Oligonuclear Actinoid Complexes with Schiff Bases as Ligands—Older Achievements and Recent Progress

 ,
, 


Abstract
1. Scope and Organization of this Review
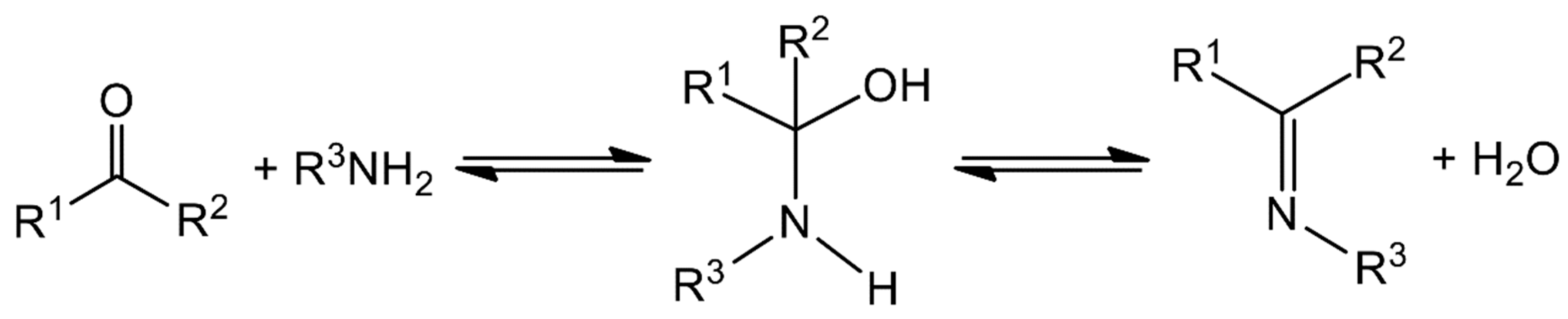
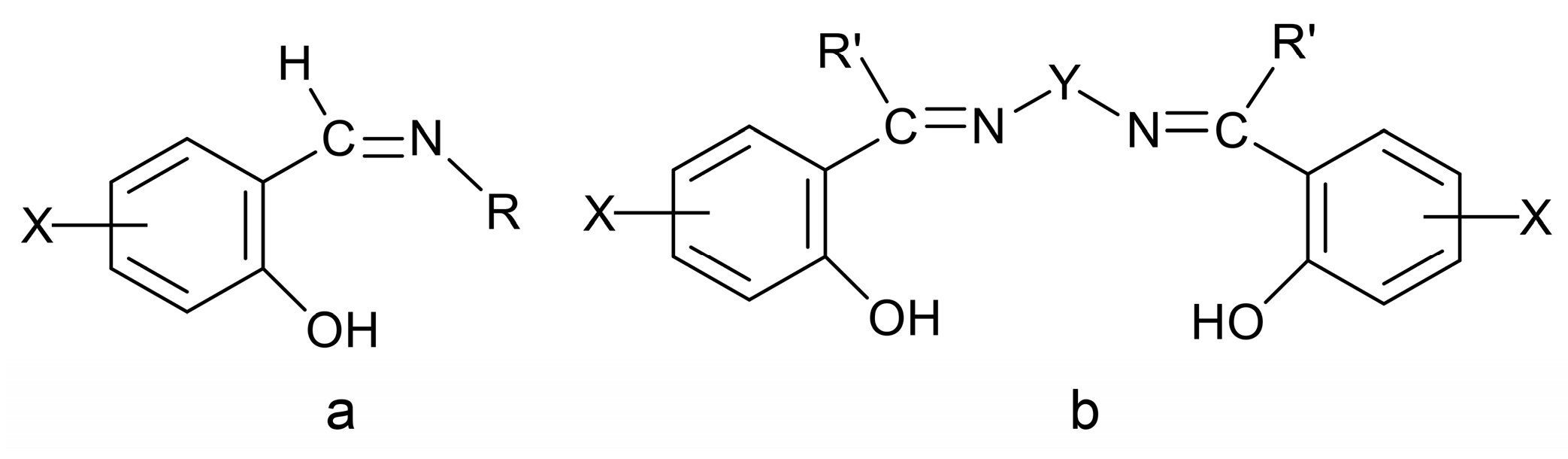
2. Schiff Bases and Their Coordination Chemistry
3. A General Overview of the Chemistry of Actinoids
4. Scientific Interest in the Chemistry of Actinoid-Schiff Base Complexes
5. Oligonuclear Schiff-Base Complexes with Actinoids Other Than Thorium and Uranium
5.1. Tetranuclear Neptunyl(V) Clusters Supported by Salen2− Ligands

5.2. Unique Np(III)/U(VI) Complexes

6. Dinuclear and Oligonuclear Thorium(IV)—Schiff Base Complexes
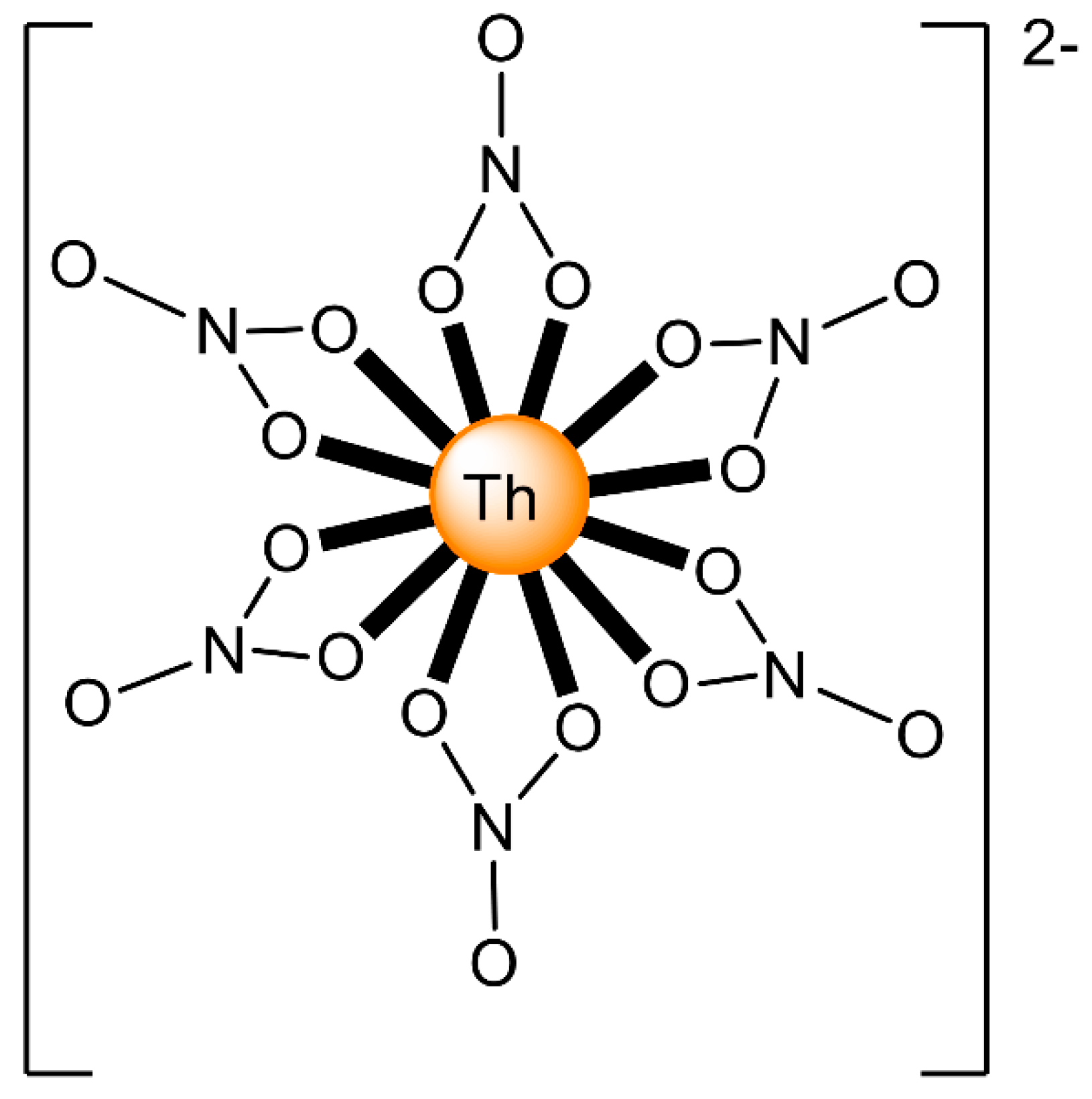
6.1. Dinuclear Thorium(IV) Complexes


6.2. Two Tetranuclear Thorium(IV) Complexes

7. Dinuclear and Oligonuclear Uranium-Schiff Base Complexes
7.1. Dinuclear Uranyl(VI) Complexes





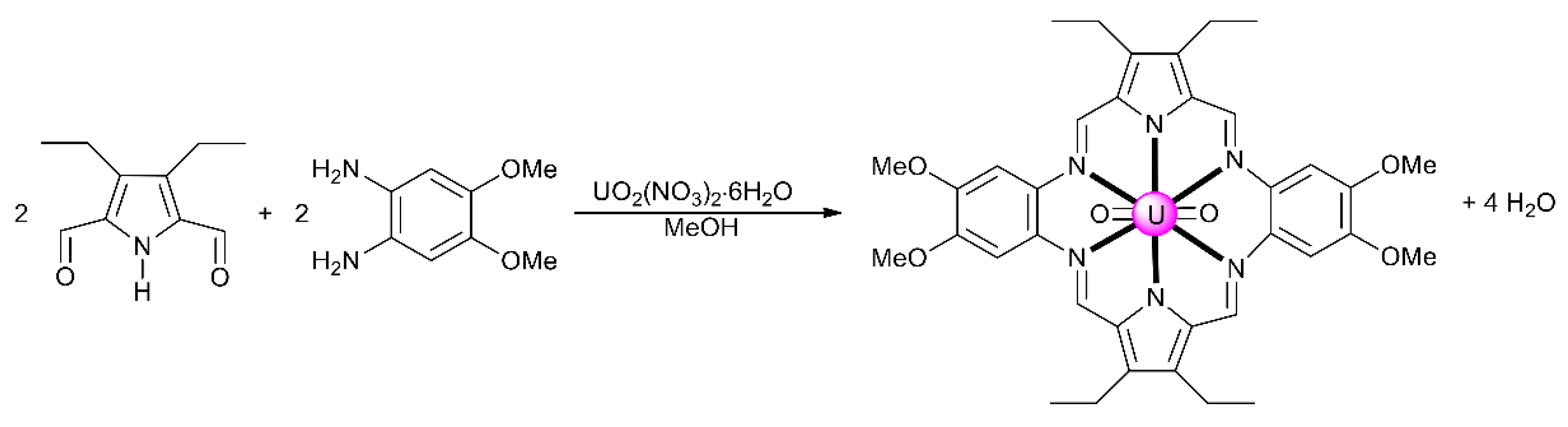
7.2. Trinuclear and Tetranuclear Uranyl(VI) Clusters



7.3. Uranium Complexes at the Oxidation States III, IV and V
7.4. Dinuclear and Oligonuclear Uranyl(V) Complexes




7.5. Dinuclear and Oligonuclear Uranium(IV) Clusters









7.6. Dinuclear Uranium(III) Complexes

7.7. Dinuclear and Oligonuclear Mixed-Valence Uranium Complexes





8. Concluding Comments in Brief and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rigamonti, L.; Forni, A.; Righetto, S.; Pasini, A. Push–pull unsymmetrical substitution in nickel(II) complexes with tetradentate N2O2 Schiff base ligands: Synthesis, structures and linear–nonlinear optical studies. Dalton Trans. 2019, 48, 11217–11234. [Google Scholar] [CrossRef] [PubMed]
- Kindra, D.R.; Evans, W.J. Magnetic susceptibility of uranium complexes. Chem. Rev. 2014, 114, 8865–8882. [Google Scholar] [CrossRef] [PubMed]
- Casellato, U.; Vidali, M.; Vigato, P.A. Actinide complexes with the most common organic ligands forming five- and six-membered chelating rings. Inorg. Chim. Acta 1976, 18, 77–112. [Google Scholar] [CrossRef]
- Cattalini, L.; Croatto, U.; Degetto, S.; Tondello, E. Uranyl chelate complexes. Inorg. Chim. Acta Rev. 1971, 5, 19–43. [Google Scholar] [CrossRef]
- Costisor, O.; Linert, W. 4f and 5f metal ion directed Schiff condensation. Rev. Inorg. Chem. 2004, 24, 61–95. [Google Scholar] [CrossRef]
- Sessler, J.L.; Melfi, P.J.; Pantos, G.D. Uranium complexes of multidentate N-donor ligands. Coord. Chem. Rev. 2006, 250, 816–843. [Google Scholar] [CrossRef]
- Hawkins, C.A.; Bustillos, C.G.; Copping, R.; May, I.; Nilsson, M. Investigations of hydrophilic Schiff base ligands for the separation of actinyl and lanthanide cations. NEA/NSC/R 2015, 2, 315–323. [Google Scholar]
- Burns, C.J.; Neu, M.P.; Boukhalfa, H.; Gutowski, K.E.; Bridges, N.J.; Rogers, R.D. The Actinides. In Comprehensive Coordination Chemistry II; McCleverty, J.A., Meyer, T.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2003; Volume 3, pp. 189–330. [Google Scholar]
- Cowie, B.E.; Purkis, J.M.; Austin, J.; Love, J.B.; Arnold, P.L. Thermal and photochemical reduction and functionalization chemistry of the uranyl dication, [UVIO2]2+. Chem. Rev. 2019, 119, 10595–10637. [Google Scholar] [CrossRef]
- Maurya, M.R.; Maurya, R.C. Coordination chemistry of Schiff base complexes of uranium. Rev. Inorg. Chem. 1995, 15, 1–107. [Google Scholar] [CrossRef]
- Liu, X.; Hamon, J.-R. Recent developments in penta-, hexa- and heptadentate Schiff base ligands and their metal complexes. Coord. Chem. Rev. 2019, 389, 94–118. [Google Scholar] [CrossRef]
- Coxall, R.A.; Harris, S.G.; Henderson, D.K.; Parsons, S.; Tasker, P.A.; Winpenny, R.E.P. Inter-ligand reactions: In situ formation of new polydentate ligands. J. Chem. Soc. Dalton Trans. 2000, 2349–2356. [Google Scholar] [CrossRef]
- Tsantis, S.T.; Mouzakitis, M.; Savvidou, A.; Raptopoulou, C.P.; Psycharis, V.; Perlepes, S.P. The “periodic table” of benzotriazoles: Uranium(VI) complexes. Inorg. Chem. Commun. 2015, 59, 57–60. [Google Scholar] [CrossRef]
- Tsantis, S.T.; Zagoraiou, E.; Savvidou, A.; Raptopoulou, C.P.; Psycharis, V.; Szyrwiel, L.; Hołyńska, M.; Perlepes, S.P. Binding of oxime group to uranyl ion. Dalton Trans. 2016, 45, 9307–9319. [Google Scholar] [CrossRef] [PubMed]
- Tsantis, S.T.; Bekiari, V.; Raptopoulou, C.P.; Tzimopoulos, D.I.; Psycharis, V.; Perlepes, S.P. Dioxidouranium(VI) complexes with Schiff bases possessing an ONO donor set: Synthetic, structural and spectroscopic studies. Polyhedron 2018, 152, 172–178. [Google Scholar] [CrossRef]
- Tsantis, S.T.; Lagou-Rekka, A.; Konidaris, K.F.; Raptopoulou, C.P.; Bekiari, V.; Psycharis, V.; Perlepes, S.P. Tetranuclear oxido-bridged thorium(IV) clusters obtained using tridentate Schiff bases. Dalton Trans. 2019, 48, 15668–15678. [Google Scholar] [CrossRef] [PubMed]
- Mylonas-Margaritis, I.; Perlepe, P.S.; Masič, M.; Raptopoulou, C.P.; Psycharis, V.; Escuer, A.; Perlepes, S.P. Carbonato- and methanediolato(-2)-bridged nickel(II) coordination clusters from the use of N-salicylidene-4-methyl-o-aminophenol. Inorg. Chem. Commun. 2017, 83, 113–117. [Google Scholar] [CrossRef]
- Alexopoulou, K.I.; Terzis, A.; Raptopoulou, C.P.; Psycharis, V.; Escuer, A.; Perlepes, S.P. NiII20 “bowls” from the use of tridentate Schiff bases. Inorg. Chem. 2015, 54, 5615–5617. [Google Scholar] [CrossRef]
- Anastasiadis, N.C.; Granadeiro, C.M.; Mayans, J.; Raptopoulou, C.P.; Bekiari, V.; Cunha-Silva, L.; Psycharis, V.; Escuer, A.; Balula, S.S.; Konidaris, K.F.; et al. Multifunctionality in two families of dinuclear lanthanide(III) complexes with a tridentate Schiff-base ligand. Inorg. Chem. 2019, 58, 9581–9585. [Google Scholar] [CrossRef]
- Anastasiadis, N.C.; Kalofolias, D.A.; Philippidis, A.; Tzani, S.; Raptopoulou, C.P.; Psycharis, V.; Milios, C.J.; Escuer, A.; Perlepes, S.P. A family of dinuclear lanthanide(III) complexes from the use of a tridentate Schiff base. Dalton Trans. 2015, 44, 10200–10209. [Google Scholar] [CrossRef]
- Anastasiadis, N.C.; Granadeiro, C.M.; Klouras, N.; Cunha-Silva, L.; Raptopoulou, C.P.; Psycharis, V.; Bekiari, V.; Balula, S.S.; Escuer, A.; Perlepes, S.P. Dinuclear lanthanide(III) complexes by metal-ion-assisted hydration of di-2-pyridyl ketone azine. Inorg. Chem. 2013, 52, 4145–4147. [Google Scholar] [CrossRef]
- Hernández-Molina, R.; Mederos, A. Acyclic and macrocyclic Schiff base ligands. In Comprehensive Coordination Chemistry II; McCleverty, J.A., Meyer, T.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2004; Volume 1, pp. 411–446. [Google Scholar]
- Hadjoudis, E.; Mavridis, I.M. Photochromism and thermochromism of Schiff bases in the solid state: Structural aspects. Chem. Soc. Rev. 2004, 33, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S. Advancement in stereochemical aspects of Schiff base metal complexes. Coord. Chem. Rev. 1999, 190–192, 537–555. [Google Scholar] [CrossRef]
- Costamagna, J.; Vargas, J.; Latorre, R.; Alvarado, A.; Mena, G. Coordination compounds of copper, nickel and iron with Schiff bases derived from hydroxynaphthaldehydes and salicylaldehydes. Coord. Chem. Rev. 1992, 119, 67–88. [Google Scholar] [CrossRef]
- Gupta, K.C.; Sutar, A.K. Catalytic activities of Schiff base transition metal complexes. Coord. Chem. Rev. 2008, 252, 1420–1450. [Google Scholar] [CrossRef]
- Cozzi, P.G. Metal-salen Schiff base complexes in catalysis: Practical aspects. Chem. Soc. Rev. 2004, 33, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Long, J. Luminescent Schiff-base lanthanide single-molecule magnets: The association between optical and magnetic properties. Front. Chem. 2019, 7, 63. [Google Scholar] [CrossRef]
- Camp, C.; Guidal, V.; Biswas, B.; Pécaut, J.; Dubois, L.; Mazzanti, M. Multielectron redox chemistry of lanthanide Schiff-base complexes. Chem. Sci. 2012, 3, 2433–2448. [Google Scholar] [CrossRef]
- Constable, E.C. Metals and Ligand Reactivity; VCH: Weinheim, Germany, 1996; pp. 72–78, 135–182. [Google Scholar]
- Ribas Gisbert, J. Coordination Chemistry; Wiley-VCH: Weinheim, Germany, 2008; pp. 132–133. [Google Scholar]
- Miessler, G.L.; Fischer, P.J.; Tarr, D.A. Inorganic Chemistry, 5th ed.; Pearson: Upper Saddle River, NJ, USA, 2014; pp. 469–470. [Google Scholar]
- Cotton, F.A.; Wilkinson, G.; Murillo, C.A.; Bochmann, M. Advanced Inorganic Chemistry, 6th ed.; Wiley: New York, NY, USA, 1999; pp. 348, 375, 376, 1130–1164. [Google Scholar]
- Housecroft, C.E.; Sharpe, A.G. Inorganic Chemistry, 5th ed.; Pearson: Harlow, UK, 2018; pp. 1033–1064. [Google Scholar]
- Knope, K.E.; Soderholm, L. Solution and solid-state structural chemistry of actinide hydrates and their hydrolysis and condensation products. Chem. Rev. 2013, 113, 944–994. [Google Scholar] [CrossRef]
- Liddle, S.T. The renaissance of non-aqueous uranium chemistry. Angew. Chem. Int. Ed. 2015, 54, 8604–8641. [Google Scholar] [CrossRef]
- Harrowfield, J.; Thuéry, P. Element 92—Uranium. Aust. J. Chem. 2019, 72, 329–333. [Google Scholar] [CrossRef]
- Abney, C.W.; Mayes, R.T.; Saito, T.; Dai, S. Materials for the recovery of uranium from seawater. Chem. Rev. 2017, 117, 13935–14013. [Google Scholar] [CrossRef] [PubMed]
- Kaltsoyannis, N.; Liddle, S.T. Catalyst: Nuclear power in the 21st century. Chem 2016, 1, 659–662. [Google Scholar] [CrossRef]
- Natrajan, L.S.; Swinburne, A.N.; Andrews, M.B.; Randall, S.; Heath, S.L. Redox and environmentally relevant aspects of actinide(IV) coordination chemistry. Coord. Chem. Rev. 2014, 266–267, 171–193. [Google Scholar] [CrossRef]
- Tutson, C.D.; Gorden, A.E.V. Thorium coordination: A comprehensive review based on coordination number. Coord. Chem. Rev. 2017, 333, 27–43. [Google Scholar] [CrossRef]
- Mohapatra, P.K. Actinide ion extraction using room temperature ionic liquids: Opportunities and challenges for nuclear fuel cycle applications. Dalton Trans. 2017, 46, 1730–1747. [Google Scholar] [CrossRef] [PubMed]
- Leoncini, A.; Huskens, J.; Verboom, W. Ligands for f-element extraction used in the nuclear fuel cycle. Chem. Soc. Rev. 2017, 46, 7229–7273. [Google Scholar] [CrossRef]
- Kaltsoyannis, N. Does covalency increase or decrease across the actinide series? Implications for minor actinide partitioning. Inorg. Chem. 2013, 52, 3407–3413. [Google Scholar] [CrossRef]
- Franczyk, T.S.; Czerwinski, K.R.; Raymond, K.N. Stereognostic coordination chemistry. 1. The design and synthesis of chelators for the uranyl ion. J. Am. Chem. Soc. 1992, 114, 8138–8146. [Google Scholar] [CrossRef]
- Loiseau, T.; Mihalcea, I.; Henry, N.; Volkringer, C. The crystal chemistry of uranium carboxylates. Coord. Chem. Rev. 2014, 266–267, 69–109. [Google Scholar] [CrossRef]
- Bart, S. Bonding with actinides. Nat. Chem. 2017, 9, 832. [Google Scholar]
- Rudkevich, D.M.; Verboom, W.; Brzozka, Z.; Palys, M.J.; Stauthamer, W.P.R.V.; van Hummel, G.J.; Franken, S.M.; Harkema, S.; Engbersen, J.F.J.; Reinhoudt, D.N. Functionalized UO2 salenes: Neutral receptors for anions. J. Am. Chem. Soc. 1994, 116, 4341–4351. [Google Scholar] [CrossRef]
- Van Axel Castelli, V.; Cacciapaglia, R.; Chiosis, G.; Van Veggel, F.C.J.M.; Mandolini, L.; Reinhoudt, D.N. The uranyl unit as electrophilic catalyst of acyl transfer reactions. Inorg. Chim. Acta 1996, 246, 181–193. [Google Scholar] [CrossRef][Green Version]
- Van Axel Castelli, V.; Dalla Cort, A.; Mandolini, L. Supramolecular catalysis of 1,4-thiol addition by salophen-uranyl complexes. J. Am. Chem. Soc. 1998, 120, 12688–12689. [Google Scholar] [CrossRef]
- Van Axel Castelli, V.; Dalla Cort, A.; Mandolini, L.; Reinhoudt, D.N.; Schiaffino, L. New insight into the mechanism of the conjugate addition of benzenethiol to cyclic and acyclic enones and of the corresponding uranyl−salophen-catalysed version. Eur. J. Org. Chem. 2003, 627–633. [Google Scholar] [CrossRef]
- Herbst, R.S.; Baron, P.; Nilsson, M. Standard and advanced separation: PUREX processes for nuclear fuel reprocessing. In Advanced Separation Techniques for Nuclear Fuel Reprocessing and Radioactive Waste Treatment; Nash, K.L., Lumetta, G.J., Eds.; Woodhead Publishing Series in Energy: Cambridge, MA, USA, 2011; Chapter 6; pp. 141–175. [Google Scholar]
- Nilsson, M.; Nash, K.L. A review of the development and operational characteristics of the TALSPEAK process. Solv. Extr. Ion Exch. 2007, 25, 665–701. [Google Scholar] [CrossRef]
- Geist, A.; Müllich, U.; Magnusson, D.; Kaden, P.; Modolo, G.; Wilden, A.; Zevaco, T. Actinide(III)/lanthanide(III) separation via selective aqueous complexation of actinides(III) using a hydrophilic 2,6-bis(1,2,4-triazin-3-yl)-pyridine in nitric acid. Solv. Extr. Ion Exch. 2012, 30, 433–444. [Google Scholar] [CrossRef]
- Takao, K.; Kato, M.; Takao, S.; Nagasawa, A.; Bernhard, G.; Hennig, C.; Ikeda, Y. Molecular structure and electrochemical behavior of uranyl(VI) complex with pentadentate Schiff base ligand: Prevention of uranyl(V) cation-cation interaction by fully chelating equatorial coordination sites. Inorg. Chem. 2010, 49, 2349–2359. [Google Scholar] [CrossRef]
- Hawkins, C.A.; Bustillos, C.G.; Copping, R.; Scott, B.L.; May, I.; Nilsson, M. Challenging conventional f-element separation chemistry-reversing uranyl(VI)/lanthanide(III) solvent extraction selectivity. Chem. Commun. 2014, 50, 8670–8673. [Google Scholar] [CrossRef]
- Back, D.F.; Bonfada, E.; de Oliveira, G.M.; Schulz-Lang, E. Chelation of ThIV, EuIII and NdIII by dianionic N,N′-bis(pyridoxylideneiminato)ethylene, (Pyr2en)2−. On the search of feasible modelings for heavy metals damage inhibition in living beings. J. Inorg. Biochem. 2007, 101, 709–714. [Google Scholar] [CrossRef]
- Kumar, S.K.A.; Vijayakrishna, K.; Sivaramakrishna, A.; Brahmmananda, C.R.; Sivaraman, N.; Sahoo, S.K. Development of the smartphone-assisted colorimetric detection of thorium by using new Schiff’s base and its applications to real time samples. Inorg. Chem. 2018, 57, 15270–15279. [Google Scholar]
- Klamm, B.E.; Windorff, C.J.; Celis-Barros, C.; Marsh, M.L.; Meeker, D.S.; Albrecht-Schmitt, T.E. Experimental and theoretical comparison of transition-metal and actinide tetravalent Schiff base coordination complexes. Inorg. Chem. 2018, 57, 15389–15398. [Google Scholar] [CrossRef] [PubMed]
- Klamm, B.E.; Windorff, C.J.; Celis-Barros, C.; Marsh, M.L.; Albrecht-Schmitt, T.E. Synthesis, spectroscopy, and theoretical details of uranyl Schiff-base coordination complexes. Inorg. Chem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Schnaars, D.D.; Batista, E.R.; Gaunt, A.J.; Hayton, T.W.; May, I.; Reilly, S.D.; Scott, B.L.; Wu, G. Differences in actinide metal-ligand orbital interactions: Comparison of U(IV) and Pu(IV) β-ketoiminate N.,O donor complexes. Chem. Commun. 2011, 47, 7647–7649. [Google Scholar] [CrossRef]
- Chuguryan, D.G.; Dzyubenko, V.I. Complex formation between neptunium(IV) and neptunium(V) with thiosemicarbazidodiacetic acid in solution. Sov. Radiochem. (Engl. Transl.) 1988, 30, 158–164. [Google Scholar]
- Copping, R.; Mougel, V.; Den Auwer, C.; Berthon, C.; Moisy, P.; Mazzanti, M. A tetrameric neptunyl(V) cluster supported by a Schiff base ligand. Dalton Trans. 2012, 41, 10900–10902. [Google Scholar] [CrossRef]
- Arnold, P.L.; Dutkiewicz, M.S.; Zegke, M.; Walter, O.; Apostolidis, C.; Hollis, E.; Pécharman, A.-F.; Magnani, N.; Griveau, J.C.; Colineau, E.; et al. Subtle interactions and electron transfer between UIII, NpIII, or PuIII and uranyl mediated by the oxo group. Angew. Chem. Int. Ed. 2016, 55, 12797–12801. [Google Scholar] [CrossRef]
- Aghabozorg, H.; Palenik, R.C.; Palenik, G.J. A ten-coordinate oxo-bridged thorium complex with an unusual coordination polyhedron. Inorg. Chim. Acta 1983, 76, L1259–L1260. [Google Scholar] [CrossRef]
- Casellato, U.; Guerriero, P.; Tamburini, S.; Vigato, P.A.; Graziani, A. Preparation, characterization and crystal structure of a binuclear thorium(IV) complex with a pentadentate compartmental Schiff base. Inorg. Chim. Acta 1987, 134, 165–174. [Google Scholar] [CrossRef]
- Bino, A.; Chayat, R. A new hydroxo-bridged thorium(IV) dimer: Preparation and structure of di-μ-hydroxo-bis[aquanitrato(2,6-diacetylpyridinedisemicarbazone)thorium(IV)] nitrate tetrahydrate. Inorg. Chim. Acta 1987, 129, 273–276. [Google Scholar] [CrossRef]
- Arnold, J.; Gianetti, T.L.; Kashtan, Y. Thorium lends a fiery hand. Nat. Chem. 2014, 6, 554. [Google Scholar] [CrossRef]
- Inman, C.J.; Cloke, F.G.N. The experimental determination of Th(IV)/Th(III) redox potentials in organometallic thorium complexes. Dalton Trans. 2019, 48, 10782–10784. [Google Scholar] [CrossRef]
- Huh, D.N.; Roy, S.; Ziller, J.W.; Furche, F.; Evans, W.J. Isolation of a square-planar Th(III) complex: Synthesis and structure of [Th(OC6H2tBu2-2,6-Me-4)4]1–. J. Am. Chem. Soc. 2019, 141, 12458–12463. [Google Scholar] [CrossRef]
- Lung, M.; Gremm, O. Perspectives of the thorium fuel cycle. Nucl. Eng. Des. 1998, 180, 133–146. [Google Scholar] [CrossRef]
- McSkimming, A.; Su, J.; Cheisson, T.; Gau, M.R.; Carroll, P.J.; Batista, E.R.; Yang, P.; Schelter, E.J. Coordination chemistry of a strongly-donating hydroxylamine with early actinides: An investigation of redox properties and electronic structure. Inorg. Chem. 2018, 57, 4387–4394. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Brown, A.T.; Tan, K.; Chabal, Y.J.; Balkus, J.K.J. Selective extraction of thorium from rare earth elements using wrinkled mesoporous carbon. J. Am. Chem. Soc. 2018, 140, 14735–14739. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Burns, P.C. Clusters of actinides with oxide, peroxide, or hydroxide bridges. Chem. Rev. 2013, 113, 1097–1120. [Google Scholar] [CrossRef] [PubMed]
- Salmon, L.; Thuéry, P.; Rivière, E.; Ephritikhine, M. Synthesis, structure, and magnetic behavior of a series of trinuclear Schiff base complexes of 5f (UIV, ThIV) and 3d (CuII, ZnII) ions. Inorg. Chem. 2006, 45, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Wildman, E.P.; Balázs, G.; Wooles, A.J.; Scheer, M.; Liddle, S.T. Thorium-phosphorus triamidoamine complexes containing Th-P single-and multiple-bond interactions. Nat. Commun. 2016, 7, 12884. [Google Scholar] [CrossRef] [PubMed]
- Cheisson, T.; Kersey, K.D.; Mahieu, N.; McSkimming, A.; Gau, M.R.; Carroll, P.J.; Schelter, E.J. Multiple bonding in lanthanides and actinides: Direct comparison of covalency in thorium(IV)- and cerium(IV)-imido complexes. J. Am. Chem. Soc. 2019, 141, 9185–9190. [Google Scholar] [CrossRef]
- Li, P.; Goswami, S.; Otake, K.-i.; Wang, X.; Chen, Z.; Hanna, S.L.; Farha, O.K. Stabilization of an unprecedented hexanuclear secondary building unit in a thorium-based metal–organic framework. Inorg. Chem. 2019, 58, 3586–3590. [Google Scholar] [CrossRef]
- Ejegbavwo, O.A.; Martin, C.R.; Olorunfemi, O.A.; Leith, G.A.; Ly, R.T.; Rice, A.M.; Dolgopolova, E.A.; Smith, M.D.; Karakalos, S.G.; Birkner, N.; et al. Thermodynamics and electronic properties of heterometallic multinuclear actinide-containing metal–organic frameworks with “structural memory”. J. Am. Chem. Soc. 2019, 141, 11628–11640. [Google Scholar] [CrossRef]
- Hardy, E.E.; Wyss, K.M.; Gorden, J.D.; Ariyarantha, I.R.; Miliordos, E.; Gorden, A.E.V. Th(IV) and Ce(IV) napthylsalophen sandwich complexes: Characterization of unusual thorium fluorescence in solution and solid-state. Chem. Commun. 2017, 53, 11984–11987. [Google Scholar] [CrossRef]
- Casellato, U.; Guerriero, P.; Tamburini, S.; Vigato, P.A.; Graziani, R. Synthesis, properties, and crystal structures of new mono- and homo-binuclear uranyl(VI) complexes with compartmental Schiff bases. J. Chem. Soc. Dalton Trans. 1990, 1533–1541. [Google Scholar] [CrossRef]
- Tamburini, S.; Vigato, P.A.; Guerriero, P.; Casellato, U.; Aguiari, A. Mononuclear and polynuclear complexes containing f ions. Inorg. Chim. Acta 1991, 183, 81–90. [Google Scholar] [CrossRef]
- Rao, P.V.; Rao, C.P.; Sreecdhara, A.; Wcgelius, E.K.; Rissanen, K.; Kolehmainen, E. Synthesis, structure and reactivity of trans-UO22+ complexes of OH-containing ligands. J. Chem. Soc. Dalton Trans. 2000, 7, 1213–1218. [Google Scholar]
- Sah, A.K.; Rao, C.P.; Saarenketo, P.K.; Wegelius, E.K.; Kolehmainen, E.; Rissanen, K. First crystallographic investigation of complexes of cis-VO2+, cis-MoO22+, and trans-UO22+ species with Schiff-base molecules derived from 4,6-O-ethylidene-β-d-glucopyranosylamine. Eur. J. Inorg. Chem. 2001, 2001, 2773–2781. [Google Scholar] [CrossRef]
- Kannappan, R.; Tooke, D.M.; Spek, A.L.; Reedijk, J. Separation of actinides and lanthanides: Synthesis and molecular structure of a new di-μ-phenoxo-bridged dinuclear bis(dioxouranium(VI)) complex. Inorg. Chim. Acta 2006, 359, 334–338. [Google Scholar] [CrossRef]
- Bharara, M.S.; Tonks, S.A.; Gorden, A.E.V. Uranyl stabilized Schiff base complex. Chem. Commun. 2007, 39, 4006–4008. [Google Scholar] [CrossRef]
- Bharara, M.S.; Strawbridge, K.; Vilsek, J.Z.; Bray, T.H.; Gorden, A.E.V. Novel dinuclear uranyl complexes with asymmetric Schiff base ligands: Synthesis, structural characterization, reactivity, and extraction studies. Inorg. Chem. 2007, 46, 8309–8315. [Google Scholar] [CrossRef]
- Takao, K.; Ikeda, Y. Structural characterization and reactivity of UO2(salophen)L and [UO2(salophen)]2: Dimerization of UO2(salophen) fragments in noncoordinating solvents (salophen = N,N′-disalicylidene-o-phenylenediaminate, L = N,N-dimethylformamide, dimethyl sulfoxide). Inorg. Chem. 2007, 46, 1550–1562. [Google Scholar] [CrossRef]
- Hazra, S.; Majumder, S.; Fleck, M.; Mohanta, S. Synthesis, molecular and supramolecular structure, spectroscopy and electrochemistry of a dialkoxo-bridged diuranyl(VI) compound. Polyhedron 2008, 27, 1408–1414. [Google Scholar] [CrossRef]
- Fleck, M.; Hazra, S.; Majumder, S.; Mohanta, S. Syntheses, structures, and electrochemistry of a dinuclear compound and a mononuclear-mononuclear cocrystalline compound of uranyl(VI). Cryst. Res. Technol. 2008, 43, 1220–1229. [Google Scholar] [CrossRef]
- Bharara, M.S.; Heflin, K.; Tonks, S.; Strawbridge, K.L.; Gorden, A.E.V. Hydroxy- and alkoxy-bridged dinuclear uranyl–Schiff base complexes: Hydrolysis, transamination and extraction studies. Dalton Trans. 2008, 2966–2973. [Google Scholar] [CrossRef] [PubMed]
- Cametti, M.; Ilander, L.; Rissanen, K. Recognition of Li+ by a salophen-UO2 homodimeric complex. Inorg. Chem. 2009, 48, 8632–8637. [Google Scholar] [CrossRef] [PubMed]
- Arnold, P.L.; Patel, D.; Pécharman, A.F.; Wilson, C.; Love, J.B. Equatorial ligand substitution by hydroxide in uranyl Pacman complexes of a Schiff-base pyrrole macrocycle. Dalton Trans. 2010, 39, 3501–3508. [Google Scholar] [CrossRef][Green Version]
- Popov, L.D.; Tupolova, Y.P.; Shcherbakov, I.N.; Levchenkov, S.I.; Suponitsky, K.Y.; Maevskiy, O.V.; Kogan, V.A. A novel binuclear uranyl(VI) complex of bis(N,N′-3-carboxysalicylidene)-1,3-diaminopropan-2-ol: Synthesis, structure, and properties. Russ. J. Coord. Chem. 2012, 38, 651–655. [Google Scholar] [CrossRef]
- Arnold, P.L.; Jones, G.M.; Pan, Q.J.; Schreckenbach, G.; Love, J.B. Co-linear, double-uranyl coordination by an expanded Schiff-base polypyrrole macrocycle. Dalton Trans. 2012, 41, 6595–6597. [Google Scholar] [CrossRef]
- Jones, G.M.; Arnold, P.L.; Love, J.B. Controlled deprotection and reorganization of uranyl oxo groups in a binuclear macrocyclic environment. Angew. Chem. Int. Ed. 2012, 51, 12584–12587. [Google Scholar] [CrossRef]
- Tamasi, A.L.; Barnes, C.L.; Walensky, J.R. Structure and spectroscopy of uranyl salicylaldiminate complexes. Radiochim. Acta 2013, 101, 631–636. [Google Scholar]
- Brancatelli, G.; Pappalardo, A.; Trusso Sfrazzetto, G.; Notti, A.; Geremia, S. Mono- and dinuclear uranyl(VI) complexes with chiral Schiff base ligand. Inorg. Chim. Acta 2013, 396, 25–29. [Google Scholar] [CrossRef]
- Schnorr, R.; Handke, M.; Kersting, B. Synthesis, characterization and molecular structure of a dinuclear uranyl complex supported by N,N′,N″,N′″-tetra-(3,5-di-tert-butylsalicylidene)-1,2,4,5-phenylene-tetraamine. Z. Naturforsch. Sect. B J. Chem. Sci. 2015, 70, 757–763. [Google Scholar] [CrossRef]
- Mäkelä, T.; Minkkinen, M.E.; Rissanen, K. Ion pair binding in the solid-state with ditopic crown ether uranyl salophen receptors. Inorg. Chem. 2016, 55, 1339–1346. [Google Scholar] [CrossRef] [PubMed]
- Majumder, I.; Chatterjee, S.; Fischer, R.C.; Neogi, S.K.; Mautner, F.A.; Chattopadhyay, T. Syntheses of U3O8 nanoparticles from four different uranyl complexes: Their catalytic performance for various alcohol oxidations. Inorg. Chim. Acta 2017, 462, 112–122. [Google Scholar] [CrossRef]
- Assefa, M.K.; Pedrick, E.A.; Wakefield, M.E.; Wu, G.; Hayton, T.W. Oxidation of the 14-membered macrocycle dibenzotetramethyltetraaza[14] annulene upon ligation to the uranyl ion. Inorg. Chem. 2018, 57, 8317–8324. [Google Scholar] [CrossRef]
- Mahjoobizadeh, M.; Takjoo, R.; Farhadipour, A.; Mague, J.T. Fe(III), Cu(II) and U(VI) binuclear complexes with a new isothiosemicarbazone ligand: Syntheses, characterization, crystal structures, thermal behavior and theoretical investigations. Inorg. Chim. Acta 2018, 482, 643–653. [Google Scholar] [CrossRef]
- Ghosh, S.; Srivastava, A.K.; Govu, R.; Pal, U.; Pal, S. A diuranyl(VI) complex and its application in electrocatalytic and photocatalytic hydrogen evolution from neutral aqueous medium. Inorg. Chem. 2019, 58, 14410–14419. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, T.; Nakaguchi, M.; Morimoto, K. Synthesis, structures, and proton self-exchange reaction of μ3-oxido/hydroxido bridged trinuclear uranyl(VI) complexes with tridentate Schiff-base ligands. Inorg. Chem. 2017, 56, 4057–4064. [Google Scholar] [CrossRef] [PubMed]
- Mougel, V.; Horeglad, P.; Nocton, G.; Pécaut, J.; Mazzanti, M. Cation-cation complexes of pentavalent uranyl: From disproportionation intermediates to stable clusters. Chem. Eur. J. 2010, 16, 14365–14377. [Google Scholar] [CrossRef]
- Mougel, V.; Horeglad, P.; Nocton, G.; Pécaut, J.; Mazzanti, M. Stable pentavalent uranyl species and selective assembly of a polymetallic mixed-valent uranyl complex by cation-cation interactions. Angew. Chem. Int. Ed. 2009, 48, 8477–8480. [Google Scholar] [CrossRef]
- Arnold, P.L.; Jones, G.M.; Odoh, S.O.; Schreckenbach, G.; Magnani, N.; Love, J.B. Strongly coupled binuclear uranium-oxo complexes from uranyl oxo rearrangement and reductive silylation. Nat. Chem. 2012, 4, 221–227. [Google Scholar] [CrossRef]
- Jones, G.M.; Arnold, P.L.; Love, J.B. Oxo-group-14-element bond formation in binuclear uranium(V) Pacman complexes. Chem. Eur. J. 2013, 19, 10287–10294. [Google Scholar] [CrossRef] [PubMed]
- Zegke, M.; Nichol, G.S.; Arnold, P.L.; Love, J.B. Catalytic one-electron reduction of uranyl(VI) to Group 1 uranyl(V) complexes via Al(III) coordination. Chem. Commun. 2015, 51, 5876–5879. [Google Scholar] [CrossRef] [PubMed]
- Bell, N.L.; Shaw, B.; Arnold, P.L.; Love, J.B. Uranyl to uranium(IV) conversion through manipulation of axial and equatorial ligands. J. Am. Chem. Soc. 2018, 140, 3378–3384. [Google Scholar] [CrossRef] [PubMed]
- Zegke, M.; Zhang, X.; Pidchenko, I.; Hlina, J.A.; Lord, R.M.; Purkis, J.; Nichol, G.S.; Magnani, N.; Schreckenbach, G.; Vitova, T.; et al. Differential uranyl(V) oxo-group bonding between the uranium and metal cations from groups 1, 2, 4, and 12; A high energy resolution X-ray absorption, computational, and synthetic study. Chem. Sci. 2019, 10, 9740–9751. [Google Scholar] [CrossRef]
- Salmon, L.; Thuéry, P.; Ephritikhine, M. Polynuclear uranium(IV) compounds with (μ3-oxo)U3 or (μ4-oxo)U4 cores and compartmental Schiff base ligands. Polyhedron 2006, 25, 1537–1542. [Google Scholar] [CrossRef]
- Salmon, L.; Thuéry, P.; Ephritikhine, M. Crystal structure of the first octanuclear uranium(IV) complex with compartmental Schiff base ligands. Polyhedron 2004, 23, 623–627. [Google Scholar] [CrossRef]
- Salmon, L.; Thuéry, P.; Ephritikhine, M. Synthesis and crystal structure of uranium(IV) complexes with compartmental Schiff bases: From mononuclear species to tri- and tetranuclear clusters. J. Chem. Soc. Dalton Trans. 2004, 4, 1635–1643. [Google Scholar] [CrossRef]
- Salmon, L.; Thuéry, P.; Ephritikhine, M. Synthesis and crystal structure of tetra- and hexanuclear uranium(IV) complexes with hexadentate compartmental Schiff-base ligands. J. Chem. Soc. Dalton Trans. 2004, 4139–4145. [Google Scholar] [CrossRef]
- Salmon, L.; Thuéry, P.; Rivière, E.; Miyamoto, S.; Yamato, T.; Ephritikhine, M. Synthesis, structure and magnetic behaviour of dinuclear uranium(IV) complexes with a “calixsalophen” type macrocycle. New J. Chem. 2006, 30, 1220–1227. [Google Scholar] [CrossRef]
- Camp, C.; Mougel, V.; Horeglad, P.; Pécaut, J.; Mazzanti, M. Multielectron redox reactions involving C-C coupling and cleavage in uranium Schiff base complexes. J. Am. Chem. Soc. 2010, 132, 17374–17377. [Google Scholar] [CrossRef]
- Camp, C.; Andrez, J.; Pécaut, J.; Mazzanti, M. Synthesis of electron-rich uranium(IV) complexes supported by tridentate Schiff base ligands and their multi-electron redox chemistry. Inorg. Chem. 2013, 52, 7078–7086. [Google Scholar] [CrossRef] [PubMed]
- Camp, C.; Toniolo, D.; Andrez, J.; Pécaut, J.; Mazzanti, M. A versatile route to homo- and hetero-bimetallic 5f-5f and 3d-5f complexes supported by a redox active ligand framework. Dalton Trans. 2017, 46, 11145–11148. [Google Scholar] [CrossRef]
- Arnold, P.L.; Stevens, C.J.; Bell, N.L.; Lord, R.M.; Goldberg, J.M.; Nichol, G.S.; Love, J.B. Multi-electron reduction of sulfur and carbon disulfide using binuclear uranium(III) borohydride complexes. Chem. Sci. 2017, 8, 3609–3617. [Google Scholar] [CrossRef]
- Cowie, B.E.; Nichol, G.S.; Love, J.B.; Arnold, P.L. Double uranium oxo cations derived from uranyl by borane or silane reduction. Chem. Commun. 2018, 54, 3839–3842. [Google Scholar] [CrossRef] [PubMed]
- Arnold, P.L.; Stevens, C.J.; Farnaby, J.H.; Gardiner, M.G.; Nichol, G.S.; Love, J.B. New chemistry from an old reagent: Mono- and dinuclear macrocyclic uranium(III) complexes from [U(BH4)3(THF)2]. J. Am. Chem. Soc. 2014, 136, 10218–10221. [Google Scholar] [CrossRef] [PubMed]
- Mougel, V.; Pécaut, J.; Mazzanti, M. New polynuclear U(IV)-U(V) complexes from U(IV) mediated uranyl(V) disproportionation. Chem. Commun. 2012, 48, 868–870. [Google Scholar] [CrossRef] [PubMed]







































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
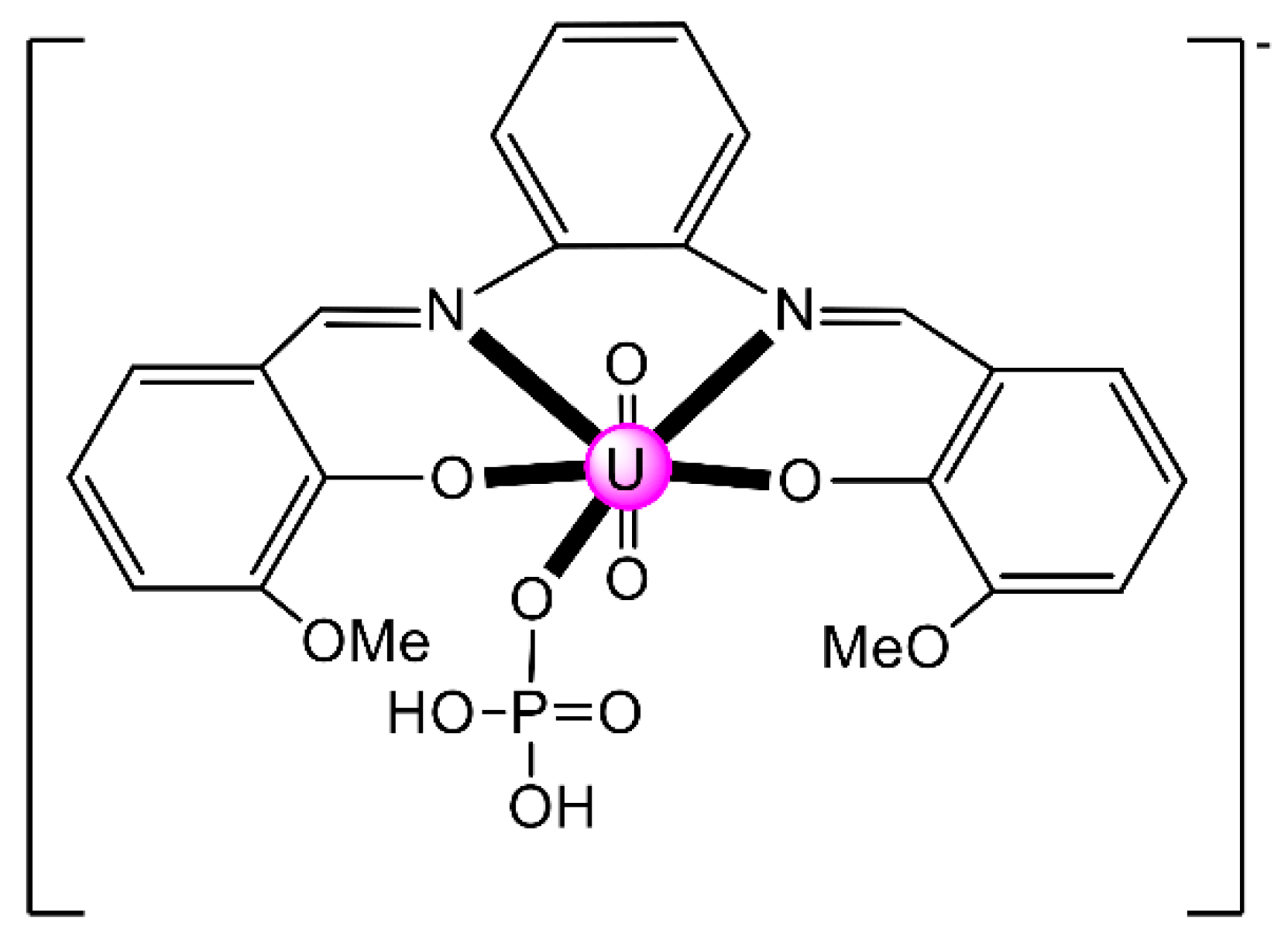{kind=link}
{kind=link}
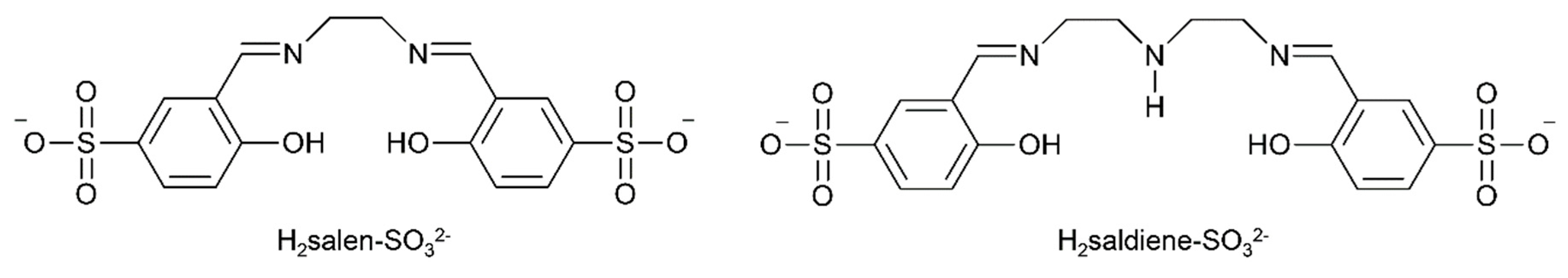{kind=link}
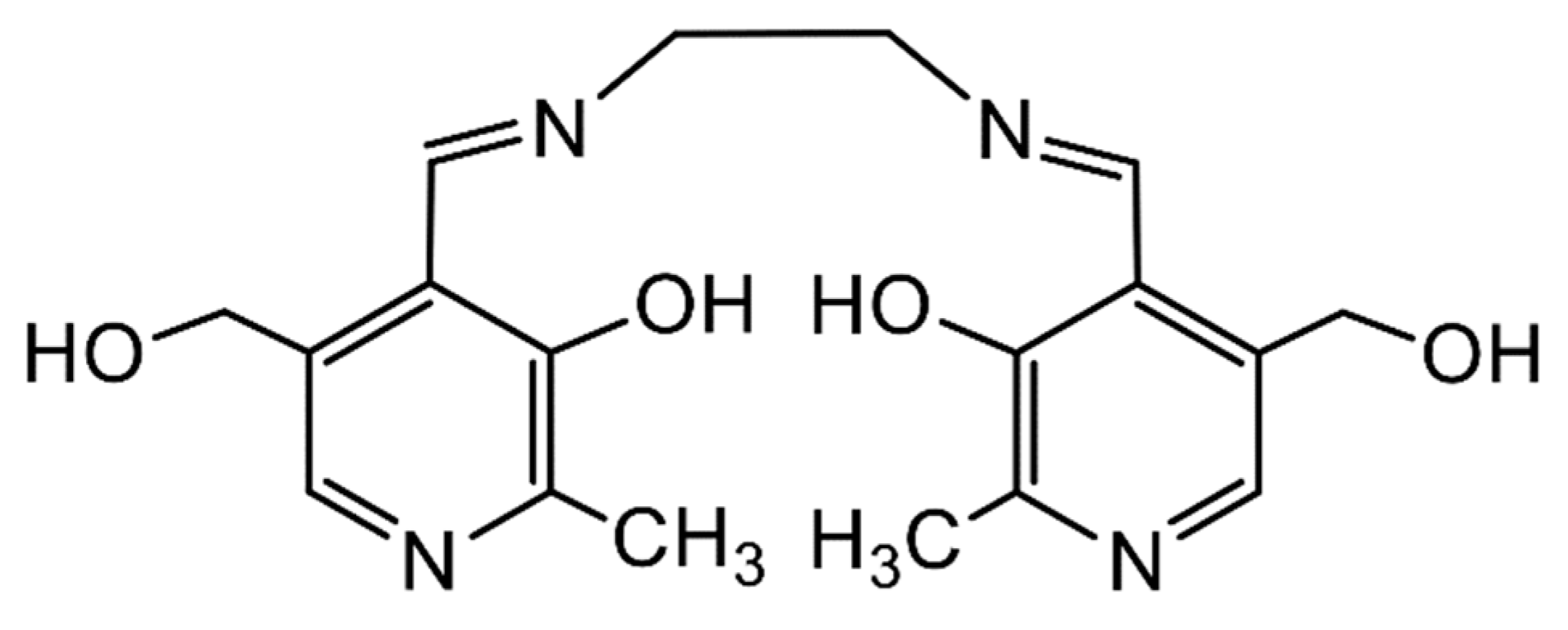{kind=link}
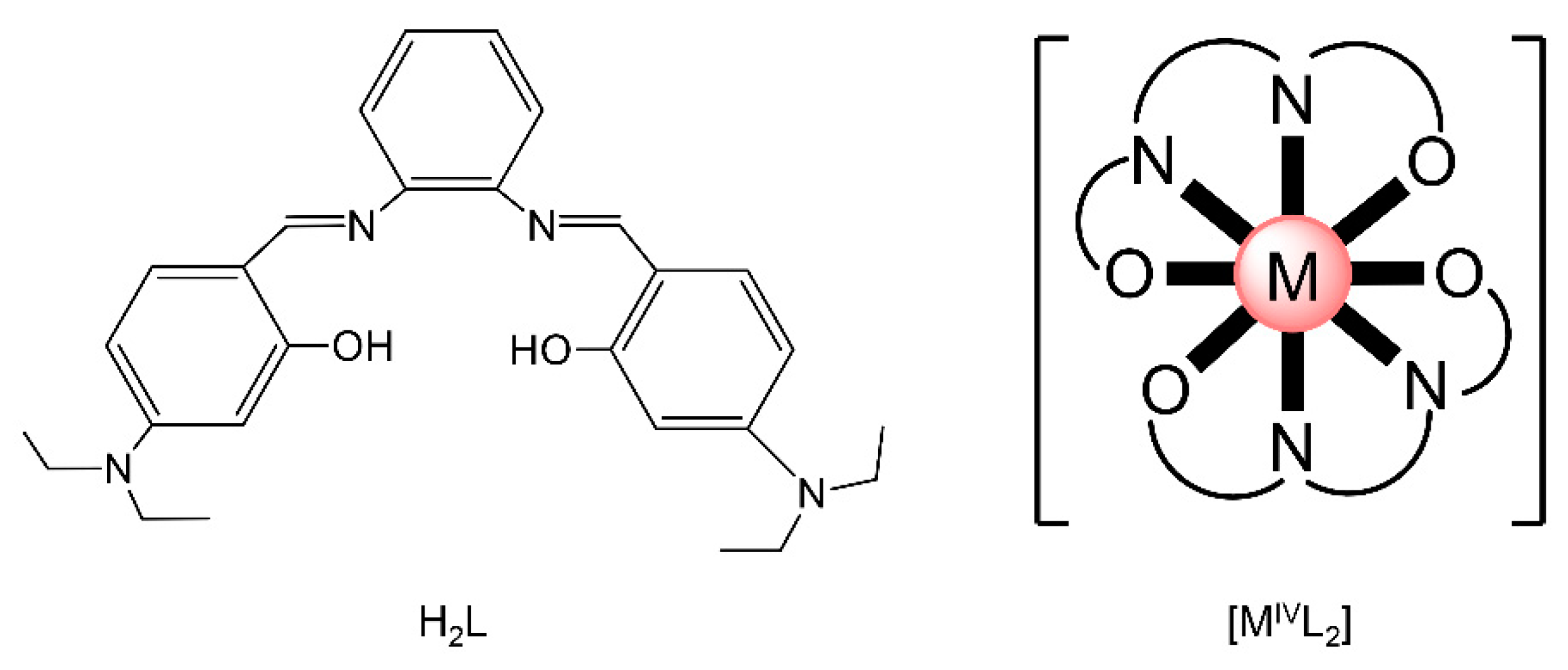{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Element | Z | Electronic Configuration in the Ground State | Radius/Å | Oxidation State [b] | |||
|---|---|---|---|---|---|---|---|
| An | An3+ | An4+ | An3+ [a] | An4+ [a] | |||
| Ac | 89 | [Rn]6d17s2 | [Rn]5fo | 1.11 | 0.99 | III | |
| Th | 90 | [Rn]6d27s2 | [Rn]5f1 | [Rn]5fo | 0.94 | III, IV | |
| Pa | 91 | [Rn]5f27s26d1 | [Rn]5f2 | [Rn]5f1 | 1.04 | 0.90 | IV, V |
| U | 92 | [Rn]5f37s26d1 | [Rn]5f3 | [Rn]5f2 | 1.03 | 0.89 | II, III, IV, V, VI |
| Np | 93 | [Rn]5f47s26d1 | [Rn]5f4 | [Rn]5f3 | 1.01 | 0.87 | III, IV, V, VI, VII |
| Pu | 94 | [Rn]5f67s2 | [Rn]5f5 | [Rn]5f4 | 1.00 | 0.86 | III, IV, V, VI, VII |
| Am | 95 | [Rn]5f77s2 | [Rn]5f6 | [Rn]5f5 | 0.98 | 0.85 | II, III, IV, V, VI |
| Cm | 96 | [Rn]5f77s26d1 | [Rn]5f7 | [Rn]5f6 | 0.97 | 0.85 | III, IV |
| Bk | 97 | [Rn]5f97s2 | [Rn]5f8 | [Rn]5f7 | 0.96 | 0.83 | III, IV |
| Cf | 98 | [Rn]5f107s2 | [Rn]5f9 | [Rn]5f8 | 0.95 | 0.82 | II, III, IV |
| Es | 99 | [Rn]5f117s2 | [Rn]5f10 | [Rn]5f9 | n.k. | II, III | |
| Fm | 100 | [Rn]5f127s2 | [Rn]5f11 | [Rn]5f10 | n.k. | II, III | |
| Md | 101 | [Rn]5f137s2 | [Rn]5f12 | [Rn]5f11 | n.k. | II, III | |
| No | 102 | [Rn]5f147s2 | [Rn]5f13 | [Rn]5f12 | n.k. | II, III | |
| Lr | 103 | [Rn]5f147s26d1 | [Rn]5f14 | [Rn]5f13 | n.k. | III | |
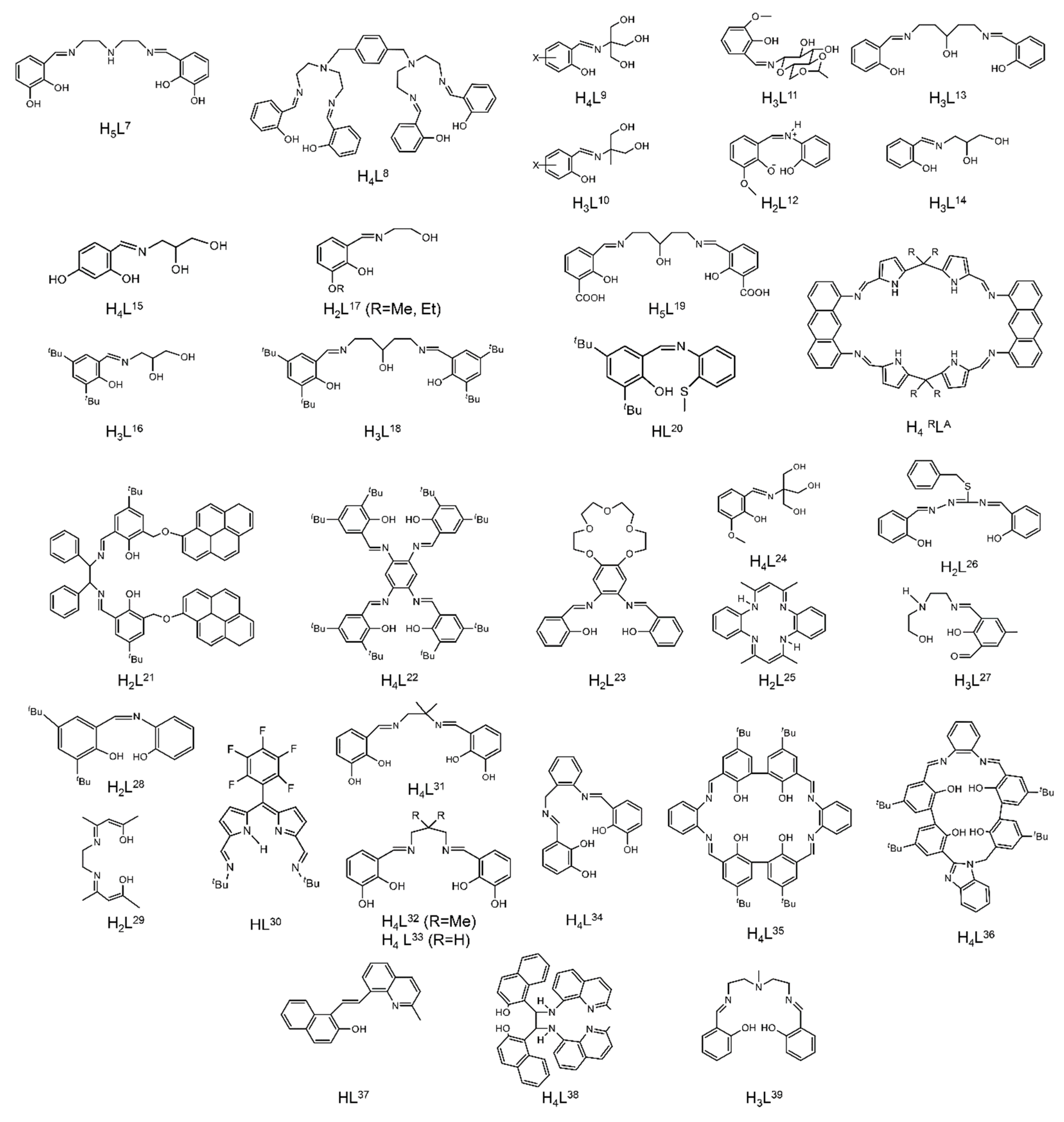
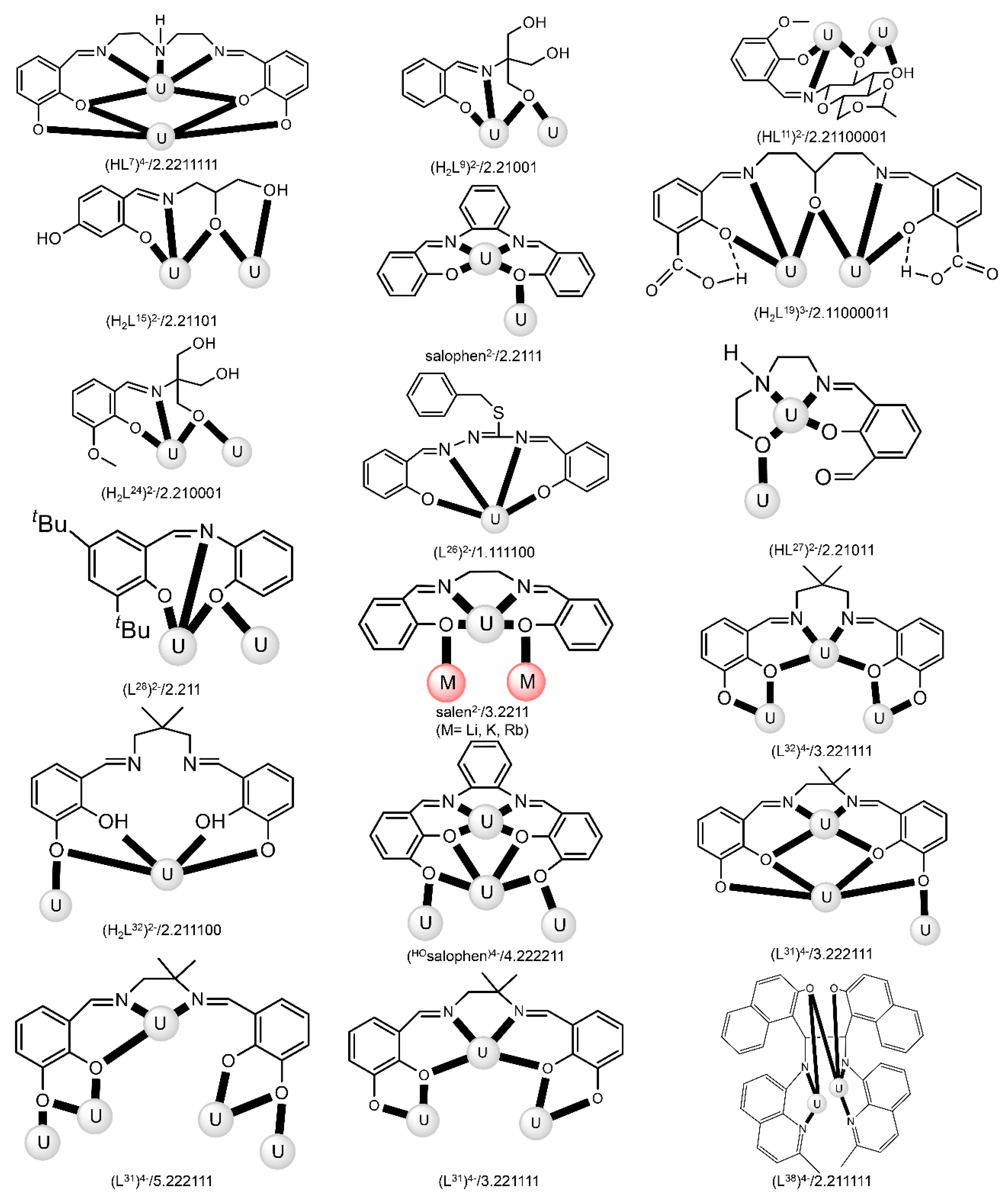
| Complex [a],[b] | Coordination Mode of the Schiff Base Ligand(s) [c] | Ref. |
|---|---|---|
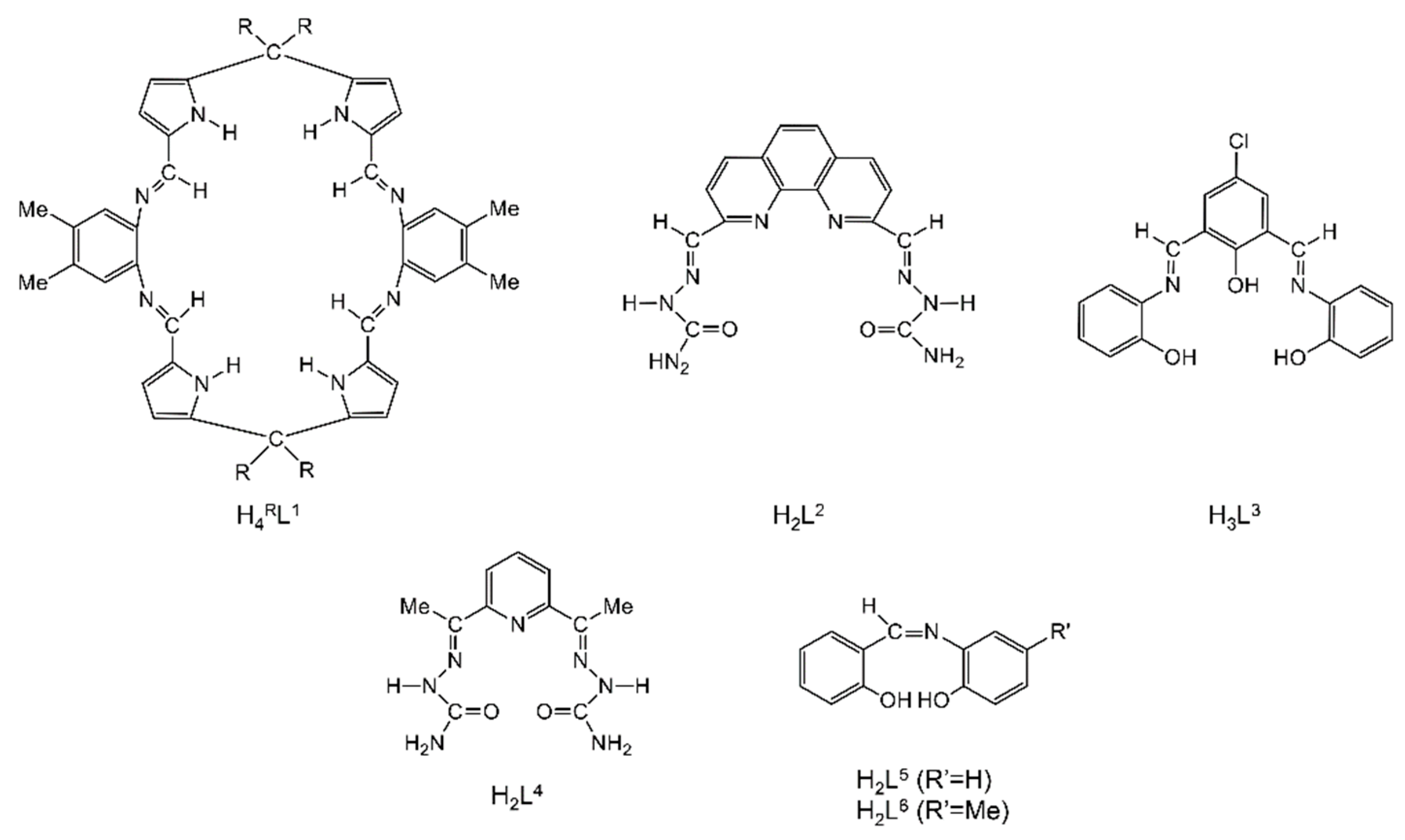
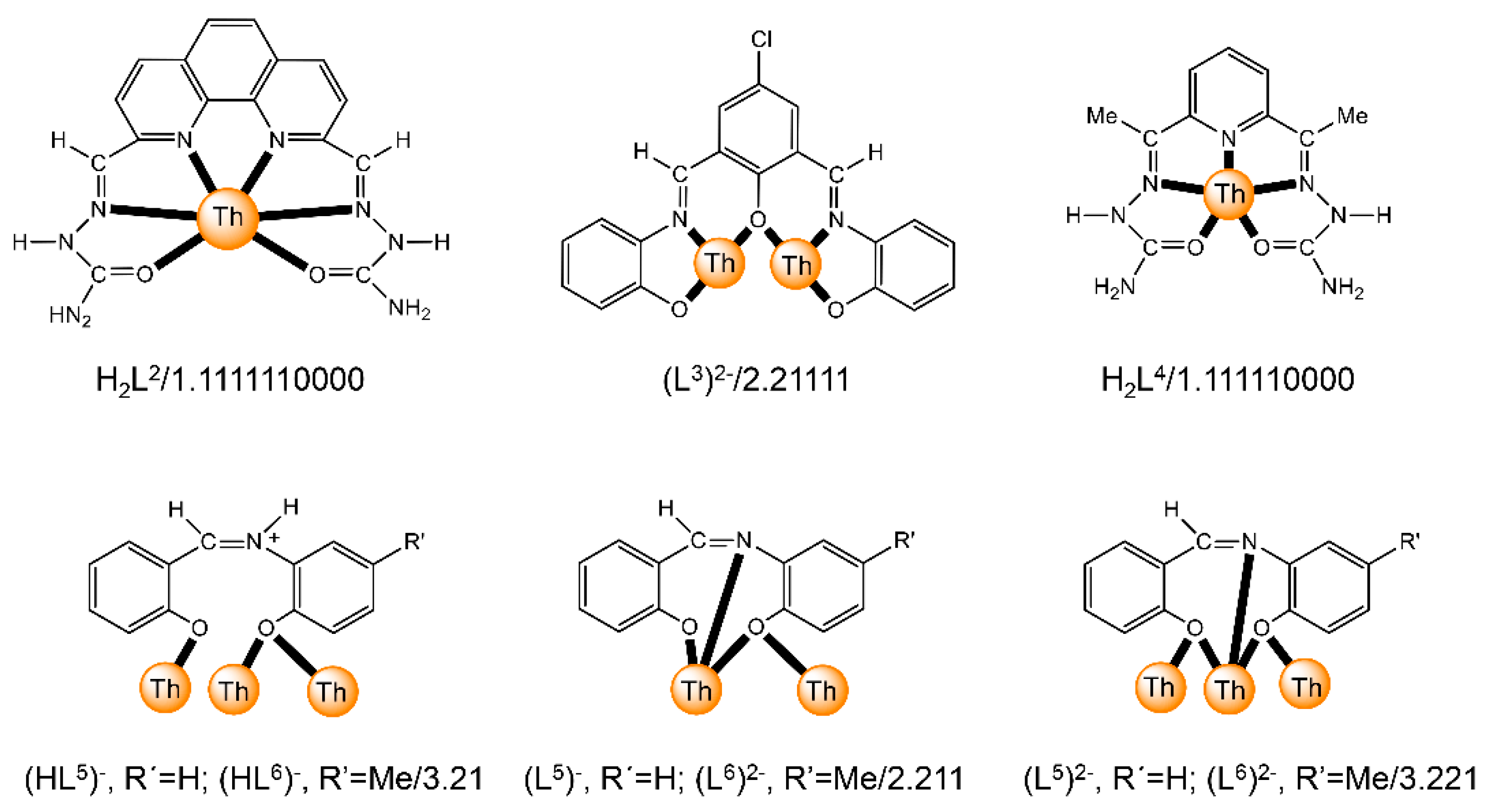
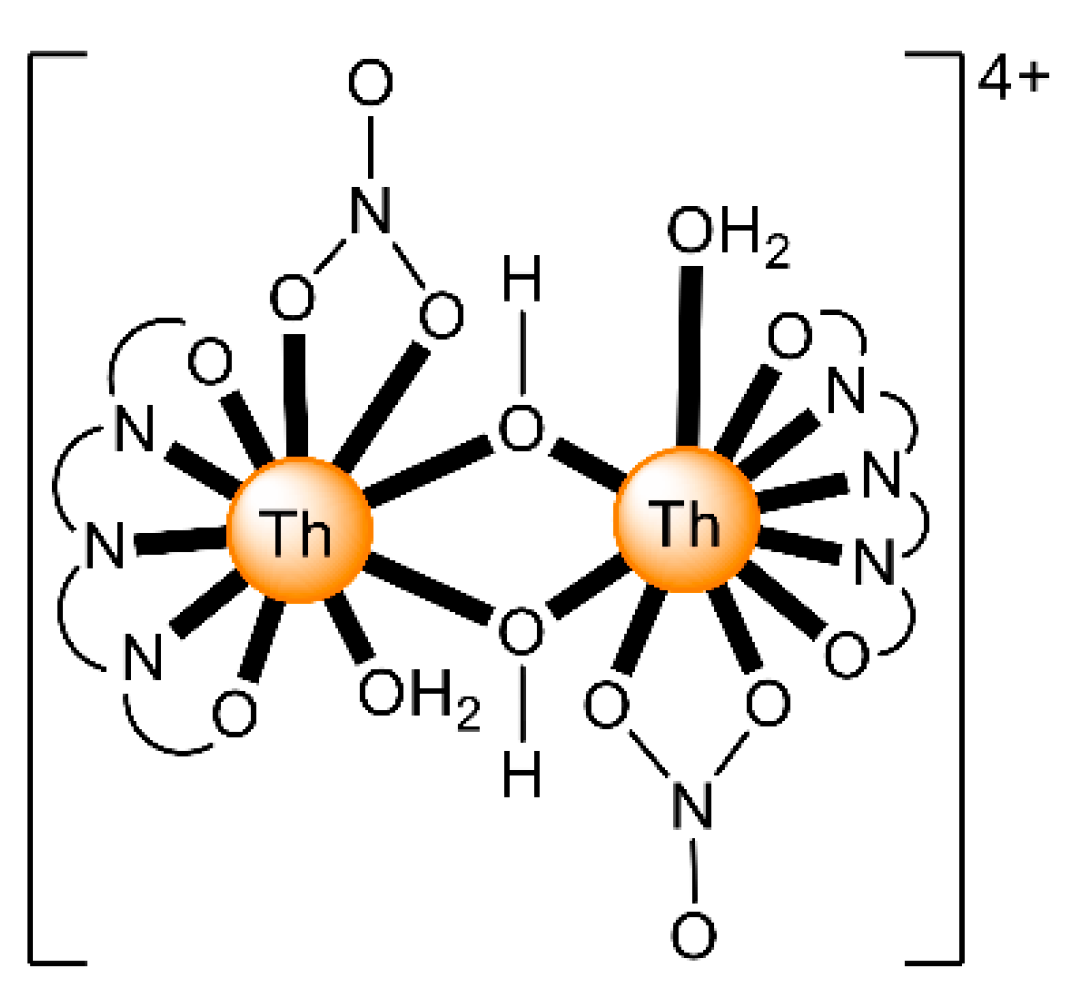
| [Th2O(NO3)2(H2L2)2(H2O)2](NO3)4 (4) | 1.1111110000 | [65] |
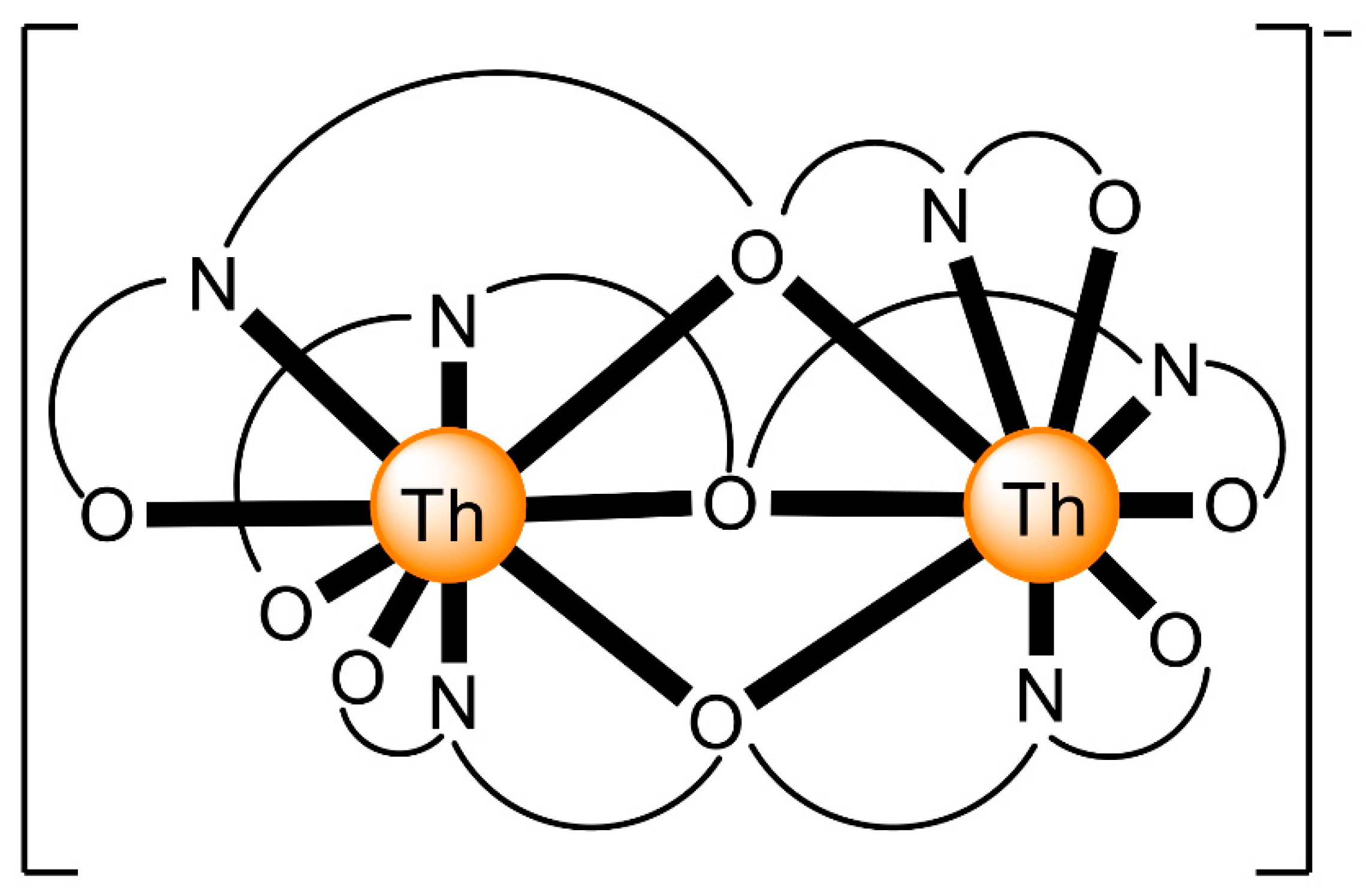
| [Mg(H2O)6][Th2(L3)3]2 (5) | 2.21111 | [66] |
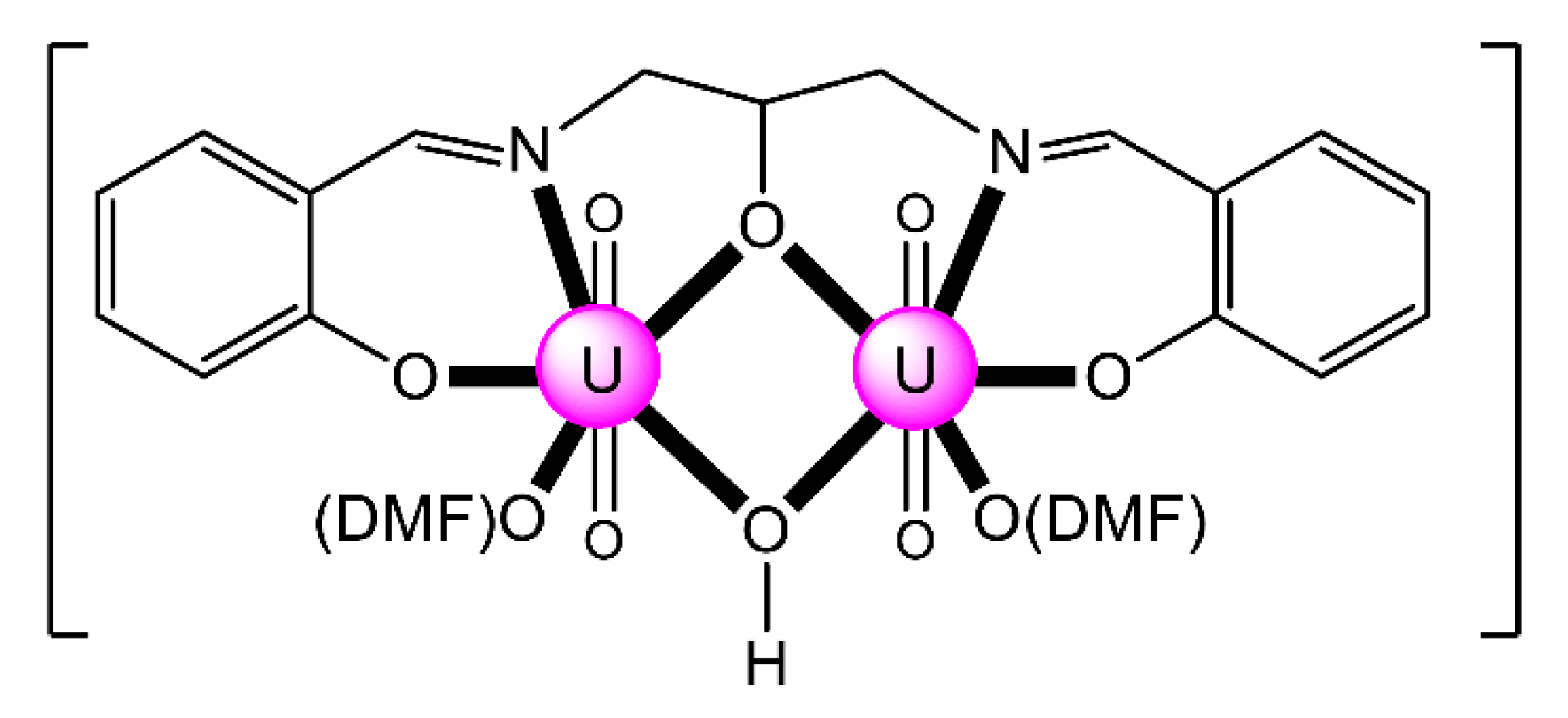
| [Th2(OH)2(NO3)2(H2L4)2(H2O)2](NO3)4 (6) | 1.1111110000 | [67] |
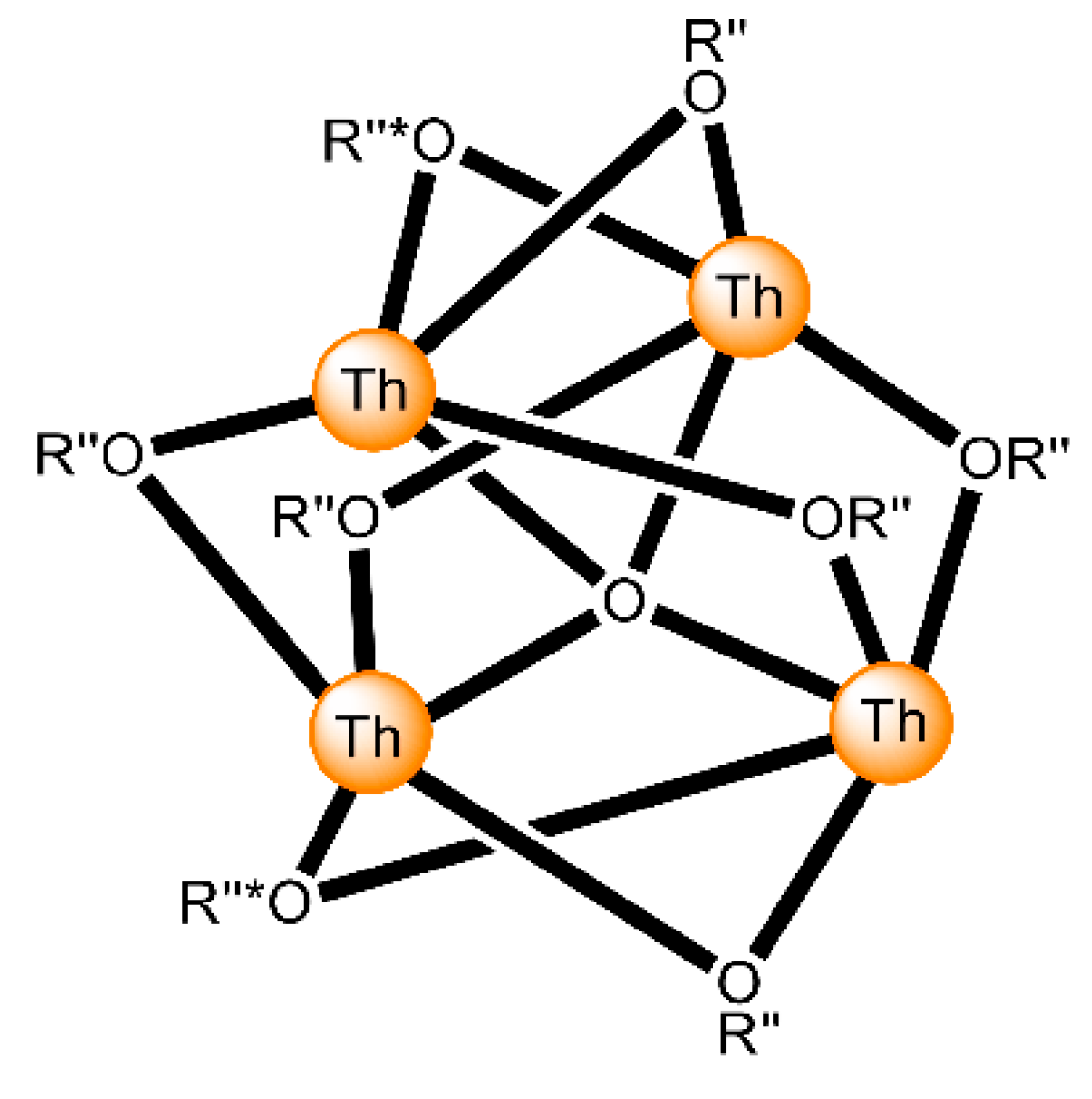
| [Th4O(NO3)2(HL5)2(L5)5] (7) | 3.21/(HL5)−, 2.211(L5)2−, 3.221/(L5)2− | [16] |
| [Th4O(NO3)2(HL6)2(L6)5] (8) | 3.21/(HL6)−, 2.211(L6)2−, 3.221/(L6)2− | [16] |
| Complex [a],[b] | Coordination Mode of the Schiff-Ligand [c] | Ref. |
|---|---|---|
| [(UO2)2(HL7)(S)] (9) [d] | 2.2211111 | [81] |
| [(UO2)2(L8)] (10) | 2.1111111111 | [82] |
| [(UO2)2(H2L9)2(H2O)2] (11) | 2.21001 | [83] |
| [(UO2)2(HL10)2(H2O)2] (12) | 2.2101 | [83] |
| [(UO2)2(HL11)2] (13) | 2.11100001 | [84] |
| [(UO2)2(L12)2(THF)2] (14) | 2.2101 | [85] |
| [(UO2)2(OH)(L13)(DMF)2] (15) | 2.21111 | [86] |
| [(UO2)2(HL14)2] (16) | 2.2111 | [87] |
| [(UO2)2(H2L15)2] (17) | 2.21101 | [87] |
| [(UO2)2(HL16)2] (18) | 2.2111 | [87] |
| [(UO2)2(salophen)2] (19) | 2.2111 | [88] |
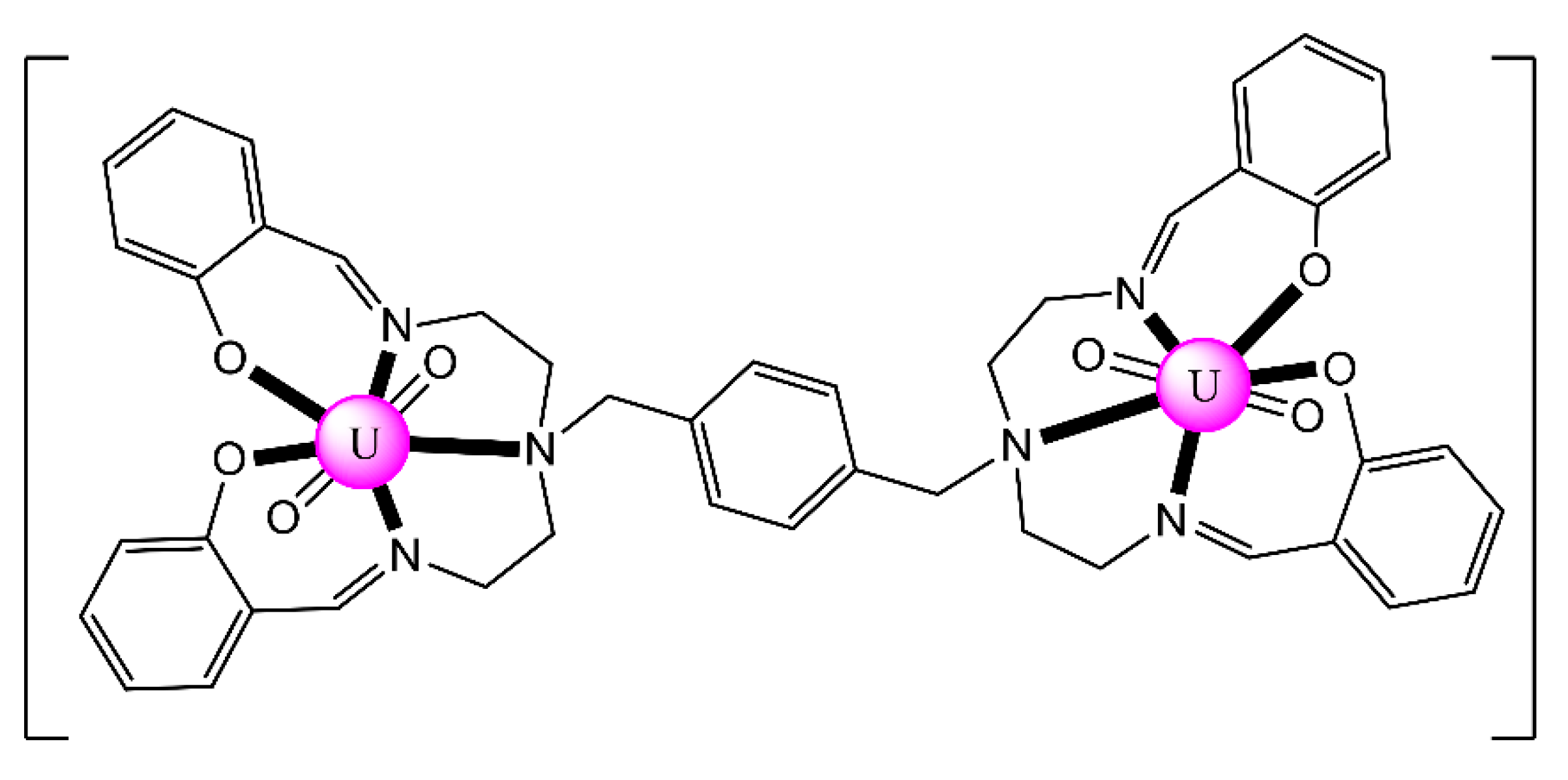
| [(UO2)2(L17)2(DMF)2] (20) [e] | 2.2101 | [89] |
| [(UO2)2(L17)2(DMF)2] (21) [f] | 2.2101 | [90] |
| [(UO2)2(OH)(L18)(DMF)2] (22) | 2.21111 | [91] |
| (Et3NH)2[(UO2)2(HHOsalophen)2] (23) | 2.211011 | [92] |
| [K2(UO2)2(OH)2(H2MeL1)2(C6H6)2] (24) | 1.11110000 | [93] |
| [(UO2)2(OH)(H2L19)(DMSO)2] (25) | 2.211000011 | [94] |
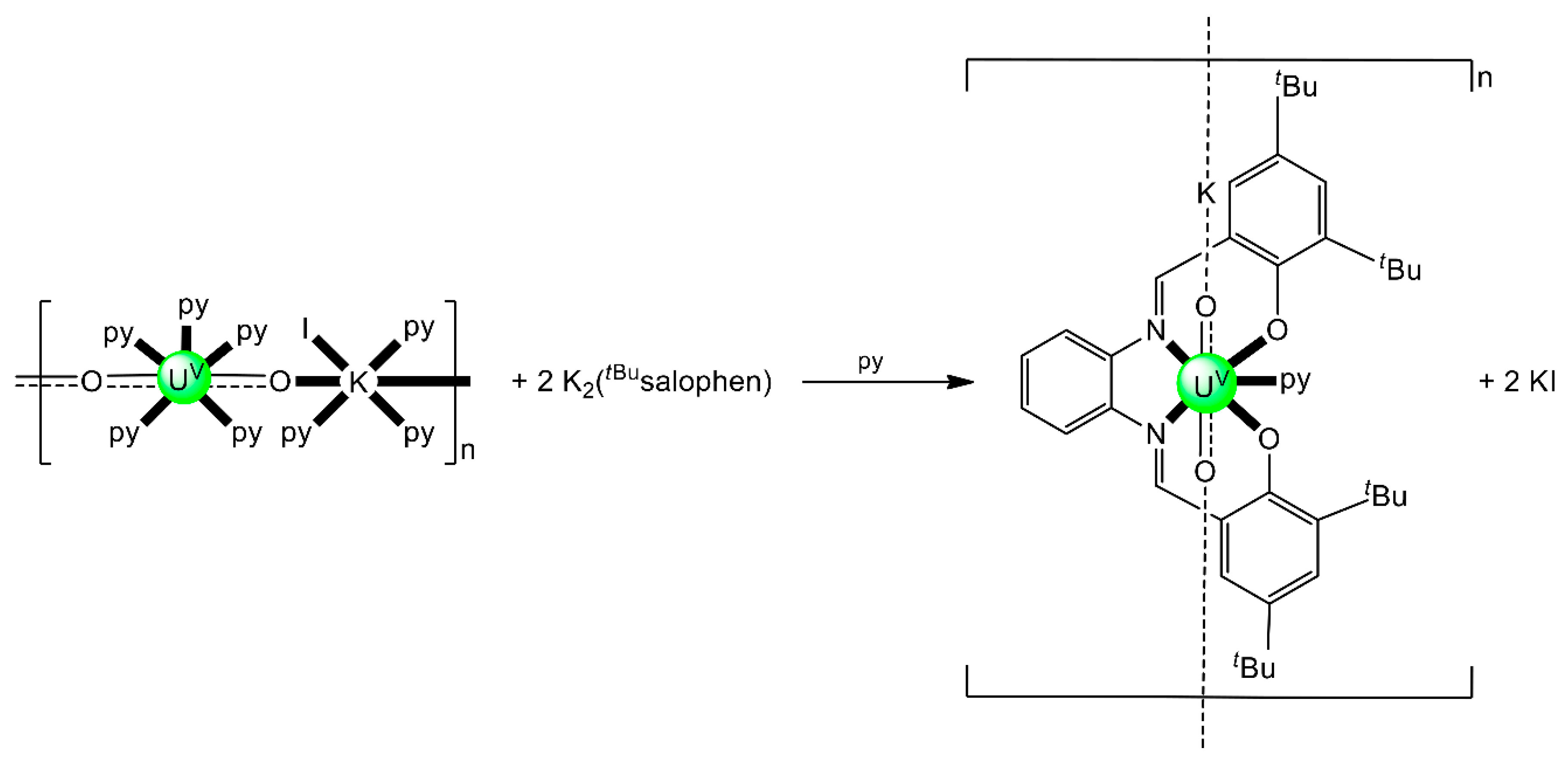
| [(UO2)2(EtLA)(py)2] (26) | 2.11111111 | [95] |
| [K2(UO2)2(O2)(MeL1)] (27) | 2.11111111 | [96] |
| [K2(UO2)2(O)(MeL1)] (28) | 2.11111111 | [96] |
| [(UO2)2Cl2(L20)2] (29) | 1.111 | [97] |
| (Me4N)[(UO2)2(OH)(L21)2] (30) | 1.110011 | [98] |
| [(UO2)2(L22)(Me2CO)2] (31) | 2.11111111 | [99] |
| [Li2(UO2)2Cl2(L23)2(H2O)2] (32) | 2.111111011 | [100] |
| [Na2(UO2)2Br2(L23)2(H2O)(MeOH] (33) | 2.111111111 | [100] |
| [(UO2)2(H2L24)2(DMSO)2] (34) | 2.210001 | [101] |
| [(UO2)2(L6)2(EtOH)2] (35) | 2.211 | [15] |
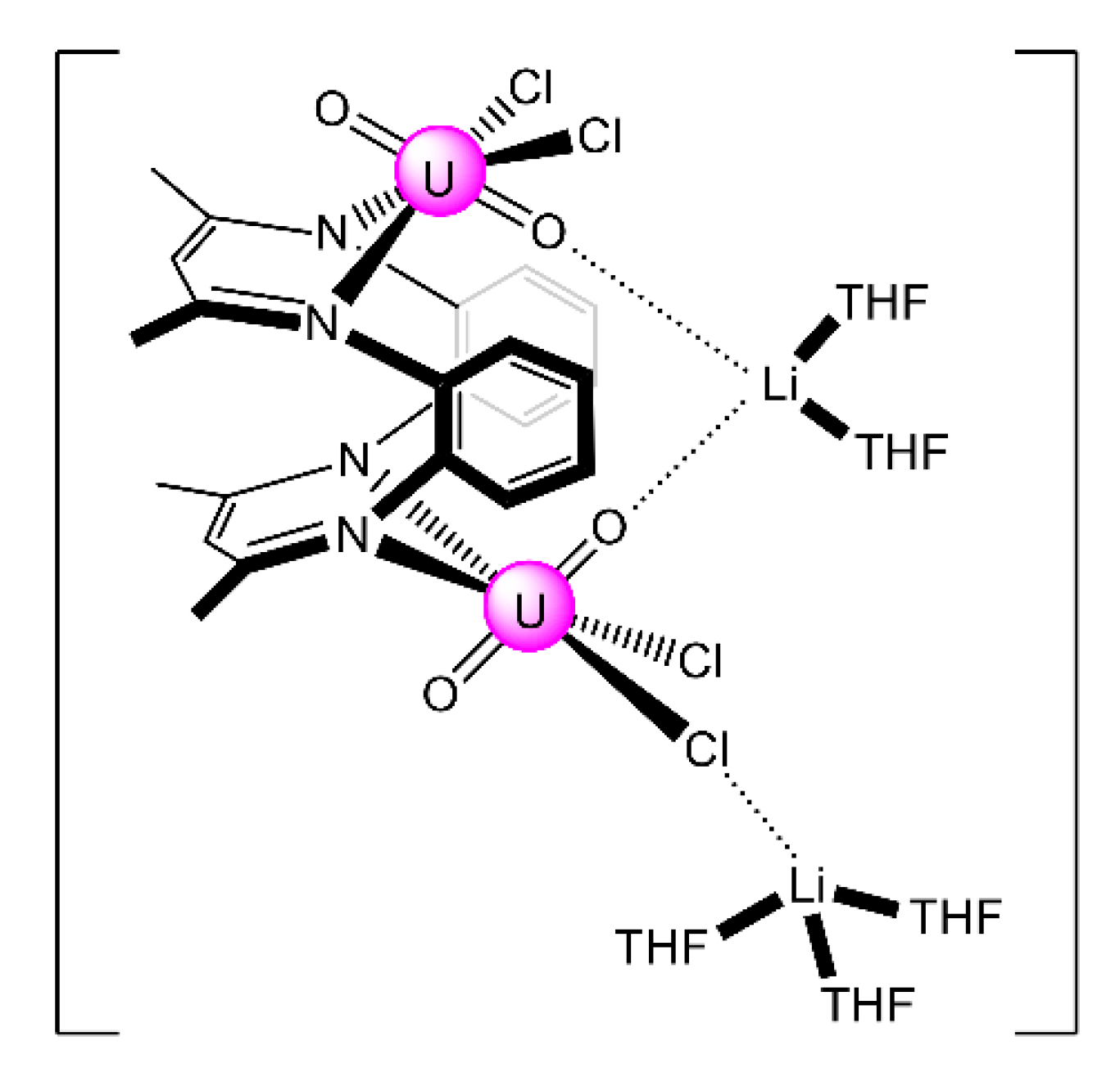
| [Li2(UO2)2Cl4(L25)(THF)5] (36) | 2.1111 | [102] |
| (Et3NH)[(UO2)2(O2CMe)(L26)2 (37) | 1.111100 | [103] |
| [(UO2)2(HL27)2] (38) | 2.21011 | [104] |
| (Et3NH)[(UO2)3(OH)(L28)3] (39) | 2.211 | [105] |
| (Et3NH)2[(UO2)3(O)(L28)3] (40) | 2.211 | [105] |
| [Li4(UO2)4(O)2(salen)4] (41) | 3.2211 | [106] |
| Complex [a],[b] | Coordination Mode of the Schiff-Base Ligand(s) [c] | Ref. |
|---|---|---|
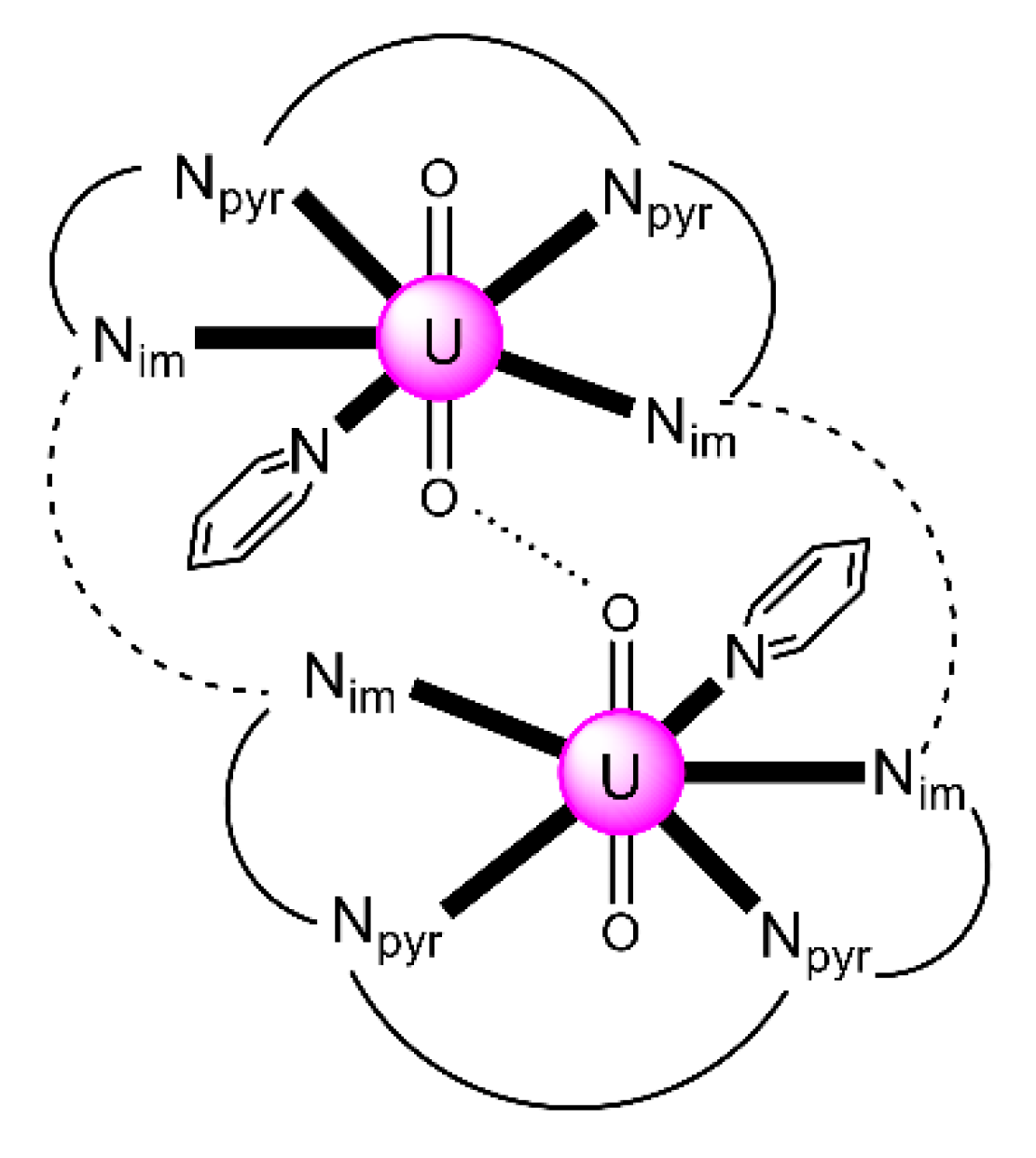
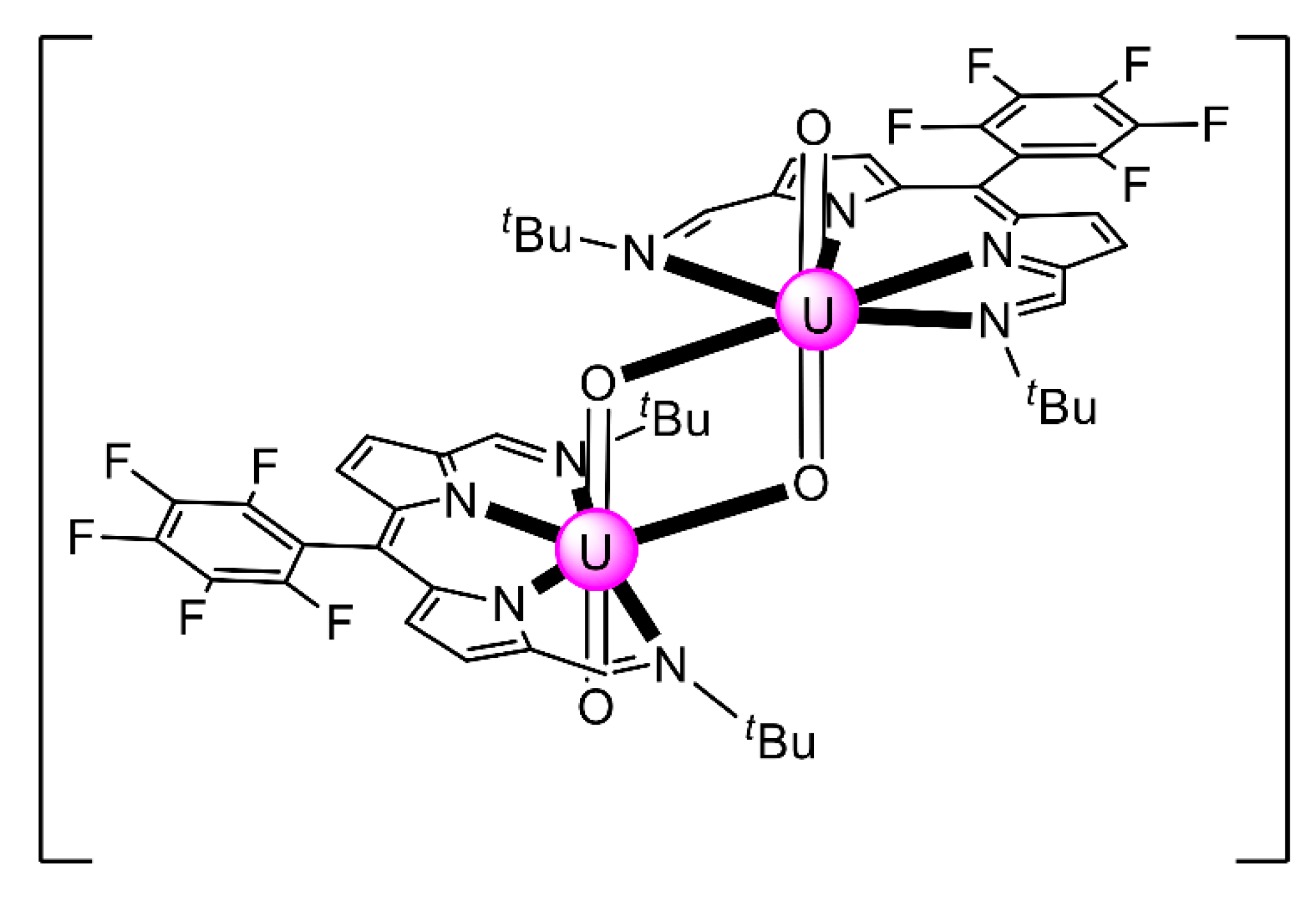
| [K(18C6)(py)]2[K2(UVO2)4(salen)4] (42) | 3.2211 | [107] |
| [K(18C6)(py)]2[K2(UVO2)4(L29)4] (43) | 3.2211 | [106] |
| [K(222)(py)]2[K2(UVO2)4(L29)4] (44) | 3.2211 | [106] |
| [K(18C6)(THF)]2[K6(UVO2)4(salopen)4I2(18C6)2]I2 (45) | 3.2211 | [106] |
| [Rb4(UVO2)4(salen)4(18C6)2] (46) | 3.2211 | [106] |
| [K2(UVO2)2(MeL1)] (47) | 2.11111111 | [96] |
| [(Me3SiOUVO)2(MeL1)] (48) | 2.11111111 | [108] |
| [Li2(UVO2)2(MeL1)(py)3] (49) | 2.11111111 | [109] |
| [Li(UVO2)(Me3SiOUVO)(MeL1)(py)3] (50) | 2.11111111 | [109] |
| [Li2(UVO2)2(H2MeL1)2(py)2] (51) | 2.11111100 | [110] |
| [(UVO2)2(L30)2] (52) | 1.1111 | [111] |
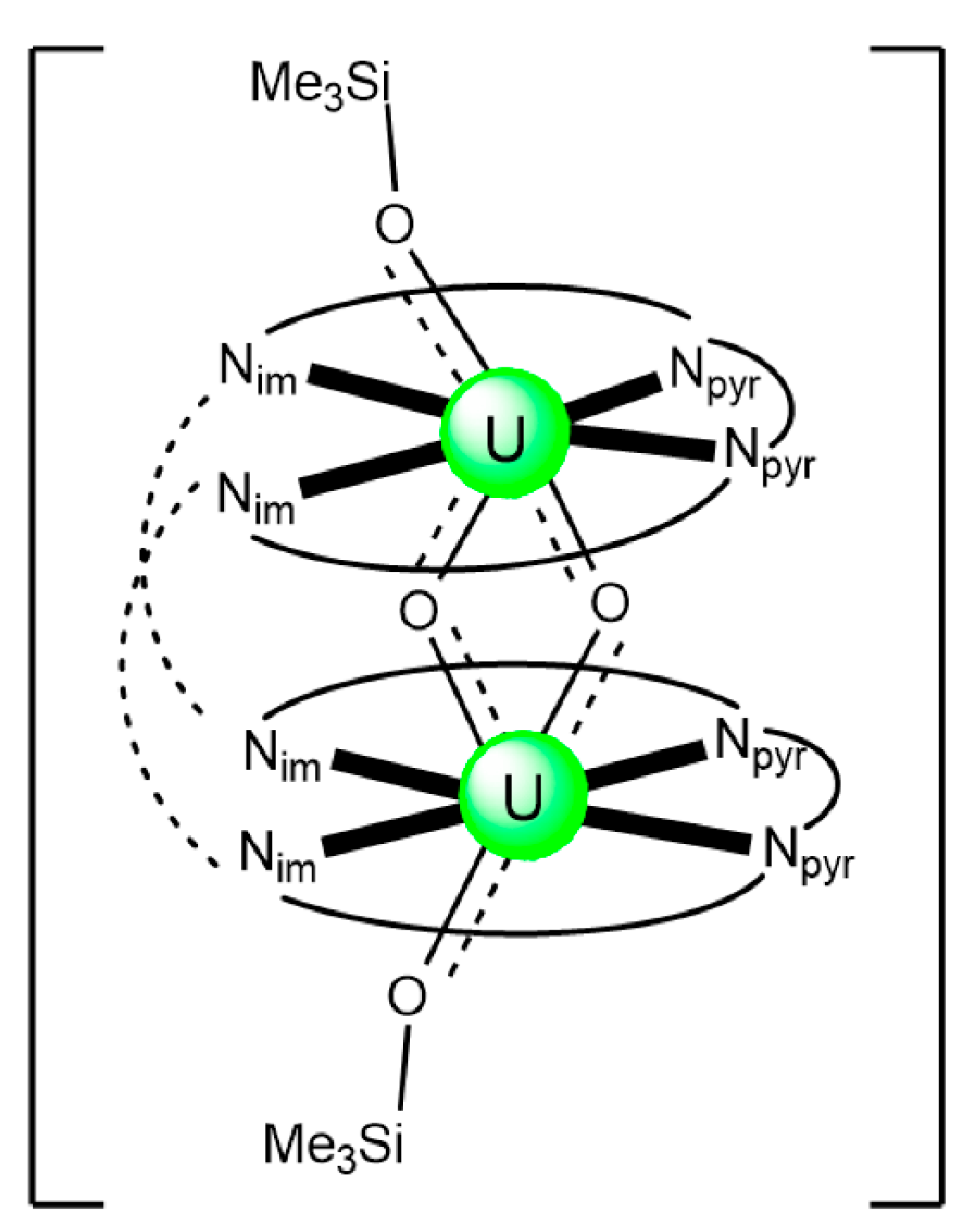
| [Rb6(UVO2)6(H2MeL1)6(py)6] (53) | 2.11111100 | [112] |
| [Cs6(UVO2)6(H2MeL1)6(py)6] (54) | 2.11111100 | [112] |
| (pyH)3[UIV3(O)Cl9(L31)] (55) | 3.221111 | [113] |
| [UIV4(O)(L32)2(H2L32)2(py)2](CF3SO3)2 (R = Me) (56) | 3.221111 [d], 2.211100 [e] | [113] |
| (pyH)2[UIV8(O)4Cl10(HOsalophen)4] (57) | 4.222211 | [114] |
| [UIV3(acac)2(HOsalophen)(HHOsalophen)2] (58) | 2.221111 [f], 3.221011 [g], 2.211011 [g] | [115] |
| [UIV4(HL34)4(H2L34)2] (59) | 2.211011 [h], 2.211000 [i] | [115] |
| (pyH)2[UIV4Cl6(L33)2(H2L33)2] (60) | 2.221111 [j], 3.211100 [k] | [116] |
| [UIV4Cl4(L33)2(H2L33)2(py)2] (61) | 2.221111 [j], 3.211100 [k] | [116] |
| (pyH)2[UIV4Cl6(L34)2(H2L34)2][UIV4Cl4(L34)2(H2L34)2(py)2] (62) | 2.221111 [j], 3.211100 [k] | [116] |
| (pyH)2[UIV6Cl10(L31)4(py)4] (63) | 3.222111, 5.222111 | [116] |
| [UIV2Cl4(L35)] (64) | 2.11111111 | [117] |
| [UIV2Cl4(L35)(py)2] (65) | 2.11111111 | [117] |
| [UIV2(acac)4(L35)] (66) | 2.22111100 | [117] |
| [UIV2(acac)5(HL36)] (67) | 2.11101110 | [117] |
| [UIV(cyclo-salophen)(py)4] (68) | 2.11112222 | [118] |
| [UIV2(L38)2] (69) [l] | 2.211111 | [119] |
| [UIV2(bis-Hsalophen)(py)6]I2 (70) | 2.11112211 | [120] |
| [UIV2Cl2(bis-Hsalophen)(THF)2] (71) | 2.11112211 | [120] |
| [UIV2(OAr)2(S2)(EtLA)] (72) [m] | 2.11111111 | [121] |
| [UIV2(OAr)2(S)(EtLA)] (73) [m] | 2.11111111 | [121] |
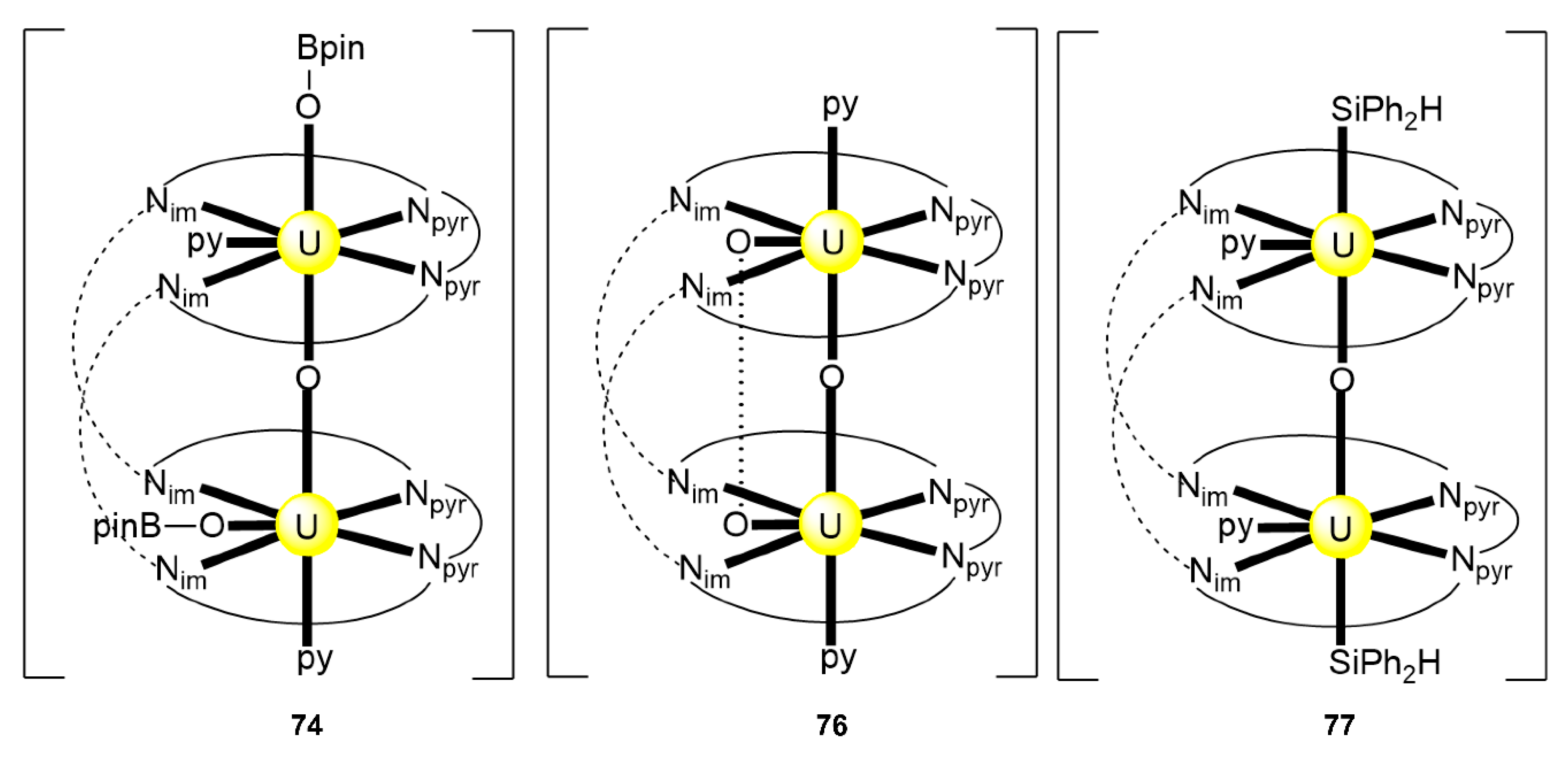
| [{pinBO)UIVOUIV(OBpin)}(EtLA)(py)2] (74) [n} | 2.11111111 | [122] |
| [{(py)catBO}UIVOUIV(OBcat)(EtLA)(py)2] (75) [o} | 2.11111111 | [122] |
| [{UIVOUIV(O2C6H4)})(EtLA)(py)2] (76) [p] | 2.11111111 | [122] |
| [{(HPh2SiO)UIVOUIV(OSiPh2H)}(EtLA)(py)2] (77) | 2.11111111 | [122] |
| [Li(THF)][UIII2(BH4)3(MeLi1)] (78) | 2.11111111 | [123] |
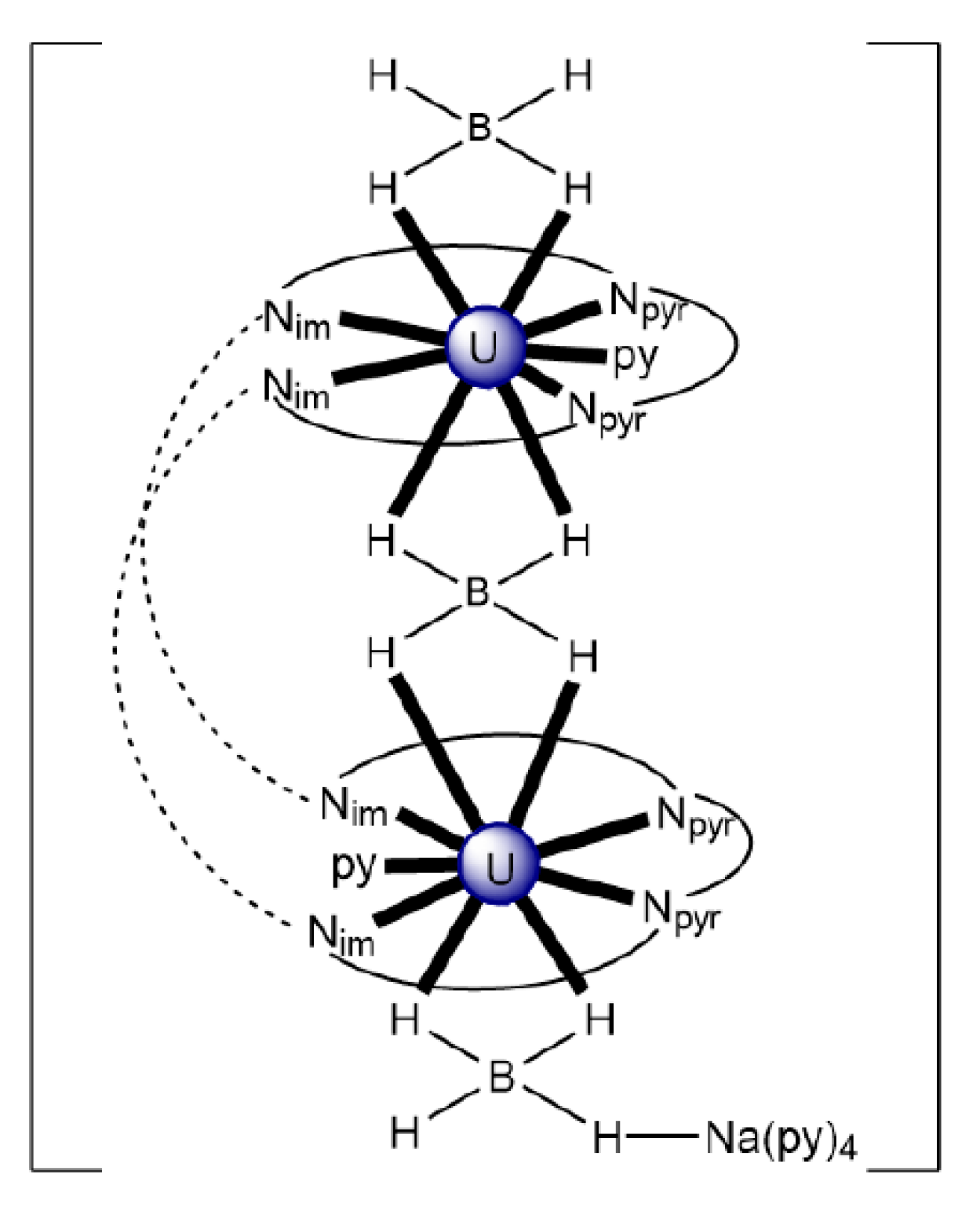
| [NaUIII2(BH4)3(EtLA)(py)6] (79) | 2.11111111 | [123] |
| [NaUIII2(OAr)2(BH4)(EtLA)(THF)2] (80) [m] | 2.11111111 | [121] |
| [KUIII2(OAr)2(BH4)(EtLA)(THF)2] (81) [m] | 2.11111111 | [121] |
| Complex [a],[b] | Coordination Mode of the Schiff-Base Ligand [c] | Ref. |
|---|---|---|
| [K3(UVIO2)(UVO2)3(salen)4(18C6)] (82) | 3.2211 | [107] |
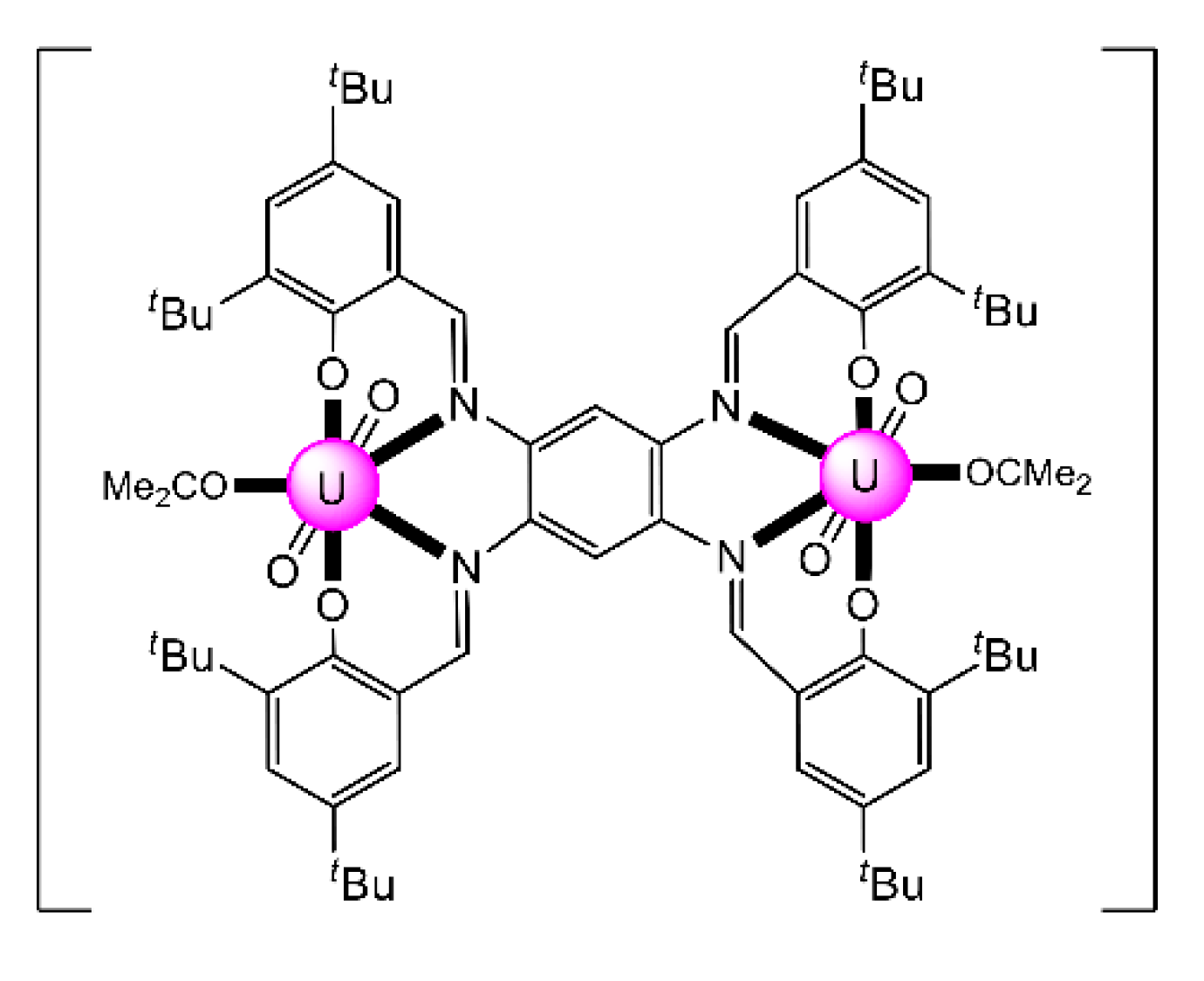
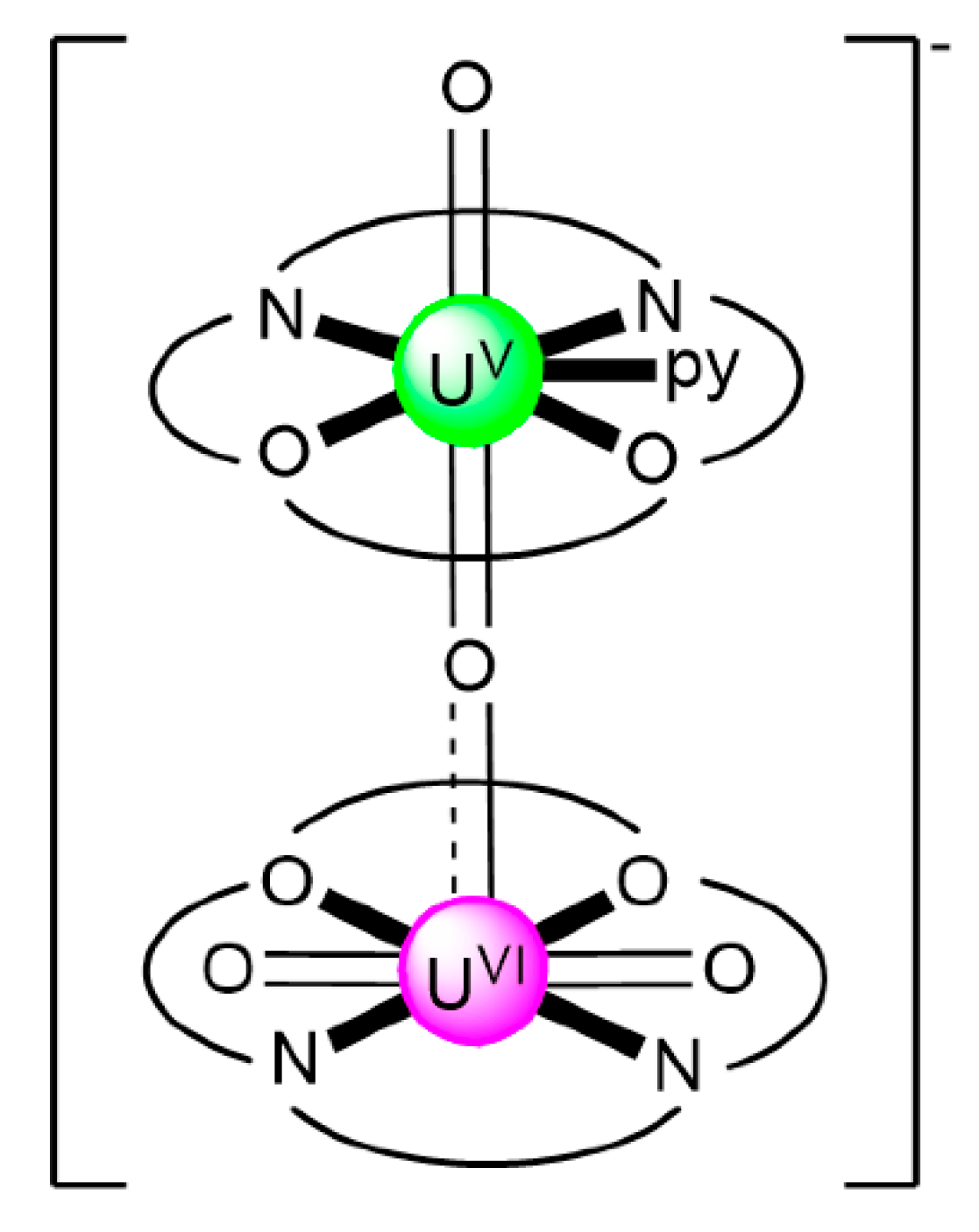
| [CoIII(Cp*)2][(UVIO2)(UVO2)(salen)2(py)] (83) | 1.1111 | [106] |
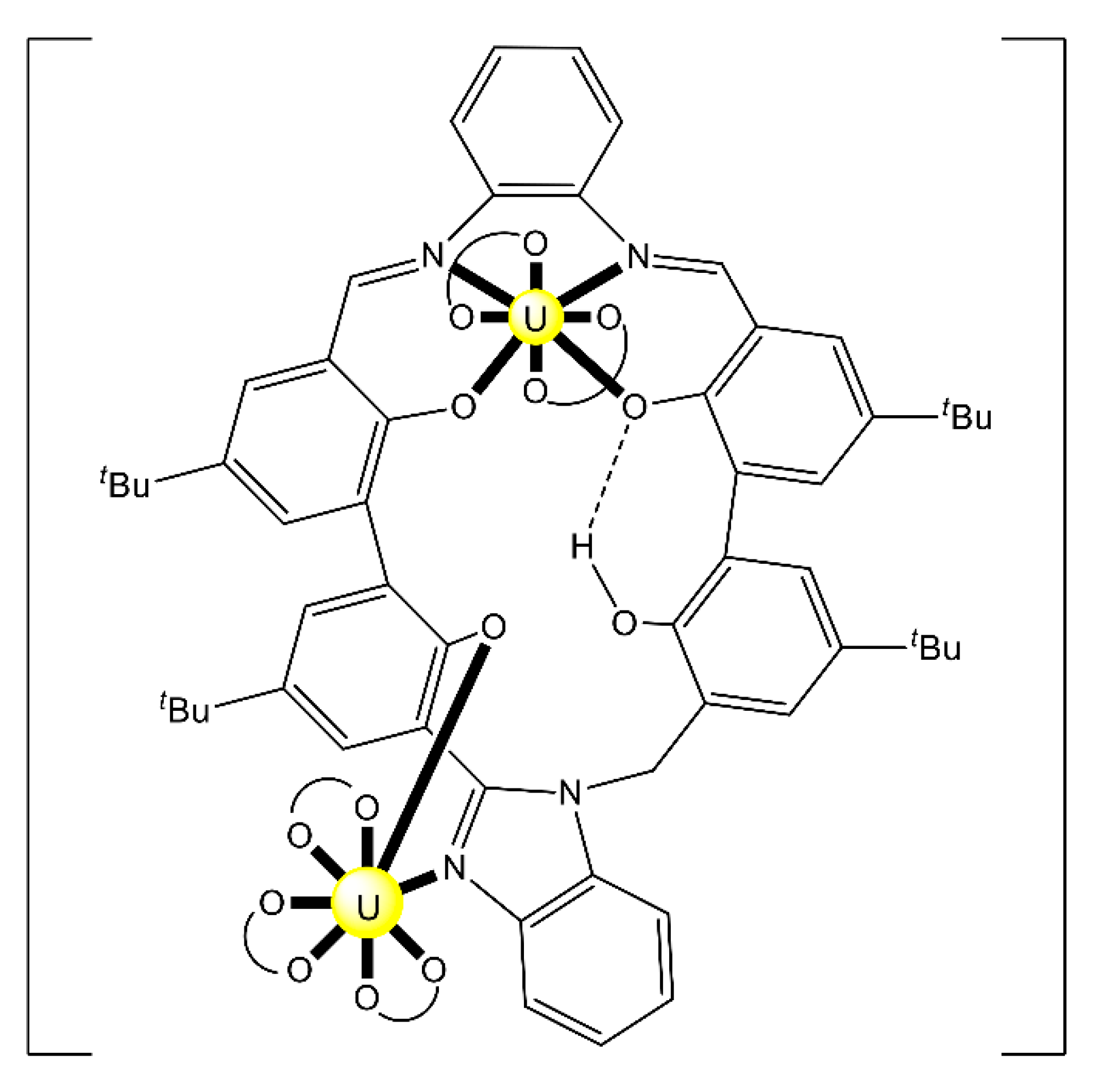
| [(UVO2)UIV2(O)(L39)4] (84) | 1.11111 | [124] |
| [(UVO2)2UIV3(O)2(tBusalophen)2(salen)3] (85) | 1.1111 [d], 2.2111 [e] | [124] |
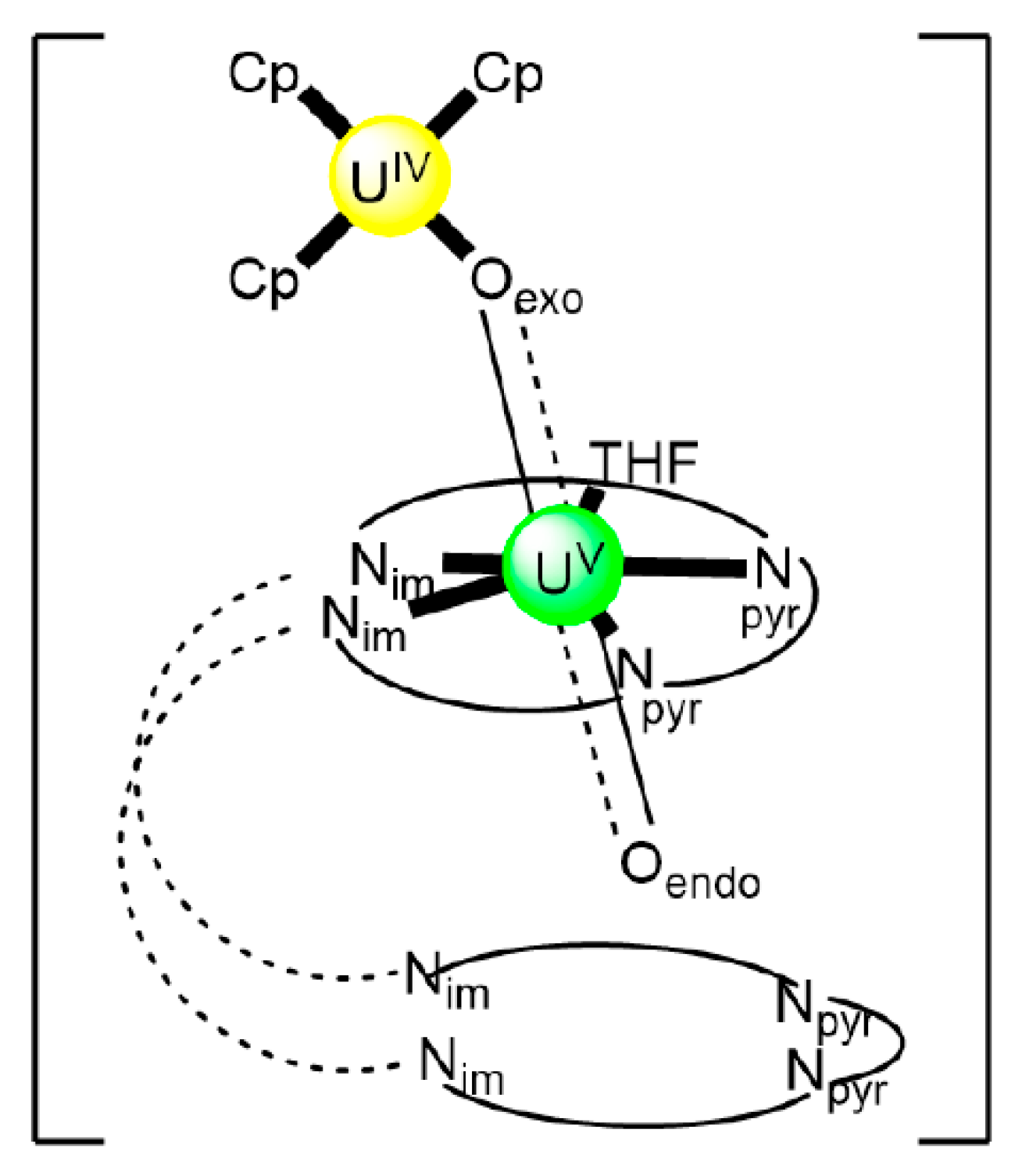
| [(UVO2)UIV(Cp)3(H2EtL1)(THF)] (85a) | 2.11110000 | [64] |
| [(UVO2)UIV(Cp)3(H2MeL1)(THF)] (85b) | 2.11110000 | [64] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsantis, S.T.; Tzimopoulos, D.I.; Holynska, M.; Perlepes, S.P. Oligonuclear Actinoid Complexes with Schiff Bases as Ligands—Older Achievements and Recent Progress. Int. J. Mol. Sci. 2020, 21, 555. https://doi.org/10.3390/ijms21020555
Tsantis ST, Tzimopoulos DI, Holynska M, Perlepes SP. Oligonuclear Actinoid Complexes with Schiff Bases as Ligands—Older Achievements and Recent Progress. International Journal of Molecular Sciences. 2020; 21(2):555. https://doi.org/10.3390/ijms21020555
Chicago/Turabian StyleTsantis, Sokratis T., Demetrios I. Tzimopoulos, Malgorzata Holynska, and Spyros P. Perlepes. 2020. "Oligonuclear Actinoid Complexes with Schiff Bases as Ligands—Older Achievements and Recent Progress" International Journal of Molecular Sciences 21, no. 2: 555. https://doi.org/10.3390/ijms21020555
APA StyleTsantis, S. T., Tzimopoulos, D. I., Holynska, M., & Perlepes, S. P. (2020). Oligonuclear Actinoid Complexes with Schiff Bases as Ligands—Older Achievements and Recent Progress. International Journal of Molecular Sciences, 21(2), 555. https://doi.org/10.3390/ijms21020555