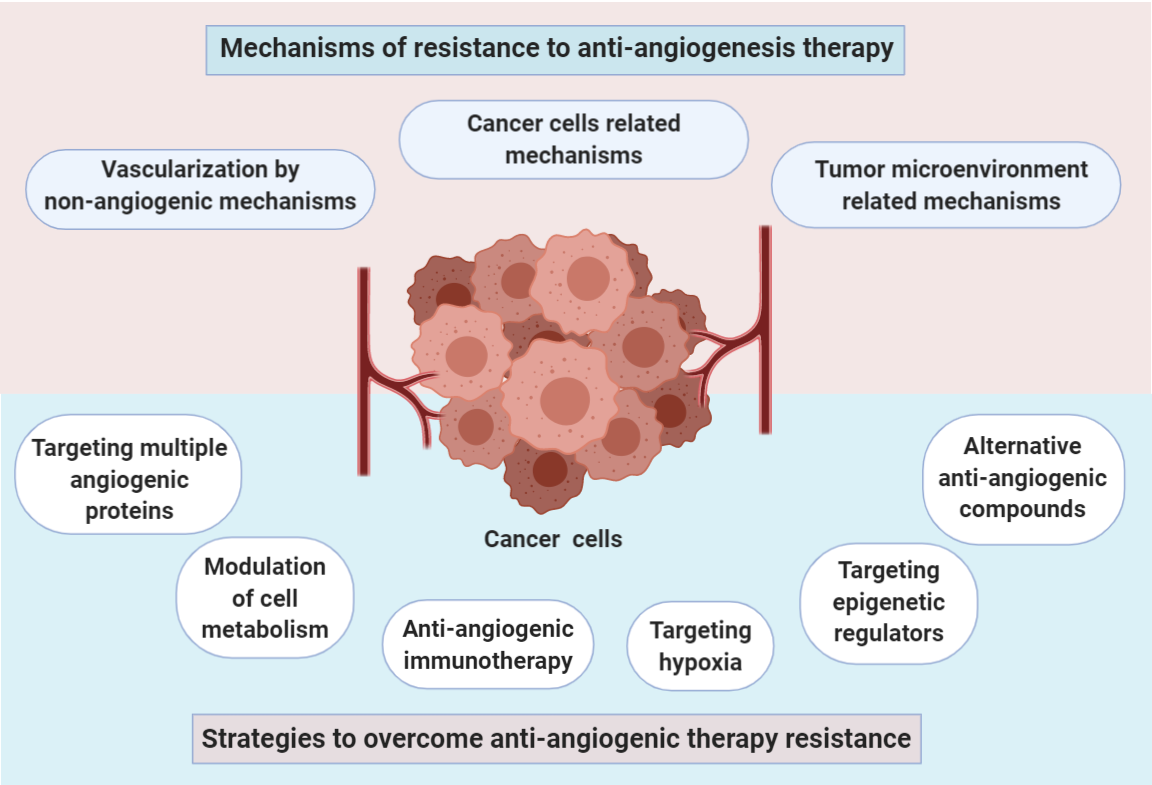
4. Strategies to Overcome Anti-Angiogenic Therapy Resistance
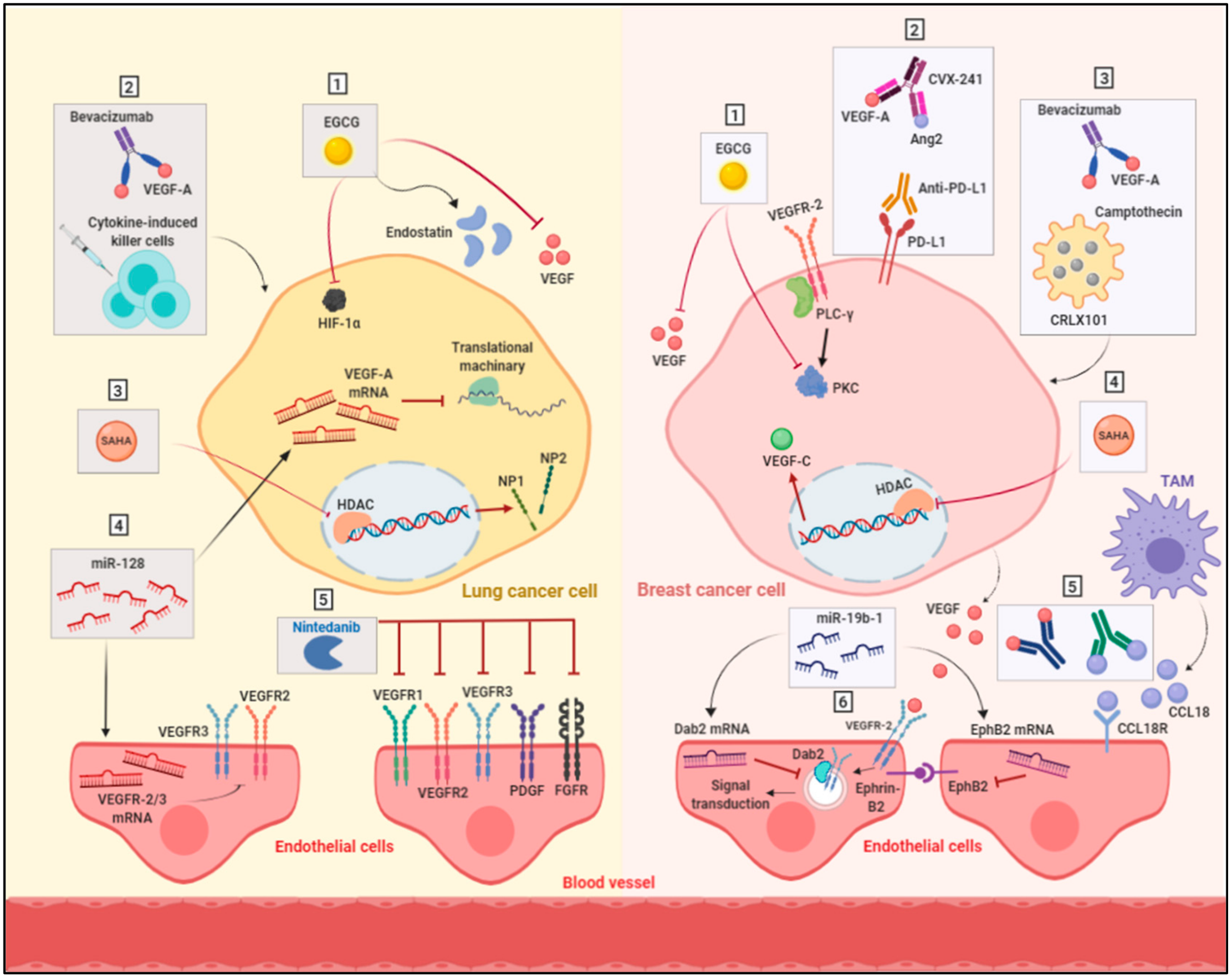
4.1. Targeting Multiple Angiogenic Proteins
4.2. Targeting Hypoxia
4.3. Anti-Angiogenic Immunotherapy
4.4. Targeting Epigenetic Regulators
4.5. Alternative Anti-Angiogenic Compounds
4.6. Modulation of Cell Metabolism
5. Conclusions
Funding
Conflicts of Interest
Abbreviations
| VEGF | Vascular endothelial growth factor |
| TSP | Thrombospondins |
| PGF | Placental growth factor |
| VEGFR | Vascular endothelial growth factor receptors |
| SCLC | Small cell lung cancer |
| NSCLC | Non-small cell lung cancer |
| TKI | Tyrosine kinase inhibitor |
| OS | Overall survival |
| PFS | Progression free survival |
| HIF-1 | Hypoxia-inducible factor |
| pCR | Pathologic complete response |
| DFS | Disease-free survival |
| PDGF | Platelet-derived growth factor receptor |
| FLT3 | Fms-related tyrosine kinase 3 |
| PD-L1 | Programmed death- ligand 1 |
| HDAC | Histone deacetylase |
| SAHA | Suberanilohydroxamic acid |
| TSA | Trichostatin A |
| NP1 | Neuropilin1 |
| NP2 | Neuropilin2 |
| miRs | MicroRNAs |
| EGCG | Epigallocatechin-3-O-gallate |
| PFKFB3 | Phosphofruktokinase-2/fructose-2,6-bisphosphatase 3 |
| MCT4 | Monocarboxylate transporter 4 |
| ATP | Adenosine triphosphate |
| MMP | Matrix metalloproteinase |
| TAM | Tumor-associated macrophages |
| TEM | TIE2 expressing macrophages |
| HSP | Heparin sulfate proteoglycan |
| bFGF | basic fibroblast growth factor |
| FGFR | Fibroblast growth factor receptor |
| IL-6 | Interleukin-6 |
| EC | Endothelial cells |
| VM | Vascular mimicry |
| VE-cadherin | Vascular endothelial cadherin |
| PECAM | Platelet EC adhesion molecule |
| PA | Plasminogen Activator |
| MCT4 | Monocarboxylate transporter 4 |
| EMT | Epithelial mesenchymal transition |
| EGFR | Epidermal growth factor receptor |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Al-Abd, A.M.; Aljehani, Z.K.; Gazzaz, R.W.; Fakhri, S.H.; Jabbad, A.H.; Alahdal, A.M.; Torchilin, V.P. Pharmacokinetic strategies to improve drug penetration and entrapment within solid tumors. J. Control. Release 2015, 219, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Khalid, E.B.; Ayman, E.-M.E.-K.; Rahman, H.; Abdelkarim, G.; Najda, A. Natural products against cancer angiogenesis. Tumour Biol. 2016, 37, 14513–14536. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Langer, R. A review of judah folkman’s remarkable achievements in biomedicine. Proc. Natl. Acad. Sci. USA 2008, 105, 13203–13205. [Google Scholar] [CrossRef]
- Folkman, J. Angiogenesis and its inhibitors. Important Adv. Oncol. 1985, 42–62. [Google Scholar]
- Ferrara, N. The role of the vegf signaling pathway in tumor angiogenesis. In Tumor Angiogenesis: A Key Target for Cancer Therapy; Marmé, D., Ed.; Springer International Publishing: Cham, Switzerland, 2019; pp. 211–226. [Google Scholar]
- Ren, B.; Yee, K.O.; Lawler, J.; Khosravi-Far, R. Regulation of tumor angiogenesis by thrombospondin-1. Biochim. Biophys. Acta 2006, 1765, 178–188. [Google Scholar] [CrossRef]
- Muppala, S.; Xiao, R.; Krukovets, I.; Verbovetsky, D.; Yendamuri, R.; Habib, N.; Raman, P.; Plow, E.; Stenina-Adognravi, O. Thrombospondin-4 mediates tgf-beta-induced angiogenesis. Oncogene 2017, 36, 5189–5198. [Google Scholar] [CrossRef]
- Folkman, J. Antiangiogenesis in cancer therapy-endostatin and its mechanisms of action. Exp. Cell Res. 2006, 312, 594–607. [Google Scholar] [CrossRef]
- Pratheeshkumar, P.; Budhraja, A.; Son, Y.-O.; Wang, X.; Zhang, Z.; Ding, S.; Wang, L.; Hitron, A.; Lee, J.-C.; Xu, M.; et al. Quercetin inhibits angiogenesis mediated human prostate tumor growth by targeting vegfr- 2 regulated akt/mtor/p70s6k signaling pathways. PLoS ONE 2012, 7, e47516. [Google Scholar] [CrossRef]
- Huang, Z.; Bao, S.D. Roles of main pro- and anti-angiogenic factors in tumor angiogenesis. World J. Gastroenterol. 2004, 10, 463–470. [Google Scholar] [CrossRef]
- Payen, V.L.; Brisson, L.; Dewhirst, M.W.; Sonveaux, P. Common responses of tumors and wounds to hypoxia. Cancer J. 2015, 21, 75. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Shibasaki, F. Hypoxia-inducible factor as an angiogenic master switch. Front. Pediatrics 2015, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Nico, B.; Crivellato, E.; Roccaro, A.M.; Vacca, A. The history of the angiogenic switch concept. Leukemia 2007, 21, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Rouhi, P.; Lee, S.L.C.; Cao, Z.; Hedlund, E.-M.; Jensen, L.D.; Cao, Y. Pathological angiogenesis facilitates tumor cell dissemination and metastasis. Cell Cycle 2010, 9, 913–917. [Google Scholar] [CrossRef]
- Folkman, J.; Shing, Y. Control of angiogenesis by heparin and other sulfated polysaccharides. Adv. Exp. Med. Biol. 1992, 313, 355–364. [Google Scholar]
- Laurenzana, A.; Fibbi, G.; Margheri, F.; Biagioni, A.; Luciani, C.; Del Rosso, M.; Chillà, A. Endothelial progenitor cells in sprouting angiogenesis: Proteases pave the way. Curr. Mol. Med. 2015, 15, 606–620. [Google Scholar] [CrossRef]
- Kirsch, M.; Schackert, G.; Black, P.M. Angiogenesis, metastasis, and endogenous inhibition. J. Neurooncol. 2000, 50, 173–180. [Google Scholar] [CrossRef]
- Bielenberg, D.R.; Zetter, B.R. The contribution of angiogenesis to the process of metastasis. Cancer J. 2015, 21, 267–273. [Google Scholar] [CrossRef]
- Yadav, L.; Puri, N.; Rastogi, V.; Satpute, P.; Sharma, V. Tumour angiogenesis and angiogenic inhibitors: A review. J. Clin. Diagn. Res. 2015, 9, XE01–XE05. [Google Scholar] [CrossRef]
- Takahashi, M.; Matsui, A.; Inao, M.; Mochida, S.; Fujiwara, K. Erk/mapk-dependent pi3k/akt phosphorylation through vegfr-1 after vegf stimulation in activated hepatic stellate cells. Hepatol. Res. 2003, 26, 232–236. [Google Scholar] [CrossRef]
- Rey, S.; Semenza, G.L. Hypoxia-inducible factor-1-dependent mechanisms of vascularization and vascular remodelling. Cardiovasc. Res. 2010, 86, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Willenborg, S.; Lucas, T.; van Loo, G.; Knipper, J.A.; Krieg, T.; Haase, I.; Brachvogel, B.; Hammerschmidt, M.; Nagy, A.; Ferrara, N.; et al. Ccr2 recruits an inflammatory macrophage subpopulation critical for angiogenesis in tissue repair. Blood 2012, 120, 613–625. [Google Scholar] [CrossRef] [PubMed]
- Romano Di Peppe, S.; Mangoni, A.; Zambruno, G.; Spinetti, G.; Melillo, G.; Napolitano, M.; Capogrossi, M.C. Adenovirus-mediated vegf(165) gene transfer enhances wound healing by promoting angiogenesis in cd1 diabetic mice. Gene Ther. 2002, 9, 1271–1277. [Google Scholar] [CrossRef] [PubMed]
- Galeano, M.; Deodato, B.; Altavilla, D.; Cucinotta, D.; Arsic, N.; Marini, H.; Torre, V.; Giacca, M.; Squadrito, F. Adeno-associated viral vector-mediated human vascular endothelial growth factor gene transfer stimulates angiogenesis and wound healing in the genetically diabetic mouse. Diabetologia 2003, 46, 546–555. [Google Scholar] [CrossRef]
- Galiano, R.D.; Tepper, O.M.; Pelo, C.R.; Bhatt, K.A.; Callaghan, M.; Bastidas, N.; Bunting, S.; Steinmetz, H.G.; Gurtner, G.C. Topical vascular endothelial growth factor accelerates diabetic wound healing through increased angiogenesis and by mobilizing and recruiting bone marrow-derived cells. Am. J. Pathol. 2004, 164, 1935–1947. [Google Scholar] [CrossRef]
- Brem, H.; Kodra, A.; Golinko, M.S.; Entero, H.; Stojadinovic, O.; Wang, V.M.; Sheahan, C.M.; Weinberg, A.D.; Woo, S.L.; Ehrlich, H.P.; et al. Mechanism of sustained release of vascular endothelial growth factor in accelerating experimental diabetic healing. J. Investig. Dermatol. 2009, 129, 2275–2287. [Google Scholar] [CrossRef]
- Corral, C.J.; Siddiqui, A.; Wu, L.; Farrell, C.L.; Lyons, D.; Mustoe, T.A. Vascular endothelial growth factor is more important than basic fibroblastic growth factor during ischemic wound healing. Arch. Surg. 1999, 134, 200–205. [Google Scholar] [CrossRef]
- Deodato, B.; Arsic, N.; Zentilin, L.; Galeano, M.; Santoro, D.; Torre, V.; Altavilla, D.; Valdembri, D.; Bussolino, F.; Squadrito, F.; et al. Recombinant aav vector encoding human vegf165 enhances wound healing. Gene Ther. 2002, 9, 777–785. [Google Scholar] [CrossRef]
- Wilgus, T.A. Vascular endothelial growth factor and cutaneous scarring. Adv. Wound Care 2019, 8, 671–678. [Google Scholar] [CrossRef]
- Ferrara, N.; Carver Moore, K.; Chen, H.; Dowd, M.; Lu, L.; O’Shea, K.S.; Powell Braxton, L.; Hillan, K.J.; Moore, M.W. Heterozygous embryonic lethality induced by targeted inactivation of the vegf gene. Nature 1996, 380, 439–442. [Google Scholar] [CrossRef]
- Carmeliet, P.; Ferreira, V.; Breier, G.; Pollefeyt, S.; Kieckens, L.; Gertsenstein, M.; Fahrig, M.; Vandenhoeck, A.; Harpal, K.; Eberhardt, C. Abnormal blood vessel development and lethality in embryos lacking a single vegf allele. Nature 1996, 380, 435. [Google Scholar] [CrossRef] [PubMed]
- Risau, W.; Flamme, I. Vasculogenesis. Ann. Rev. Cell Dev. Biol. 1995, 11, 73–91. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, H.; Golding, M.; Fruttiger, M.; Ruhrberg, C.; Lundkvist, A.; Abramsson, A.; Jeltsch, M.; Mitchell, C.; Alitalo, K.; Shima, D.; et al. Vegf guides angiogenic sprouting utilizing endothelial tip cell filopodia. J. Cell Biol. 2003, 161, 1163–1177. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Tammela, T.; Yamamoto, M.; Anisimov, A.; Holopainen, T.; Kaijalainen, S.; Karpanen, T.; Lehti, K.; Yla-Herttuala, S.; Alitalo, K. Notch restricts lymphatic vessel sprouting induced by vascular endothelial growth factor. Blood 2011, 118, 1154–1162. [Google Scholar] [CrossRef]
- Baluk, P.; Tammela, T.; Ator, E.; Lyubynska, N.; Achen, M.G.; Hicklin, D.J.; Jeltsch, M.; Petrova, T.V.; Pytowski, B.; Stacker, S.A. Pathogenesis of persistent lymphatic vessel hyperplasia in chronic airway inflammation. J. Clin. Investig. 2005, 115, 247–257. [Google Scholar] [CrossRef]
- Karkkainen, M.J.; Haiko, P.; Sainio, K.; Partanen, J.; Taipale, J.; Petrova, T.V.; Jeltsch, M.; Jackson, D.G.; Talikka, M.; Rauvala, H. Vascular endothelial growth factor c is required for sprouting of the first lymphatic vessels from embryonic veins. Nat. Immunol. 2004, 5, 74. [Google Scholar] [CrossRef]
- Hagerling, R.; Pollmann, C.; Andreas, M.; Schmidt, C.; Nurmi, H.; Adams, R.H.; Alitalo, K.; Andresen, V.; Schulte-Merker, S.; Kiefer, F. A novel multistep mechanism for initial lymphangiogenesis in mouse embryos based on ultramicroscopy. EMBO J. 2013, 32, 629–644. [Google Scholar] [CrossRef]
- Kim, K.E.; Koh, Y.J.; Jeon, B.H.; Jang, C.; Han, J.; Kataru, R.P.; Schwendener, R.A.; Kim, J.M.; Koh, G.Y. Role of cd11b+ macrophages in intraperitoneal lipopolysaccharide-induced aberrant lymphangiogenesis and lymphatic function in the diaphragm. Am. J. Pathol. 2009, 175, 1733–1745. [Google Scholar] [CrossRef]
- Aspelund, A.; Tammela, T.; Antila, S.; Nurmi, H.; Leppanen, V.M.; Zarkada, G.; Stanczuk, L.; Francois, M.; Makinen, T.; Saharinen, P.; et al. The schlemm’s canal is a vegf-c/vegfr-3-responsive lymphatic-like vessel. J. Clin. Investig. 2014, 124, 3975–3986. [Google Scholar] [CrossRef]
- Nurmi, H.; Saharinen, P.; Zarkada, G.; Zheng, W.; Robciuc, M.R.; Alitalo, K. Vegf-c is required for intestinal lymphatic vessel maintenance and lipid absorption. EMBO Mol. Med. 2015, 7, 1418–1425. [Google Scholar] [CrossRef]
- Maae, E.; Olsen, D.A.; Steffensen, K.D.; Jakobsen, E.H.; Brandslund, I.; Sørensen, F.B.; Jakobsen, A. Prognostic impact of placenta growth factor and vascular endothelial growth factor a in patients with breast cancer. Breast Cancer Res. Treat. 2012, 133, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Antiangiogenesis strategies revisited: From starving tumors to alleviating hypoxia. Cancer Cell 2014, 26, 605–622. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Heukamp, L.C.; Siobal, M.; Schöttle, J.; Wieczorek, C.; Peifer, M.; Frasca, D.; Koker, M.; König, K.; Meder, L.; et al. Tumor vegf:Vegfr2 autocrine feed-forward loop triggers angiogenesis in lung cancer. J. Clin. Investig. 2013, 123, 1732–1740. [Google Scholar] [CrossRef] [PubMed]
- Maione, F.; Capano, S.; Regano, D.; Zentilin, L.; Giacca, M.; Casanovas, O.; Bussolino, F.; Serini, G.; Giraudo, E. Semaphorin 3a overcomes cancer hypoxia and metastatic dissemination induced by antiangiogenic treatment in mice. J. Clin. Investig. 2012, 122, 1832–1848. [Google Scholar] [CrossRef]
- Goel, S.; Duda, D.G.; Xu, L.; Munn, L.L.; Boucher, Y.; Fukumura, D.; Jain, R.K. Normalization of the vasculature for treatment of cancer and other diseases. Physiol. Rev. 2011, 91, 1071–1121. [Google Scholar] [CrossRef]
- Liang, W.; Wu, X.; Hong, S.; Zhang, Y.; Kang, S.; Fang, W.; Qin, T.; Huang, Y.; Zhao, H.; Zhang, L. Multi-targeted antiangiogenic tyrosine kinase inhibitors in advanced non-small cell lung cancer: Meta-analyses of 20 randomized controlled trials and subgroup analyses. PLoS ONE 2014, 9, e109757. [Google Scholar] [CrossRef]
- Jayson, G.C.; Kerbel, R.; Ellis, L.M.; Harris, A.L. Antiangiogenic therapy in oncology: Current status and future directions. Lancet 2016, 388, 518–529. [Google Scholar] [CrossRef]
- Varella, L.; Abraham, J.; Kruse, M. Revisiting the role of bevacizumab in the treatment of breast cancer. Semin. Oncol. 2017, 44, 273–285. [Google Scholar] [CrossRef]
- Kesisis, G.; Broxterman, H.; Giaccone, G. Angiogenesis inhibitors. Drug selectivity and target specificity. Curr. Pharm. Des. 2007, 13, 2795–2809. [Google Scholar] [CrossRef]
- Siemann, D.W.; Rojiani, A.M. Enhancement of radiation therapy by the novel vascular targeting agent zd6126. Int. J. Radiat. Oncol. Biol. Phys. 2002, 53, 164–171. [Google Scholar] [CrossRef]
- Liang, W.; Ni, Y.; Chen, F. Tumor resistance to vascular disrupting agents: Mechanisms, imaging, and solutions. Oncotarget 2016, 7, 15444–15459. [Google Scholar] [CrossRef] [PubMed]
- Horsman, M.R.; Siemann, D.W. Pathophysiologic effects of vascular-targeting agents and the implications for combination with conventional therapies. Cancer Res. 2006, 66, 11520–11539. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.-P.; Bodrug, N.; Hodivala-Dilke, K.M. Exploring novel methods for modulating tumor blood vessels in cancer treatment. Curr. Biol. 2016, 26, R1161–R1166. [Google Scholar] [CrossRef]
- Bos, R.; Zhong, H.; Hanrahan, C.F.; Mommers, E.C.M.; Semenza, G.L.; Pinedo, H.M.; Abeloff, M.D.; Simons, J.W.; van Diest, P.J.; van der Wall, E. Levels of hypoxia-inducible factor-1α during breast carcinogenesis. J. Natl. Cancer Inst. 2001, 93, 309–314. [Google Scholar] [CrossRef]
- Pugh, C.W.; Ratcliffe, P.J. Regulation of angiogenesis by hypoxia: Role of the hif system. Nat. Med. 2003, 9, 677–684. [Google Scholar] [CrossRef]
- Lohela, M.; Bry, M.; Tammela, T.; Alitalo, K. Vegfs and receptors involved in angiogenesis versus lymphangiogenesis. Curr. Opin. Cell Biol. 2009, 21, 154–165. [Google Scholar] [CrossRef]
- Uzzan, B.; Nicolas, P.; Cucherat, M.; Perret, G.-Y. Microvessel density as a prognostic factor in women with breast cancer: A systematic review of the literature and meta-analysis. Cancer Res. 2004, 64, 2941–2955. [Google Scholar] [CrossRef]
- Penson, R.T.; Huang, H.Q.; Wenzel, L.B.; Monk, B.J.; Stockman, S.; Long, H.J.; Ramondetta, L.M.; Landrum, L.M.; Oaknin, A.; Reid, T.J.; et al. Bevacizumab for advanced cervical cancer: Patient-reported outcomes of a randomised, phase 3 trial (nrg oncology-gynecologic oncology group protocol 240). Lancet Oncol. 2015, 16, 301–311. [Google Scholar] [CrossRef]
- Valachis, A.; Polyzos, N.P.; Patsopoulos, N.A.; Georgoulias, V.; Mavroudis, D.; Mauri, D. Bevacizumab in metastatic breast cancer: A meta-analysis of randomized controlled trials. Breast Cancer Res. Treat. 2010, 122, 1–7. [Google Scholar] [CrossRef]
- Spring, L.; Greenup, R.; Niemierko, A.; Schapira, L.; Haddad, S.; Jimenez, R.; Coopey, S.; Taghian, A.; Hughes, K.S.; Isakoff, S.J.; et al. Pathologic complete response after neoadjuvant chemotherapy and long-term outcomes among young women with breast cancer. J. Natl. Compr. Cancer Netw. 2017, 15, 1216–1223. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Escudier, B.; Gannon, A.; Figlin, R.A. Sunitinib: Ten years of successful clinical use and study in advanced renal cell carcinoma. Oncologost 2017, 22, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xu, B.; Li, Q.; Jiang, D.; Yan, S. Anti-cancer effects of a novel pan-raf inhibitor in a hepatocellular carcinoma cell line. Mol. Med. Rep. 2018, 17, 6185–6193. [Google Scholar] [CrossRef] [PubMed]
- Bergh, J.; Bondarenko, I.M.; Lichinitser, M.R.; Liljegren, A.; Greil, R.; Voytko, N.L.; Makhson, A.N.; Cortes, J.; Lortholary, A.; Bischoff, J.; et al. First-line treatment of advanced breast cancer with sunitinib in combination with docetaxel versus docetaxel alone: Results of a prospective, randomized phase iii study. J. Clin. Oncol. 2012, 30, 921–929. [Google Scholar] [CrossRef]
- Baselga, J.; Zamagni, C.; Gómez, P.; Bermejo, B.; Nagai, S.E.; Melichar, B.; Chan, A.; Mángel, L.; Bergh, J.; Costa, F.; et al. Resilience: Phase iii randomized, double-blind trial comparing sorafenib with capecitabine versus placebo with capecitabine in locally advanced or metastatic her2-negative breast cancer. Clin. Breast Cancer 2017, 17, 585–594. [Google Scholar] [CrossRef]
- Hall, R.D.; Le, T.M.; Haggstrom, D.E.; Gentzler, R.D. Angiogenesis inhibition as a therapeutic strategy in non-small cell lung cancer (nsclc). Transl. Lung Cancer Res. 2015, 4, 515–523. [Google Scholar]
- Seto, T.; Kato, T.; Nishio, M.; Goto, K.; Atagi, S.; Hosomi, Y.; Yamamoto, N.; Hida, T.; Maemondo, M.; Nakagawa, K.; et al. Erlotinib alone or with bevacizumab as first-line therapy in patients with advanced non-squamous non-small-cell lung cancer harbouring egfr mutations (jo25567): An open-label, randomised, multicentre, phase 2 study. Lancet Oncol. 2014, 15, 1236–1244. [Google Scholar] [CrossRef]
- Masoud, V.; Pages, G. Targeted therapies in breast cancer: New challenges to fight against resistance. World J. Clin. Oncol. 2017, 8, 120–134. [Google Scholar] [CrossRef]
- Van Beijnum, J.R.; Nowak-Sliwinska, P.; Huijbers, E.J.; Thijssen, V.L.; Griffioen, A.W. The great escape; the hallmarks of resistance to antiangiogenic therapy. Pharmacol. Rev. 2015, 67, 441–461. [Google Scholar] [CrossRef]
- Shen, Y.; Quan, J.; Wang, M.; Li, S.; Yang, J.; Lv, M.; Chen, Z.; Zhang, L.; Zhao, X. Tumor vasculogenic mimicry formation as an unfavorable prognostic indicator in patients with breast cancer. Oncotarget 2017, 8, 56408–56416. [Google Scholar] [CrossRef]
- Maniotis, A.J.; Folberg, R.; Hess, A.; Seftor, E.A.; Gardner, L.M.; Pe’er, J.; Trent, J.M.; Meltzer, P.S.; Hendrix, M.J. Vascular channel formation by human melanoma cells in vivo and in vitro: Vasculogenic mimicry. Am. J. Pathol. 1999, 155, 739–752. [Google Scholar] [CrossRef]
- Pinto, M.P.; Sotomayor, P.; Carrasco-Avino, G.; Corvalan, A.H.; Owen, G.I. Escaping antiangiogenic therapy: Strategies employed by cancer cells. Int. J. Mol. Sci. 2016, 17, 1489. [Google Scholar] [CrossRef] [PubMed]
- Shirakawa, K.; Wakasugi, H.; Heike, Y.; Watanabe, I.; Yamada, S.; Saito, K.; Konishi, F. Vasculogenic mimicry and pseudo-comedo formation in breast cancer. Int. J. Cancer 2002, 99, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Williamson, S.C.; Metcalf, R.L.; Trapani, F.; Mohan, S.; Antonello, J.; Abbott, B.; Leong, H.S.; Chester, C.P.; Simms, N.; Polanski, R.; et al. Vasculogenic mimicry in small cell lung cancer. Nat. Commun. 2016, 7, 13322. [Google Scholar] [CrossRef]
- Ma, S.; Pradeep, S.; Hu, W.; Zhang, D.; Coleman, R.; Sood, A. The role of tumor microenvironment in resistance to anti-angiogenic therapy. F1000Research 2018, 7, 326. [Google Scholar] [CrossRef]
- Dey, N.; De, P.; Brian, L.J. Evading anti-angiogenic therapy: Resistance to anti-angiogenic therapy in solid tumors. Am. J. Transl. Res. 2015, 7, 1675–1698. [Google Scholar] [CrossRef]
- Sun, H.; Zhang, D.; Yao, Z.; Lin, X.; Liu, J.; Gu, Q.; Dong, X.; Liu, F.; Wang, Y.; Yao, N.; et al. Anti-angiogenic treatment promotes triple-negative breast cancer invasion via vasculogenic mimicry. Cancer Biol. Ther. 2017, 18, 205–213. [Google Scholar] [CrossRef]
- Yang, W.H.; Xu, J.; Mu, J.B.; Xie, J. Revision of the concept of anti-angiogenesis and its applications in tumor treatment. Chronic Dis. Transl. Med. 2017, 3, 33–40. [Google Scholar] [CrossRef]
- Jensen, L.D. When tumors are (co-)opting to resist anti-angiogenic treatment. Transl. Cancer Res. 2016, 5, S1433–S1436. [Google Scholar] [CrossRef]
- Kilarski, W.W.; Samolov, B.; Petersson, L.; Kvanta, A.; Gerwins, P. Biomechanical regulation of blood vessel growth during tissue vascularization. Nat. Med. 2009, 15, 657–664. [Google Scholar] [CrossRef]
- Bridgeman, V.L.; Vermeulen, P.B.; Foo, S.; Bilecz, A.; Daley, F.; Kostaras, E.; Nathan, M.R.; Wan, E.; Frentzas, S.; Schweiger, T.; et al. Vessel co-option is common in human lung metastases and mediates resistance to anti-angiogenic therapy in preclinical lung metastasis models. J. Pathol. 2017, 241, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Coelho, A.L.; Gomes, M.P.; Catarino, R.J.; Rolfo, C.; Lopes, A.M.; Medeiros, R.M.; Araujo, A.M. Angiogenesis in nsclc: Is vessel co-option the trunk that sustains the branches? Oncotarget 2017, 8, 39795–39804. [Google Scholar] [CrossRef] [PubMed]
- Kalyanaraman, B. Teaching the basics of cancer metabolism: Developing antitumor strategies by exploiting the differences between normal and cancer cell metabolism. Redox Biol. 2017, 12, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Lipstein, M.R.; Pal, I.; Bates, S.E.; Deng, C. Metabolic symbiosis in cancer and its therapeutic implication. Semin. Oncol. 2017, 44, 233–234. [Google Scholar] [CrossRef] [PubMed]
- Allen, E.; Mieville, P.; Warren, C.M.; Saghafinia, S.; Li, L.; Peng, M.W.; Hanahan, D. Metabolic symbiosis enables adaptive resistance to anti-angiogenic therapy that is dependent on mtor signaling. Cell Rep. 2016, 15, 1144–1160. [Google Scholar] [CrossRef]
- Englinger, B.; Lotsch, D.; Pirker, C.; Mohr, T.; van Schoonhoven, S.; Boidol, B.; Lardeau, C.H.; Spitzwieser, M.; Szabo, P.; Heffeter, P.; et al. Acquired nintedanib resistance in fgfr1-driven small cell lung cancer: Role of endothelin-a receptor-activated abcb1 expression. Oncotarget 2016, 7, 50161–50179. [Google Scholar] [CrossRef]
- Pisarsky, L.; Bill, R.; Fagiani, E.; Dimeloe, S.; Goosen, R.W.; Hagmann, J.; Hess, C.; Christofori, G. Targeting metabolic symbiosis to overcome resistance to anti-angiogenic therapy. Cell Rep. 2016, 15, 1161–1174. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, Z.; Xu, Y.; Xing, L.; Liu, J.; Li, J.; Tan, Q. The expression of hypoxia inducible factor 1-alpha in lung cancer and its correlation with p53 and vegf. J. Huazhong Univ. Sci. Technol. Med. Sci. 2004, 24, 124–127. [Google Scholar]
- Cao, Y. Future options of anti-angiogenic cancer therapy. Chin. J. Cancer 2016, 35, 21. [Google Scholar] [CrossRef]
- Deryugina, E.I.; Quigley, J.P. Tumor angiogenesis: Mmp-mediated induction of intravasation- and metastasis-sustaining neovasculature. Matrix Biol. 2015, 44–46, 94–112. [Google Scholar] [CrossRef]
- Merchant, N.; Nagaraju, G.P.; Rajitha, B.; Lammata, S.; Jella, K.K.; Buchwald, Z.S.; Lakka, S.S.; Ali, A.N. Matrix metalloproteinases: Their functional role in lung cancer. Carcinogenesis 2017, 38, 766–780. [Google Scholar] [CrossRef] [PubMed]
- Hwang-Verslues, W.W.; Chang, P.H.; Jeng, Y.M.; Kuo, W.H.; Chiang, P.H.; Chang, Y.C.; Hsieh, T.H.; Su, F.Y.; Lin, L.C.; Abbondante, S.; et al. Loss of corepressor per2 under hypoxia up-regulates oct1-mediated emt gene expression and enhances tumor malignancy. Proc. Natl. Acad. Sci. USA 2013, 110, 12331–12336. [Google Scholar] [CrossRef] [PubMed]
- Yeo, C.D.; Kang, N.; Choi, S.Y.; Kim, B.N.; Park, C.K.; Kim, J.W.; Kim, Y.K.; Kim, S.J. The role of hypoxia on the acquisition of epithelial-mesenchymal transition and cancer stemness: A possible link to epigenetic regulation. Korean J. Intern. Med. 2017, 32, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Xie, K.; Ding, G.; Li, J.; Chen, K.; Li, H.; Qian, J.; Jiang, C.; Fang, J. Tumor resistance to anti-vegf therapy through up-regulation of vegf-c expression. Cancer Lett. 2014, 346, 45–52. [Google Scholar] [CrossRef]
- Kubo, H.; Fujiwara, T.; Jussila, L.; Hashi, H.; Ogawa, M.; Shimizu, K.; Awane, M.; Sakai, Y.; Takabayashi, A.; Alitalo, K.; et al. Involvement of vascular endothelial growth factor receptor-3 in maintenance of integrity of endothelial cell lining during tumor angiogenesis. Blood 2000, 96, 546–553. [Google Scholar] [CrossRef]
- Laakkonen, P.; Waltari, M.; Holopainen, T.; Takahashi, T.; Pytowski, B.; Steiner, P.; Hicklin, D.; Persaud, K.; Tonra, J.R.; Witte, L.; et al. Vascular endothelial growth factor receptor 3 is involved in tumor angiogenesis and growth. Cancer Res. 2007, 67, 593–599. [Google Scholar] [CrossRef]
- Smith, N.R.; Baker, D.; James, N.H.; Ratcliffe, K.; Jenkins, M.; Ashton, S.E.; Sproat, G.; Swann, R.; Gray, N.; Ryan, A.; et al. Vascular endothelial growth factor receptors vegfr-2 and vegfr-3 are localized primarily to the vasculature in human primary solid cancers. Clin. Cancer Res. 2010, 16, 3548–3561. [Google Scholar] [CrossRef]
- Fernando, N.T.; Koch, M.; Rothrock, C.; Gollogly, L.K.; D’Amore, P.A.; Ryeom, S.; Yoon, S.S. Tumor escape from endogenous, extracellular matrix–associated angiogenesis inhibitors by up-regulation of multiple proangiogenic factors. Clin. Cancer Res. 2008, 14, 1529–1539. [Google Scholar] [CrossRef]
- De Palma, M.; Biziato, D.; Petrova, T.V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 2017, 17, 457–474. [Google Scholar] [CrossRef]
- Dvorak, H.F. Tumor stroma, tumor blood vessels, and antiangiogenesis therapy. Cancer J. 2015, 21, 237–243. [Google Scholar] [CrossRef]
- Huijbers, E.J.; van Beijnum, J.R.; Thijssen, V.L.; Sabrkhany, S.; Nowak-Sliwinska, P.; Griffioen, A.W. Role of the tumor stroma in resistance to anti-angiogenic therapy. Drug Resist. Updates 2016, 25, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Bocci, G.; Di Paolo, A.; Danesi, R. The pharmacological bases of the antiangiogenic activity of paclitaxel. Angiogenesis 2013, 16, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, K.; Ohga, N.; Hida, Y.; Kawamoto, T.; Sadamoto, Y.; Ishikawa, S.; Maishi, N.; Akino, T.; Kondoh, M.; Matsuda, A.; et al. Tumor endothelial cells acquire drug resistance by mdr1 up-regulation via vegf signaling in tumor microenvironment. Am. J. Pathol. 2012, 180, 1283–1293. [Google Scholar] [CrossRef] [PubMed]
- Cascone, T.; Herynk, M.H.; Xu, L.; Du, Z.; Kadara, H.; Nilsson, M.B.; Oborn, C.J.; Park, Y.Y.; Erez, B.; Jacoby, J.J.; et al. Upregulated stromal egfr and vascular remodeling in mouse xenograft models of angiogenesis inhibitor-resistant human lung adenocarcinoma. J. Clin. Investig. 2011, 121, 1313–1328. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, C.; Tewari, B.N.; Kanchan, R.K.; Baghel, K.S.; Nautiyal, N.; Shrivastava, R.; Kaur, H.; Bhatt, M.L.; Bhadauria, S. Macrophages are recruited to hypoxic tumor areas and acquire a pro-angiogenic m2-polarized phenotype via hypoxic cancer cell derived cytokines oncostatin m and eotaxin. Oncotarget 2014, 5, 5350–5368. [Google Scholar] [CrossRef]
- De Palma, M.; Lewis, C.E. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell 2013, 23, 277–286. [Google Scholar] [CrossRef]
- Welford, A.F.; Biziato, D.; Coffelt, S.B.; Nucera, S.; Fisher, M.; Pucci, F.; Di Serio, C.; Naldini, L.; De Palma, M.; Tozer, G.M.; et al. Tie2-expressing macrophages limit the therapeutic efficacy of the vascular-disrupting agent combretastatin a4 phosphate in mice. J. Clin. Investig. 2011, 121, 1969–1973. [Google Scholar] [CrossRef]
- Incio, J.; Ligibel, J.A.; McManus, D.T.; Suboj, P.; Jung, K.; Kawaguchi, K.; Pinter, M.; Babykutty, S.; Chin, S.M.; Vardam, T.D.; et al. Obesity promotes resistance to anti-vegf therapy in breast cancer by up-regulating il-6 and potentially fgf-2. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Shojaei, F.; Wu, X.; Malik, A.K.; Zhong, C.; Baldwin, M.E.; Schanz, S.; Fuh, G.; Gerber, H.P.; Ferrara, N. Tumor refractoriness to anti-vegf treatment is mediated by cd11b+gr1+ myeloid cells. Nat. Biotechnol. 2007, 25, 911–920. [Google Scholar] [CrossRef]
- Caglevic, C.; Grassi, M.; Raez, L.; Listi, A.; Giallombardo, M.; Bustamante, E.; Gil-Bazo, I.; Rolfo, C. Nintedanib in non-small cell lung cancer: From preclinical to approval. Ther. Adv. Respir. Dis. 2015, 9, 164–172. [Google Scholar] [CrossRef]
- Paz-Ares, L.G.; Biesma, B.; Heigener, D.; von Pawel, J.; Eisen, T.; Bennouna, J.; Zhang, L.; Liao, M.; Sun, Y.; Gans, S.; et al. Phase iii, randomized, double-blind, placebo-controlled trial of gemcitabine/cisplatin alone or with sorafenib for the first-line treatment of advanced, nonsquamous non-small-cell lung cancer. J. Clin. Oncol. 2012, 30, 3084–3092. [Google Scholar] [CrossRef] [PubMed]
- Groen, H.J.; Socinski, M.A.; Grossi, F.; Juhasz, E.; Gridelli, C.; Baas, P.; Butts, C.A.; Chmielowska, E.; Usari, T.; Selaru, P.; et al. A randomized, double-blind, phase ii study of erlotinib with or without sunitinib for the second-line treatment of metastatic non-small-cell lung cancer (nsclc). Ann. Oncol. 2013, 24, 2382–2389. [Google Scholar] [CrossRef] [PubMed]
- Aisner, J.; Manola, J.B.; Dakhil, S.R.; Stella, P.J.; Sovak, M.A.; Schiller, J.H. Vandetanib plus chemotherapy for induction followed by vandetanib or placebo as maintenance for patients with advanced non-small-cell lung cancer: A randomized phase 2 precog study (pre0501). J. Thorac. Oncol. 2013, 8, 1075–1083. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.S.; Lee, K.H.; Sun, J.M.; Park, K.; Kang, E.S.; Cho, E.K.; Lee, D.H.; Kim, S.W.; Lee, G.W.; Kang, J.H.; et al. A randomized, phase ii study of vandetanib maintenance for advanced or metastatic non-small-cell lung cancer following first-line platinum-doublet chemotherapy. Lung Cancer 2013, 82, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Bareschino, M.A.; Schettino, C.; Colantuoni, G.; Rossi, E.; Rossi, A.; Maione, P.; Ciardiello, F.; Gridelli, C. The role of antiangiogenetic agents in the treatment of breast cancer. Curr. Med. Chem. 2011, 18, 5022–5032. [Google Scholar] [CrossRef]
- Hilberg, F.; Roth, G.J.; Krssak, M.; Kautschitsch, S.; Sommergruber, W.; Tontsch-Grunt, U.; Garin-Chesa, P.; Bader, G.; Zoephel, A.; Quant, J.; et al. Bibf 1120: Triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res. 2008, 68, 4774–4782. [Google Scholar] [CrossRef]
- Wakelee, H.A.; Lee, J.W.; Hanna, N.H.; Traynor, A.M.; Carbone, D.P.; Schiller, J.H. A double-blind randomized discontinuation phase-ii study of sorafenib (bay 43-9006) in previously treated non-small-cell lung cancer patients: Eastern cooperative oncology group study e2501. J. Thorac. Oncol. 2012, 7, 1574–1582. [Google Scholar] [CrossRef]
- Conley, S.J.; Baker, T.L.; Burnett, J.P.; Theisen, R.L.; Lazarus, D.; Peters, C.G.; Clouthier, S.G.; Eliasof, S.; Wicha, M.S. Crlx101, an investigational camptothecin-containing nanoparticle-drug conjugate, targets cancer stem cells and impedes resistance to antiangiogenic therapy in mouse models of breast cancer. Breast Cancer Res. Treat. 2015, 150, 559–567. [Google Scholar] [CrossRef]
- Lin, C.J.; Lin, Y.L.; Luh, F.; Yen, Y.; Chen, R.M. Preclinical effects of crlx101, an investigational camptothecin-containing nanoparticle drug conjugate, on treating glioblastoma multiforme via apoptosis and antiangiogenesis. Oncotarget 2016, 7, 42408–42421. [Google Scholar] [CrossRef]
- Falchook, G.S.; Wheler, J.J.; Naing, A.; Jackson, E.F.; Janku, F.; Hong, D.; Ng, C.S.; Tannir, N.M.; Lawhorn, K.N.; Huang, M.; et al. Targeting hypoxia-inducible factor-1alpha (hif-1alpha) in combination with antiangiogenic therapy: A phase i trial of bortezomib plus bevacizumab. Oncotarget 2014, 5, 10280–10292. [Google Scholar] [CrossRef]
- Khan, K.A.; Kerbel, R.S. Improving immunotherapy outcomes with anti-angiogenic treatments and vice versa. Nat. Rev. Clin. Oncol. 2018, 15, 310–324. [Google Scholar] [CrossRef] [PubMed]
- Allen, E.; Jabouille, A.; Rivera, L.B.; Lodewijckx, I.; Missiaen, R.; Steri, V.; Feyen, K.; Tawney, J.; Hanahan, D.; Michael, I.P.; et al. Combined antiangiogenic and anti-pd-l1 therapy stimulates tumor immunity through hev formation. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.T.; Man, S.; Xu, P.; Chow, A.; Paez-Ribes, M.; Lee, C.R.; Pirie-Shepherd, S.R.; Emmenegger, U.; Kerbel, R.S. Efficacy of cotargeting angiopoietin-2 and the vegf pathway in the adjuvant postsurgical setting for early breast, colorectal, and renal cancers. Cancer Res. 2016, 76, 6988–7000. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Wang, R.; Chen, Y.; Song, H.; Chen, L.; Huang, G. Combining antiangiogenic therapy with adoptive cell immunotherapy exerts better antitumor effects in non-small cell lung cancer models. PLoS ONE 2013, 8, e65757. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Chen, Y.S.; Yao, Y.D.; Chen, J.Q.; Chen, J.N.; Huang, S.Y.; Zeng, Y.J.; Yao, H.R.; Zeng, S.H.; Fu, Y.S.; et al. Ccl18 from tumor-associated macrophages promotes angiogenesis in breast cancer. Oncotarget 2015, 6, 34758–34773. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Shi, J.; Du, Y.; Chen, K.; Liu, Z.; Li, B.; Li, J.; Tao, F.; Gu, H.; Jiang, C.; et al. Profiling analysis of histone modifications and gene expression in lewis lung carcinoma murine cells resistant to anti-vegf treatment. PLoS ONE 2016, 11, e0158214. [Google Scholar] [CrossRef] [PubMed]
- Barr, M.P.; O’Byrne, K.J.; Al-Sarraf, N.; Gray, S.G. Vegf-mediated cell survival in non-small-cell lung cancer: Implications for epigenetic targeting of vegf receptors as a therapeutic approach. Epigenomics 2015, 7, 897–910. [Google Scholar] [CrossRef]
- Cheng, H.T.; Hung, W.C. Inhibition of lymphangiogenic factor vegf-c expression and production by the histone deacetylase inhibitor suberoylanilide hydroxamic acid in breast cancer cells. Oncol. Rep. 2013, 29, 1238–1244. [Google Scholar] [CrossRef]
- Roybal, J.D.; Zang, Y.; Ahn, Y.H.; Yang, Y.; Gibbons, D.L.; Baird, B.N.; Alvarez, C.; Thilaganathan, N.; Liu, D.D.; Saintigny, P.; et al. Mir-200 inhibits lung adenocarcinoma cell invasion and metastasis by targeting flt1/vegfr1. Mol. Cancer Res. 2011, 9, 25–35. [Google Scholar] [CrossRef]
- Shi, L.; Zhang, S.; Wu, H.; Zhang, L.; Dai, X.; Hu, J.; Xue, J.; Liu, T.; Liang, Y.; Wu, G. Mir-200c increases the radiosensitivity of non-small-cell lung cancer cell line a549 by targeting vegf-vegfr2 pathway. PLoS ONE 2013, 8, e78344. [Google Scholar] [CrossRef]
- Hu, J.; Cheng, Y.; Li, Y.; Jin, Z.; Pan, Y.; Liu, G.; Fu, S.; Zhang, Y.; Feng, K.; Feng, Y. Microrna-128 plays a critical role in human non-small cell lung cancer tumourigenesis, angiogenesis and lymphangiogenesis by directly targeting vascular endothelial growth factor-c. Eur. J. Cancer 2014, 50, 2336–2350. [Google Scholar] [CrossRef] [PubMed]
- Yin, R.; Guo, L.; Gu, J.; Li, C.; Zhang, W. Over expressing mir-19b-1 suppress breast cancer growth by inhibiting tumor microenvironment induced angiogenesis. Int. J. Biochem. Cell Biol. 2018, 97, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Bishayee, A.; Sethi, G. Bioactive natural products in cancer prevention and therapy: Progress and promise. Semin. Cancer Biol. 2016, 40–41, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, Y.; Terashita, N.; Muraguchi, T.; Fukusato, T.; Kubota, S. Effects of epigallocatechin-3-gallate (egcg) on a549 lung cancer tumor growth and angiogenesis. Biosci. Biotechnol. Biochem. 2013, 77, 1799–1803. [Google Scholar] [CrossRef]
- Sagar, S.M.; Yance, D.; Wong, R.K. Natural health products that inhibit angiogenesis: A potential source for investigational new agents to treat cancer-part 1. Curr. Oncol. 2006, 13, 14–26. [Google Scholar]
- Shi, J.; Liu, F.; Zhang, W.; Liu, X.; Lin, B.; Tang, X. Epigallocatechin-3-gallate inhibits nicotine-induced migration and invasion by the suppression of angiogenesis and epithelial-mesenchymal transition in non-small cell lung cancer cells. Oncol. Rep. 2015, 33, 2972–2980. [Google Scholar] [CrossRef]
- Sartippour, M.R.; Shao, Z.M.; Heber, D.; Beatty, P.; Zhang, L.; Liu, C.; Ellis, L.; Liu, W.; Go, V.L.; Brooks, M.N. Green tea inhibits vascular endothelial growth factor (vegf) induction in human breast cancer cells. J. Nutr. 2002, 132, 2307–2311. [Google Scholar] [CrossRef]
- Crew, K.D.; Brown, P.; Greenlee, H.; Bevers, T.B.; Arun, B.; Hudis, C.; McArthur, H.L.; Chang, J.; Rimawi, M.; Vornik, L.; et al. Phase ib randomized, double-blinded, placebo-controlled, dose escalation study of polyphenon e in women with hormone receptor-negative breast cancer. Cancer Prev. Res. 2012, 5, 1144–1154. [Google Scholar] [CrossRef]
- Samavat, H.; Ursin, G.; Emory, T.H.; Lee, E.; Wang, R.; Torkelson, C.J.; Dostal, A.M.; Swenson, K.; Le, C.T.; Yang, C.S.; et al. A randomized controlled trial of green tea extract supplementation and mammographic density in postmenopausal women at increased risk of breast cancer. Cancer Prev. Res. 2017, 10, 710–718. [Google Scholar] [CrossRef]
- Jiao, D.; Wang, J.; Lu, W.; Tang, X.; Chen, J.; Mou, H.; Chen, Q.Y. Curcumin inhibited hgf-induced emt and angiogenesis through regulating c-met dependent pi3k/akt/mtor signaling pathways in lung cancer. Mol. Ther. Oncolytics 2016, 3, 16018. [Google Scholar] [CrossRef]
- Ferreira, L.C.; Arbab, A.S.; Jardim-Perassi, B.V.; Borin, T.F.; Varma, N.R.; Iskander, A.S.; Shankar, A.; Ali, M.M.; Zuccari, D.A. Effect of curcumin on pro-angiogenic factors in the xenograft model of breast cancer. Anticancer Agents Med. Chem. 2015, 15, 1285–1296. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, N.; Aggarwal, B.B.; Newman, R.A.; Wolff, R.A.; Kunnumakkara, A.B.; Abbruzzese, J.L.; Ng, C.S.; Badmaev, V.; Kurzrock, R. Phase ii trial of curcumin in patients with advanced pancreatic cancer. Clin. Cancer Res. 2008, 14, 4491–4499. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.L.; Hsu, C.H.; Lin, J.K.; Hsu, M.M.; Ho, Y.F.; Shen, T.S.; Ko, J.Y.; Lin, J.T.; Lin, B.R.; Ming-Shiang, W.; et al. Phase i clinical trial of curcumin, a chemopreventive agent, in patients with high-risk or pre-malignant lesions. Anticancer Res. 2001, 21, 2895–2900. [Google Scholar] [PubMed]
- Wang, L.Q.; Shi, H.S.; Wang, Y.S. [liposomal curcumin inhibits tumor growth and angiogenesis in lewis lung cancer]. Sichuan Da Xue Xue Bao Yi Xue Ban 2013, 44, 46–48. [Google Scholar]
- Shi, L.; Pan, H.; Liu, Z.; Xie, J.; Han, W. Roles of pfkfb3 in cancer. Signal Transduct. Target. Ther. 2017, 2, 17044. [Google Scholar] [CrossRef]
- Cantelmo, A.R.; Conradi, L.C.; Brajic, A.; Goveia, J.; Kalucka, J.; Pircher, A.; Chaturvedi, P.; Hol, J.; Thienpont, B.; Teuwen, L.A.; et al. Inhibition of the glycolytic activator pfkfb3 in endothelium induces tumor vessel normalization, impairs metastasis, and improves chemotherapy. Cancer Cell 2016, 30, 968–985. [Google Scholar] [CrossRef]
- Chowdhury, N.; Vhora, I.; Patel, K.; Doddapaneni, R.; Mondal, A.; Singh, M. Liposomes co-loaded with 6-phosphofructo-2-kinase/fructose-2, 6-biphosphatase 3 (pfkfb3) shrna plasmid and docetaxel for the treatment of non-small cell lung cancer. Pharm. Res. 2017, 34, 2371–2384. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Preclinical Studies | |||
|---|---|---|---|
| Strategy | Targets | Main Conclusion | |
| Targeting multiple angiogenic proteins | VEGFRs (1, 2 & 3), PDGFa & b, FGFRs (1–4), FLT3 and SRC family kinases via Nintedanib | Nintedanib showed promising inhibition in the tumor growth of NSCLC cells | |
| Targeting hypoxia | HIF-1α via camptothecin | Combination of camptothecin with bevacizumab showed improved response of breast cancer cells and decreased the cancer stem cells population | |
| Anti-angiogenic immunotherapy | VEGF and angiopoietin 2 | Targeting VEGF and angiopoietin 2 in combination with anti-PD-L1 antibody improved the survival of mouse breast cancer model | |
| Targeting epigenetic regulators | VEGF-C via miR-128 | Overexpression of miR-128 resulted in reducing the expression of VEGF-C and subsequent suppression of angiogenesis in NSCLC tumor xenograft model | |
| Alternative anti-angiogenic compounds | HIF-1α, VEGF, VEGFR, PKC and endostatin via EGCG | EGCG reduced the density of tumor vessels and inhibited angiogenesis in breast and lung cancer xenograft models | |
| Clinical Studies | |||
| Clinical Trial ID | Phase No. | Treatment | Condition or Disease |
| NCT03377023 | Phase I/II | Combination of Nintedanib with nivolumab and ipilimumab | Advanced or Metastatic Non-small Cell Lung Cancer |
| NCT02299141 | Phase I | Nintedanib | Metastatic Non-small Cell Lung Cancer That Cannot Be Removed by Surgery and Mutations in Nintedanib-Targeted Genes |
| NCT00428545 | Phase I | Combination of Bevacizumab and Bortezomib | Advanced Malignancies including breast and lung cancer |
| NCT01454102 | Phase I | Combination of bevacizumab with anti-PD-1 nivolumab | Non-small Cell Lung cancer |
| NCT03072992 | Phase II | Combination of curcumin and paclitaxel | Advanced and metastatic breast cancer |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramadan, W.S.; Zaher, D.M.; Altaie, A.M.; Talaat, I.M.; Elmoselhi, A. Potential Therapeutic Strategies for Lung and Breast Cancers through Understanding the Anti-Angiogenesis Resistance Mechanisms. Int. J. Mol. Sci. 2020, 21, 565. https://doi.org/10.3390/ijms21020565
Ramadan WS, Zaher DM, Altaie AM, Talaat IM, Elmoselhi A. Potential Therapeutic Strategies for Lung and Breast Cancers through Understanding the Anti-Angiogenesis Resistance Mechanisms. International Journal of Molecular Sciences. 2020; 21(2):565. https://doi.org/10.3390/ijms21020565
Chicago/Turabian StyleRamadan, Wafaa S., Dana M. Zaher, Alaa M. Altaie, Iman M. Talaat, and Adel Elmoselhi. 2020. "Potential Therapeutic Strategies for Lung and Breast Cancers through Understanding the Anti-Angiogenesis Resistance Mechanisms" International Journal of Molecular Sciences 21, no. 2: 565. https://doi.org/10.3390/ijms21020565
APA StyleRamadan, W. S., Zaher, D. M., Altaie, A. M., Talaat, I. M., & Elmoselhi, A. (2020). Potential Therapeutic Strategies for Lung and Breast Cancers through Understanding the Anti-Angiogenesis Resistance Mechanisms. International Journal of Molecular Sciences, 21(2), 565. https://doi.org/10.3390/ijms21020565