Schiff Base Ancillary Ligands in Bis(diimine) Copper(I) Dye-Sensitized Solar Cells

 , ,
, ,  and
and 
Abstract

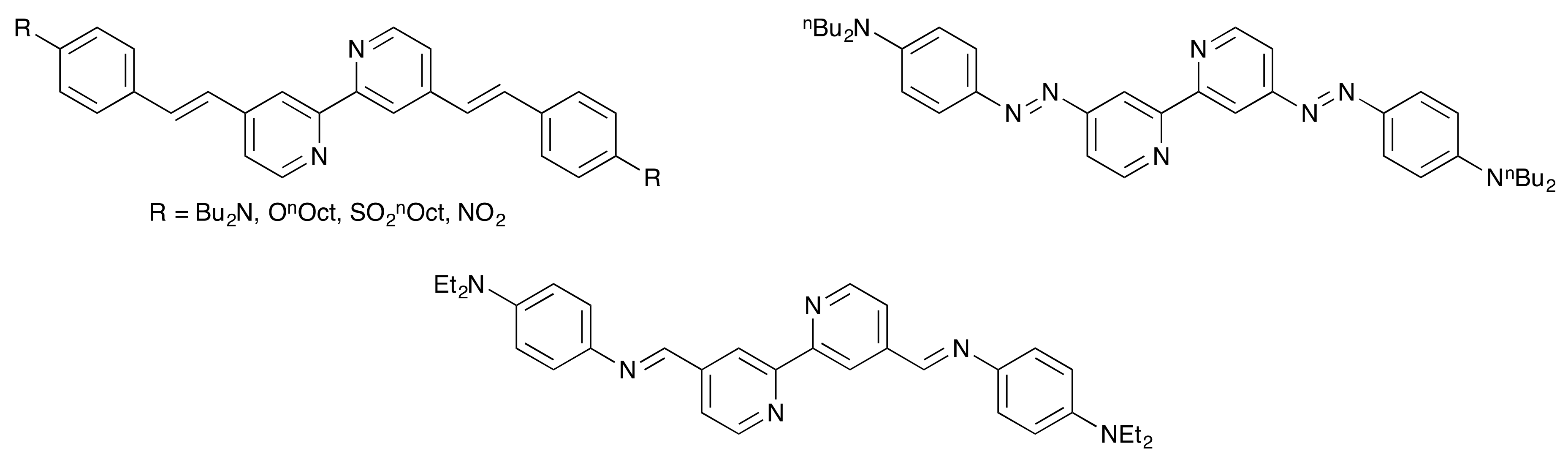
1. Introduction
2. Results and Discussion
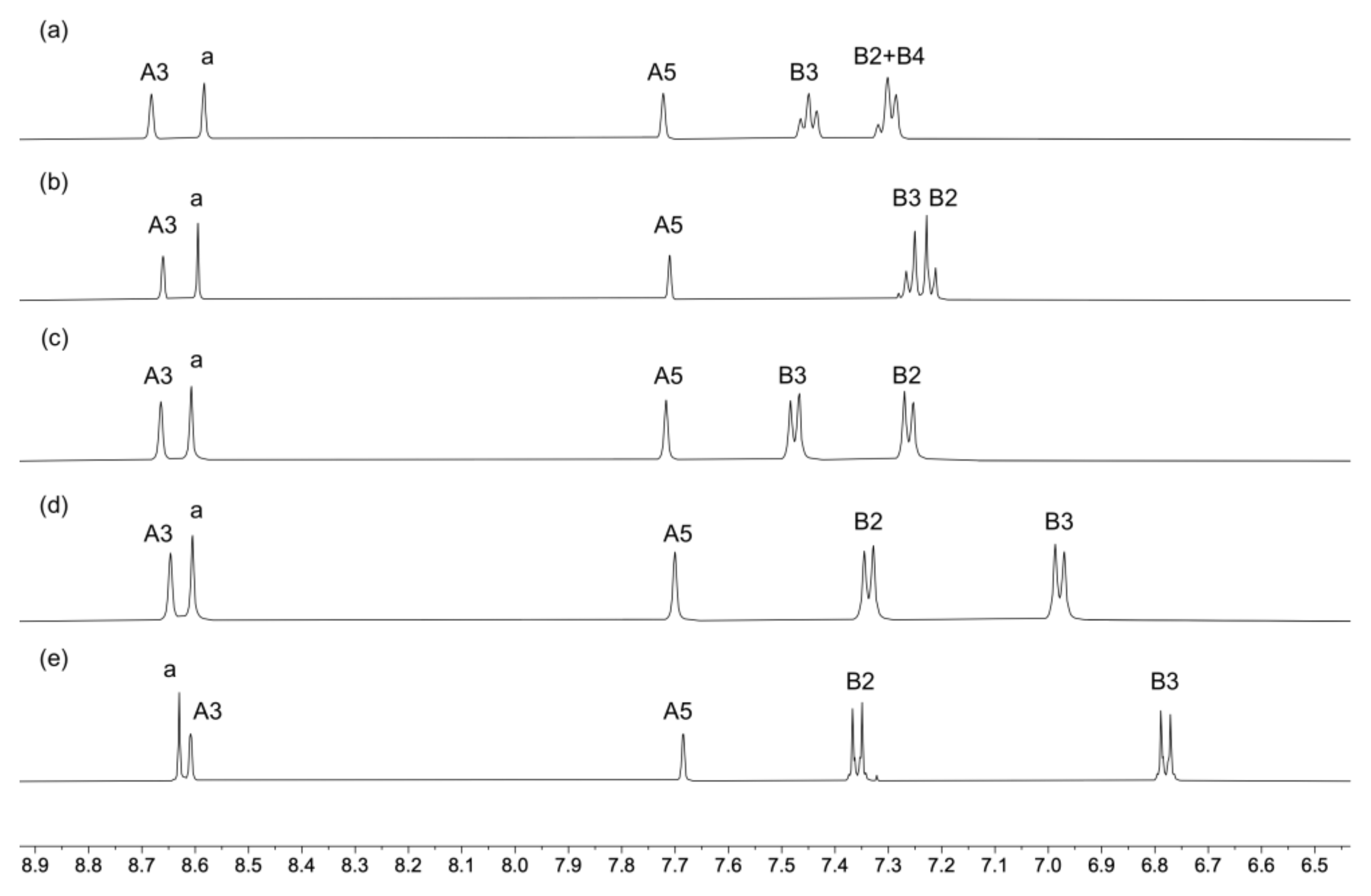
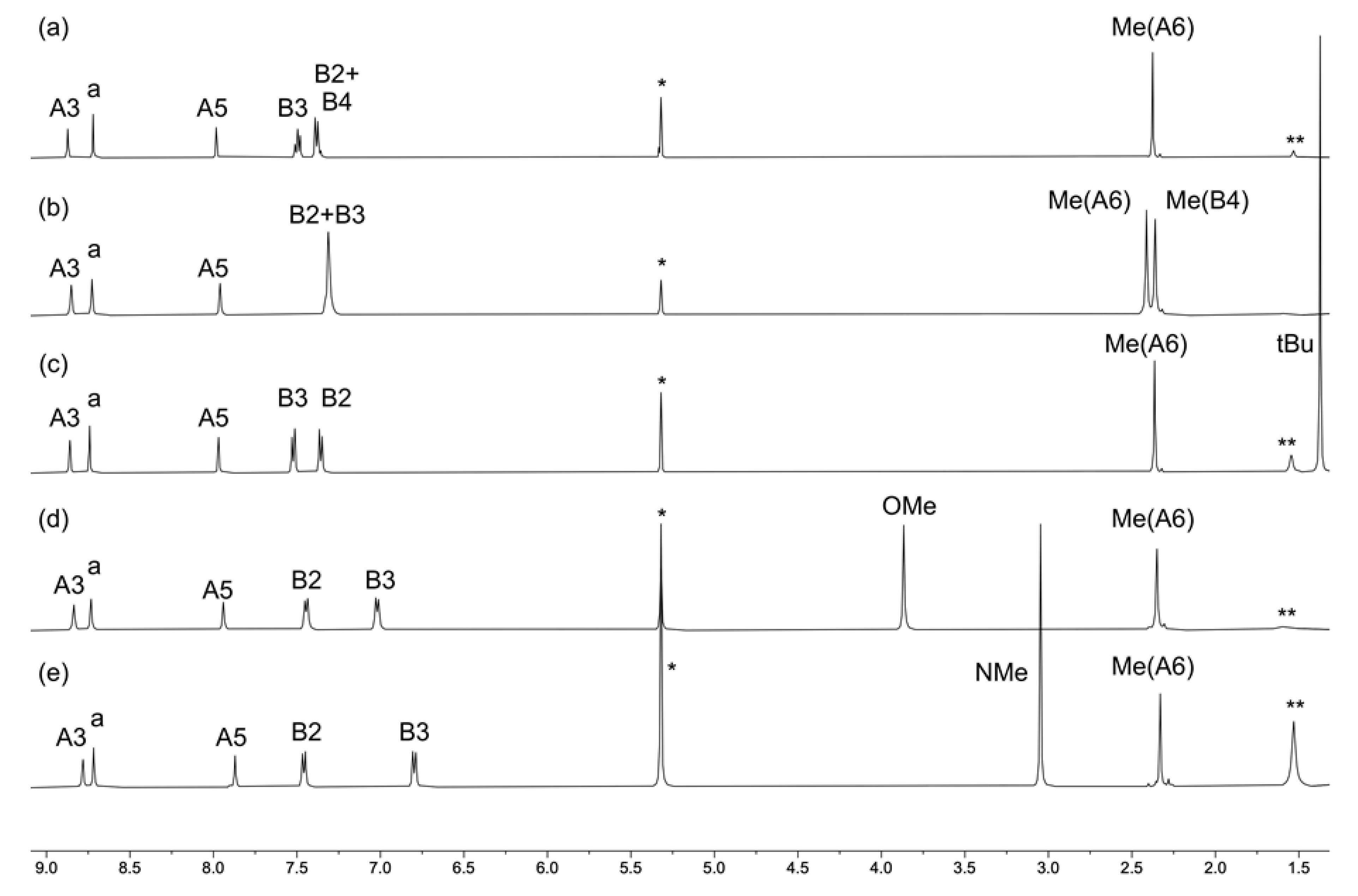
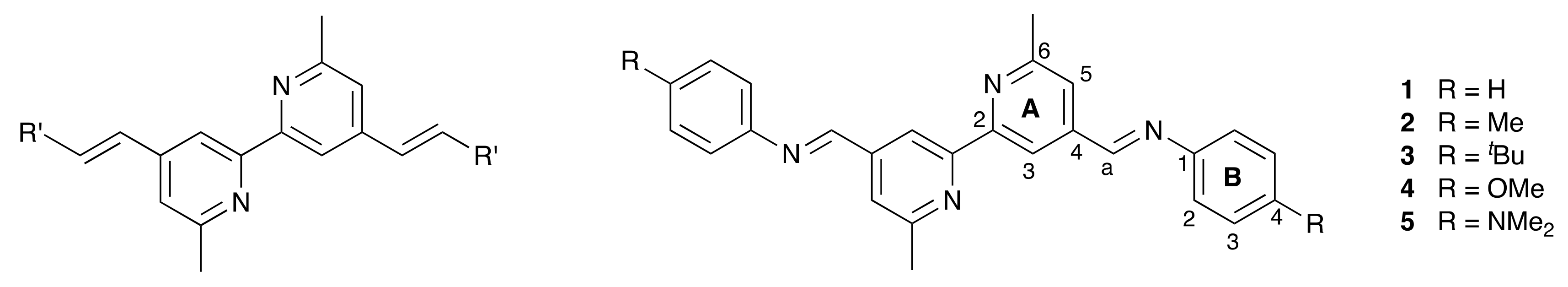
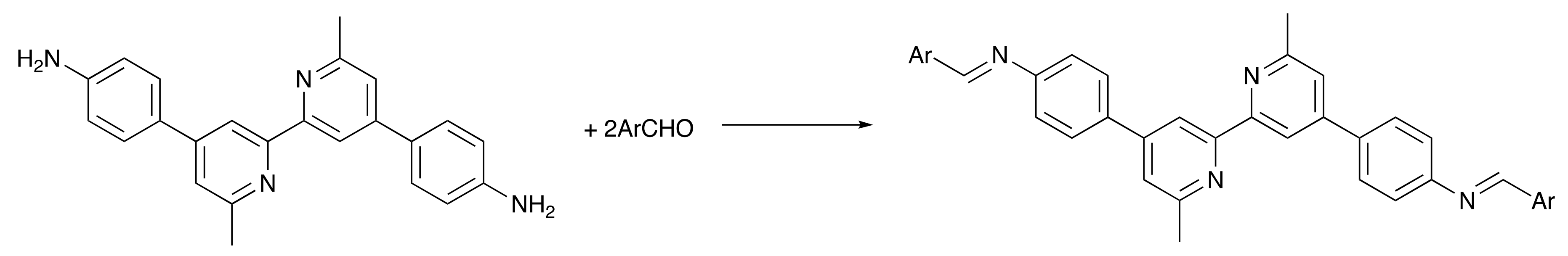
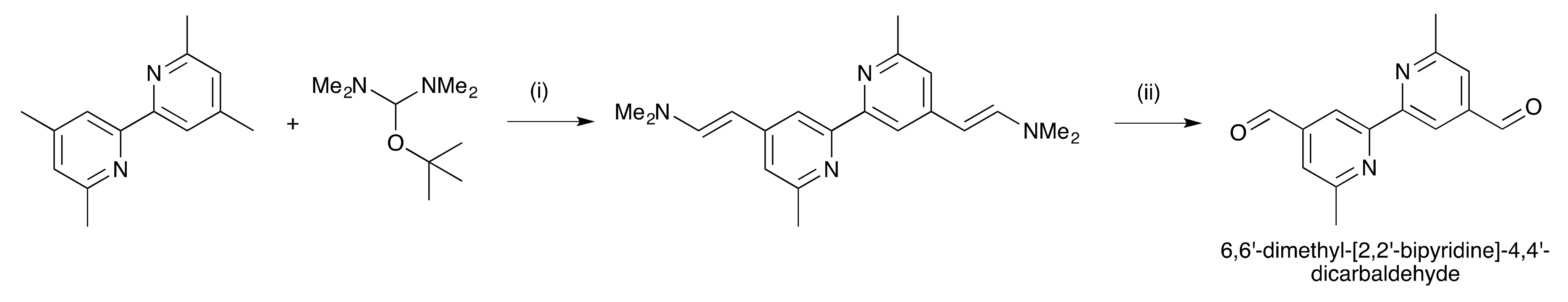
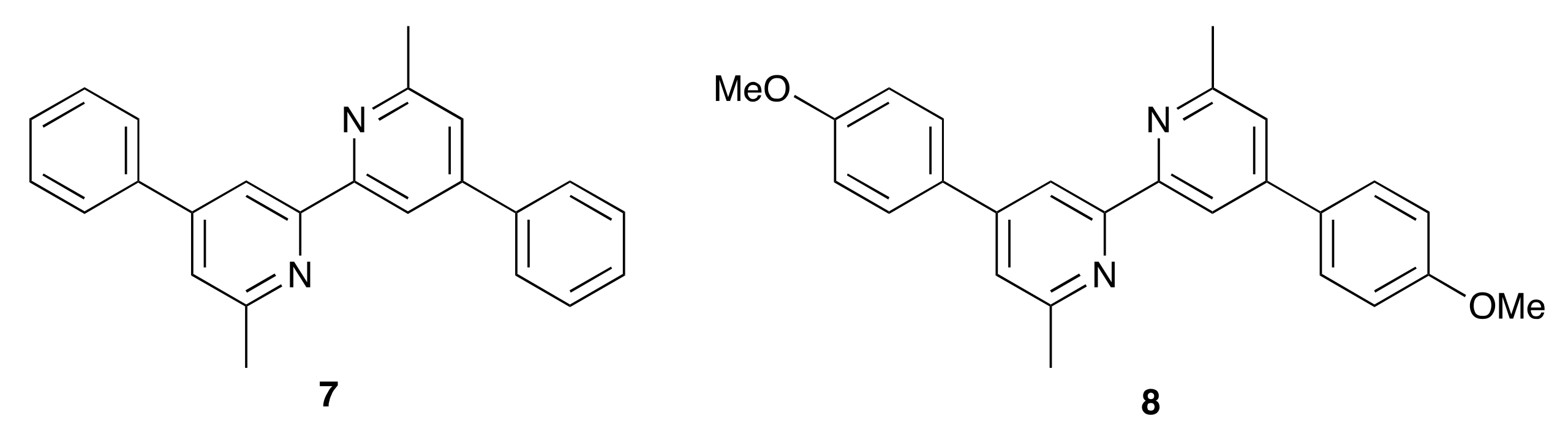
2.1. Ligand Synthesis and Characterization
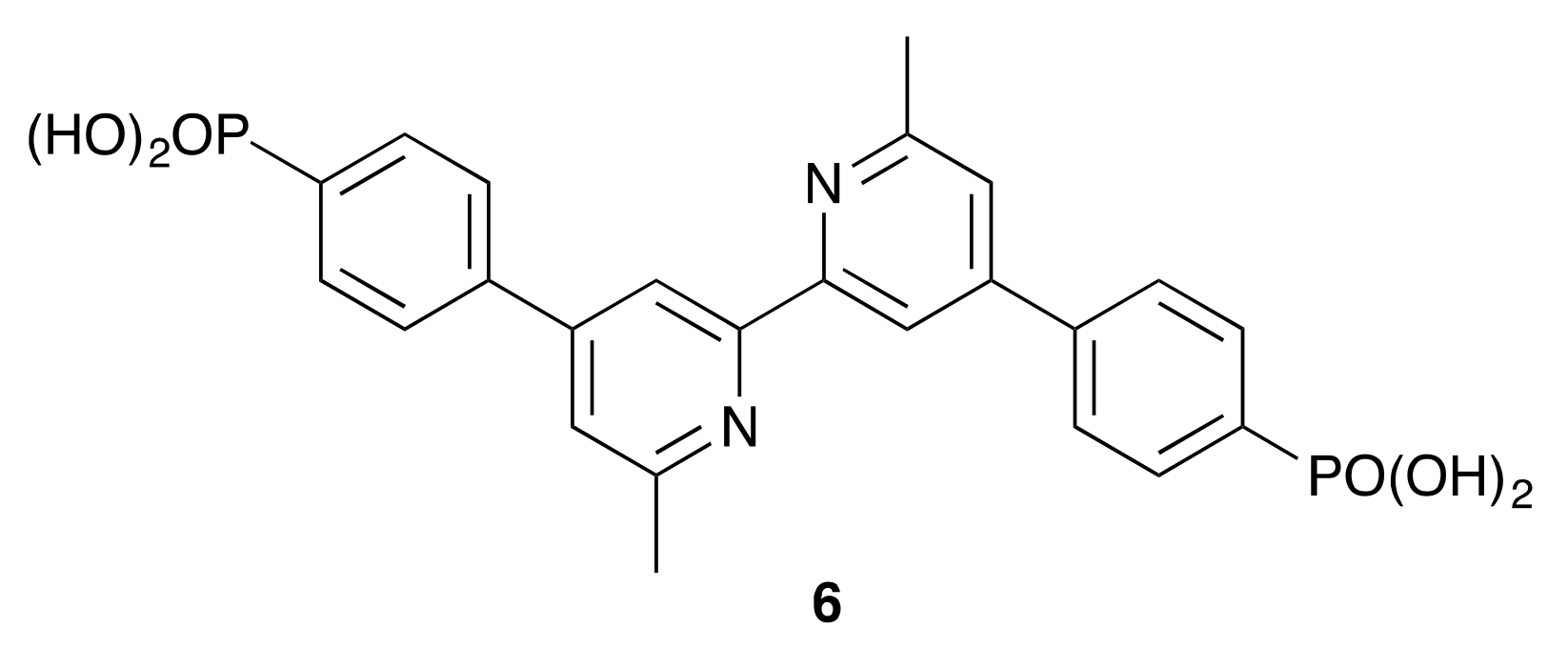
2.2. Synthesis and Characterization of the Homoleptic Copper(I) Complexes
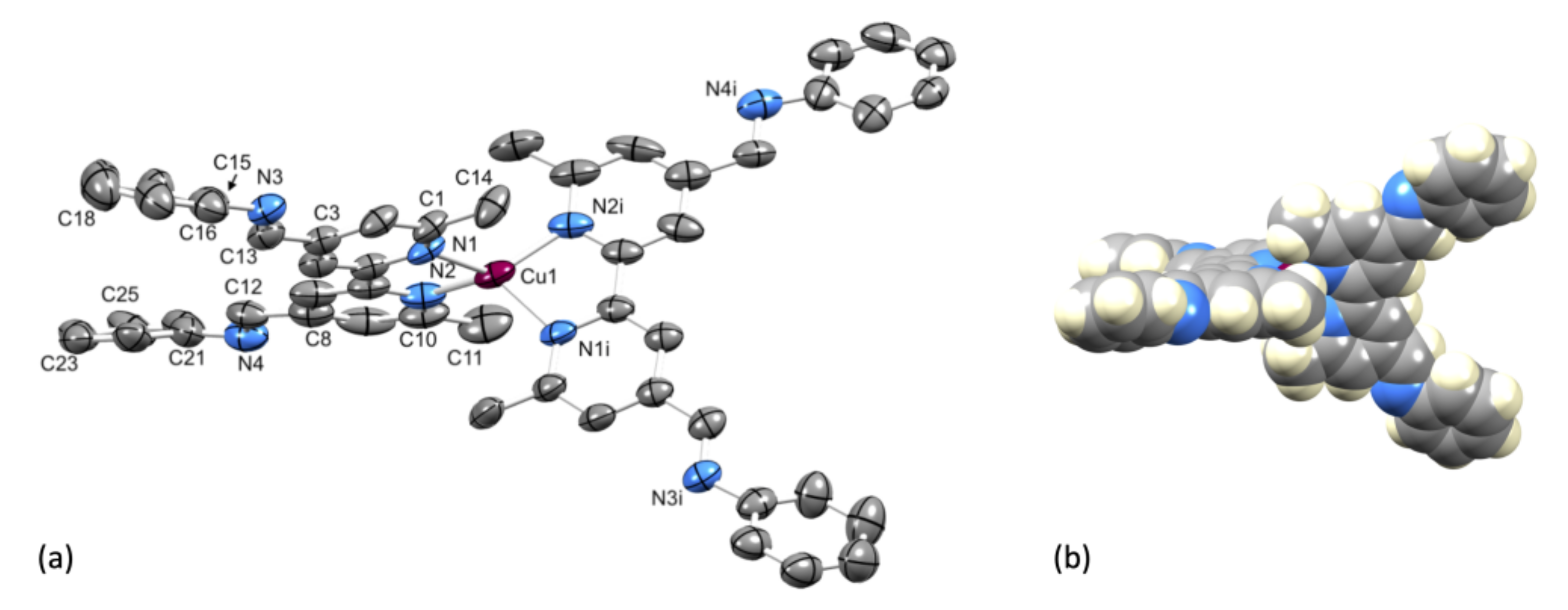
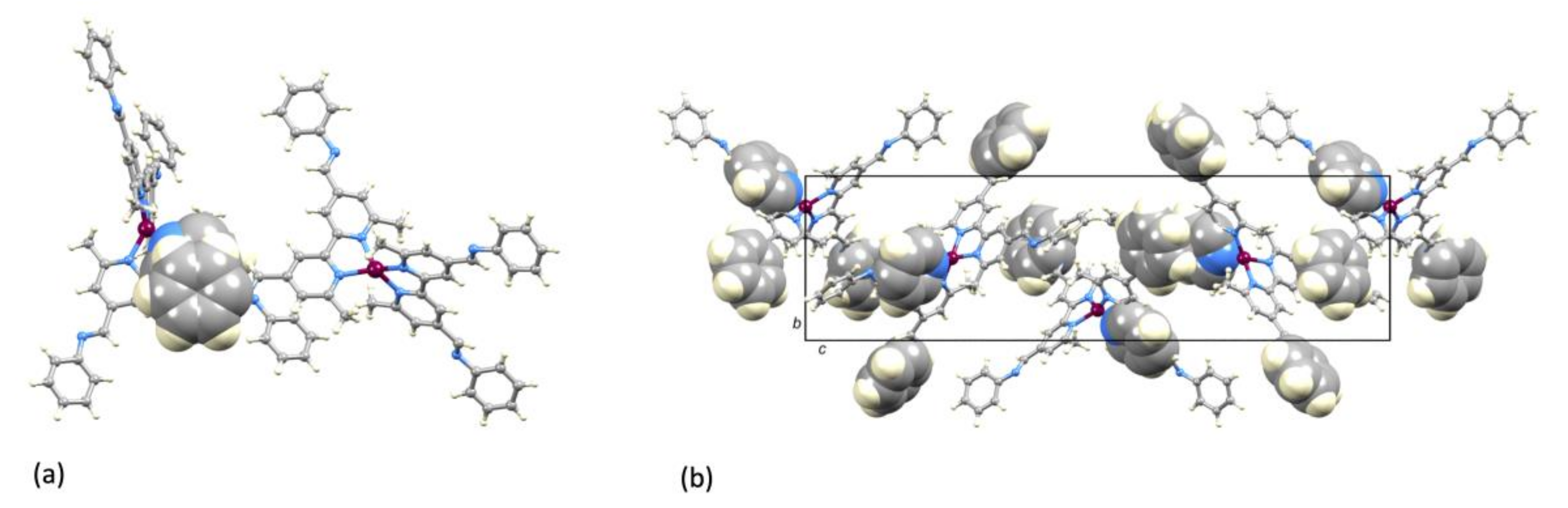
2.3. Crystal Structure of [Cu(1)2][PF6]·Et2O.
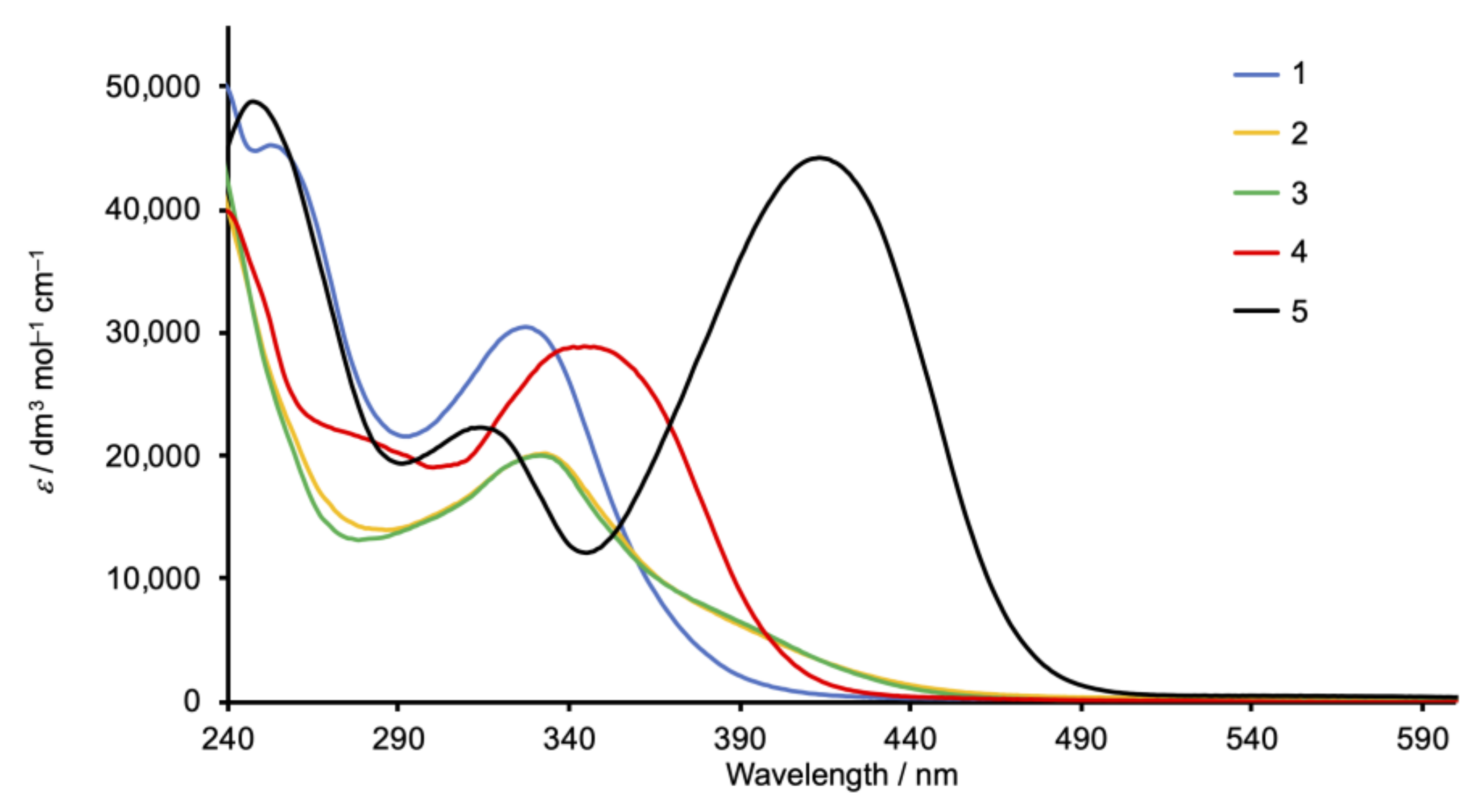
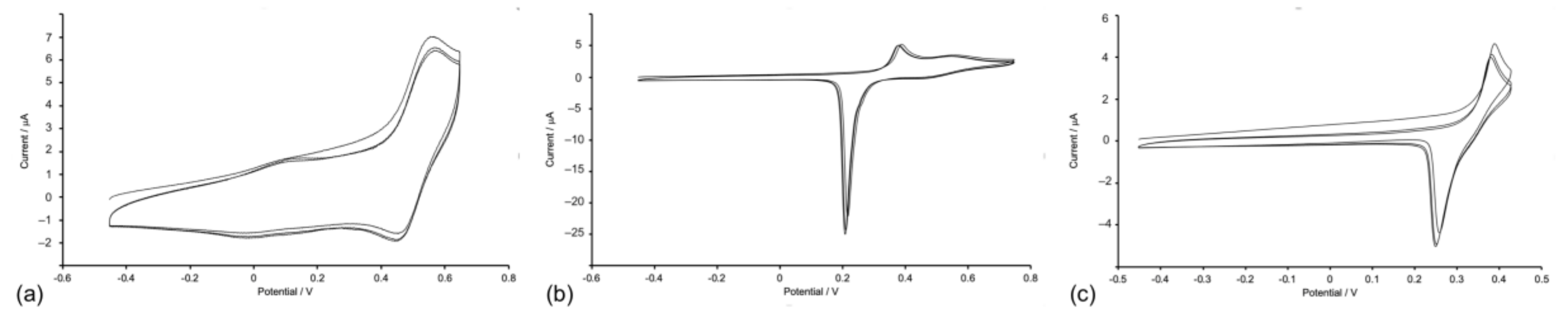
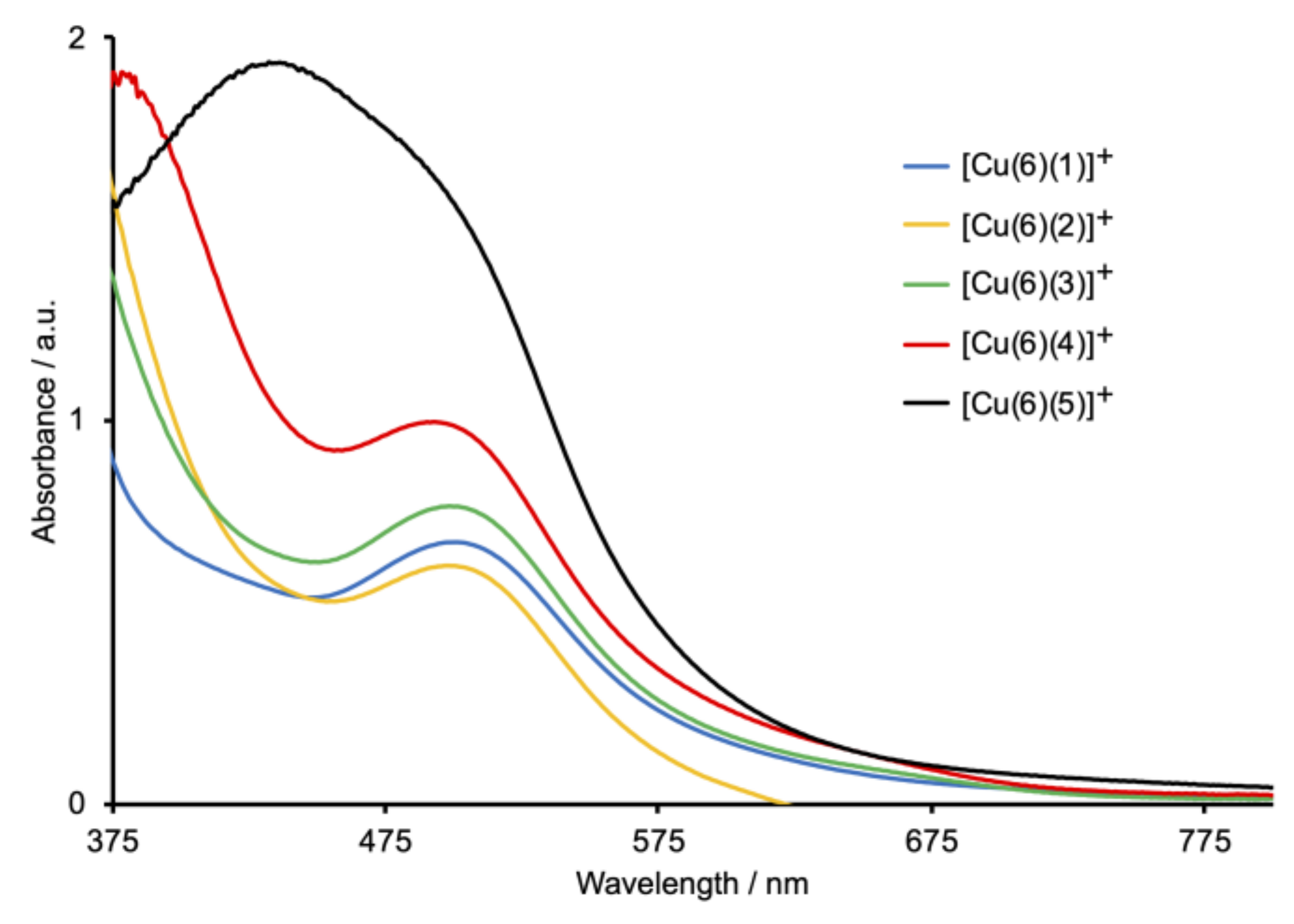
2.4. Absorption Spectra and Electrochemical Properties of the Homoleptic Copper(I) Complexes
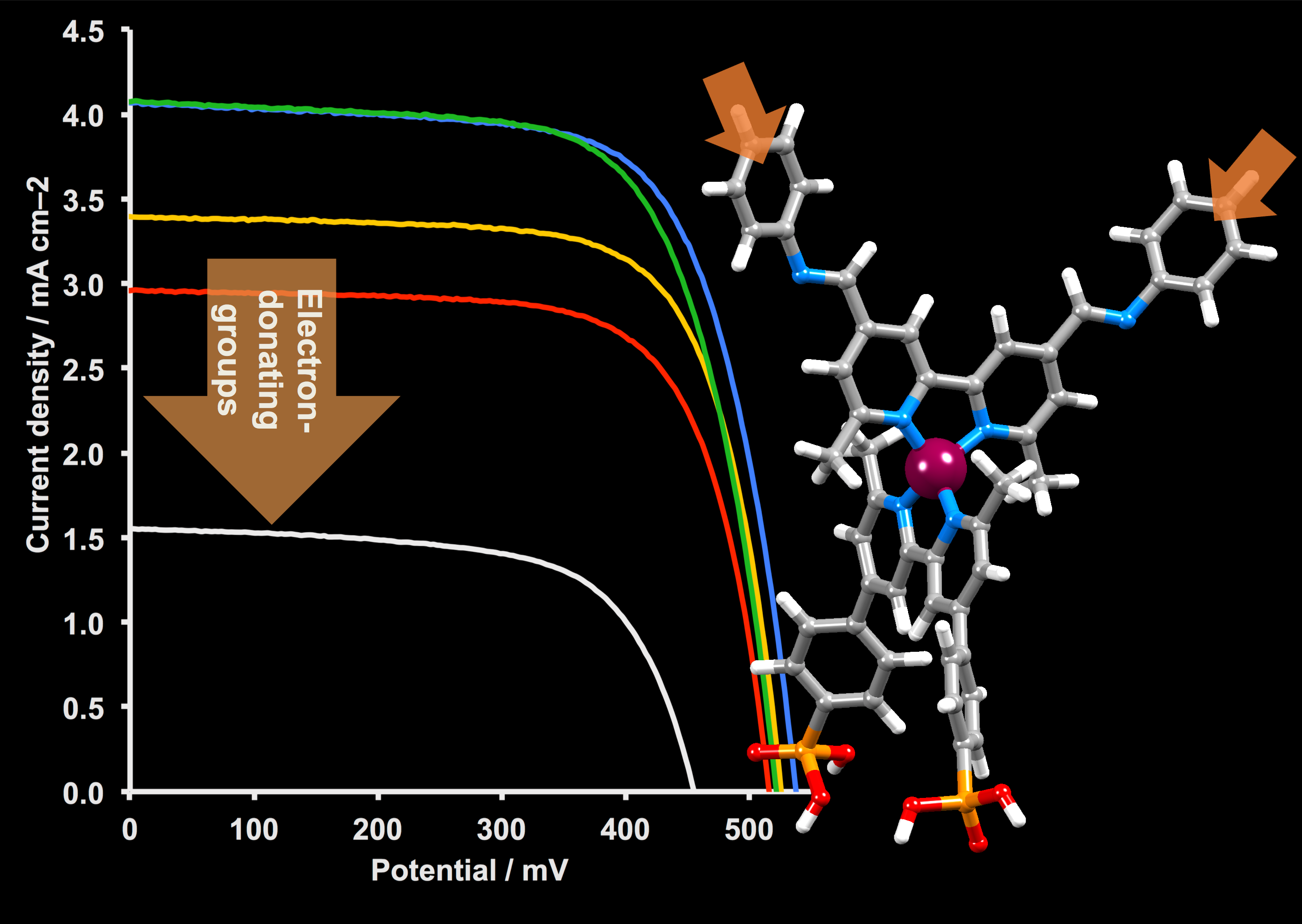
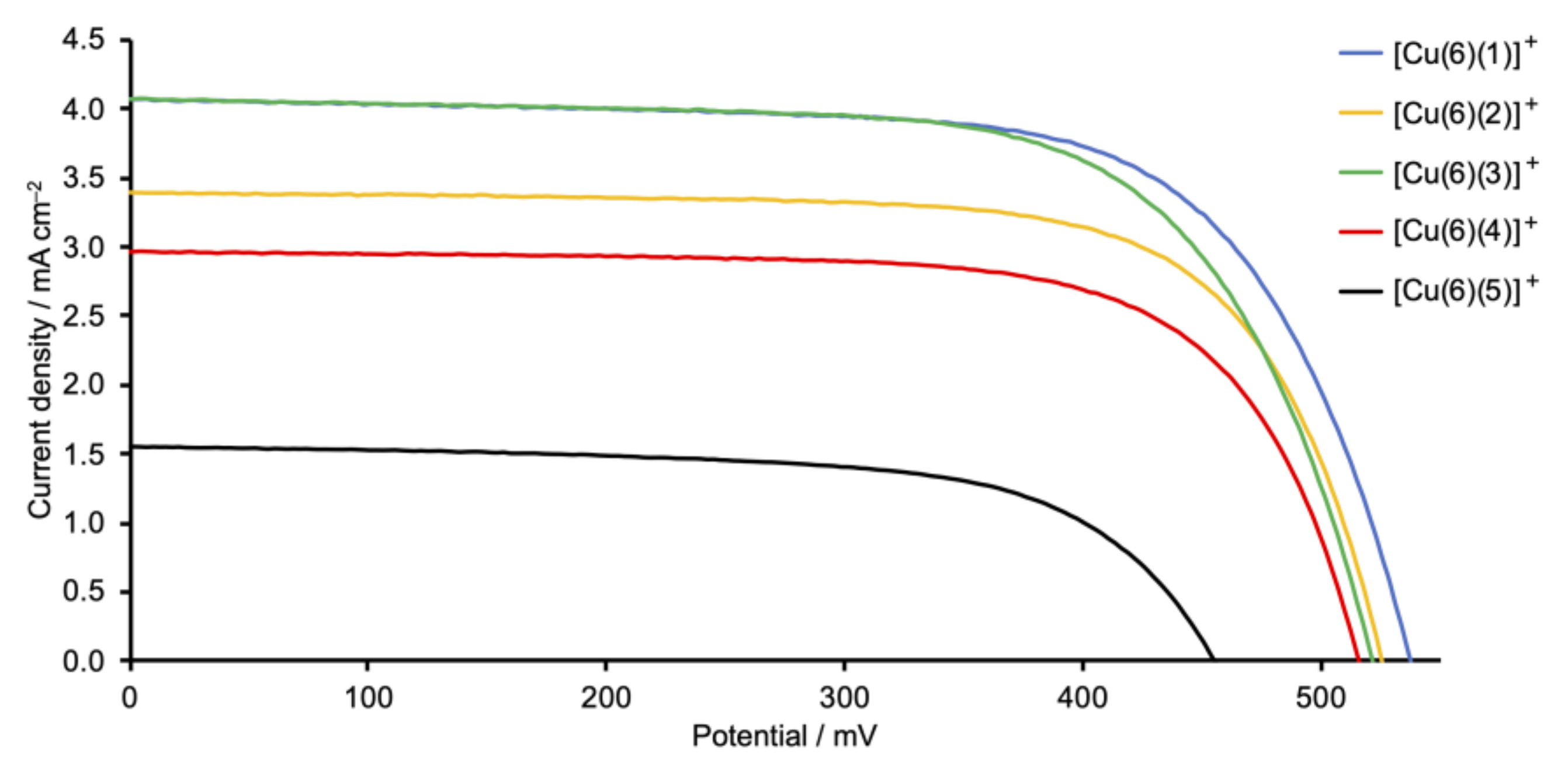
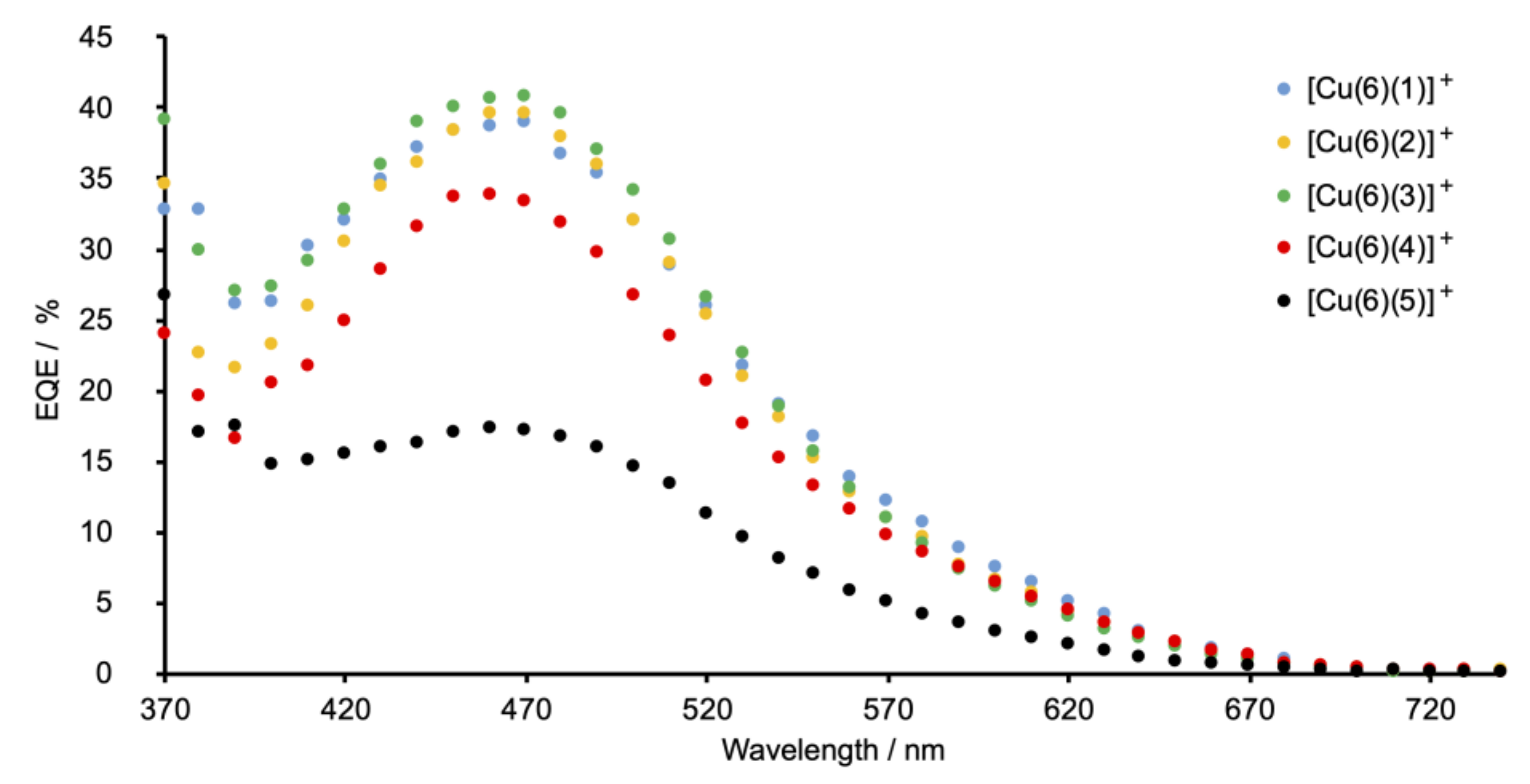
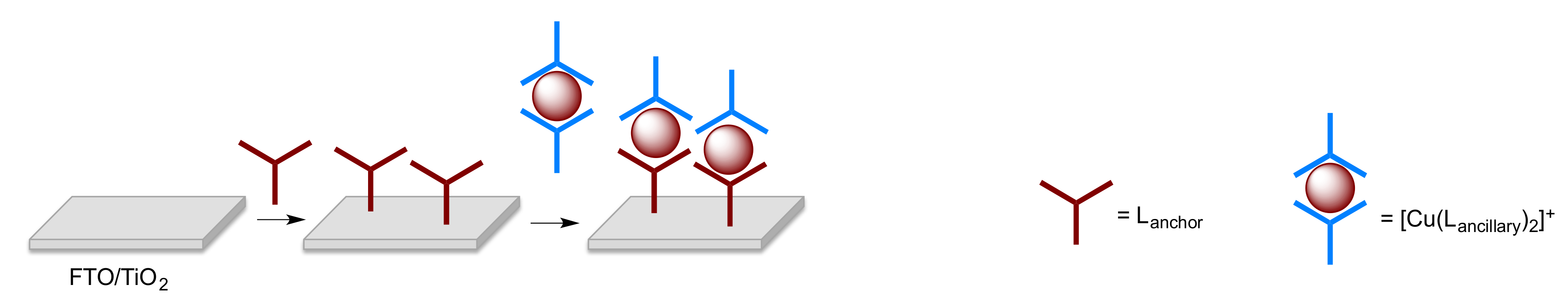
2.5. Solar Cell Fabrication and Performances
3. Materials and Methods
3.1. General
3.2. 4,4′,6,6-Tetramethyl-2,2′-Bipyridine
3.3. Synthesis of (1E,1′E)-2,2′-(6,6′-Dimethyl[2,2′-bipyridine]-4,4′-diyl)bis(N,N-dimethylethen-1-amine)
3.4. Synthesis of 6,6′-Dimethyl-[2,2′-Bipyridine]-4,4′-Dicarbaldehyde
3.5. General Procedure for the Synthesis of the Schiff Base Ligands
3.6. Compound 1
3.7. Compound 2
3.8. Compound 3
3.9. Compound 4
3.10. Compound 5
3.11. General Procedure for the Synthesis of Copper Complexes
3.12. [Cu(1)2][PF6]
3.13. [Cu(2)2][PF6]
3.14. [Cu(3)2][PF6]
3.15. [Cu(4)2][PF6]
3.16. [Cu(5)2][PF6]
3.17. Crystallography
3.18. DSC Fabrication
3.19. Electrodes for Solid-State Absorption Spectroscopy.
3.20. DSC and EQE Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- O’Reagan, B.; Grätzel, M. A low-cost, high-efficiency solar cell based on dye-sensitized colloidal TiO2 films. Nature 1991, 353, 737–740. [Google Scholar] [CrossRef]
- Grätzel, M. Solar energy conversion by dye-sensitized photovoltaic cells. Inorg. Chem. 2005, 44, 6841–6851. [Google Scholar] [CrossRef] [PubMed]
- Grätzel, M. Dye-sensitized Solar Cells. J. Photochem. Photobiol. C 2003, 4, 145–153. [Google Scholar] [CrossRef]
- Grätzel, M. Recent Advances in Sensitized Mesoscopic Solar Cells. Acc. Chem. Res. 2009, 42, 1788–1798. [Google Scholar] [CrossRef] [PubMed]
- Yella, A.; Lee, H.W.; Tsao, H.N.; Yi, C.; Chandiran, A.K.; Nazeeruddin, M.K.; Diau, E.W.; Yeh, C.Y.; Zakeeruddin, S.M.; Grätzel, M. Porphyrin-sensitized solar cells with cobalt (II/III)-based redox electrolyte exceed 12 percent efficiency. Science 2011, 334, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Higashino, T.; Imahori, H. Porphyrins as excellent dyes for dye-sensitized solar cells: Recent developments and insights. Dalton Trans. 2015, 44, 448–463. [Google Scholar] [CrossRef] [PubMed]
- Mathew, S.; Yella, A.; Gao, P.; Humphry-Baker, R.; Curchod, B.F.; Ashari-Astani, N.; Tavernelli, I.; Rothlisberger, U.; Nazeeruddin, M.K.; Grätzel, M. Dye-sensitized solar cells with 13% efficiency achieved through the molecular engineering of porphyrin sensitizers. Nat. Chem. 2014, 6, 242–247. [Google Scholar] [CrossRef]
- Kakiage, K.; Aoyama, Y.; Yano, T.; Oya, K.; Kyomen, T.; Hanaya, M. Fabrication of a high-performance dye-sensitized solar cell with 12.8% conversion efficiency using organic silyl-anchor dyes. Chem. Commun. 2015, 51, 6315–6317. [Google Scholar] [CrossRef]
- Kakiage, K.; Aoyama, Y.; Yano, T.; Oya, K.; Fujisawa, J.-I.; Hanaya, M. Highly-efficient dye-sensitized solar cells with collaborative sensitization by silyl-anchor and carboxy-anchor dyes. Chem. Commun. 2015, 51, 15894–15897. [Google Scholar] [CrossRef]
- Housecroft, C.E.; Constable, E.C. The emergence of copper(I)-based dye sensitized solar cells. Chem. Soc. Rev. 2015, 44, 8386–8398. [Google Scholar] [CrossRef]
- Lazorski, M.S.; Castellano, F.N. Advances in the light conversion properties of Cu(I)-based photosensitizers. Polyhedron 2014, 82, 57–70. [Google Scholar] [CrossRef]
- Sandroni, M.; Pellegrin, Y.; Odobel, F. Heteroleptic bis-diimine copper(I) complexes for applications in solar energy conversion. Compt. Rendus Chim. 2016, 19, 79–93. [Google Scholar] [CrossRef]
- Liu, Y.; Yiu, S.-C.; Ho, C.-L.; Wong, W.-Y. Recent Advances in Copper Complexes for Electrical/Light Energy Conversion. Coord. Chem. Rev. 2018, 375, 514–557. [Google Scholar] [CrossRef]
- Wenger, O.S. Is Iron the New Ruthenium? Chem. Eur. J. 2019, 25, 6043–6052. [Google Scholar] [CrossRef] [PubMed]
- Duchanois, T.; Liu, L.; Pastore, M.; Monari, A.; Cebrián, C.; Trolez, Y.; Darari, M.; Magra, K.; Francés-Monerris, A.; Domenichini, E.; et al. NHC-Based Iron Sensitizers for DSSCs. Inorganics 2019, 6, 63. [Google Scholar] [CrossRef]
- Jakubikova, E.; Bowman, D.N. Fe(II)-Polypyridines as Chromophores in Dye-Sensitized Solar Cells: A Computational Perspective. Acc. Chem. Res. 2015, 48, 1441–1449. [Google Scholar] [CrossRef]
- Liu, Y.; Persson, P.; Sundström, V.; Wärnmark, K. Fe N-Heterocyclic Carbene Complexes as Promising Photosensitizers. Acc. Chem. Res. 2016, 49, 1477–1485. [Google Scholar] [CrossRef]
- Mishra, A.; Fischer, M.K.R.; Bäuerle, P. Metal-Free Organic Dyes fro Dye-Sensitized Solar Cells: From Structure:Property Relationships to Design Rules. Angew. Chem. Int. Ed. 2009, 48, 2464–2499. [Google Scholar] [CrossRef]
- Lu, J.; Liu, S.; Wang, M. Push–Pull Zinc Porphyrins as Light-Harvesters for Efficient Dye-Sensitized Solar Cells. Front. Chem. 2018, 6, 541. [Google Scholar] [CrossRef]
- Wu, Y.; Zhu, W. Organic sensitizers from D-π-A to D-A-π-A: Effect of the internal electron-withdrawing units on molecular absorption, energy levels and photovoltaic performances. Chem. Soc. Rev. 2013, 42, 2039–2058. [Google Scholar] [CrossRef]
- Sandroni, M.; Kayanuma, M.; Planchat, A.; Szuwarski, N.; Blart, E.; Pellegrin, Y.; Daniel, C.; Boujtita, M.; Odobel, F. First application of the HETPHEN concept to new heteroleptic bis(diimine) copper(I) complexes as sensitizers in dye sensitized solar cells. Dalton. Trans. 2013, 42, 10818–10827. [Google Scholar] [CrossRef] [PubMed]
- Sandroni, M.; Favereau, L.; Planchat, A.; Akdas-Kilig, H.; Szuwarski, N.; Pellegrin, Y.; Blart, E.; Le Bozec, H.; Boujtita, M.; Odobel, F. Heteroleptic copper(I)–polypyridine complexes as efficient sensitizers for dye sensitized solar cells. J. Mater. Chem. A 2014, 2, 9944–9947. [Google Scholar] [CrossRef]
- Malzner, F.J.; Housecroft, C.E.; Constable, E.C. The versatile SALSAC approach to heteroleptic copper(I) dye assembly in dye-sensitized solar cells. Inorganics 2018, 6, 57. [Google Scholar] [CrossRef]
- Freimann, S.A.; Zare, D.; Housecroft, C.E.; Constable, E.C. The SALSAC Approach: Comparing the reactivity of solvent-dispersed nanoparticles with nanoparticulate surfaces. Nanoscale Adv. 2020, 2, 679–690. [Google Scholar] [CrossRef]
- Renouard, T.; Le Bozec, H.; Brasselet, S.; Ledoux, I.; Zyss, J. Tetrahedral bipyridyl copper(I) complexes: A new class of non-dipolar chromophore for non-linear optics. Chem. Commun. 1999, 871–872. [Google Scholar] [CrossRef]
- Bessho, T.; Constable, E.C.; Graetzel, M.; Hernandez Redondo, A.; Housecroft, C.E.; Kylberg, W.; Nazeeruddin, M.K.; Neuburger, M.; Schaffner, S. An element of surprise—Efficient copper-functionalized dye-sensitized solar cells. Chem. Chem. Commun. 2008, 3717–3719. [Google Scholar] [CrossRef]
- Sandroni, M.; Kayanuma, M.; Rebarz, M.; Akdas-Kilig, H.; Pellegrin, Y.; Blart, E.; Le Bozec, H.; Daniel, C.; Odobel, F. Heteroleptic diimine copper(I) complexes with large extinction coefficients: Synthesis, quantum chemistry calculations and physico-chemical properties. Dalton Trans. 2013, 42, 14628–14638. [Google Scholar] [CrossRef]
- Zhang, L.; Cole, J.M.; Waddell, P.G.; Low, K.S.; Liu, X. Relating Electron Donor and Carboxylic Acid Anchoring Substitution Effects in Azo Dyes to Dye-Sensitized Solar Cell Performance. ACS Sustain. Chem. Eng. 2013, 1, 1440–1452. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, L.; Wong, W.-Y. Energy materials based on metal Schiff base complexes. Coord. Chem. Rev. 2018, 355, 180–198. [Google Scholar] [CrossRef]
- Büttner, A. Dye Sensitized Solar Cells–Investigation of Bis(diimine)copper(I) Dyes. Ph.D. Thesis, University of Basel, Basel, Switzerland, 2018. Available online: https://edoc.unibas.ch/65143/ (accessed on 3 March 2020).
- Maury, O.; Guégan, J.-P.; Renouard, T.; Hilton, A.; Dupau, P.; Sandon, N.; Toupet, L.; Le Bozec, H. Design and synthesis of 4,4′-π-conjugated[2,2′]-bipyridines: A versatile class of tunable chromophores and fluorophores. New J. Chem. 2001, 25, 1553–1566. [Google Scholar] [CrossRef]
- Dupau, P.; Renouard, T.; Bozec, H.L. Straightforward Synthesis of 4-Formyl- and 4,4′-Diformyl-2,2′-Bipyridines: Access to New Dialkenyl Substituted Bipyridyl Ligands. Tetrahedron Lett. 1996, 37, 7503–7506. [Google Scholar] [CrossRef]
- Janiak, C. A critical account on π–π stacking in metal complexes with aromatic nitrogen-containing ligands. J. Chem. Soc. Dalton Trans. 2000, 3885–3896. [Google Scholar] [CrossRef]
- Bozic-Weber, B.; Brauchli, S.Y.; Constable, E.C.; Fürer, S.O.; Housecroft, C.E.; Malzner, F.J.; Wright, I.A.; Zampese, J.A. Improving the photoresponse of copper(I) dyes in dye-sensitized solar cells by tuning ancillary and anchoring ligand modules. Dalton Trans. 2013, 42, 12293–12308. [Google Scholar] [CrossRef] [PubMed]
- Saygili, Y.; Söderberg, M.; Pellet, N.; Giordano, F.; Cao, Y.; Belen Muñoz-García, A.; Zakeeruddin, S.M.; Vlachopoulos, N.; Pavone, M.; Boschloo, G.; et al. Copper Bipyridyl Redox Mediators for Dye-Sensitized Solar Cells with High Photovoltage. J. Am. Chem. Soc. 2016, 138, 15087–15096. [Google Scholar] [CrossRef]
- Pavlishchuk, V.V.; Addison, A.W. Conversion constants for redox potentials measured versus different reference electrodes in acetonitrile solutions at 25 °C. Inorg. Chim. Acta 2000, 298, 97–102. [Google Scholar] [CrossRef]
- Stephens, A.J.; Malzner, F.J.; Constable, E.C.; Housecroft, C.E. The influence of phosphonic acid protonation state on the efficiency of bis(diimine)copper(I) dye-sensitized solar cells. Sustain. Ener. Fuels 2018, 2, 786–794. [Google Scholar] [CrossRef]
- Klein, Y.M.; Willgert, M.; Prescimone, A.; Constable, E.C.; Housecroft, C.E. Positional isomerism makes a difference: Phosphonic acid anchoring ligands with thienyl spacers in copper(I)-based dye-sensitized solar cells. Dalton Trans. 2016, 45, 4659–4672. [Google Scholar] [CrossRef]
- Baumgartner, Y.; Klein, Y.M.; Constable, E.C.; Housecroft, C.E.; Willgert, M. Cyanoacrylic- and (1-cyanovinyl)phosphonic acid anchoring ligands for application in copper-based dye-sensitized solar cells. RSC Adv. 2016, 6, 86220–86231. [Google Scholar] [CrossRef]
- Malzner, F.J.; Brauchli, S.Y.; Schönhofer, E.; Constable, E.C.; Housecroft, C.E. To deprotect or not to deprotect: Phosphonate ester versus phosphonic acid anchor ligands in copper(I)-based dye-sensitized solar cells. Polyhedron 2014, 82, 116–121. [Google Scholar] [CrossRef]
- Büttner, A.; Brauchli, S.Y.; Constable, E.C.; Housecroft, C.E. Effects of introducing methoxy groups into the ancillary ligands in bis(diimine)copper(I) dyes for dye-sensitized solar cells. Inorganics 2018, 6, 40. [Google Scholar] [CrossRef]
- Kubas, G.J.; Monzyk, B.; Crumbliss, A.L. Tetrakis(acetonitrile)copper(I) hexafluorophosphate. Inorg. Synth. 1979, 19, 90–92. [Google Scholar] [CrossRef]
- Mukkala, V.-M.; Kankare, J.J. New 2,2′-Bipyridine Derivatives and Their Luminescence Properties with Europium(III) and Terbium(III) Ions. Helv. Chim. Acta 1992, 75, 1578–1592. [Google Scholar] [CrossRef]
- Harding, M.M.; Koert, U.; Lehn, J.-M.; Marquis-Rigault, A.; Piguet, C.; Siegel, J. Synthesis of Unsubstituted and 4,4′-Substituted Oligobipyridines as Ligand Strands for Helicate Self-Assembly. Helv. Chim. Acta 1991, 74, 594–610. [Google Scholar] [CrossRef]
- Boyer, N.; Gloanec, P.; De Nanteuil, G.; Jubault, P.; Quirion, J.-C. Chemoselective and stereoselective synthesis of gem-difluoro-β-aminoesters or gem-difluoro-β-lactams from ethylbromodifluoroacetate and imines during Reformatsky reaction. Tetrahedron 2007, 63, 12352–12366. [Google Scholar] [CrossRef]
- Software for the Integration of CCD Detector System Bruker Analytical X-ray Systems; Bruker axs: Madison, WI, USA, (after 2013).
- Sheldrick, G.M. ShelXT-Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. Olex2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with ShelXL. Acta Cryst. 2015, C27, 3–8. [Google Scholar] [CrossRef]
- Spek, A.L. PLATON SQUEEZE: A Tool for the Calculation of the Disordered Solvent Contribution to the Calculated Structure Factors. Acta Crystallogr. 2015, C71, 9–18. [Google Scholar] [CrossRef]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: Visualization and Analysis of Crystal Structures. J. Appl. Cryst. 2006, 39, 453–457. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0 - New Features for the Visualization and Investigation of Crystal Structures. J. Appl. Cryst. 2008, 41, 466–470. [Google Scholar] [CrossRef]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | E1/2ox/V | Epc – Epa/mV | Epca | Epaa,c/V |
|---|---|---|---|---|
| 2 | −2.11 | |||
| 5 | +0.39 | −2.17 | ||
| [Cu(1)2][PF6] | +0.49b | 130 | −1.68, –1.89, –2.04 | |
| [Cu(2)2][PF6] | +0.49b | 70 | −1.69, −1.96, −2.04 | |
| [Cu(3)2][PF6] | +0.48b | 70 | −1.71, −1.92, −2.07, −2.21 | |
| [Cu(4)2][PF6] | +0.49b | 90 | −1.74, −1.95, −2.12 | |
| [Cu(5)2][PF6] | +0.38,c +0.54b | −1.78, −2.01, −2.11, −2.20 |
| Dye | Cell Number | JSC/mA cm−2 | VOC/mV | ff/% | η/% | Relative η/% |
|---|---|---|---|---|---|---|
| [Cu(6)(1)]+ | 1 | 4.08 | 538 | 69 | 1.51 | 26 |
| [Cu(6)(1)]+ | 2 | 3.64 | 520 | 69 | 1.32 | 23 |
| [Cu(6)(1)]+ | 3 | 3.87 | 534 | 70 | 1.44 | 25 |
| [Cu(6)(2)]+ | 1 | 3.40 | 526 | 71 | 1.28 | 22 |
| [Cu(6)(2)]+ | 2 | 3.01 | 530 | 72 | 1.14 | 20 |
| [Cu(6)(2)]+ | 3 | 3.44 | 524 | 70 | 1.26 | 22 |
| [Cu(6)(3)]+ | 1 | 4.08 | 522 | 68 | 1.45 | 25 |
| [Cu(6)(3)]+ | 2 | 3.51 | 522 | 70 | 1.28 | 22 |
| [Cu(6)(3)]+ | 3 | 3.40 | 518 | 71 | 1.26 | 22 |
| [Cu(6)(4)]+ | 1 | 2.83 | 508 | 70 | 1.01 | 18 |
| [Cu(6)(4)]+ | 2 | 2.96 | 516 | 71 | 1.08 | 19 |
| [Cu(6)(4)]+ | 3 | 2.74 | 514 | 70 | 0.99 | 17 |
| [Cu(6)(5)]+ | 1 | 1.56 | 455 | 65 | 0.46 | 8 |
| [Cu(6)(5)]+ | 2 | 1.49 | 461 | 66 | 0.45 | 8 |
| [Cu(6)(5)]+ | 3 | 1.55 | 455 | 65 | 0.46 | 8 |
| N719 | 13.42 | 640 | 67 | 5.74 | 100 |
| Dye | Imine Spacer in Lancillary | JSC/mA cm−2 | VOC/mV | ff/% | η/% | Reference |
|---|---|---|---|---|---|---|
| [Cu(6)(1)]+ | yes | 4.08 | 538 | 69 | 1.51 | This work |
| [Cu(6)(4)]+ | yes | 2.96 | 516 | 71 | 1.08 | This work |
| N719 | 13.42 | 640 | 67 | 5.74 | This work | |
| [Cu(6)(7)]+ | no | 4.27 | 545 | 71 | 1.66 | [41] |
| N719 | 13.29 | 647 | 67 | 5.79 | [41] | |
| [Cu(6)(8)]+ | no | 4.87 | 528 | 71 | 1.82 | [41] |
| N719 | 13.91 | 635 | 68 | 6.04 | [41] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lüthi, E.; Forero Cortés, P.A.; Prescimone, A.; Constable, E.C.; Housecroft, C.E. Schiff Base Ancillary Ligands in Bis(diimine) Copper(I) Dye-Sensitized Solar Cells. Int. J. Mol. Sci. 2020, 21, 1735. https://doi.org/10.3390/ijms21051735
Lüthi E, Forero Cortés PA, Prescimone A, Constable EC, Housecroft CE. Schiff Base Ancillary Ligands in Bis(diimine) Copper(I) Dye-Sensitized Solar Cells. International Journal of Molecular Sciences. 2020; 21(5):1735. https://doi.org/10.3390/ijms21051735
Chicago/Turabian StyleLüthi, Elias, Paola Andrea Forero Cortés, Alessandro Prescimone, Edwin C. Constable, and Catherine E. Housecroft. 2020. "Schiff Base Ancillary Ligands in Bis(diimine) Copper(I) Dye-Sensitized Solar Cells" International Journal of Molecular Sciences 21, no. 5: 1735. https://doi.org/10.3390/ijms21051735
APA StyleLüthi, E., Forero Cortés, P. A., Prescimone, A., Constable, E. C., & Housecroft, C. E. (2020). Schiff Base Ancillary Ligands in Bis(diimine) Copper(I) Dye-Sensitized Solar Cells. International Journal of Molecular Sciences, 21(5), 1735. https://doi.org/10.3390/ijms21051735