Molecular Profile of Phage Infection: A Novel Approach for the Characterization of Erwinia Phages through qPCR


Abstract
1. Introduction
2. Results
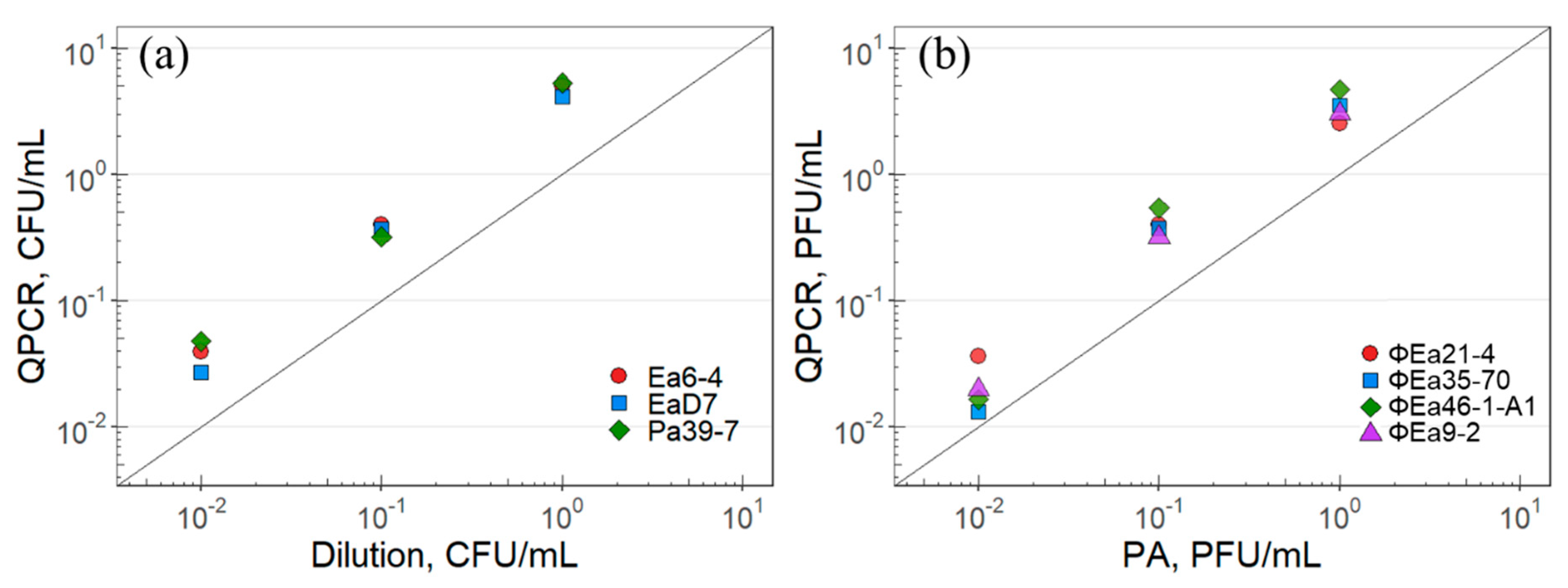
2.1. Correlation of Molecular Quantification of Plating Techniques
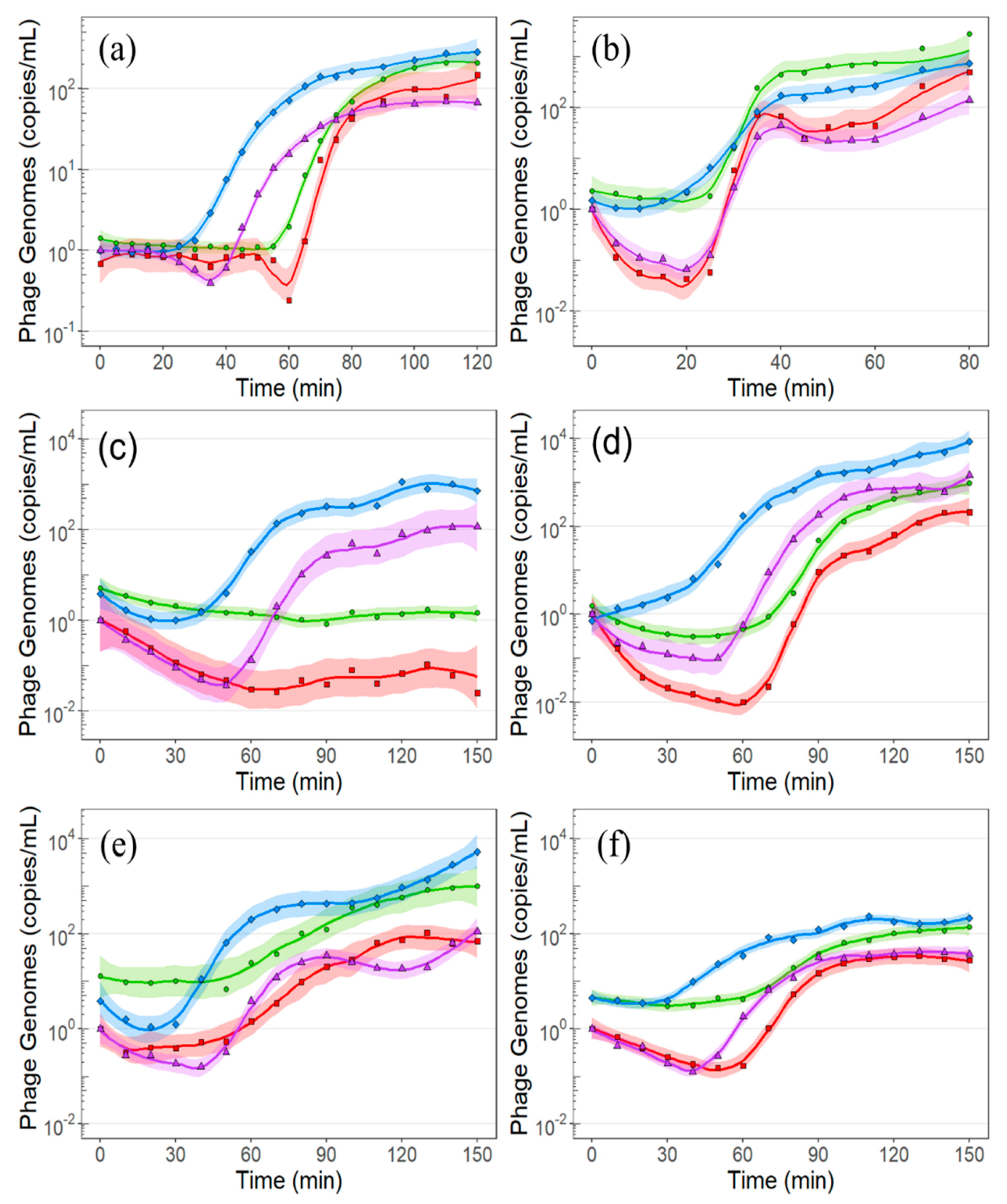
2.2. E. amylovora Infection by ΦEa21-4 and ΦEa46-1-A1
2.3. E. amylovora Infection by Phage ΦEa9-2
2.4. P. agglomerans Infection by Phage ΦEa35-70
3. Discussion
3.1. Molecular Quantification and Plating Techniques
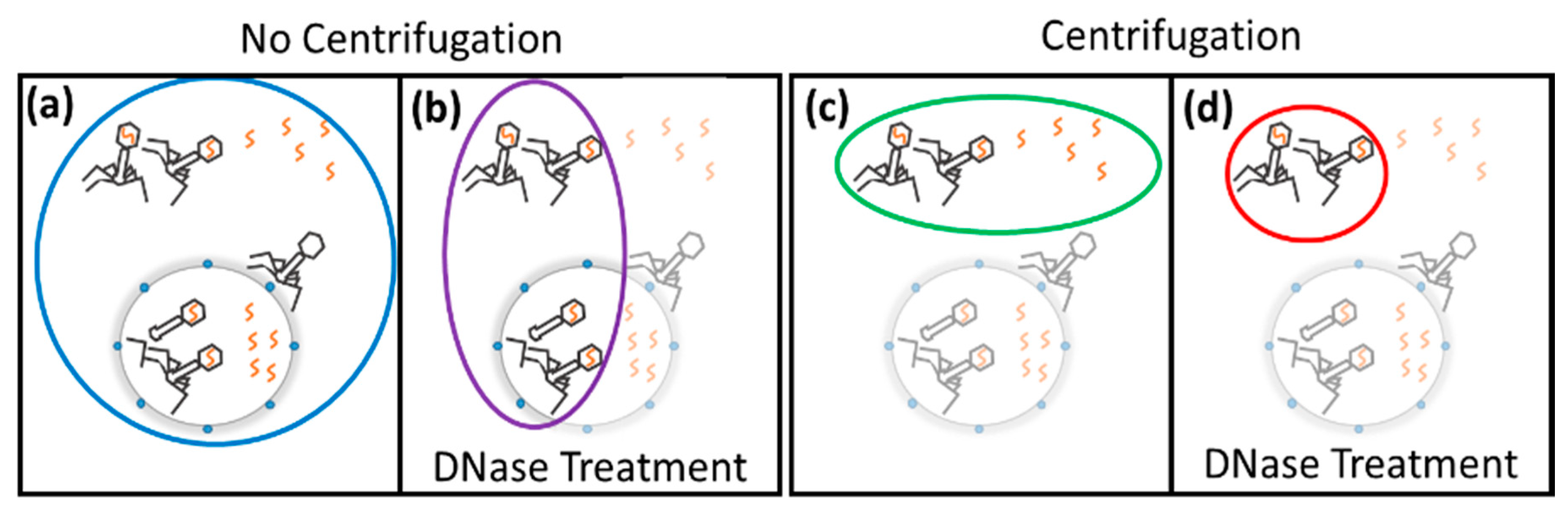
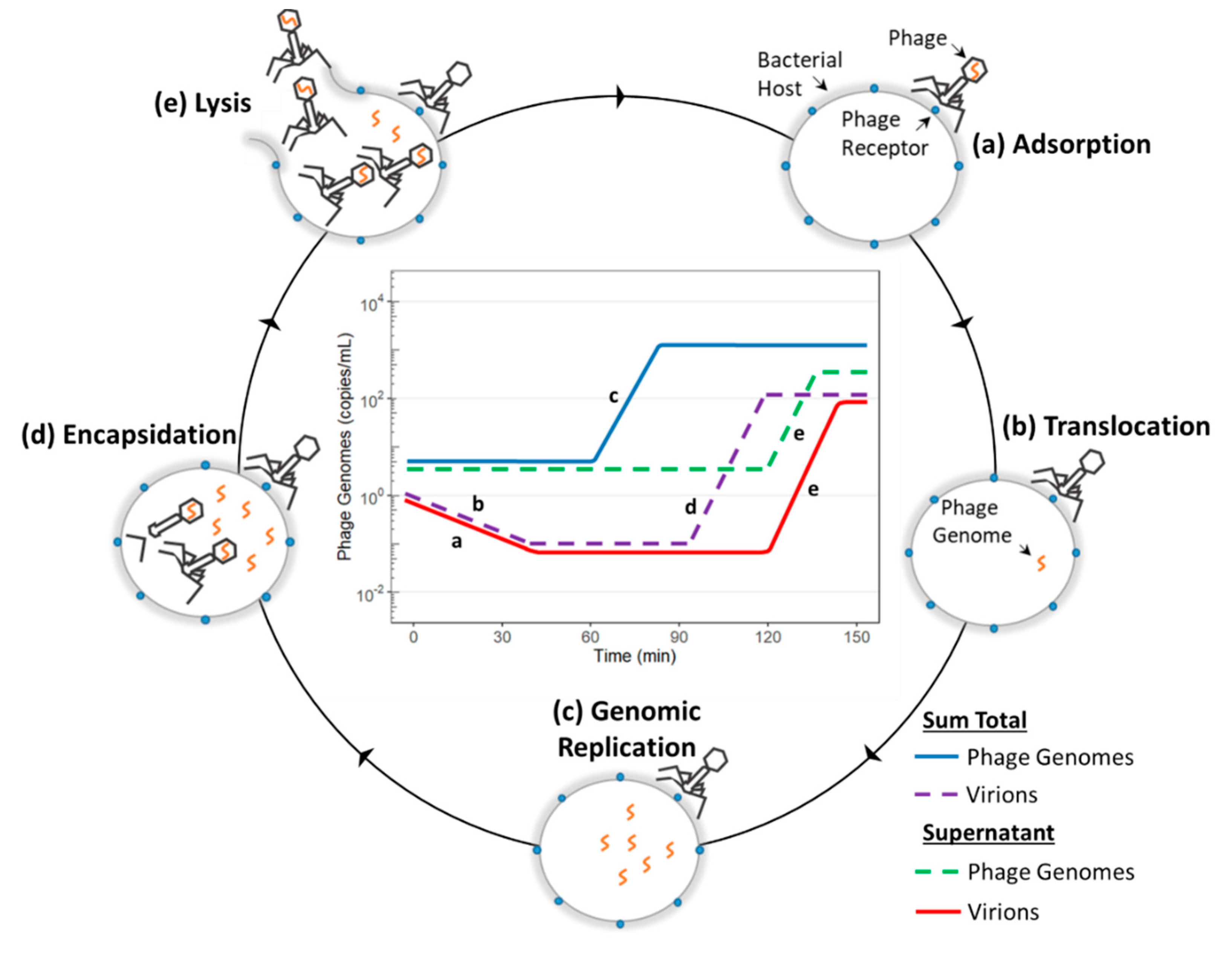
3.2. Adsorption, Infection, and Genomic Replication
3.3. Latent Period, Burst Size, and Impact of MOI
3.4. Erwinia Phages as Biocontrol Agents
3.5. Strengths and Limitations of MPI
4. Materials and Methods
4.1. Phage Propagation and Bacterial Growth Conditions
4.2. Standardization of Dilution Plating to qPCR
4.3. Standardization of Plaque Assay to qPCR
4.4. Generating a Molecular Profile of Phage Infection
4.5. Data Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Van der Zwet, T.; Orolaza-Halbrendt, N.; Zeller, W. Losses Due to Fire Blight and Economic Importance of the Disease. In Fire Blight: History, Biology, and Management; The American Phytopathological Society: St. Paul, MN, USA, 2016; pp. 37–41. [Google Scholar]
- Nemeth, J. Practice of applying streptomycin to control fireblight in Hungary. EPPO Bull. 2004, 34, 381–382. [Google Scholar] [CrossRef]
- McManus, P.S.; Stockwell, V.O.; Sundin, G.W.; Jones, A.L. Antibiotic use in plant agriculture. Annu. Rev. Phytopathol. 2002, 40, 443–465. [Google Scholar] [CrossRef] [PubMed]
- Tancos, K.A.; Cox, K.D. Exploring diversity and origins of streptomycin-resistant Erwinia amylovora isolates in New York through CRISPR spacer arrays. Plant Dis. 2016, 100, 1307–1313. [Google Scholar] [CrossRef] [PubMed]
- Förster, H.; McGhee, G.C.; Sundin, G.W.; Adaskaveg, J.E. Characterization of streptomycin resistance in Isolates of Erwinia amylovora in California. Phytopathology 2015, 105, 1302–1310. [Google Scholar] [CrossRef]
- Thomson, S.V.; Gouk, S.C.; Vanneste, J.L.; Hale, C.N.; Clark, R.G. The presence of streptomycin resistant strains of Erwinia amylovora in New Zealand. Acta Hortic. 1993, 338, 223–230. [Google Scholar] [CrossRef]
- Lehman, S.M. Development of a Bacteriophage-Based Biopesticide for Fire Blight. Ph.D. Thesis, Biological Sciences, Brock University, St. Catharines, ON, Canada, 2007. [Google Scholar]
- Svircev, A.; Roach, D.; Castle, A. Framing the future with bacteriophages in agriculture. Viruses 2018, 10, 218. [Google Scholar] [CrossRef]
- Roach, D.R.; Sjaarda, D.; Castle, A.J.; Svircev, A.M. Bacteriophages as biopesticides: Role of bacterial exopolysaccharides. Acta Hortic. 2011, 896, 449–455. [Google Scholar] [CrossRef]
- Jones, J.B.; Svircev, A.M.; Obradović, A.Ž. Crop use of bacteriophages. In Bacteriophages: Biology, Technology, Therapy; Harper, D.R., Abedon, S.T., Burrowes, B.H., McConville, M.L., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 1–18. [Google Scholar]
- Gill, J.J.; Svircev, A.M.; Smith, R.; Castle, A.J. Bacteriophages of Erwinia amylovora. Appl. Environ. Microbiol. 2003, 69, 2133–2138. [Google Scholar] [CrossRef]
- Pusey, P.L.; Stockwell, V.O.; Reardon, C.L.; Smits, T.H.; Duffy, B. Antibiosis activity of Pantoea agglomerans biocontrol strain E325 against Erwinia amylovora on apple flower stigmas. Phytopathology 2011, 101, 1234–1241. [Google Scholar] [CrossRef]
- Gayder, S.; Parcey, M.; Castle, A.J.; Svircev, A.M. Host range of bacteriophages against a world-wide collection of Erwinia amylovora determined using a quantitative PCR assay. Viruses 2019, 11, 910. [Google Scholar] [CrossRef]
- Roach, D.R.; Sjaarda, D.R.; Castle, A.J.; Svircev, A.M. Host exopolysaccharide quantity and composition impact Erwinia amylovora bacteriophage pathogenesis. Appl. Environ. Microbiol. 2013, 79, 3249–3256. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.H.; Wade, E. Classification of bacterial viruses: The relationship of two serratia phages to coli-dysentery phages T3, T7, and D44. J. Bacteriol. 1954, 68, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.H. Bacteriophages; Interscience Publishers Inc.: New York, NY, USA, 1959. [Google Scholar]
- Jacob, F.; Fuerst, C.R. The mechanism of lysis by phage studied with defective lysogenic bacteria. J. Gen. Microbiol. 1958, 18, 518–526. [Google Scholar] [CrossRef]
- Xie, Y.; Wahab, L.; Gill, J.J. Development and validation of a microtiter plate-based assay for determination of bacteriophage host range and virulence. Viruses 2018, 10, 189. [Google Scholar] [CrossRef]
- Bryan, D.; El-Shibiny, A.; Hobbs, Z.; Porter, J.; Kutter, E.M. Bacteriophage T4 infection of stationary phase E. coli: Life after log from a phage perspective. Front. Microbiol. 2016, 7, 1391. [Google Scholar] [CrossRef]
- Breidt, F.; Romick, T.L.; Fleming, H.P. A rapid method for the determination of bacterial growth kinetics. J. Rapid Methods Automat. Microbiol. 1994, 3, 59–68. [Google Scholar] [CrossRef]
- Anderson, B.; Rashid, M.H.; Carter, C.; Pasternack, G.; Rajanna, C.; Revazishvili, T.; Dean, T.; Senecal, A.; Sulakvelidze, A. Enumeration of bacteriophage particles: Comparative analysis of the traditional plaque assay and real-time QPCR- and nanosight-based assays. Bacteriophage 2011, 1, 86–93. [Google Scholar] [CrossRef]
- Carlson, K. Working with Bacteriophages: Common Techniques and Methodological Approaches; CRC Press: Boca Raton, FL, USA, 2005; pp. 437–494. [Google Scholar]
- Nabergoj, D.; Modic, P.; Podgornik, A. Effect of bacterial growth rate on bacteriophage population growth rate. MicrobiologyOpen 2018, 7, e00558. [Google Scholar] [CrossRef]
- Jurač, K.; Nabergoj, D.; Podgornik, A. Bacteriophage production processes. Appl. Microbiol. Biotechnol. 2019, 103, 685–694. [Google Scholar] [CrossRef]
- Edelman, D.C.; Barletta, J. Real-time PCR provides improved detection and titer determination of bacteriophage. Biotechniques 2003, 35, 368–375. [Google Scholar] [CrossRef]
- Lehman, S.M.; Kim, W.S.; Castle, A.J.; Svircev, A.M. Duplex real-time polymerase chain reaction reveals competition between Erwinia amylovora and E. pyrifoliae on pear blossoms. Phytopathology 2008, 98, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Roach, D.R.; Sjaarda, D.R.; Sjaarda, C.P.; Ayala, C.J.; Howcroft, B.; Castle, A.J.; Svircev, A.M. Absence of lysogeny in wild populations of Erwinia amylovora and Pantoea agglomerans. Microb. Biotechnol. 2015, 8, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Refardt, D. Real-time quantitative PCR to discriminate and quantify lambdoid bacteriophages of Escherichia coli K-12. Bacteriophage 2012, 2, 98–104. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Imamovic, L.; Serra-Moreno, R.; Jofre, J.; Muniesa, M. Quantification of Shiga toxin 2-encoding bacteriophages, by real-time PCR and correlation with phage infectivity. J. Appl. Microbiol. 2010, 108, 1105–1114. [Google Scholar] [CrossRef] [PubMed]
- Morella, N.M.; Yang, S.C.; Hernandez, C.A.; Koskella, B. Rapid quantification of bacteriophages and their bacterial hosts in vitro and in vivo using droplet digital PCR. J. Virol. Methods 2018, 259, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Kropinski, A.M. Practical advice on the one-step growth curve. Methods Mol. Biol. 2018, 1681, 41–47. [Google Scholar]
- Hyman, P.; Abedon, S.T. Practical methods for determining phage growth parameters. Methods Mol. Biol. 2009, 501, 175–202. [Google Scholar]
- Yagubi, A.I.; Castle, A.J.; Kropinski, A.M.; Banks, T.W.; Svircev, A.M. Complete genome sequence of Erwinia amylovora bacteriophage vB_EamM_Ea35-70. Genome Announc. 2013, 2, e00413–e00414. [Google Scholar] [CrossRef]
- Vandenbergh, P.A.; Wright, A.M.; Vidaver, A.K. Partial purification and characterization of a polysaccharide depolymerase associated with phage-infected Erwinia amylovora. Appl. Environ. Microbiol. 1985, 49, 994–996. [Google Scholar] [CrossRef]
- Sebaihia, M.; Bocsanczy, A.M.; Biehl, B.S.; Quail, M.A.; Perna, N.T.; Glasner, J.D.; DeClerck, G.A.; Cartinhour, S.; Schneider, D.J.; Bentley, S.D.; et al. Complete genome sequence of the plant pathogen Erwinia amylovora strain ATCC 49946. J. Bacteriol. 2010, 192, 2020–2021. [Google Scholar] [CrossRef]
- Choi, C.; Kuatsjah, E.; Wu, E.; Yuan, S. The effect of cell size on the burst size of T4 bacteriophage infections of Escherichia coli B23. J. Exp. Microbiol. Immunol. 2010, 14, 85–91. [Google Scholar]
- Yagubi, A.I. Phage-Mediated Biological Control of Erwinia Amylovora: The Role of CRISPRs and Exopolysaccharide; Brock University: St. Catharines, ON, Canada, 2016. [Google Scholar]
- Storms, Z.J.; Sauvageau, D. Modeling tailed bacteriophage adsorption: Insight into mechanisms. Virology 2015, 485, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Washizaki, A.; Yonesaki, T.; Otsuka, Y. Characterization of the interactions between Escherichia coli receptors, LPS and OmpC, and bacteriophage T4 long tail fibers. MicrobiologyOpen 2016, 5, 1003–1015. [Google Scholar] [CrossRef]
- Puck, T.T. The first steps of virus invasion. Cold Spring Harb. Symp. Quant. Biol. 1953, 18, 149–154. [Google Scholar] [CrossRef]
- Bertozzi Silva, J.; Storms, Z.; Sauvageau, D. Host receptors for bacteriophage adsorption. FEMS Microbiol. Lett. 2016, 363, fnw002. [Google Scholar] [CrossRef]
- Dressman, H.K.; Drake, J.W. Lysis and lysis inhibition in bacteriophage T4: rV mutations reside in the holin t gene. J. Bacteriol. 1999, 181, 4391–4396. [Google Scholar] [CrossRef]
- Delbrück, M. The burst size distribution in the growth of bacterial viruses (bacteriophages). J. Bacteriol. 1945, 50, 131–135. [Google Scholar] [CrossRef]
- Hadas, H.; Einav, M.; Fishov, I.; Zaritsky, A. Bacteriophage T4 development depends on the physiology of its host Escherichia coli. Microbiology 1997, 143, 179–185. [Google Scholar] [CrossRef]
- Abedon, S.T.; Herschler, T.D.; Stopar, D. Bacteriophage latent-period evolution as a response to resource availability. Appl. Environ. Microbiol. 2001, 67, 4233–4241. [Google Scholar] [CrossRef]
- Moldovan, R.; Chapman-McQuiston, E.; Wu, X.L. On kinetics of phage adsorption. Biophys. J. 2007, 93, 303–315. [Google Scholar] [CrossRef]
- Ellis, E.; Delbrück, M. The growth of bacteriophage. J. Gen. Physiol. 1939, 22, 365–384. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.B. Fire blight of apple and pear. Plant Health Instr. 2000. [Google Scholar] [CrossRef]
- Delbrück, M. Adsorption of bacteriophage under various physiological coditions of the host. J. Gen. Physiol. 1940, 23, 631–642. [Google Scholar] [CrossRef] [PubMed]
- Abedon, S.; Thomas-Abedon, C. Phage therapy pharmacology. Curr. Pharm. Biotechnol. 2010, 11, 28–47. [Google Scholar] [CrossRef] [PubMed]
- Abedon, S.T. Phage therapy dosing: The problem(s) with multiplicity of infection (MOI). Bacteriophage 2016, 6, e1220348. [Google Scholar] [CrossRef]
- Team, R.C. R: A Language and Environment for Statistcal Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Wickman, H. Ggplot2: Elegant Graphics for Data Analysis; R Package; Springer: New York, NY, USA, 2016. [Google Scholar]
- Skowron, P.M.; Kropinski, A.M.; Zebrowska, J.; Janus, L.; Szemiako, K.; Czajkowska, E.; Maciejewska, N.; Skowron, M.; Los, J.; Los, M.; et al. Sequence, genome organization, annotation and proteomics of the thermophilic, 47.7-kb Geobacillus stearothermophilus bacteriophage TP-84 and its classification in the new Tp84virus genus. PLoS ONE 2018, 13, e0195449. [Google Scholar]
- Shkoporov, A.N.; Hill, C. Bacteriophages of the human gut: The “known unknown” of the microbiome. Cell Host Microbe 2019, 25, 195–209. [Google Scholar] [CrossRef]



 (blue diamond) = total number of phage genomes;
(blue diamond) = total number of phage genomes;  (green circle) = total number of phage genomes in the supernatant;
(green circle) = total number of phage genomes in the supernatant;  (purple triangle = total number of virions;
(purple triangle = total number of virions;  (red square = total number of virions in the supernatant.
(blue diamond) = total number of phage genomes; (green circle) = total number of phage genomes in the supernatant; (purple triangle = total number of virions; (red square = total number of virions in the supernatant.
(red square = total number of virions in the supernatant.
(blue diamond) = total number of phage genomes; (green circle) = total number of phage genomes in the supernatant; (purple triangle = total number of virions; (red square = total number of virions in the supernatant.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lytic Cycle Stage | ||||||
|---|---|---|---|---|---|---|
| Adsorption A | Infection B | Genomic Rep. | Encapsidation | Lysis | ||
| Phage (Host: MOI) | Rate (min−1): Max (%) | Rate (min−1): Max (%) | Start (min): Rate (min−1) | Start (min): Rate (min−1): Virions C | Start (min): Rate (min−1): Burst Size | End (min) |
| ΦEa21-4 (Ea6-4: 0.45) | ND | 0.052: 60.7 | 29: 0.166 | 37: 0.191: 63 | 57: 0.318: 185 | 98 |
| ΦEa46-1-A1 (EaD7: 0.625) | 0.290: 95.7 | 0.221: 93.4 | 21: 0.173 | 23: 0.537: 45 | 23: 0.712: 57 | 38 |
| ΦEa9-2 (Ea17-1-1: 0.015) | 0.071: 97.6 | 0.074: 96.3 | 39: 0.154 | 52: 0.196: 49 | ND: ND: 51 | ND |
| ΦEa9-2 (Ea273: 0.015) | 0.167: 99.0 | 0.152: 90.3 | 35: 0.139 | 50: 0.215: 768 | 65: 0.301: 850 | 137 |
| ΦEa35-70 (Pa39-7: 0.7) | 0.044: 87.8 | 0.051: 87.7 | 28: 0.090 | 42: 0.181: 31 | 56: 0.174: 43 | 108 |
| ΦEa35-70 (Pa39-7: 0.03) | ND | 0.074: 84.3 | 28: 0.174 | 43: 0.160: 35 | 52: 0.090: 42 | 116 |
| Family | Strain | Genus species | Accession | Reference |
|---|---|---|---|---|
| Myoviridae | ΦEa21-4 | Kolesnikvirus Erwinia virus Ea214 | NC_011811.1 | [11] |
| ΦEa35-70 | Agricanvirus Erwinia virus Ea35-70 | NC_023557.1 | [11] | |
| Podoviridae | ΦEa46-1-A1 | Zindervirus Erwinia virus Era103 | Unpublished | [11] |
| ΦEa9-2 | Johnsonvirus Erwinia virus Ea9-2 | KF806588.1 | [11] | |
| Enterobacteriaceae | Ea6-4 | Erwinia amylovora | Unpublished | [13] |
| EaD7 | Erwinia amylovora | Unpublished | [13] | |
| Ea17-1-1 | Erwinia amylovora | Unpublished | [13] | |
| Ea273 | Erwinia amylovora | NC_013971.1 | [35] | |
| Pa39-7 | Pantoea agglomerans | Unpublished | [27] |
| Target | Name | Sequences (5’-3’) | Reference |
|---|---|---|---|
| ΦEa21-4 | END37-F | TTCAGCTTTAGCGGCTTCGAGA | [13] |
| END37-R | AGCAAGCCCTTGAGGTAATGGA | ||
| END37-P | /56-ROXN/AGTCGGTACACCTGCAACGTCAAGAT/3IAbRQSp/ | ||
| ΦEa35-70 | RDH311-F | TGGAAGGTCTTCTTCGAGAC | [9] |
| RDH311-R | GACTACCTGGGGATGTTCAG | ||
| RDH311-P | /56-ROX/GACGGAAAAGATCACGGTACTCTT/3IAbRQSp/ | ||
| ΦEa46-1-A1 | STS3-F | GACAAACAAGAACGCGGCAACTGA | [13] |
| STS3-R | ATACCCAGCAAGGCGTCAACCTTA | ||
| STS3-P | /56-FAM/AGATGAAGTAGGTTATCTTCACAGTGCCCT/3BHQ_1/ | ||
| ΦEa9-2 | N14-F | CATTGGGTAATCCCTTTGAG | [13] |
| N14-R | GATAGACTGGTTCCCCTGTG | ||
| N14-P | /56-FAM/TCTGGTGGA/ZEN/CAGAGACGATGTAAT/3IABkFQ/ | ||
| E. amylovora | Pa-Gnd-F | TGGATGAAGCAGCGAACA | [7] |
| Pa-Gnd-R | GACAGAGGTTCGCCGAGA | ||
| Pa-Gnd-P | /5HEX/AAATGGACCAGCCAGAGCTCACTG/3BHQ-1/ | ||
| P. agglomerans | Ea-Lsc-F | CGCTAACAGCAGATCGCA | [7] |
| Ea-Lsc-R | AAATACGCGCACGACCAT | ||
| Ea-Lsc-P | /5Cy5/CTGATAATCCGCAATTCCAGGATG/3IAbRQsp/ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parcey, M.; Gayder, S.; Castle, A.J.; Svircev, A.M. Molecular Profile of Phage Infection: A Novel Approach for the Characterization of Erwinia Phages through qPCR. Int. J. Mol. Sci. 2020, 21, 553. https://doi.org/10.3390/ijms21020553
Parcey M, Gayder S, Castle AJ, Svircev AM. Molecular Profile of Phage Infection: A Novel Approach for the Characterization of Erwinia Phages through qPCR. International Journal of Molecular Sciences. 2020; 21(2):553. https://doi.org/10.3390/ijms21020553
Chicago/Turabian StyleParcey, Michael, Steven Gayder, Alan J. Castle, and Antonet M. Svircev. 2020. "Molecular Profile of Phage Infection: A Novel Approach for the Characterization of Erwinia Phages through qPCR" International Journal of Molecular Sciences 21, no. 2: 553. https://doi.org/10.3390/ijms21020553
APA StyleParcey, M., Gayder, S., Castle, A. J., & Svircev, A. M. (2020). Molecular Profile of Phage Infection: A Novel Approach for the Characterization of Erwinia Phages through qPCR. International Journal of Molecular Sciences, 21(2), 553. https://doi.org/10.3390/ijms21020553